
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReverse engineering: transaminase biocatalyst development using ancestral sequence reconstruction†
Matthew
Wilding
 *a,
Thomas S.
Peat
b,
Subha
Kalyaanamoorthy
a,
Janet
Newman
b,
Colin
Scott
a and
Lars S.
Jermiin
ac
*a,
Thomas S.
Peat
b,
Subha
Kalyaanamoorthy
a,
Janet
Newman
b,
Colin
Scott
a and
Lars S.
Jermiin
ac
aCSIRO Land and Water, Black Mountain, Canberra, ACT 2601, Australia. E-mail: matt.wilding@csiro.au
bCSIRO Manufacturing, Parkville, VIC 3052, Australia
cResearch School of Biology, Australian National University, Canberra, ACT 2601, Australia
First published on 24th October 2017
Abstract
The development of new biocatalysts using ancestral sequence reconstruction is reported. When applied to an ω-transaminase, the ancestral proteins demonstrated novel and superior activities with eighty percent of the forty compounds tested compared to the modern day protein, and improvements in activity of up to twenty fold. These included a range of compounds pertinent as feedstocks in polyamide manufacture.
Biocatalytic approaches to chemical manufacture offer numerous advantages over conventional chemical methods including excellent activity, selectivity and catalytic stability. They are often facilitated under ambient temperature and pressure, requiring no organic solvents or heavy metal additives, and the biocatalyst can be recovered and recycled with relative ease. Transaminase-mediated reactions are among the best characterised and most widely utilised in industrial biocatalysis, with established applications in the chemical and pharmaceutical industries.1,2 The enzymes boast an attractive repertoire of products, including chiral building blocks such as amino acids (α, β and ω), aliphatic and aromatic amines, polymer feedstocks and complex pharmaceuticals. However, there are limitations associated with transaminase-catalysed reactions. Firstly, equilibria are often unfavourable. This has led to the development of numerous strategies to circumvent the problem (for an excellent overview see Guo et al., 2017
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3) and in many cases resolve the issue. Secondly, the catalytic efficiency of transaminase reactions is often low compared with that of other enzyme-catalysed reactions. The enzymes can be inhibited by both the reaction substrates and/or products, a mechanistic element which remains unresolved, and protein engineering studies often yield only modest improvements in activity.
3) and in many cases resolve the issue. Secondly, the catalytic efficiency of transaminase reactions is often low compared with that of other enzyme-catalysed reactions. The enzymes can be inhibited by both the reaction substrates and/or products, a mechanistic element which remains unresolved, and protein engineering studies often yield only modest improvements in activity.
Where successful transaminase engineering has been reported, the predominant approaches have been random or structure-guided mutagenesis.3 In addition, a small number of sequence alignment based approaches have also been described. Here, we report an alternative method of engineering transaminases using ancestral sequence reconstruction (ASR). ASR is a term used to describe the process of inferring ancestral genes or proteins from multiple sequence alignments (MSAs) of nucleotides or amino acids. The technique has been used in biocatalyst development previously, but in almost all cases, used in a similar context to the sequence alignment based methods previously described. Typically, ASR has been used to infer ancestral states at sites that are likely to be important for activity, and replace those residues in modern proteins with the ancestral residues by site-directed mutagenesis.4–8 Although this approach is quick, and can be used to generate small libraries, it may be limited to proteins for which the structure:function relationships are well understood.9 It also places ancestral residues within the context of the other residues of a modern protein and overlooks the concepts of co-evolution of residues and intramolecular epistasis (i.e., where the functional effect of one amino-acid substitution may be augmented or countered by another).10,11
There are additional advantages to using ASR in biocatalyst development. One of the virtues of ASR is that it can lead to new insights into the evolutionary pathways of individual nucleotides or amino acids within a sequence,12,13 and the inferred changes can be related to functional changes in the corresponding gene or protein. In addition, several studies have suggested that ancestral proteins may possess greater thermotolerance and/or acid-tolerance than their extant descendants. Whilst the reasoning for this is still a topic of debate,14–20 increased thermostability is often a desirable trait in biocatalysis. Given these potential benefits, it is perhaps surprising that ASR has not been employed more often as an alternative method of directed evolution, especially in biocatalyst development. Previously, this may have been partly due to cost, but as the cost of gene synthesis has continued to decline, the study of ancestral proteins has also become increasingly viable.
Herein, we report the use of ASR to generate a family of evolutionarily related ancestral transaminases with potential as industrial biocatalysts, as well as a detailed characterisation of those proteins. The ancestors in question were derived from a previously characterised ω-transaminase, KES23360, from Pseudomonas sp. strain AAC.21 KES23360 is a 4-aminobutyrate transaminase with promiscuous activity towards 12-aminododecanoic acid, a non-natural compound with industrial significance as the constituent building block of nylon-12. Very few proteins have been confirmed to turn over this compound,22,23 and as such, this activity is unusual. Furthermore, 12-aminododecanoic acid is an attractive biocatalytic target because of the hazards associated with conventional chemical routes to polyamides (including nylon-12), which have previously led to global shortages of the material.24 Indeed, a range of polyamide feedstocks including ω-amino acids and α,ω-diamines are typically accessed from 1,3-butadiene or other crude oil products, and as such alternative green routes to these materials are sought. Hence, we decided to investigate whether ASR could be used to produce novel biocatalysts with superior catalytic properties to KES23360 for 12-aminododecanoic acid as well as other similar substrates.
Results and discussion
An ancestral sequence reconstruction was carried out based on the amino acid sequence of KES23360 and the closest 192 unique homologues as described in ESI.† From the phylogeny, we focussed on a subtree within the full phylogenetic tree (shown in Fig. 1; the complete phylogenetic tree is shown in Fig. S1†) that contained KES23360 and forty-seven other protein sequences. From the ancestral reconstruction, six nodes (N6, 15, 16, 17, 43 and 48; highlighted in Fig. 1) were chosen as target sequences for gene synthesis. The target sequences were selected from the subtree and were expressed in E. coli from synthetic genes for further characterisation. | ||
| Fig. 1 The subtree containing KES23360 (shown in blue). All other tips are labelled using their Uniprot KB reference number. Ancestral nodes selected for gene synthesis are labelled. Unless shown, all calculated bootstrap values were 100. A complete phylogenetic tree is shown in Fig. S1.† | ||
Transaminases are often highly promiscuous, and the limited characterisation of KES23360 had already revealed some promiscuity.21,25 However, the substrate range of KES23360 had not been investigated rigorously, so we sought to fully characterise KES23360 before comparing it with the ancestral transaminases. Specific and relative activities were determined using an alanine dehydrogenase coupled assay, as previously reported (Fig. 2).21 A library of 40 amines, which included a range of feedstock molecules for homo- and hetero-nylon (ω-amino acids and α,ω-diamines) was tested against KES23360 with pyruvate as cosubstrate. For KES23360, 27 of the 40 compounds were found to be substrates. The compounds were converted with varying levels of efficiency (subset shown in Table 1 and Fig. 3, the complete dataset shown in Table S3†) but it was observed that all the polyamide feedstock molecules tested, ω-amino acids and α,ω-diamines, were converted by KES23360 (C3–C12 and C3–C10 respectively) to some degree. Despite a high degree of promiscuity, it was clear from the observed rates that KES23360 had a strong preference for ω-amino acid substrates, whilst detectable but significantly lower rates were observed with α,ω-diamines, α-amino acids and other compounds. Interestingly, the highest specific activity was not observed with 4-aminobutyrate, the previously predicted physiological substrate, but rather an analogue of that compound, 4-amino-2-(S)-hydroxybutyrate (AHBA). To our knowledge, this is the first reported example of an AHBA transamination reaction, with one alternative multi-step biosynthetic route to AHBA confirmed to date.26 AHBA is not utilised in polyamide manufacture, but is useful in the synthesis of chiral building blocks,27 and has also been shown to improve the pharmacological properties of antibiotics.26 The molecule is therefore an attractive synthetic target, and another example of an industrially relevant substrate that can be accessed using this biocatalyst.
 | ||
| Fig. 2 Schematic representation of the alanine dehydrogenase coupled assay used in screening. | ||
 | ||
| Fig. 3 A comparison of relative activity for KES23360 (centre) and the ancestral proteins (clockwise around KES23360) with a subset of the substrates screened. Each spoke represents a compound, with the node position on the spoke for each compound illustrating the relative activity for that protein:substrate pair. Activity data for each substrate is shown as a percentage, relative to the highest observed activity for each individual protein (treated as 100%). Substrates are grouped by type, with ω-amino acids (dark grey), α,ω-diamines (light grey) and α-amino acids (white) highlighted accordingly. For the ω-amino acids and α,ω-diamines, the substrates are annotated by the length of the carbon chain in the substrate, so for example C6 represents 6-aminohexanoate in the ω-amino acids and 1,6-hexamethylenediamine in the α,ω-diamines. Analysis of the data shows that whilst KES23360 has apparently specialised towards the ω-amino acids, many of the ancestors exhibit improved substrate preference towards α,ω-diamines, and to a lesser extent the α-amino acids. | ||
| Substrate | KES23360 | N6 | N15 | N16 | N17 | N43 | N48 |
|---|---|---|---|---|---|---|---|
| β-Alanine | 3.2 (3) | 7.8 (13) | 4.3 (19) | 0.4 (7) | 1.2 (12) | 1.8 (2) | 0.1 (3) |
| 4-Aminobutyrate | 48.1 (51) | 60.9 (100) | 21.9 (100) | 4.2 (77) | 9.0 (94) | 49.7 (65) | 1.4 (63) |
| 5-Aminopentanoate | 81.4 (86) | 56.3 (92) | 19.5 (89) | 5.5 (100) | 9.6 (100) | 76.5 (100) | 2.1 (96) |
| 6-Aminohexanoate | 81.4 (86) | 57.7 (95) | 20.5 (94) | 4.5 (83) | 9.1 (95) | 74.7 (98) | 1.7 (78) |
| 7-Aminoheptanoate | 78.6 (83) | 56.5 (93) | 18.0 (82) | 4.2 (78) | 8.5 (88) | 70.6 (92) | 1.9 (85) |
| 8-Aminooctanoate | 64.9 (68) | 37.9 (62) | 17.5 (80) | 3.5 (64) | 7.6 (79) | 61.0 (80) | 1.9 (85) |
| 12-Aminododecanoate | 8.2 (9) | 10.2 (17) | 4.6 (21) | 0.6 (10) | 1.8 (18) | 18.7 (24) | 0.2 (11) |
| 4-NH2-(S)-2-hydroxybutyrate | 95.1 (100) | 27.0 (44) | 14.9 (68) | 4.6 (84) | 7.5 (78) | 50.6 (66) | 2.2 (100) |
| 2,4-(S)-Diaminobutyrate | 24.0 (25) | 6.8 (11) | 5.3 (24) | 1.5 (28) | 3.7 (38) | 15.9 (21) | 0.5 (22) |
| 1,3-Diaminopropane | 0.7 (1) | 1.4 (2) | 1.1 (5) | 0.1 (3) | 0.5 (5) | 1.8 (2) | 0.02 (1) |
| Putrescine | 15.1 (16) | 38.8 (64) | 17.9 (82) | 2.8 (51) | 7.9 (82) | 32.3 (42) | 0.3 (15) |
| Cadaverine | 4.7 (5) | 48.6 (80) | 21.5 (98) | 2.6 (47) | 3.5 (36) | 62.0 (81) | 0.3 (15) |
| 1,6-Hexamethylenediamine | 5.4 (6) | 17.9 (29) | 13.8 (63) | 2.0 (36) | 6.7 (70) | 37.8 (49) | 0.5 (22) |
| 1,7-Heptamethylenediamine | 5.7 (6) | 29.6 (49) | 14.0 (64) | 2.1 (38) | 6.8 (70) | 48.7 (64) | 0.6 (26) |
| 1,8-Octamethylenediamine | 6.8 (7) | 24.2 (40) | 13.9 (64) | 1.7 (32) | 4.6 (48) | 37.4 (49) | 0.3 (15) |
| 1,9-Nonamethylenediamine | 12.2 (13) | 20.6 (34) | 12.1 (55) | 1.7 (31) | 6.0 (62) | 43.7 (57) | 0.5 (22) |
| 1,10-Decamethylenediamine | 7.2 (8) | 47.3 (78) | 13.7 (63) | 2.0 (36) | 6.1 (63) | 38.3 (50) | 0.6 (26) |
The 2-hydroxy substituent was probed further by testing with 2,4-(S)-diaminobutyrate, but the observed rate was only 25% relative to the hydroxylated compound, suggesting that the enzyme has a strong preference for the α-hydroxy acid. Although there is insufficient evidence to support that this is a physiologically relevant reaction, it is to our knowledge the first report transamination of AHBA and the specific activity is almost double that of 4-aminobutyrate (95.1 nmol min−1 mg−1vs. 48.1 nmol min−1 mg−1). As such, this may warrant further investigation to determine if a novel metabolic pathway is present in the Pseudomonas sp. strain AAC.
It is often assumed that ancient enzymes were more promiscuous than their modern day descendants.28 These proteins are thought to have enabled ancient organisms to survive or even flourish with a small, versatile proteome, and that proteome subsequently evolved and expanded, enabling protein specialisation.25 As such, we anticipated that the promiscuity exhibited by KES23360 would at least be mirrored by its ancestral counterparts. On analysis, we observed that the ancestral proteins not only possessed the broad substrate scope of KES23360, but exceeded it, accepting up to 36 of the 40 compounds tested (N15 and N16; Table 1). Notably, N48, the most ancient of the node sequences tested, only accepted 29 of the 40 substrates tested, more than the KES23360, but fewer than the other ancestral peptides. However, all observed activities with N48 were significantly lower than the other enzymes and it is possible that N48 is able to turnover a greater number of substrates, albeit at undetectable rates (due to the conditions in which the assays were conducted).
Although the maximal rates achieved by KES23360 were higher, those rates were only observed with the C5–C8 ω-amino acids, and significant decreases in activity were measured with the majority of the other substrates. For the ancestral peptides, the lower specific activities with some of the ω-amino acids were compensated for by higher activities towards a broader range of compounds (Fig. 3). This included an improved activity against 12-aminododecanoic acid (the monomer used in nylon-12 production). N43 exhibited a modest but improved specific activity of 18.7 nmol min−1 mg−1 compared to 8.2 nmol min−1 mg−1 for KES23360, a 2-fold increase in activity. In addition, >10-fold improvements in activity were observed with a range of α,ω-diamines (up to 13-fold improvement in specific activity for N43 vs. KES23360 with cadaverine). Overall, for the 40 substrates tested, improvements in specific activity were observed in 32 cases (80%) with at least one ancestral peptide. Up to 20-fold improvements were observed (observed with the amphetamine precursor 2-aminoindane), and in many cases, multiple ancestral peptides showed superior catalytic activity over KES23360. With respect to the polyamide feedstocks, improvements in activity were observed with all the α,ω-diamines (C3–C10), as well as β-alanine, 4-aminobutyrate and 12-aminododecanoic acid. The N43 ancestor in particular showed excellent, broad activity under the conditions tested, retaining >90% activity with the remaining ω-amino acids (C5–C8) vs. KES23360, while surpassing it with 12-aminododecanoic acid and the diamines.
As described previously, many of the compounds tested have applications as feedstocks in polyamide manufacture. In many cases, such as the C6 and C9 α,ω-diamines (1,6-hexamethylenediamine and 1,9-nonamethylenediamine), the compounds are chemically synthesised from 1,3-butadiene, and are dependent on crude oil sources. Alleviating this dependence has afforded new opportunities in green chemistry, and a small number of examples of microbial routes to polyamide feedstocks have now been reported. For example, the C4 diamine (putrescine) can be synthesised from propylene, but an alternative fermentation system has also been reported that produces the platform chemical from glucose.29 Metabolic engineering approaches have also been reported for the C5 diamine (cadaverine)30 and 12-aminododecanoic acid.22,31 However, analogous routes do not exist for all these chemicals and there is still a need to expand the repertoire of fermentation approaches. This is conceptually easier with shorter chain compounds that exist naturally, but as chain length increases the number of reported enzymes available to facilitate biotransformations decreases markedly and the complexity of the metabolic engineering increases. To that end, identifying biocatalysts with desirable substrate ranges that can plug in to pathways will enable new platforms for green chemical production. With respect to this work, the biocatalysts reported here are the first reported transaminases capable of catalysing the conversion of 1,9-nonamethylenediamine and could therefore be used in a pathway to produce the diamine as a platform chemical and polyamide feedstock.
Given the relationship between ancestral reconstructions and increased thermostability, we also tested for protein stability over a range of temperatures to determine thermotolerance. This can be a desirable trait, but it is dependent on the application. To assess thermostability, we analysed all of the proteins by differential scanning fluorimetry (DSF). Analysis of these data (shown in Table S1†) revealed that the ancestral peptides showed increases in thermostability (up to 10 °C), relative to KES23360. However, the most ancient of the sequences (N48) showed the lowest melting temperature of all proteins tested, a contradiction of the common perception that ancestral peptides exhibit greater thermotolerance than their modern counterparts.
Finally, we sought to analyse the proteins at sequence and structural levels to try to rationalise the observed catalytic differences between the proteins. This could potentially assist in the development of subsequent biocatalysts from the ancestral peptides towards specific targets, but also more broadly contribute to the understanding of the sequence:structure:function relationships in ω-transaminases. Thus, structural characterisation of one or more of the proteins was required to give context to the protein sequences and if necessary, allow for homology modelling and comparison. Each of the proteins was prepared for crystallographic studies, and crystals were successfully obtained for all seven proteins as described in ESI (crystallographic conditions and datasets outlined in Table S2†). Full X-ray datasets and high-resolution structures (1.95 Å–2.62 Å) were obtained for each protein and comparison of the structures revealed that they were all characteristic α/β-fold dimers (KES23360 structure shown in Fig. S3†) with high structural conservation to one-another. Despite the high structural similarities between the proteins, identification of the residues that contributed to substrate preference or turnover was challenging. Examination of the non-conserved residues in the structures revealed that their positions were scattered throughout the protein, but interestingly the residues comprising the active site cavity were strictly conserved in all seven proteins (Fig. 4).
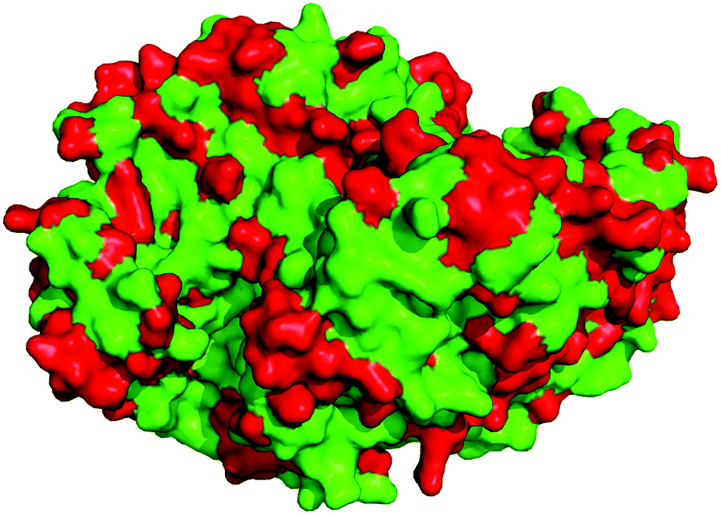 | ||
| Fig. 4 The X-ray structure of KES23360 as a dimer, highlighting the positions varied on the template using ASR. The locations of the 302 amino acid substitutions that differentiate the proteins are shown in red. | ||
Considering our current understanding of transaminase reaction mechanisms,32 we would therefore expect that substrate binding in the active site would be conserved. However the apparent differences in substrate preference and activity between each enzyme are suggestive that the local environment of the active site may make a minimal contribution to substrate selectivity. To further investigate this we conducted molecular dynamics simulations to compare changes in protein dynamics (as described in ESI†). Analysis of the simulations showed no significant differences in protein conformation or dynamics. Given the conserved active sites and similar behaviour of the proteins, this would suggest that other regions of the protein surface may contribute to catalysis. However, with 302 varied sites across the transaminase dimers, identifying the relevant residues is not trivial. These findings may go towards explaining the promiscuity associated with transaminases, and further, why the rational design of ω-transaminases has been challenging in the past.33,34 Despite our advancements in protein engineering and computational science, there are still significant gaps in our understanding with respect to how biocatalysts function. This is highlighted by many reported engineering strategies, which are frequently either random or site-directed, and in the latter case often focus on enzyme active sites. ASR however presents an opportunity to explore much more of a protein sequence in a sequence-directed manner, as demonstrated by these investigations. Whilst we do not yet appreciate the consequences of the changes being incorporated, one of the advantages of ASR is that it does not require an empirical understanding of the changes being made, and it therefore allows us to probe and utilise sequence space more effectively than other comparable methods. One of the challenges for the future will be to develop computational techniques that can rationalise and ideally predict these changes, and aid in the engineering of the next generation of biocatalysts.
Conclusions
The original aim of these investigations was to assess whether ASR could generate proteins that are suitable for biocatalytic applications, preferably with superior activity to the modern day biocatalyst. The ancestral enzymes generated here were highly promiscuous, accepting up to 36 of the 40 compounds screened. Under the conditions tested they exhibited higher activity with 80% of the substrates tested and improvements in specific activity of up to 20-fold. In most cases, the ancestral proteins were also easier to overproduce, and demonstrated comparable or improved thermostability. When screened against a range of polymer feedstock molecules widely synthesised from petrochemical sources, improvements in specific activity were demonstrated with our initial substrate of interest, 12-aminododecanoic acid, as well as a range of other ω-amino acids and α,ω-diamines. Particularly, in the case of the diamines, significant improvements in activity were observed with several ancestral proteins. As metabolic engineering approaches continue to improve, these enzymes may have utility for the biocatalytic production of α,ω-diamines and ultimately provide alternative green routes to a range of polyamides.As well as improved biocatalysts in their own right, the ancestral proteins also represent new templates for biocatalyst development, and furthermore, may be useful to identify and characterise putative enzymes in the phylogenetic tree with desirable substrate ranges. Furthermore, given that random and structure-guided approaches to transaminase engineering can be carried out downstream, the ancient proteins may better serve as biocatalytic scaffolds, which can be engineered further towards specific targets. ASR therefore offers a complementary tool to existing methods in biocatalyst development and engineering, and is capable of delivering comparable results to the more widely used approaches.3 As the cost of gene synthesis continues to decline, the utility of ASR in the manner demonstrated herein should continue to increase.
Finally, the exploration of the sequence:structure:function relationship of KES23360 and its ancestors is perhaps where ASR will broadly prove most valuable with respect to transaminase development. The generation of highly homologous ancestral protein sequences (sequence similarity 76–92%) with new or different functions provided new insights into ω-transaminase catalysis. The newly determined crystal structures and activity data preliminarily suggest that the determinants of specificity may lie away from the active site, and given the overall structural similarities across the ω-transaminase family, the implications for these findings could be significant.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Carol Hartley and John Oakeshott for helpful discussions in the preparation of this manuscript. MW and SK were supported by CSIRO Research Office Postdoctoral Fellowships.References
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Green Chem., 2017, 19, 361–366 RSC.
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- U. Alcolombri, M. Elias and D. S. Tawfik, J. Mol. Biol., 2011, 411, 837–853 CrossRef CAS PubMed.
- G. Conti, L. Pollegioni, G. Molla and E. Rosini, FEBS J., 2014, 281, 2443 CrossRef CAS PubMed.
- D. Gonzalez, J. Hiblot, N. Darbinian, J. C. Miller, G. Gotthard, S. Amini, E. Chabriere and M. Elias, FEBS Open Bio, 2014, 4, 121–127 CrossRef CAS PubMed.
- J. Miyazaki, S. Nakaya and T. Suzuki, J. Biochem., 2001, 129, 777–782 CrossRef CAS PubMed.
- K. Watanabe, T. Ohkuri, S.-i. Yokobori and A. Yamagishi, J. Mol. Biol., 2006, 355, 664–674 CrossRef CAS PubMed.
- B. G. Hall, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5431–5436 CrossRef CAS PubMed.
- E. A. Ortlund, J. T. Bridgham, M. R. Redinbo and J. W. Thornton, Science, 2007, 317, 1544–1548 CrossRef CAS PubMed.
- S. Noor, M. Taylor, R. Russell, L. S. Jermiin, C. Jackson, J. Oakeshott and C. Scott, PLoS One, 2012, 7 CAS , e39822.
- C. B. Stewart, J. W. Schilling and A. C. Wilson, Nature, 1987, 330, 401–404 CrossRef CAS PubMed.
- Z. Yang, S. Kumar and M. Nei, Genetics, 1995, 141, 1641–1650 CAS.
- E. A. Gaucher, S. Govindarajan and O. K. Ganesh, Nature, 2008, 451, 704–707 CrossRef CAS PubMed.
- D. M. Giulio, J. Theor. Biol., 2003, 221, 425–436 CrossRef PubMed.
- J. K. Hobbs, C. Shepherd, D. J. Saul, N. J. Demetras, S. Haaning, C. R. Monk, R. M. Daniel and V. L. Arcus, Mol. Biol. Evol., 2012, 29, 825–835 CrossRef CAS PubMed.
- R. Perez-Jimenez, A. Inglés-Prieto, Z.-M. Zhao, I. Sanchez-Romero, J. Alegre-Cebollada, P. Kosuri, S. Garcia-Manyes, T. J. Kappock, M. Tanokura, A. Holmgren, J. M. Sanchez-Ruiz, E. A. Gaucher and J. M. Fernandez, Nat. Struct. Mol. Biol., 2011, 18, 592–596 CAS.
- V. A. Risso, F. Manssour-Triedo, A. Delgado-Delgado, R. Arco, A. Barroso-delJesus, A. Ingles-Prieto, R. Godoy-Ruiz, J. A. Gavira, E. A. Gaucher, B. Ibarra-Molero and J. M. Sanchez-Ruiz, Mol. Biol. Evol., 2015, 32, 440–455 CrossRef PubMed.
- D. L. Trudeau, M. Kaltenbach and D. S. Tawfik, Mol. Biol. Evol., 2016, 33, 2633–2641 CrossRef CAS PubMed.
- P. D. Williams, D. D. Pollock, B. P. Blackburne and R. A. Goldstein, Assessing the accuracy of ancestral protein reconstruction methods, 2006 Search PubMed.
- M. Wilding, E. F. A. Walsh, S. J. Dorrian and C. Scott, Microb. Biotechnol., 2015, 8, 665–672 CrossRef CAS PubMed.
- M. Schrewe, N. Ladkau, B. Bühler and A. Schmid, Adv. Synth. Catal., 2013, 355, 1693–1697 CrossRef CAS.
- M. Wilding, T. S. Peat, J. Newman and C. Scott, Appl. Environ. Microbiol., 2016, 82, 3846–3856 CrossRef CAS PubMed.
- Advisen, Chemical Companies: Minimizing the Risks of Supply Chain Disruptions, 2013 Search PubMed.
- R. A. Jensen, Annu. Rev. Microbiol., 1976, 30, 409–425 CrossRef CAS PubMed.
- Y. Li, N. M. Llewellyn, R. Giri, F. Huang and J. B. Spencer, Chem. Biol., 2005, 12, 665–675 CrossRef CAS PubMed.
- J. M. Bentley, H. J. Wadsworth and C. L. Willis, J. Chem. Soc., Chem. Commun., 1995, 231–232 RSC.
- O. K. Tawfik and S. Dan, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef PubMed.
- Z.-G. Qian, X.-X. Xia and S. Y. Lee, Biotechnol. Bioeng., 2009, 104, 651–662 CAS.
- T. Mimitsuka, H. Sawai, M. Hatsu and K. Yamada, Biosci., Biotechnol., Biochem., 2007, 71, 2130–2135 CrossRef CAS PubMed.
- N. Ladkau, M. Assmann, M. Schrewe, M. K. Julsing, A. Schmid and B. Bühler, Metab. Eng., 2016, 36, 1–9 CrossRef CAS PubMed.
- K. E. Cassimjee, B. Manta and F. Himo, Org. Biomol. Chem., 2015, 13, 8453–8464 CAS.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- T. Yano, S. Oue and H. Kagamiyama, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5511–5515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02343j |
| This journal is © The Royal Society of Chemistry 2017 |