
Metal organic frameworks as catalysts in solvent-free or ionic liquid assisted conditions
Amarajothi
Dhakshinamoorthy
 *ab,
Abdullah M.
Asiri
c,
Mercedes
Alvaro
b and
Hermenegildo
Garcia
*bc
*ab,
Abdullah M.
Asiri
c,
Mercedes
Alvaro
b and
Hermenegildo
Garcia
*bc
aSchool of Chemistry, Madurai Kamaraj University, Madurai-625 021, Tamil Nadu, India. E-mail: admguru@gmail.com
bDepartamento de Quimica and Instituto Universitario de Tecnologia Quimica (CSIC-UPV), Universitat Politecnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
cCenter of Excellence in Materials Research, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 9th October 2017
Abstract
Metal organic frameworks (MOFs) are being intensively studied as solid catalysts for organic reactions in liquid media. This review focuses on those reports in which these materials have been used as catalysts in the absence of solvents or embedding ionic liquids (ILs). One of the major roles of solvents in liquid phase reactions is to desorb reagents and products from the active sites, facilitating the turnover of the active sites. For this reason, it is a general observation that most solid catalysts undergo strong deactivation and poisoning in the absence of solvents. In the present review, examples are presented showing that, due to their large porosity and framework flexibility, MOFs can be used as catalysts in the absence of solvents for several reaction types including cyanosilylations, condensations, cyloadditions and CO2 insertions, among others, and that they show even better performance than in the presence of conventional organic solvents. This review also describes the synergy that arises from the combination of ILs, frequently with suitable task-specific chains, and MOFs due to the cooperation with the catalysis when two centres in MOF and ionic liquid are present and due to the change in the microenvironment of the active sites. By embedding an ionic liquid in the MOF pores or by synthesising the ionic liquid covalently attached to the ligand in satellite positions, reusable and efficient catalysts requiring the minimum amount of ionic liquid can be obtained. Both complementary strategies increase the greenness of MOFs as heterogeneous catalysts and have advantages from the environmental point of view. Finally, the last section describes the catalytic activity of hierarchical porous MOFs in some selected reactions.
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy received his PhD degree in 2009 from Madurai Kamaraj University, Madurai, India. Later, he worked as a postdoctoral researcher with Prof. Hermenegildo Garcia at the Technical University of Valencia for four years. Then he returned to India and joined as UGC-Assistant Professor in June 2013 at the School of Chemistry, Madurai Kamaraj University. His research interests include catalysis by MOFs and graphene-related materials. He is the recipient of the Young Scientist Award 2014 for Chemical Sciences from The Academy of Sciences, India. He was also awarded an INSA-DFG international bilateral exchange award to visit Germany for three months in 2015. He has co-authored over ninety five publications, two book chapters and one international patent. |
 Abdullah M. Asiri | Abdullah M. Asiri received his PhD from the University of Wales, College of Cardiff, UK, in 1995. He has been the Head of the Chemistry Department at King Abdulaziz University since October 2009 and he is the founder and the director of the Center of Excellence for Advanced Materials Research. He is a Professor of Organic Photochemistry. His research interests cover color chemistry, the synthesis of novel photochromic, thermochromic systems, the synthesis of novel coloring matters and dyeing of textiles, materials chemistry, nanochemistry and nanotechnology, polymers and plastics. He is the Editor-in-Chief of King Abdulaziz University Journal of Science. He is also a member of the Editorial Board of Pigments and Resin Technology (UK), Organic Chemistry in Sight (New Zealand), Designed Monomers & Polymers and Journal of Single Molecule Research. He is the Vice-President of the Saudi Chemical Society (Western Province Branch). He has co-authored over 500 papers and three book chapters and holds three international patents. |
 Mercedes Alvaro | Mercedes Alvaro is full professor at the Chemistry Department of the Technical University of Valencia. She has co-authored over 150 papers and has supervised 9 theses. Her research interests include advanced oxidation techniques for water treatment, applied photochemistry for environmental remediation and organic synthesis. |
 Hermenegildo Garcia | Hermenegildo Garcia is full professor at the Instituto de Tecnologıa Quimica of the Technical University of Valencia, a joint center of the Technical University of Valencia and the Spanish National Research Council, and Honorary Adjunct Professor at the Center of Excellence in Advanced Materials Research of King Abdulaziz University. He made a postdoctoral stay at the University of Reading with Professor Andrew Gilbert and has taken several sabbatical leaves with the group of Professor J. C. Scaiano at the University of Ottawa. Prof. Garcia has been active in the field of heterogeneous catalysis, working with porous catalysts and nanoparticles, has published over 550 papers and has filed over 25 patents. Prof. Garcia is Doctor Honoris Causa from the University of Bucharest and the recipient of the 2011 Janssen-Cilag award given by the Spanish Royal Society of Chemistry and the 2008 Alpha Gold of the Spanish Society of Glass and Ceramics. |
1. Introduction
Green chemistry is aimed at improving the environmental benignity of chemical processes by reducing the amount of byproducts and wastes formed in a chemical process, by using less toxic and tolerable substrates and reagents and by minimizing, as much as possible, work-up and energy consumption.1–6 To achieve these general goals, one of the most powerful tools is the use of catalysts that should increase the efficiency of chemical reactions and selectively produce the desired products.6 Compared to homogeneous catalysts, heterogeneous catalysts have the following advantages: easier recovery of the catalysts from the reaction mixture, the possibility to reuse the catalyst in consecutive runs and their adaptability to continuous flow processes that are better suited for large scale production. Heterogeneous catalysis is mainly based on the use of insoluble solids that should exhibit a high surface area to favour the contact of the active sites with substrates and reagents. Among the various types of solid catalysts, MOFs are the ones that are currently attracting considerable attention.7–9MOFs are porous crystalline materials whose lattice is constituted by metallic nodes coordinated with rigid multipodal organic linkers, mostly aromatic polycarboxylates.10–12 The catalytic activity of these materials derives from the presence of coordinatively unsaturated sites (CUS) around the metal ions at the nodes and structural defects, from the presence of acidic or basic substituents at the ligands or from the presence of guest species located within the internal voids of the structure.13–15 The extensive use of MOFs as catalysts is understandable considering that there is a wide choice in the nature of the metal that can form a part of the MOF and the possibility to adapt the properties of the MOF after the synthesis. Other attractive features of MOFs as catalysts are their large surface area and high pore volume and the flexibility in their synthesis. In this way, MOFs have become one of the preferred solid catalysts for liquid phase reactions including oxidations,16,17 condensations,18 and C–C couplings,9 among others.19 The use of MOFs as solid catalysts can contribute to the greenness of the process, but other important factors to be considered regardless of the catalyst are the nature of the reagents and solvents employed in the reaction. Particularly, volatile organic solvents are one of the main sources of undesirable environmental impact, especially those containing halogens which have been shown to have adverse effects on the ozone layer in the atmosphere as well as on human health. For this reason, research is being carried out in one of the lines in green chemistry aimed at finding suitable alternatives to conventional low boiling point organic solvents that can be emitted into the atmosphere or can become a waste product after the reaction.
One of the simplest approaches to avoid the use of volatile organic solvents is to perform the reaction in the absence of any solvent, just by using substrates/reagents that are in liquid form under the reaction conditions. While solvent-free conditions are favourable from the point of view of avoiding the emission of vapours into the atmosphere, the absence of solvents generates considerable stress on the catalyst, particularly when using solid porous materials. This is because one of the main roles of solvents is to assist in the desorption of reagents, products or byproducts from active sites by competing with them. In other words, in the absence of solvents, it is a general observation that solid catalysts become easily deactivated by strong adsorption onto the active sites of chemicals, including reagents, products and byproducts.
The use of solvent-free conditions on a solid catalyst is, as previously commented, very convenient from the green chemistry point of view; however, it is not very practical due to the rapid deactivation of the catalyst. In the present review, examples will be shown which illustrate that there are several general reaction types that have been proved to be successful under solvent-free conditions in the presence of MOFs. The present state of the art has shown that silylations, condensations, including aldolic reactions and formation of heterocycles, as well as selective hydrogenations and CO2 insertions can be carried out by MOFs in the absence of organic solvents. Although solventfree conditions are not exclusive to MOFs and other classes of catalysts have also been used in the absence of solvents, the purpose of this review is to show that some general reaction types can be carried out advantageously using MOFs as catalysts without solvents.
Besides solvent-free conditions, another alternative to the use of volatile organic compounds that has also become a research front in catalysis is the use of ILs.20–22 They have a negligible vapour pressure and it has been shown that they can dissolve a large variety of organic compounds and can also be a convenient liquid media for organic reactions. One of the main problems of ILs is their viscosity that considerably complicates the extraction of products and chemicals after the reaction as well as the recovery of the catalysts. In this context, one of the strategies that has been developed to take advantage of the benefits of the use of ILs as the reaction medium as well as to diminish as much as possible the volume of ILs and simplify the reaction work-up has been to deposit these ILs as a thin film on the surface of large surface area materials.23 The presence of porosity in MOFs makes it possible to incorporate ILs inside these solid catalysts and in this way, the advantages of heterogeneous catalysis are combined with the benefits of ILs in terms of cooperation with the reaction mechanism, while still being recoverable together with the solid after the reaction. For these reasons, this review also covers those reports combining MOFs and ILs in minimal amounts. Particularly relevant are those examples in which task-specific ILs, i.e. those ILs that have been designed having functional groups to promote organic reactions while maintaining their solubility properties, will also be commented on. As will be shown, the combination of ILs that have Brønsted acidity/basicity with the active sites of MOFs can serve to preserve the catalytic activity of MOFs in Knoevenagel condensations and CO2 insertions and also to avoid the particle size increase that is a common deactivation mechanism observed when supported metal nanoparticles (NPs) are used as catalysts. In addition, another possibility to increase the catalytic activity of MOFs is the use of hierarchical structures in which the diffusion of reagents and substrates is facilitated by the presence of meso- and micropores.
The aim of the present review is to show the current state of the art of the use of MOFs as catalysts in the absence of solvents, incorporating ILs within the pores in such a way that they form a system with the MOF or exhibiting a hierarchical structure with the presence of meso- and micropores. While there is abundant literature reporting on the use of MOFs as catalysts in the presence of solvents, this review aims at demonstrating that MOFs can also perform with high activity and selectivity in the absence of solvents without compromising on their stability, thus fulfilling one of the principles of “Green Chemistry”. In addition, the present review also shows the utility of ILs attached to MOFs to provide a microenvironment to the active sites or to provide additional sites to enhance the activity of these materials compared to the activity of the parent MOFs. A final aspect included in the present review is hierarchical structuration to facilitate the accessibility of substrates and reagents to the active sites.
Table 1 lists the reactions and the MOF catalysts that will be included in the present review. Emphasis is placed on the evidence provided on the stability of the MOF structure under different reaction conditions.
| Catalyst | Reaction | Stability evidences | Ref. |
|---|---|---|---|
| InPF: indium polymeric framework; hfipbb: hexafluoroisopropylidene bisbenzoate; bipy: bipyridine; BTT: 1,3,5-benzene tristetrazolate; RPF: rare earth polymer framework; DSB: 3,5-disulfobenzoate; Phen: 1,10-phenathroline; tipp = 5,10,15,20-tetrakis(4-(imidazol-1-yl)phenyl)-porphyrin; bpdc: biphenyl-4,4′-dicarboxylic acid; DMA: N,N′-dimethylacetamide; H2LOMe: 5,5′-(2,3,6,7-tetramethoxyanthracene-9,10-diyl)diisophthalic acid; NMP: N-methylpyrrolidine; 4,4′-bpe = 1,2-bis(4-pyridyl)ethylene; m-bds = 1,3-benzenedisulfonic acid; PTA: phosphotungstic acid; TPA: tetrapropylammonium; BDC: 1,4-benzenedicarboxylate; DMA: dimethylamine; BTC: 1,3,5-benzenetricarboxylate; ptz: 2,4,6-tri-(4-pyridyl)-1,3,5-triazine; nsa: sodium 2,6-naphthalene disulfonate; BTB: 1,3,5-tris(4-carboxyphenyl)benzene; NDC: naphthalenedicarboxylic acid; ABIL: amino-functionalized basic ionic liquid; DAIL: dual amino-functionalized ionic liquid. | |||
| Vanadium–salen based Cd-MOF | Cyanosilylation of benzaldehyde | XRD, UV-Vis | 24 |
| In-PF-6 | -do- | XRD, reuse | 25 |
| [In4(OH)4(hfipbb)4(4,4′-bipy)] | -do- | Reuse, hot-filtration | 26 |
| [(Cu4O0.27Cl0.73)3(H0.5BTT)8] | -do- | XRD, reuse | 27 |
| [Nd(3,5-DSB)(Phen)] (RPF-19) | -do- | XRD, reuse | 28 |
| [Cd3(tipp)(bpdc)2]·DMA·9H2O | -do- | Leaching, reuse | 29 |
| Ba2(H2LOMe)2(NMP)·NMP | -do- | Reuse | 30 |
| M(4,4′-bpe)2(H2O)4(m-bds) (M: Zn2+ or Cd2+) | Synthesis of 2-amino-6-(arylthio)pyridine-3,5-dicarbonitriles | Reuse | 31 |
| IRMOF-3 | Synthesis of dihydropyrimidinones and dihydropyridines | — | 32 |
| IRMOF-3 | Synthesis of polyhydroquinoline | — | 33 |
| PTA@MIL-101 | Synthesis of 3,4-dihydropyrimidin-2(1H)-one | Hot-filtration, reuse, FT-IR, FESEM | 34 |
| [TPA]2[Zn2-(BDC)3(DMA)2] | Michael addition of pyrrole to β-nitrostyrenes | XRD, reuse | 35 |
| Cu3(BTC)2 | Michael addition between β-nitrostyrene and indole | XRD, FT-IR and reuse | 36 |
| [TPA]2[Zn2-(BDC)3(DMA)2] | Knoevenagel condensation reaction between benzaldehyde and malononitrile | XRD, reuse | 37 |
| Cu(ptz)(nsa)0.5·H2O | Azide–alkyne cycloaddition | XRD, reuse | 38 |
| Cu(I)-MOF | Synthesis of propargylamine | XRD, reuse, leaching | 39 |
| MIL-101(Cr) | Coupling of CO2 with styrene oxide | Reuse, XRD | 40 |
| F-IRMOF-3 | Coupling of CO2 and propylene oxide | XRD | 41 |
| [Zn4O(2,6-NDC)(BTB)4/3] | -do- | XRD, reuse | 42 |
| ABIL-OH/HKUST-1 | Knoevenagel condensation between benzaldehyde and malononitrile | XRD, hot-filtration | 43 |
| PdCl2-ILs/Cu3(BTC)2 | Oxidation of cyclohexene | Reuse, XPS, XRD | 44 |
| IL@UiO-66 | Oxidative desulfurization | Reuse | 45 |
| PW/DAIL/MIL-101(Cr) | Oxidation of benzyl alcohol | XRD, reuse, leaching | 46 |
| PW4/DAIL/MIL-100(Fe) | Oxidation of benzyl alcohol | XRD, reuse, leaching | 47 |
| DAIL-Fe3O4@NH2-MIL-88B(Fe) | Esterification of oleic acid with ethanol | Reuse | 48 |
| [SO3H-(CH2)3-HIM]3PW12O40@MIL-100(Fe) | -do- | Reuse | 49 |
| [NMP]+CH3SO3−@MIL-101(Cr) | Esterification of acetic acid with amyl alcohol | Reuse, leaching, TEM | 50 |
| TEDA-BAIL/MIL-101 | Acetalization of benzaldehyde with glycerol | Reuse, leaching | 51 |
| Pd/IL/Cu3(BTC)2 | Hydrogenation of acetylene | Reuse, XRD, TEM | 52 |
| Cu-MOF | Hydrocarboxylation of cyclohexane | XRD, leaching, reuse | 53 |
| CuI@UiO-67-IM | Azide–alkyne cycloaddition | Reuse, leaching, XPS | 54 |
| MIL-101(Cr)-X(n-Bu)3Br (X: N or P) | Coupling of CO2 and propylene oxide | ICP, FT-IR, XRD | 55 |
| UiO-67-IL | Coupling of CO2 to epichlorohydrin | XRD, leaching | 56 |
| IL-ZIF-90 | Coupling of CO2 with propylene oxide | Reuse, XRD, leaching | 57 |
2. MOFs as catalysts under solvent-free conditions
2.1 Addition of trimethylsilyl cyanide (TMSCN) to carbonyl compounds
Nucleophilic additions to carbonylic compounds are general organic reactions with a large versatility for the preparation of alcohol derivatives. Cyanohydrins can be easily obtained by the addition of cyanide to carbonyl groups. In this regard, TMSCN is one of the most useful and safe reagents for the nucleophilic addition of cyanide to carbonyl compounds to give the corresponding cyanohydrin trimethylsilyl ethers. Hence, the development of efficient catalysts for the cyanosilylation of carbonyl compounds by TMSCN is an important subject in the context of the preparation of cyanohydrins. Several efficient Lewis acid catalysts have been developed so far for cyanosilylation.58–62 Solvent-free conditions can be suitable for the synthesis of various chiral products of interest in biomedicinal chemistry such as α-hydroxy acids, α-hydroxy aldehydes and β-amino alcohols61,63 through the corresponding cyanohydrin.Recently, the synthesis of a chiral vanadium–salen based Cd-MOF with a chiral ligand (R,R)-(−)-1,2-cyclohexanediamino-N,N′-bis(3-tert-butyl-5-(4-pyridyl)salicylidene) has been reported under solvothermal conditions and its catalytic activity has been examined in the solvent-free cyanosilylation of benzaldehyde (Scheme 1).24 Chiral VO–salen complexes were used as homogeneous asymmetric catalysts for various types of organic reactions.64,65 Under optimized conditions, the reaction of benzaldehyde with TMSCN catalyzed by the chiral VO–salen attached to Cd-MOF gave 95% yield with 78% ee. In contrast, the same reaction afforded 91% yield with 57% ee when VO–salen was used as the homogeneous catalyst. These data clearly indicate the accessibility of substrates to the chiral centers located inside the pores of the Cd-MOF. On the other hand, the observed activity with this Cd-MOF under solvent-free conditions is comparable with that of other related chiral MOFs in the presence of solvents.66,67 Furthermore, control experiments performed in the presence of chloroform and acetonitrile detected the occurrence of leaching, thus showing the convenience of carrying out organic reactions under solvent-free conditions, although the exact reasons why leaching does not occur in the absence of solvents remain unknown. The activity of this catalyst was maintained in two runs, although a decrease in the yield and ee was noticed in the third run. This deterioration in the catalytic activity was believed to be due to the modification of the surrounding environment of chiral centers or/and the undesirable adsorption of organic compounds. However, in spite of the decrease in activity, the catalyst maintained its crystallinity after the third run and the vanadium oxidation state did not show remarkable changes.
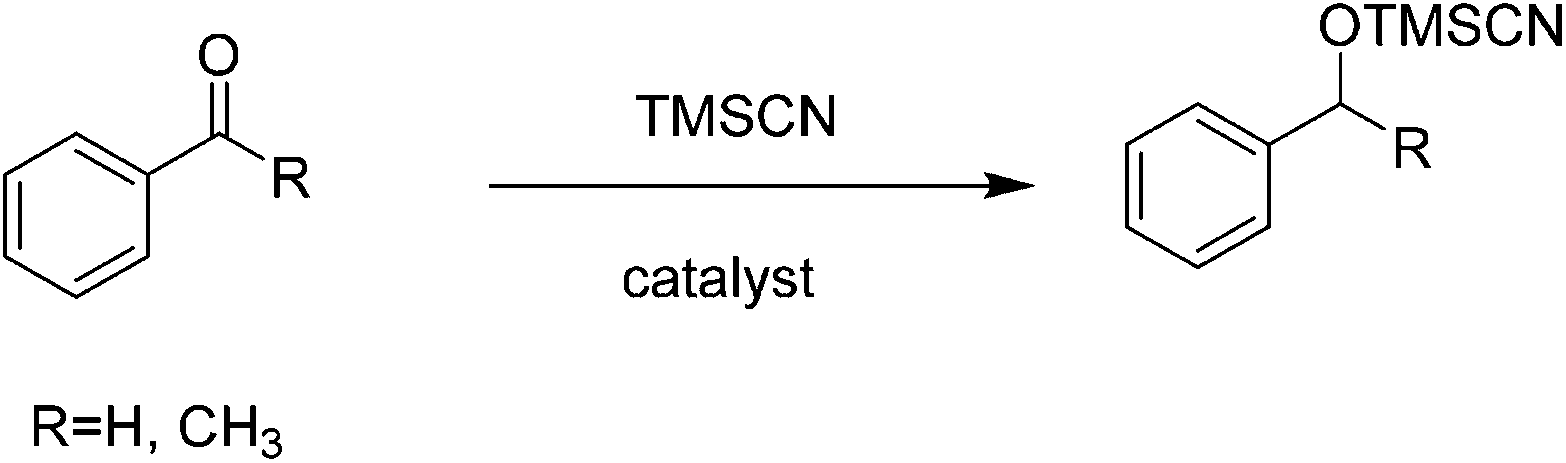 | ||
| Scheme 1 Cyanosilylation of benzaldehyde/acetophenone with TMSCN. | ||
The use of a new flexible ligand in the synthesis of two In-MOFs with ancillary coordination resulted in one one-dimensional and two isoreticular 2D structures, their catalytic activity being evaluated in the cyanosilylation of carbonyl compounds.25 It was found that those In-MOFs with donor ancillary ligands in their structures show the best activity, the one having 1,10-phenanthroline being the most active. The turnover frequency (TOF) value reached in the cyanosilylation of benzaldehyde with the most active In-MOF was 2149 h−1 with 99% yield at 50 °C under solvent-free conditions. Furthermore, the cyanosilylation of acetophenone at 80 °C with the best-performing In-MOF resulted in 39% yield with a TOF value of 19 h−1. The recyclability data indicated that the In-MOF could be reused at least four times, maintaining, according to powder XRD patterns, its crystallinity, exhibiting, however, a small loss in its catalytic activity.
Later, four new indium MOFs, namely [In2(hfipbb)3(1,10-phen)2]·2H2O (phen: phenanthroline) (InPF-12), [In2(hfipbb)3(2,2′-bipy)2]·2H2O (InPF-13), [In2(hfipbb)3(4,4′-bipy)] (InPF-14) and [In4(OH)4(hfipbb)4(4,4′-bipy)] (InPF-15), were hydrothermally synthesised and their activity in the solvent-free cyanosilylation of carbonyl compounds was also studied.26 Among these catalysts, InPF-15 exhibited better activity in the cyanosilylation of benzaldehyde and acetophenone compared to other analogous catalysts (Table 2). As an example, the cyanosilylation of cyclohexanone using TMSCN in the presence of In-PF-15 resulted in a TOF value of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 048 h−1 at 80 °C with 0.1 mol% catalyst loading under a nitrogen atmosphere. InPF-15 maintains its crystallinity after seven catalytic cycles and hot filtration experiments, supporting the heterogeneity of the process.
048 h−1 at 80 °C with 0.1 mol% catalyst loading under a nitrogen atmosphere. InPF-15 maintains its crystallinity after seven catalytic cycles and hot filtration experiments, supporting the heterogeneity of the process.
| Catalyst | Benzaldehyde | Acetophenonec | ||
|---|---|---|---|---|
| Yieldb (%), (h) | TOFd (h−1) | Yieldb (%), (h) | TOFd (h−1) | |
| a Without solvent, 2.5 mol% of catalyst under a N2 atmosphere, 50 °C (benzaldehyde) or 80 °C (acetophenone). b Based on GC-MS. c All reactions were completed at 7 h. d TOF: mmol substrate/mmol catalyst × h. | ||||
| InPF-11 | 99 (0.67) | 105 | 65 (4) | 109 |
| InPF-12 | 99 (2) | 284 | 24 (4) | 1 |
| InPF-13 | 93 (2) | 298 | 41 (2) | 38 |
| InPF-14 | 99 (0.75) | 332 | 45 (1) | 50 |
| InPF-15 | 80 (1.5) | 265 | 77 (4) | 140 |
Recently, the solvothermal reaction between Cu(OAc)2 and H3BTT·2HCl produced a sodalite-type MOF with the following structural formula: [(Cu4O0.27Cl0.73)3(H0.5BTT)8(H2O)12]·3MeOH·9DMF.27 This MOF exhibits a porous 3D (3,8)-connected framework constructed by square [Cu4(μ4-O/Cl)] units and triangular BTT ligands. Furthermore, this material can be dehydrated by removing solvent molecules to form [(Cu4O0.27Cl0.73)3(H0.5BTT)8], possessing coordinatively unsaturated Cu2+ centres. Interestingly, the activity of [(Cu4O0.27Cl0.73)3(H0.5BTT)8] at 1 mol% for the cyanosilylation of benzaldehyde led to a 96% conversion at 40 °C under a N2 atmosphere, which is one-eleventh of the loading used in the related Mn–BTT catalyst.68 On the other hand, the reactivity of [(Cu4O0.27Cl0.73)3(H0.5BTT)8] was higher than that of Sc-MOF and Ln-MOFs in which the loading of catalysts was 5 mol%.28,69 Leaching tests under optimized reaction conditions demonstrated the heterogeneity of the process. Notably, the use of 1-naphthaldehyde as the substrate gave only 78% of the product, while the use of 9-anthracenecarboxaldehyde did not afford any conversion. These experimental data are in clear agreement with the expectations of size selective catalysis, implying that the cyanosilylation occurs within the pores of [(Cu4O0.27Cl0.73)3(H0.5BTT)8]. The catalyst could be reused five times for the reaction of furfuraldehyde and TMSCN without noticeable loss of activity.
Three Ln-based 2D MOFs with the formula [Ln(3,5-DSB)(Phen)] (Ln = La, Pr (RPF-18); Ln = Nd (RPF-19)) were hydrothermally synthesized and their activity in the cyanosilylation of carbonyl compounds was examined.28 RPF-19-Nd exhibits the highest activity for the cyanosilylation of benzaldehyde with 5 mol% catalyst loading at 50 °C under a N2 atmosphere, showing a TOF value of 12.94 h−1. Furthermore, the reactivity of RPF-19-Nd (benzaldehyde, 2 h, 94.8% yield) was higher than that of Mn-MOF (benzaldehyde, 9 h, 98% yield),68 with the latter requiring a solvent. In contrast, the activity of RPF-19-Nd was slightly lower than that of Ln-MOF (benzaldehyde, 1 h, 88% yield);70 however, the latter requires a pre-activation treatment. The RPF-19-Nd catalyst was reused in four consecutive runs without observing appreciable changes. The synthesis of a porous 4-fold interpenetrated MOF, namely [Cd3(tipp)(bpdc)2]·DMA·9H2O, has been reported and its activity tested for the solvent-free cyanosilylation of carbonyl compounds.29 The reaction of benzaldehyde with TMSCN afforded 94% yield of the desired product with 6 mol% of [Cd3(tipp)(bpdc)2]·DMA·9H2O at room temperature with a turnover number (TON) value of 156.7. Moreover, no catalytic activity was observed for acetophenone and 4-methylacetophenone in the cyanosilylation reactions with [Cd3(tipp)(bpdc)2]·DMA·9H2O. The solvothermal reaction of 5,5′-(2,3,6,7-tetramethoxyanthracene-9,10-diyl)diisophthalic acid (H4L, Scheme 2) and Ba(NO3)2 resulted in the formation of a microporous MOF, namely Ba2(H2LOMe)2(NMP)·NMP, with high thermal stability. Upon heating at 325 °C, Ba2(H2LOMe)2(NMP)·NMP transformed to [Ba(H2LOMe)0.5(H2O)·4H2O]n through a single-crystal-to-single-crystal conversion.30 Furthermore, the formation of an intermediate was observed and its catalytic activity studied in the cyanosilylation of carbonyl compounds with TMSCN. The intermediate Ba MOF catalyst showed 100% yields for acetophenone, p-methylacetophenone, p-chloroacetophenone, p-nitroacetophenone, and 4-phenylbut-3-en-2-one at room temperature. The intermediate Ba MOF exhibited higher activity than Sc-MOF, Cu-DDQ and CPO-27-Mn catalysts for the addition of TMSCN with acetophenone.71–73 Moreover, the TON and TOF for the cyanosilylation of benzaldehyde were 192 and 384 h−1, respectively, which was much higher than those achieved for other MOF catalysts mentioned above. Also, the activity of this intermediate catalyst was higher than for polymer-supported metal complexes and SbCl3-based catalysts.74,75 The Ba MOF catalyst maintained its activity in the cyanosilylation of benzaldehyde in three consecutive cycles.
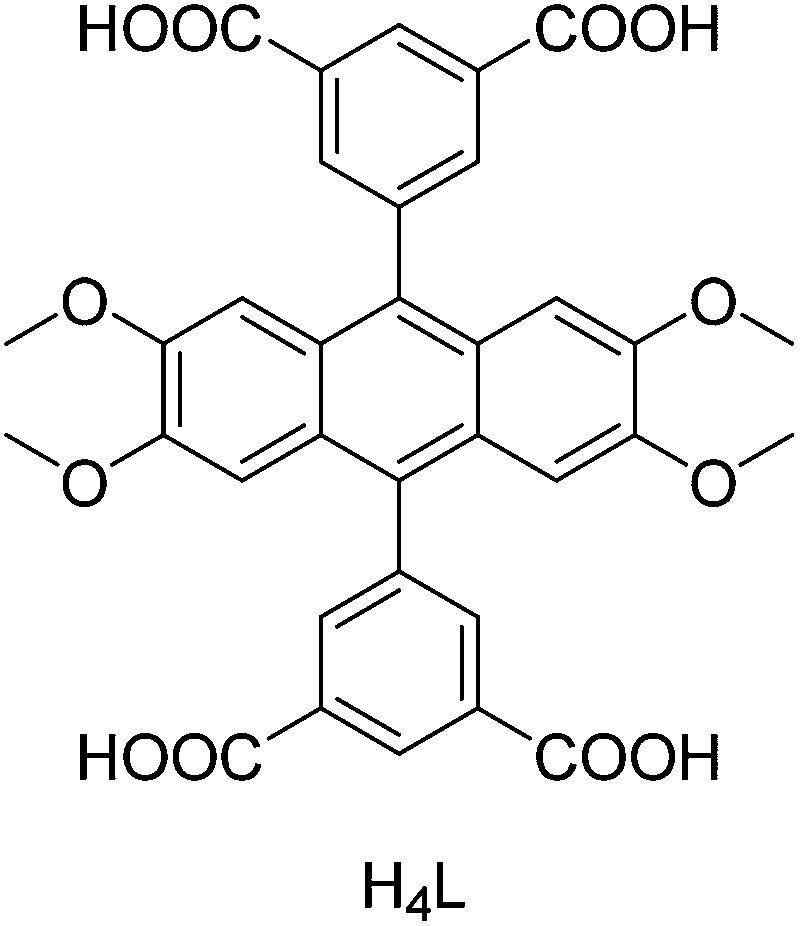 | ||
| Scheme 2 Structure of the ligand H4L. | ||
2.2 Heterocycle synthesis
Condensation reactions of carbonylic compounds are widely used for the synthesis of six- and five-membered heterocycles, including N-containing ones with pyridine, dihydropyridine or the substituted analogues. MOFs have been used as catalysts for the synthesis of these compounds under solvent-free conditions.The activity of M(4,4′-bpe)2(H2O)4(m-bds) (M = Zn2+ or Cd2+) was investigated in the synthesis of 2-amino-6-(arylthio)pyridine-3,5-dicarbonitriles under solvent-free conditions using benzaldehyde, malononitrile, and thiophenol as reagents, as shown in Scheme 3.31 The use of Zn and Cd MOFs as catalysts gave 86 and 87% yield, respectively, under solvent-free conditions, while the same MOFs required twenty times longer time to achieve comparable yields using toluene under identical reaction conditions. These experiments clearly demonstrate the advantage of performing organic transformations under solvent-free conditions by reducing the time as well as energy consumption associated with the process. It could be that the excellent packing already documented for alkyl aromatics inside the cavities of certain MOFs disfavors the accessibility for substrates inside MOFs,76,77 just the opposite role that the solvents are supposed to play. Furthermore, these catalysts also afforded the desired product with substituted thiophenols in high yields (81–87%) under solvent-free conditions.
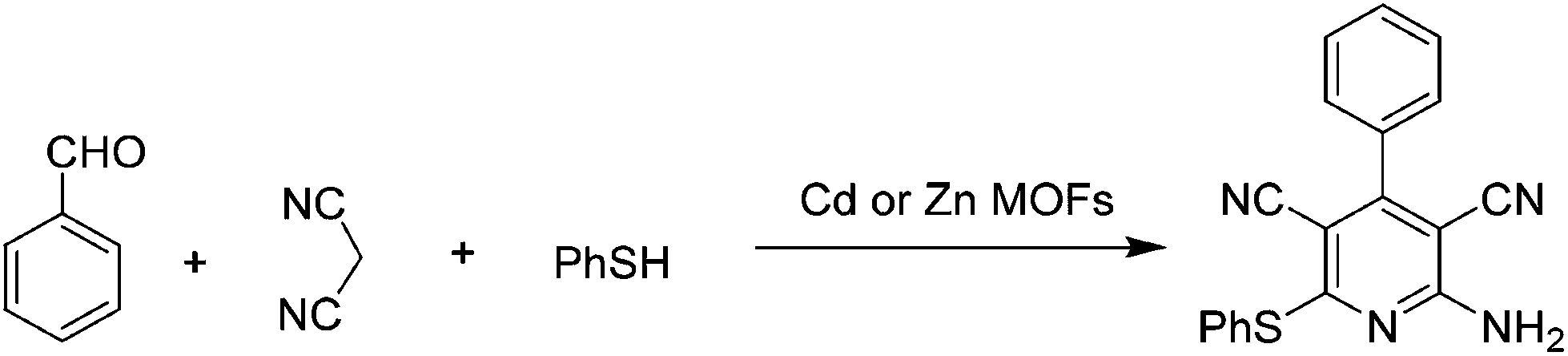 | ||
| Scheme 3 Synthesis of pyridine derivatives using Cd or Zn MOFs. | ||
Dihydropyrimidinones (DHPMs) exhibit a variety of important biological activities, acting as calcium channel blockers, antihypertensives, adrenergic antagonists, and so forth. On the other hand, dihydropyridines (DHPs) are among the most widely used drugs, some relevant examples being shown in Scheme 4. These heterocyclic rings are also known as neuroprotectants because they exhibit anti-platelet aggregation properties and, therefore, they are important in the treatment of Alzheimer's disease and as anti-ischemic agents.78–81
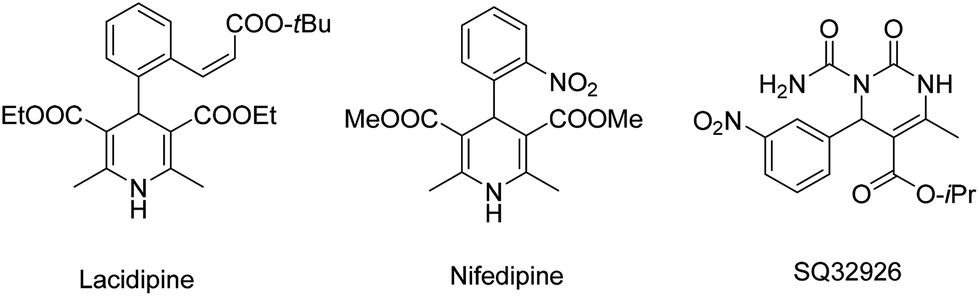 | ||
| Scheme 4 Examples of biologically important DHPM and DHP derivatives. | ||
Most of these nitrogen heterocycles are synthesised using a large variety of homogeneous catalysts. In this context, the use of isoreticular MOFs (IRMOF-3) as heterogeneous catalysts to promote the Hantzsch and Biginelli coupling towards DHPMs and DHPs has been reported.32 The reaction of benzaldehyde, methyl acetoacetate and ammonium acetate using IRMOF-3 as a catalyst under solvent-free conditions afforded DHP with 90% yield (Scheme 5). This catalyst was also tested for the synthesis of DHPMs, and high yields of the desired products were observed here also (Scheme 5). It is however, worth noting that leaching and reusability data, necessary to support the heterogeneity of the process as well as catalyst stability, were not studied.
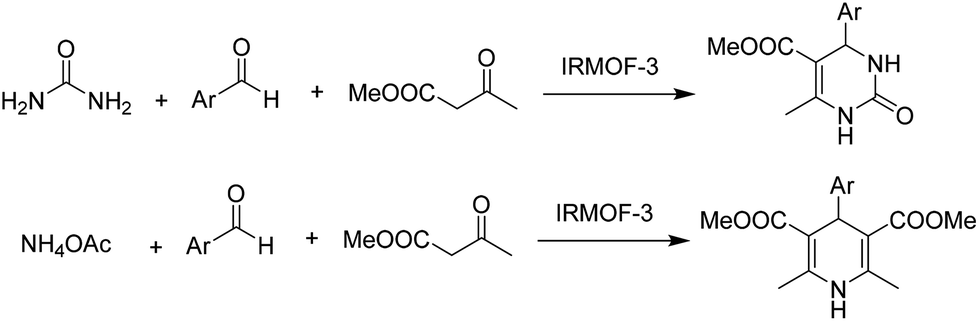 | ||
| Scheme 5 Synthesis of DHPs and DHPMs using IRMOF-3 as a catalyst. | ||
In another precedent, IRMOF-3 was reported as a catalyst for the synthesis of hydroquinoline (Scheme 6) with moderate to high yields under solvent-free conditions.33 The catalyst was reused four times without any change in the yield for the first three runs; however, the yield from the third to the fourth run decreased from 90 to 72%. Unfortunately, there was no data available on the used catalyst that could shed light on the reasons for the catalyst deactivation.
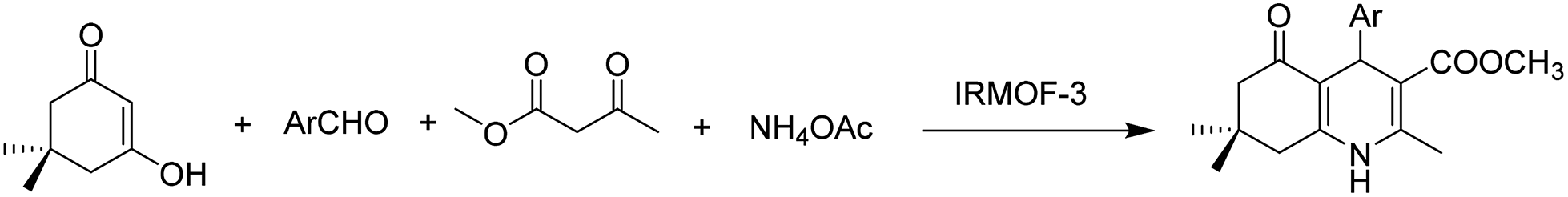 | ||
| Scheme 6 IRMOF-3 catalyzed synthesis of polyhydroquinoline. | ||
MIL-101(Cr) is a robust MOF whose crystal structure defines two types of cages of 29 and 34 Å diameter that can be accessed through five- and six-membered ring windows. Keggin-type phosphotungstic acid (H3PW12O40, PTA) was encapsulated in the mesocages of MIL-101 (PTA@MIL-101) and its catalytic activity was studied in the three-component Biginelli condensation between aldehydes, 1,3-dicarbonyl compounds and urea or thiourea under solvent-free conditions.34 Interestingly, PTA@MIL-101 was extremely active in promoting the reaction of the substituted benzaldehydes with methyl acetoacetate and thiourea to provide various DHPM derivatives. However, a hot filtration test revealed the presence of PTA in the liquid phase, thus contributing to the yield increase after the removal of the catalyst. Nevertheless, the reusability data indicated that there was no decline in the efficiency of the catalyst for up to three runs.
2.3 Friedel–Crafts alkylation
A series of anionic MOFs in which the lattice has an excess of negative charge that has to be compensated by the presence of a charge balancing cation in the internal pores were reported as efficient heterogeneous catalysts for the Michael addition of pyrrole to β-nitrostyrenes (Scheme 7) under solvent-free conditions to afford the corresponding adducts in good to excellent yields.35 Initially, the catalytic activity of an anionic MOF, [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF (HDMA+: dimethylammonium and DMF: N,N-dimethylformamide), was evaluated. The structure of [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF is defined by 1D channels along the crystallographic b axis. These channels with rectangular shapes (width and length of about 7.7 and 12.5 Å, respectively) are occupied by guest DMF molecules and HDMA+ cations. This MOF was subjected to cation exchange with larger organic cations of tetraethylammonium (TEA+) and tetrapropylammonium (TPA+), respectively, to obtain other anionic MOFs with [TEA]2[Zn2(BDC)3(DMA)2] and [TPA]2[Zn2-(BDC)3(DMA)2]. It was expected that the acidic properties of these MOFs would decrease by exchanging the HDMA with the larger organic cations such as TEA+ and TPA+ which are lacking in ammonium protons, thus, enhancing the basicity of the anionic framework. The catalytic activity decreased in the order of [TPA]2[Zn2-(BDC)3(DMA)2] > [TEA]2[Zn2(BDC)3(DMA)2] > [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF by giving 100, 70 and 54% yield of the Michael adduct under solvent-free conditions at 70 °C. This work clearly demonstrates how MOFs can be tuned to increase their activity, following strategies that have been used in zeolites.82 Using these anionic Zn MOFs, a series of β-nitrostyrenes were reacted with pyrrole to achieve the target adduct in high yields (>90%) under solvent-free conditions, reaching 100% yield. It was proposed that the high reaction rate was due to the high concentration and availability of reactants inside the MOF pores. [TPA]2[Zn2(BDC)3(DMA)2] was reused for six runs without any loss in its activity.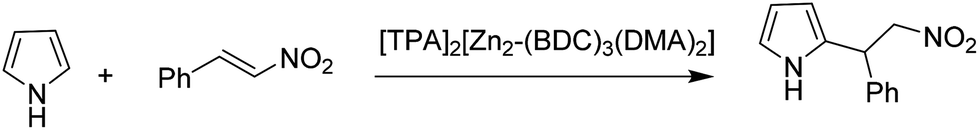 | ||
| Scheme 7 Michael addition between pyrrole and β-nitrostyrene catalyzed by [TPA]2[Zn2(BDC)3(DMA)2]. | ||
In this context, in another precedent, Cu3(BTC)2 has been reported as a heterogeneous catalyst for the Michael addition between β-nitrostyrene and indole, achieving 98% yield of the adduct under solvent-free conditions (Scheme 8).36 This activity of Cu3(BTC)2 is comparable with those of UiO-67-Squar/bpdc MOF83 and Cu(dbda) using a squaramide ligand84 in chloroform.
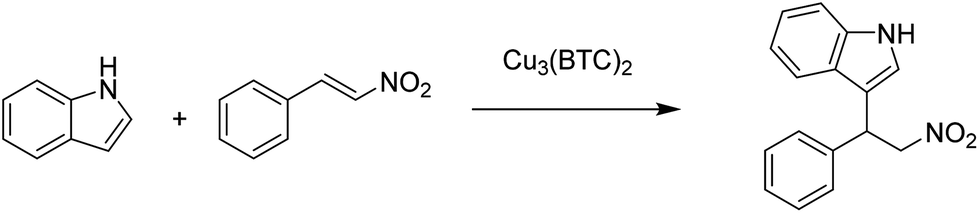 | ||
| Scheme 8 Michael addition of indole to β-nitrostyrene catalyzed by Cu3(BTC)2. | ||
2.4 Knoevenagel condensation
The catalytic activity of anionic MOFs, namely [TPA]2[Zn2-(BDC)3(DMA)2], [TEA]2[Zn2(BDC)3(DMA)2] and [HDMA]2[Zn2(BDC)3(DMA)2]·6DMF, was also investigated in the Knoevenagel condensation reaction between benzaldehyde and malononitrile (Scheme 9), observing 100% yield of the condensation product in 10, 35 and 60 min, respectively, under solvent-free conditions at room temperature.37 It is interesting to note that low to moderate yields were observed for this reaction in the presence of solvents like acetonitrile, toluene and dichloromethane; however, 100% yield is achieved under solvent-free conditions. In addition, [TPA]2[Zn2(BDC)3-(DMA)2] could be reused in nine runs without any loss in its activity. On the other hand, the reaction of benzaldehyde with ethyl cyanoacetate led to the formation of the desired product in 3% yield. These activity data can be easily rationalized assuming that the reaction occurred predominantly within the pores of MOFs.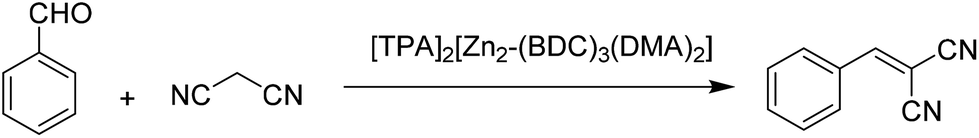 | ||
| Scheme 9 Knoevenagel condensation reaction between benzaldehyde and malononitrile using [TPA]2[Zn2-(BDC)3(DMA)2] as a catalyst. | ||
2.5 Huisgen 1,3-dipolar cycloaddition
Huisgen 1,3-dipolar cycloaddition of organic azides to terminal alkynes is one of the most convenient synthetic routes to obtain triazole derivatives. The conventional Huisgen uncatalyzed thermal methods result in the formation of a 1:1 mixture of 1,4- and 1,5-regioisomers.85 A highly regioselective Huisgen 1,3-dipolar cycloaddition between azides and alkynes in the presence of Cu(I) species has been reported independently by Sharpless86 and Meldal87 under mild conditions to afford 1,4-disubstituted 1,2,3-triazoles. Later, this reaction has been recognized as a Cu(I)-mediated click reaction which has found a myriad of applications in modern synthesis.88–92 Although 1,2,3-triazoles by themselves have generated great interest due to their wide range of biological activities, finding various applications as pharmaceuticals and agrochemicals, a large part of the interest in these pentacyclic heterocycles derives from their organic synthesis and the possibility to join two independent units to form smart functional molecules and materials under mild conditions.93–95 In this aspect, a new Cu-MOF was reported as a heterogeneous catalyst for the azide–alkyne cycloaddition under solvent-free conditions.38 This Cu-MOF was synthesized from copper acetate and ptz and nsa as ligands under hydrothermal conditions. The molecular formula of the resulting Cu-MOF was Cu(ptz)(nsa)0.5·H2O. The oxidation state of Cu in this MOF was ascertained to be +1, as evidenced by XPS. Under the optimized reaction conditions at room temperature (8 h) or at 50 °C (2.5 h), the reaction of phenylacetylene and benzyl azide resulted in 92 and 94% yields of the triazole under solvent-free conditions (Scheme 10). The catalyst was reused for five cycles with no significant loss in activity, although the 4th and 5th cycles required slightly longer time to reach complete conversion compared to the previous cycles. This longer time needed to complete the reaction is, however, a clear sign of catalyst deactivation although the origin was not addressed.
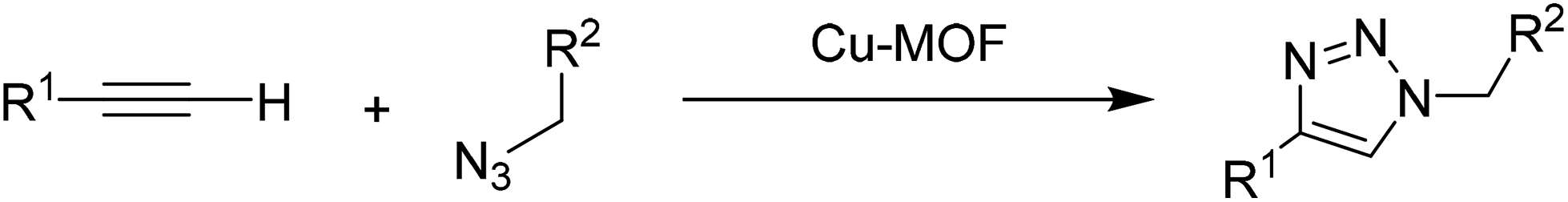 | ||
| Scheme 10 Synthesis of substituted 1,2,3-triazoles using Cu-MOF as a solid catalyst. | ||
2.6 Propargylamine synthesis
Propargylamine is considered as an important motif that can be found in many natural products and therapeutic drugs.96 In addition, they are also used as one of the key intermediates for the synthesis of many nitrogen-containing natural products and biologically active compounds, such as β-lactams97 and tremorine analogues.98 Due to the interest in this type of nitrogen compounds, a new Cu(I)-MOF has been developed for the synthesis of propargylamine via the three-component coupling of secondary amines, alkynes, and aromatic aldehydes under solvent-free conditions (Scheme 11).39 A heterogeneity test confirmed the absence of leaching of the relevant catalytic species. The catalyst was reused for five cycles with no considerable decrease in the final yield.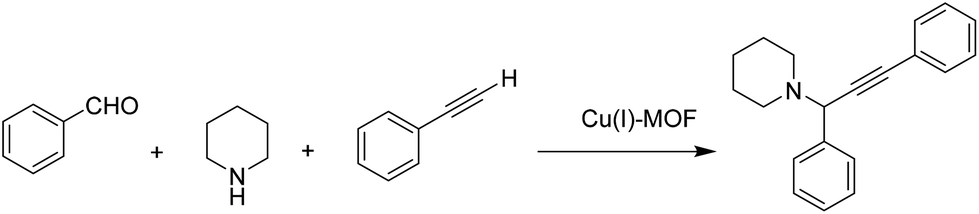 | ||
| Scheme 11 Cu(I) MOF catalyzed propargylamine. | ||
2.7 CO2 cycloaddition to epoxides
CO2 can be considered as a cheap, nontoxic, non-flammable and abundant feedstock in the synthesis of some important chemical products.99–102 In the context of CO2 fixation, one of the most convenient synthetic intermediates are cyclic carbonates obtained by the cycloaddition of CO2 to epoxides.103,104 Cyclic carbonates can be adequate precursors for producing polycarbonates and other fine chemicals. Although many homogeneous and heterogeneous catalytic systems have been reported for the cycloaddition of CO2 to epoxides,105–110 the use of solid catalysts is advantageous from the point of view of simplification of the work-up in the separation process.111The next section describes the incorporation of ILs inside the pores of the MOF. Organic tetraalkylammonium halides have been used in combination with MOFs to promote CO2 insertion in epoxides. Thus, MIL-101(Cr) has been reported as a heterogeneous catalyst promoting the solvent-free coupling of CO2 with styrene oxide to produce styrene carbonate.40 The presence of tetrabutylammonium bromide as a co-catalyst was essential for the formation of the styrene carbonate. On the other hand, the cycloaddition of CO2 to propylene oxide resulted in the formation of propylene carbonate. The catalyst stability was, however, insufficient and the catalyst lost its crystallinity after being reused twice, as evidenced from powder XRD patterns.
An amino group functionalized IRMOF-3 (F-IRMOF-3; Scheme 12) was reported as a heterogeneous catalyst for the solvent-free synthesis of cyclic carbonates without any co-catalyst, reaching TOF values of 778 mmol g−1 h−1.41 Furthermore, F-IRMOF-3-4d resulted in better catalytic activity than other reported catalysts like MIXMOF/Et4NBr (499 mmol g−1 h−1),112 MgO (0.9 mmol g−1 h−1),113 SmOCl (4.1 mmol g−1 h−1),114 cross-linked polymeric nanoparticles (127 mmol g−1 h−1),115 hydroxyl IL grafted onto cross-linked divinylbenzene polymer (70 mmol g−1 h−1)116 and polystyrene-supported threonine (16 mmol g−1 h−1).117 F-IRMOF-3-4d was reused for four runs without much change in its catalytic activity.
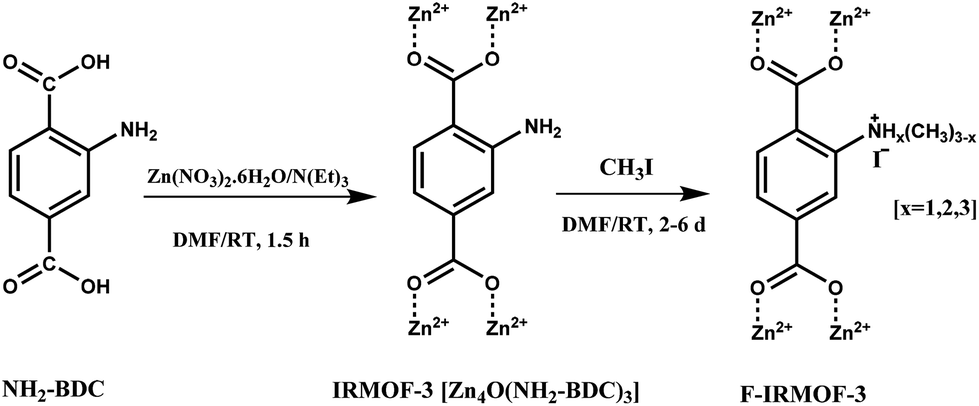 | ||
| Scheme 12 Synthesis of F-IRMOF-3 from NH2-BDC. | ||
A 3D MOF, [Zn4O(2,6-NDC)(BTB)4/3] (MOF-205, BET = 4200 m2 g−1), was synthesised by microwave heating and its catalytic activity was examined in the cycloaddition of CO2 to propylene oxide to produce the corresponding five-membered cyclic carbonates under solvent-free conditions.42
All the above data show that MOFs can frequently be used as solid catalysts in the absence of solvents for condensations, Michael addition, cycloadditions and couplings, giving yields that are even better than in the presence of solvents. Although it should be remarked that catalysis in the absence of solvent is not specific to MOFs and most of the above discussed reactions have also been conducted with other solid catalysts, the present review aims to illustrate that under solvent-free conditions, MOFs can exhibit even higher catalytic activity than in the presence of solvents. An additional advantage is the environmental benefits derived from the diminution of wastes associated with the use of solvents.
3. IL@MOFs
Due to their low vapor pressure, ILs are considered as green solvents for various applications including reaction media,118,119 extractions,120 catalysis,121,122 gas absorption123 and electrolytes in electrochemical devices.124 Although the use of ILs as alternative media to conventional organic solvents is well documented,125 an appealing approach is to impregnate ILs onto porous materials like MOFs forming a thin, micrometric film coating the walls of the solid. Due to Coulombic and polar interactions, the IL film remains on the surface of the material, modifying the environment of the surface active sites, even if a bulk liquid solvent, immiscible with the IL, is present in the system. In this context, a recent review has summarized the different strategies employed for the incorporation of ILs over MOFs and their applications including gas absorption.23 Hence, the present review restricts itself to the catalytic applications of ILs supported on MOFs. The following section will describe those reactions catalysed by MOFs embedding ILs, frequently having acidic or basic groups to cooperate in the reaction mechanism.3.1 Knoevenagel condensation
Task-specific ionic liquids (TSILs) have been reported to be environmentally benign solvents and highly active co-catalysts in Knoevenagel condensation.126,127 However, the use of TSILs in homogeneous catalysis or liquid–liquid biphasic systems is hampered by the difficulty in the separation and recycling of the catalyst. Another problem is that the reaction mixture becomes also inevitably contaminated with byproducts formed from the consecutive reaction of the primary product in contact with the homogeneous catalyst. In this regard, these limitations together with an economy in the amount of TSIL needed can be achieved by coating a porous surface with a thin film of the IL.As an example to illustrate this approach, a size- and shape-selective porous catalyst embedding IL, namely HKUST-1 incorporating an amino-functionalized basic ionic liquid (ABIL-OH/HKUST-1), was synthesized by impregnation (Scheme 13).43 The characterization of this catalyst revealed that the ABIL-OH is likely confined within the nanocavities of HKUST-1 via a Cu–NH2 coordination bond. The fact that the ABIL-OH/HKUST-1 catalyst exhibits a pale blue colour that can be visually perceived was taken as evidence of the change in the coordination of copper ions as the colour is associated with the d–d transition of Cu2+ ions. Furthermore, XPS studies of ABIL-OH/HKUST-1 indicate that the Cu 2p3/2 and Cu 2p1/2 binding energy shifted from 933.8 eV to 933.3 eV and from 953.5 eV to 953.0 eV, respectively. This slight reduction of binding energy is in agreement with the occurrence of an electron transfer from the NH2 to the Cu2+ centre. The loading of ABIL-OH in ABIL-OH/HKUST-1 was determined to be 1.07 mmol g−1 by elemental analysis.
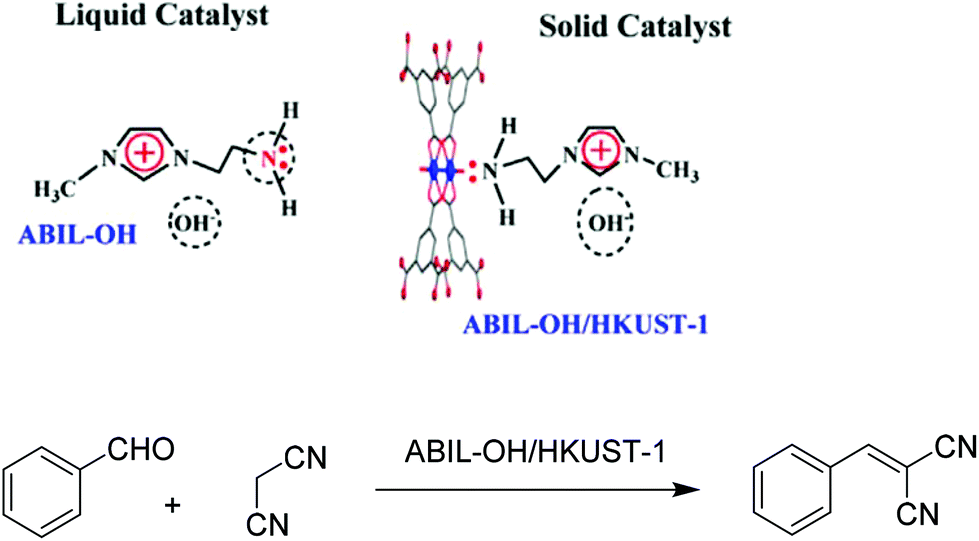 | ||
| Scheme 13 Structure and proposed interaction of ABIL-OH with HKUST-1 and Knoevenagel condensation catalyzed by ABIL-OH/HKUST-1. Reproduced from ref. 43 with permission from Elsevier. | ||
The catalytic activity of ABIL-OH/HKUST-1 was evaluated in the Knoevenagel condensation between benzaldehyde and malononitrile. The as-synthesized HKUST-1 resulted in 55.3% benzaldehyde conversion after 240 min. On the other hand, the ABIL-OH/HKUST-1 catalyst exhibited complete benzaldehyde conversion after 210 min under the same conditions. Meanwhile, the use of ABIL-OH as a homogeneous organocatalyst resulted in complete benzaldehyde conversion after 120 min. This high activity of ABIL-OH could be due to the availability of the free NH2 group in ABIL-OH, thus acting as a Lewis base catalyst. ABIL-OH/HKUST-1 showed high catalytic activity (>90% conversion) with 100% selectivity after being reused for five recycles; however, a slight decline in conversion is observed, again indicative of the occurrence of some deactivation. On the other hand, comparison of the powder XRD patterns between the fresh and reused catalysts revealed that the main diffraction peaks are still observed in the reused catalyst, although the disappearance of several peaks at high angles was observed, indicating that there were some changes in the MOF structure. It has frequently been observed that HKUST-1 has an unstable structure that can collapse due to the strong binding of Cu2+ with substrates, reagents or solvents.128,129 However, a hot filtration test indicated the heterogeneity of the process without clues of Cu leaching. No catalytic activity was observed for the reaction between o-hydroxybenzaldehyde (6.65 × 5.41 Å) and 3-methoxy-4-hydroxybenzaldehyde (7.60 × 6.43 Å) with malononitrile in the presence of ABIL-OH/HKUST-1, whereas these substrates were completely converted to their respective products with ABIL-OH as the homogeneous catalyst under identical reaction conditions. This difference in activity was attributed to the confinement of the active site within the microenvironment of HKUST-1.
3.2 Oxidation reactions
ILs have been one among the preferred green solvents to carry out catalytic reactions promoted by transition metals and metal nanoparticles.130 In this regard, Cu3(BTC)2 has been employed as a support to immobilize PdCl2 with the aid of ILs such as 1-carboxyethyl-3-methylimidazolium chloride (PdCl2-ILs/Cu3(BTC)2).44 The catalytic activity of the as-synthesized PdCl2-ILs/Cu3(BTC)2 catalyst was studied in the selective oxidation of cyclohexene, using molecular oxygen as the oxidant and t-butylhydroperoxide (TBHP) as the initiator. The use of the PdCl2-ILs/Cu3(BTC)2 catalyst afforded 24% conversion of cyclohexene to 2-cyclohexene-1-one, with a maximum selectivity of 53.2%. The other products formed in this reaction are 2-cyclohexene-1-ol and t-butyl-2-cyclohexenyl-1-peroxide. The corresponding epoxide was not detected. Furthermore, it was observed that the selectivity towards 2-cyclohexene-1-one achieved with PdCl2-ILs/CNTs (14%) and PdCl2-ILs/SBA-15 (10%) is lower than that achieved with the PdCl2-ILs/Cu3(BTC)2 catalyst. This enhanced activity of the latter catalyst is believed to be due to the Pd–Cu cooperative effect. Electron paramagnetic resonance (ESR) suggested that the most probable radical species should be a TBHP-derived oxygen centred radical according to an earlier report.131 This proposal of the involvement of a oxygen radical initiator was further verified by a strong inhibition in cyclohexene conversion when the oxidation of cyclohexene is performed using 2,6-di-t-butyl-methylphenol as the radical scavenger.132 Inductively coupled plasma (ICP) analysis showed the existence of copper and palladium in the solution at 127 and 223 ppm, respectively, indicating that the metal leaching occurs to a very limited extent. The catalyst was recycled three times without any appreciable change in conversion and selectivity of 2-cyclohexene-1-one. Powder XRD patterns showed the collapse of the crystalline structure of the Cu-MOF. The catalytic activity of PdCl2-ILs/Cu3(BTC)2 was superior to that of PdCl2/Cu3(BTC)2 in terms of the reaction rate and the TOF value. The reaction rate and TOF value for the latter catalyst was 0.4 mol g−1 h−1 and 6.7 h−1, respectively, while the reaction rate and TOF value achieved for the former catalyst were 0.54 mol g−1 h−1 and 8.1 h−1, respectively. These activity data indicate the benefits of using ILs probably because they favour the uniform dispersion of the PdCl2 species in the interior of the MOF. On the other hand, a significant decrease in conversion was noticed when PdCl2/Cu3(BTC)2 was used as the catalyst during reusability experiments.Recently, UiO-66 was loaded with different percentages of 1-methylimidazolium-3-propylsulfonate hydrogensulfate and its activity was studied in the oxidative desulfurization using hydrogen peroxide as the oxidant.45 It was observed that at 40 °C the removal of sulfur is higher than 94%, with an O/S molar ratio of 7:1 using 40% 1-methylimidazolium-3-propylsulfonate hydrogen sulfate supported on UiO-66. Increasing the content of 1-methylimidazolium-3-propylsulfonate hydrogen sulfate lowered the activity to 92.9% under identical conditions. Reusability experiments showed that the removal of sulfur was 90.6% after the sixth cycle, thus, showing the stability of the catalytic activity under the experimental conditions used. However, no data were given on leaching or the structural integrity of the reused sample compared to the fresh catalyst.
Recently, phosphotungstic acid (H3PW12O40, PW) was immobilized within the nanocages of DAIL-modified MIL-101(Cr) to obtain PW/DAIL/MIL-101(Cr) and its activity in the oxidation of benzyl alcohol using TBHP was examined.46 An analogous catalyst, namely PW/MIL-101(Cr), was also prepared as a control and its activity was compared with that of PW/DAIL/MIL-101(Cr) to gain information on the role played by DAIL during this oxidation reaction. Activated MIL-101(Cr) was treated with DAIL to obtain DAIL/MIL-101(Cr) in which a strong coordination bond between the metal centres of the MOF and the amino groups of the DAIL was proposed to occur. The immobilization of PW onto DAIL/MIL-101(Cr) was performed through anion exchange of the corresponding polyoxometal and the DAIL. The PW cluster has an appropriate dimension to diffuse through the larger12 Å windows of MIL-101(Cr). The preparation procedure followed to obtain PW/DAIL/MIL-101(Cr) and the chemical structure of DAIL are illustrated in Fig. 1. The powder XRD pattern of DAIL/MIL-101(Cr) showed good agreement with that of MIL-101(Cr), confirming that the crystal structure remains mostly intact after the functionalization of the MOF with DAIL cations. It was observed that the thermal stability of PW/DAIL/MIL-101(Cr) increased compared to that of DAIL/MIL-101(Cr) and this enhanced stability was proposed to be due to the additional interactions between DAIL and PW molecules. The XRF measurements indicated that the loading values of PW were 0.18 mmol g−1 and 0.23 mmol g−1 for HPW/MIL-101(Cr) and PW/DAIL/MIL-101(Cr), respectively. PW/DAIL/MIL-101(Cr) exhibited higher catalytic activity (95% conversion and 1900 TON) than HPW/MIL-101(Cr) (70% conversion and 1400 TON) in the oxidation of benzyl alcohol to benzaldehyde. On the other hand, the conversion of benzyl alcohol was influenced by the loading of DAIL, as was observed from the fact that benzyl alcohol conversion gradually increased when DAIL loading increased from 0.3 to 0.9 mmol g−1. These data suggest that the presence of the DAIL group is essential to achieve high activity. In contrast, MIL-101(Cr) gave 43% benzyl alcohol conversion under identical conditions. The hot filtration test proved the heterogeneity of the process. The catalyst was reused five times with no significant decrease in the catalytic activity. The similar powder XRD patterns of the fresh material and reused PW/DAIL/MIL-101(Cr) catalyst indicate that the system is stable under the reaction conditions. Although high conversion of benzyl alcohol was observed with this catalytic system in chloroform, it is clear that the use of volatile organic solvents, particularly chlorinated solvents, do not comply with the principles of green chemistry and further work is necessary to replace these liquids with more environmentally tolerable solvents. In another precedent, the catalytic performance of PW4/DAIL/MIL-100(Fe) was compared with that of the DAIL free catalyst (HPW4/MIL-100(Fe)) in the oxidation of benzyl alcohol using TBHP as an oxidant.47 It was interesting to note that the TON value of the former catalyst is 613, which is superior to that of the latter catalyst, with the value of 433, under identical conditions and this higher activity is ascribed to the ability of the DAIL groups to form hydrogen bonds, enhancing the accessibility of TBHP.
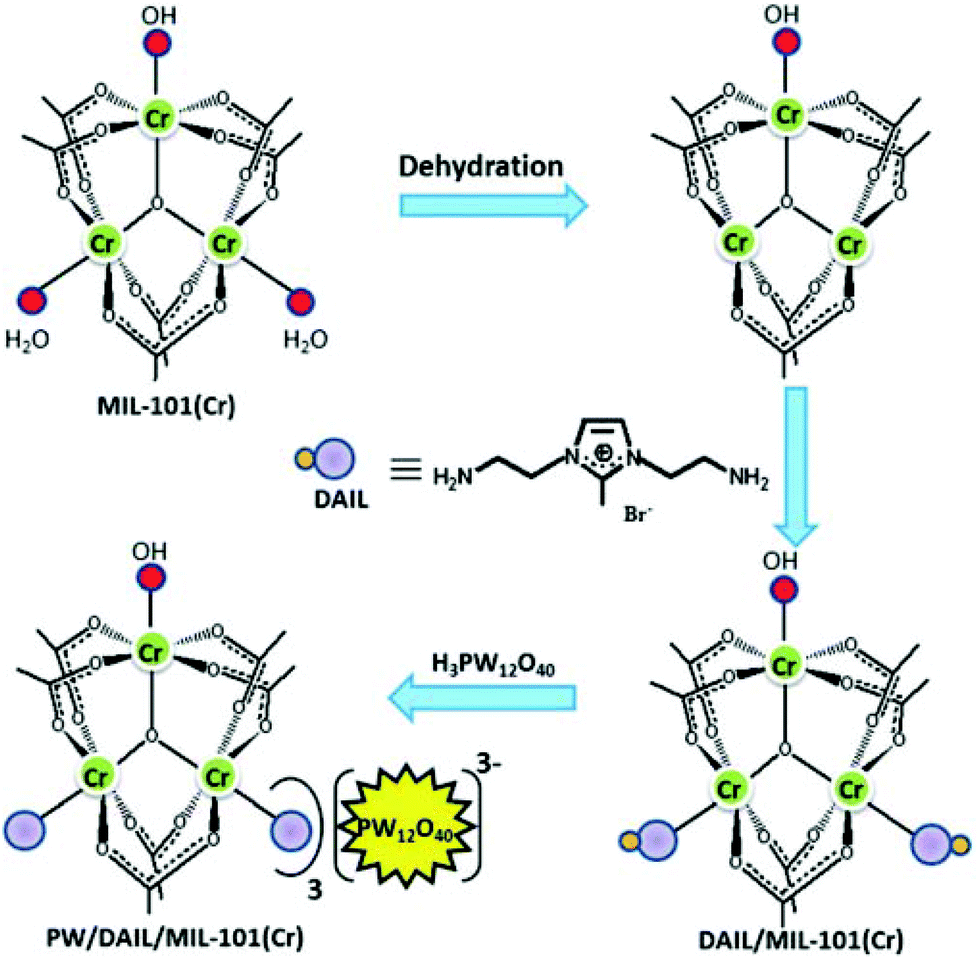 | ||
| Fig. 1 Structure of DAIL and synthesis of PW/DAIL/MIL-101(Cr) catalyst. Reproduced from ref. 46 with permission from the Royal Society of Chemistry. | ||
3.3 Esterification reactions
A magnetically recoverable and efficient catalyst was prepared for biodiesel synthesis (esterification of oleic acid with ethanol) by confining the Brønsted ionic liquid 1,4-butanediyl-3,3′-bis(3-sulfopropyl)imidazolium bis(hydrogensulfate) through the amino groups within an amino-functionalized magnetic MOF composite (Fe3O4@NH2-MIL-88B(Fe)), as shown in Fig. 2.48 The use of Fe3O4@NH2-MIL-88B(Fe) as catalyst for the esterification of oleic acid with ethanol resulted in 8.1% yield, which was believed to be due to the presence of unsaturated FeIII metal centres acting as Lewis acid sites.133 On the other hand, the use of the homogeneous counterpart, namely IL DAIL, showed 90.3% yield that is similar, but, lower, than that of the magnetic heterogeneous catalyst DAIL-Fe3O4@NH2-MIL-88B(Fe) (acidity of 1.76 mmol H+ g−1; DAIL loading amount of 0.58 mmol g−1) that afforded 93.2% yield. Interestingly, the catalytic activity of DAIL-Fe3O4@NH2-MIL-88B(Fe) was comparable to that of earlier reported acidic catalysts (80–93% yields).134–139 The advantage of the present magnetic MOF catalyst is not only that DAIL-Fe3O4@NH2-MIL-88B(Fe) showed slightly higher catalytic activity but also its convenient and efficient separation by applying a weak magnetic field. However, the catalytic activity of DAIL-Fe3O4@NH2-MIL-88B(Fe) is still far from perfect since reusability tests showed that it decreased after six consecutive cycles, something that is not surprising considering the tendency of larger molecules to poison porous materials. The catalysis was nevertheless found to be heterogeneous in nature.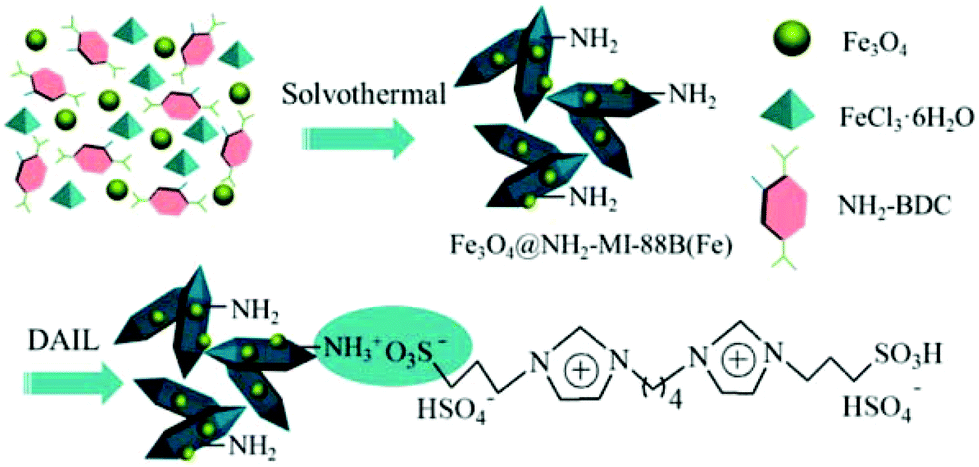 | ||
| Fig. 2 Preparation of functionalized magnetic MOF incorporating the acid IL DAIL-Fe3O4@NH2-MIL-88B(Fe). Reproduced from ref. 48 with permission from the American Chemical Society. | ||
A new strategy designed to construct the IL, POM, and MOF composite is shown in Fig. 3. In this approach, POM was first loaded within the cages of MIL-100(Fe) to play the role of a bridge. Then, the IL was introduced to encapsulate the heteropolyanion.49 The POM-IL-functionalized MOF has several advantages such as a simple preparation process, high stability, high content of active components, ease of separation and high reusability. Furthermore, the preparation method did not use toxic or harmful reagents, such as HF and DMF, thus exhibiting advantages from the environmental point of view. The catalytic activity of [SO3H-(CH2)3-HIM]3PW12O40@MIL-100(Fe) was studied as a catalyst in the esterification of oleic acid with ethanol. The pristine MIL-100(Fe) showed some catalytic activity of 15.5% conversion at 111 °C, probably due to the presence of coordinatively unsaturated Fe3+ ions as Lewis acid sites. On the other hand, encapsulated phosphotungstate HPW12O40@MIL-100(Fe) (acidity: 0.39 mmol H+ g−1) gave 40.3% conversion of oleic acid. Importantly, 94.6% conversion was achieved using [SO3H-(CH2)3-HIM]3PW12O40@MIL-100 as a catalyst (acidity: 1.74 mmol H+ g−1). This enhanced activity of the composite catalyst can be attributed to the sulfonic acid groups in the IL cation increasing the number of Brønsted acid sites compared to the heteropolyanion. Also, the existence of a cooperative effect between Lewis and Brønsted acid sites improving the activity of the catalyst was proposed.140 Although the catalytic activity of the pure IL was higher (95.1%), the heterogeneous catalyst could be easily separated from the reaction medium. In contrast, the [SO3H-(CH2)3-HIM][HSO4]@MIL-100(Fe) catalyst that lacked POM and was prepared by the direct impregnation of MIL-100(Fe) with IL exhibited 90.2% conversion under identical condition; however, its activity decreased upon reuse (Fig. 4). Overall, the [SO3H-(CH2)3-HIM]3PW12O40@MIL-100(Fe) catalyst showed optimal activity for the esterification of oleic acid compared with other catalysts.135,141–144 The catalyst was reused for six cycles without any appreciable decline in its activity.
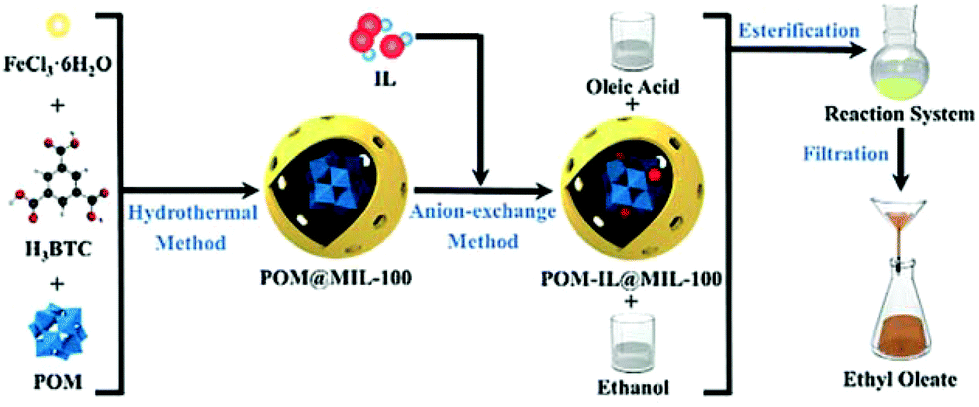 | ||
| Fig. 3 Synthesis of [SO3H-(CH2)3-HIM]3PW12O40@MIL-100(Fe) and its use as an esterification catalyst for the preparation of biodiesel. Reproduced from ref. 49 with permission from Wiley. | ||
 | ||
| Fig. 4 Activity of [SO3H-(CH2)3-HIM]3PW12O40@MIL-100(Fe) (blue) and [SO3H-(CH2)3-HIM][HSO4]@MIL-100(Fe) (red) upon recycling for the esterification of oleic acid with ethanol. Reproduced from ref. 49 with permission from Wiley. | ||
A Brønsted IL acid, namely N-methyl-2-pyrrolidonium methyl sulfonate ([NMP]+CH3SO3−), was immobilized on MIL-101(Cr) and its catalytic activity was evaluated in the esterification of acetic acid with amyl alcohol.50 This catalyst exhibited 82.4% conversion which is comparable to those of [NMP]+CH3SO3− (97%)145 and HPW/MIL-101 (92.3%)146 catalysts for the esterification reaction. Although the Brønsted IL acid loaded on MIL-101(Cr) showed slightly lower activity compared to those of [NMP]+CH3SO3− and HPW/MIL-101, the merit of MIL-1001(Cr) supported IL is to be a heterogeneous catalyst, avoiding tungstic acid. Furthermore, the catalyst was reused six times without any considerable decline in its activity.
Another Brønsted acidic IL (BAIL) confined inside the nanocages of MIL-101(Cr) was reported for the acetalization of benzaldehyde with glycerol (Scheme 14).51 The BAIL functionalized MIL-101(Cr) catalyst was prepared by heating at reflux temperature imidazole (IMIZ) or triethylene diamine (TEDA) with the dehydrated MIL-101(Cr). These precursors were further treated with 1,4-butane sultone and exchanged with H2SO4, as shown in Fig. 5. The parent MIL-101(Cr) showed 32% conversion of benzaldehyde at 90 °C after 3 h, this low conversion being probably due to its mild Lewis acidity. Interestingly, the TEDA-BAIL/MIL-101(Cr) catalyst afforded 88% conversion of benzaldehyde while 58% conversion of benzaldehyde is achieved with the IMIZ-BAIL/MIL-101(Cr) catalyst under identical conditions. Furthermore, the catalytic activity achieved by TEDA-BAIL/MIL-101(Cr) was comparable with that of the benchmark catalyst, namely p-toluenesulfonic acid, which gave 90% conversion of benzaldehyde. The catalyst was recycled six times without any significant decline in its catalytic activity or leaching of active sites.
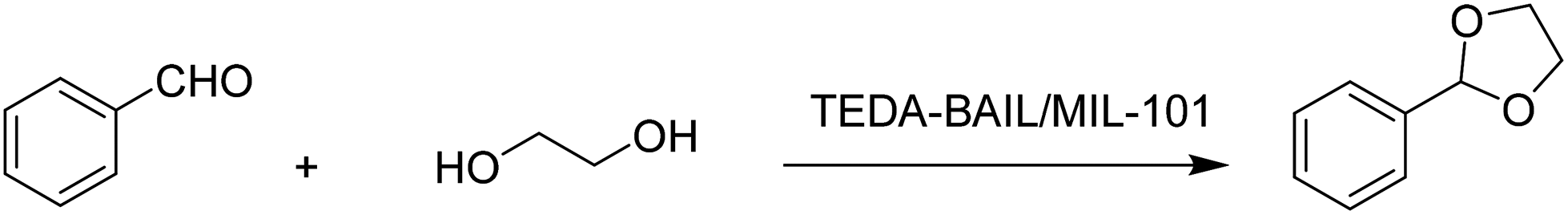 | ||
| Scheme 14 Acetalization of benzaldehyde with glycerol using TEDA-BAIL/MIL-101 as a catalyst. | ||
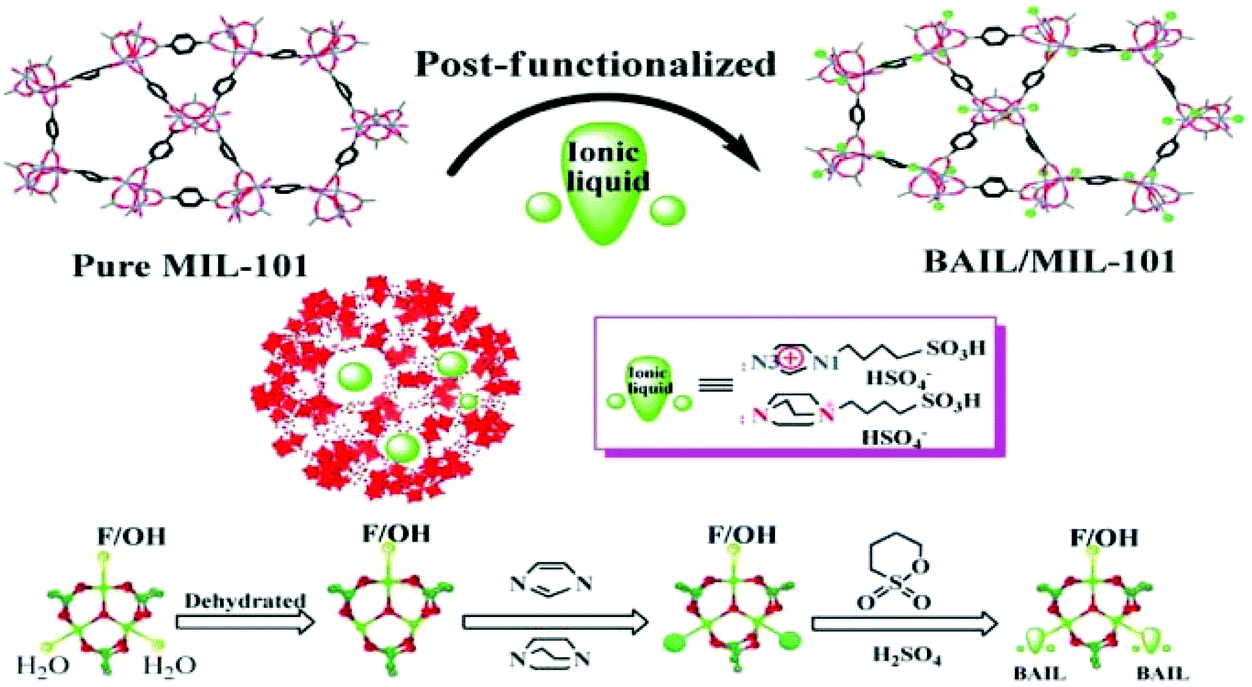 | ||
| Fig. 5 Procedure for the synthesis of BAIL confined in MIL-101 nanocages by post-synthetic modification. Reproduced from ref. 51 with permission from the Royal Society of Chemistry. | ||
3.4 Hydrogenation of acetylene
The selective hydrogenation of alkynes to alkenes without further reduction to the corresponding alkanes is of considerable interest in the areas of polymer synthesis and fine chemicals.147 In this context, the IL microphase was used for enhancing the selectivity of metal NPs supported on an MOF (Pd/IL/Cu3(BTC)2).52 It is believed that the IL microphase provides an adequate environment for stabilizing metal NPs. Typically, the precursors of Cu3(BTC)2 and Pd were loaded into the IL 1,1,3,3-tetramethylguanidinium trifluoroacetate (TMGT), followed by reduction, affording Pd/IL/Cu3(BTC)2. FT-IR spectra showed the coordination of the carboxylate groups of BTC to Cu(II) ions. The powder XRD pattern of the as-synthesized Pd/IL/Cu3(BTC)2 is in agreement with MOF formation.148 Inductively coupled plasma atomic emission spectroscopy (ICP-AES) showed 1.27 wt% of Pd in Pd/IL/Cu3(BTC)2. Furthermore, the IL content in Pd/IL/Cu3(BTC)2 was 13.2 wt%, as evidenced from TGA analysis. HRTEM images showed the existence of Pd NPs with an average size around 2 nm. Nitrogen adsorption–desorption analysis revealed that Pd/IL/Cu3(BTC)2 possesses a BET surface area of 708 m2 g−1 while the parent Cu3(BTC)2 has a higher surface area of 1026 m2 g−1. This surface area decrease should be a reflection of the incorporation of IL and Pd NPs into the micropores of Cu3(BTC)2. Phenylacetylene was completely converted to styrene upon hydrogenation (Fig. 6) catalyzed by Pd/IL/Cu3(BTC)2, with very high selectivity and with a TOF value of 2287 h−1. Under identical conditions, Pd/Cu3(BTC)2 required more than twenty times longer time to reach 83% conversion with 99% selectivity to styrene (TOF: 99 h−1). This difference in activity was attributed to the fact that the Pd NPs undergo aggregation in the absence of IL. A comparison of the catalytic activity of Pd/IL/Cu3(BTC)2 with Pd/C and other reported catalysts149 revealed that Pd/IL/Cu3(BTC)2 shows much better performance in terms of conversion and selectivity under the same reaction conditions. It has been widely reported that N-containing compounds can be used to modify the support to improve the selectivity of the catalyst for alkyne hydrogenation to alkenes.150–152 A hot filtration test proved the heterogeneity of the reaction. The catalyst was reused for four cycles with no significant decline in its activity. The TEM image of the catalyst that was used five times shows no evidence of particle aggregation and no major difference was seen between the fresh and recovered catalyst by powder XRD patterns. ICP-AES analysis indicated that the Pd loading of the recovered catalyst was 1.23 wt%, which is close to the loading of the fresh material (1.27 wt%). Therefore, all the available characterization data indicate a remarkable stability of Pd/IL/Cu3(BTC)2 under the reaction conditions.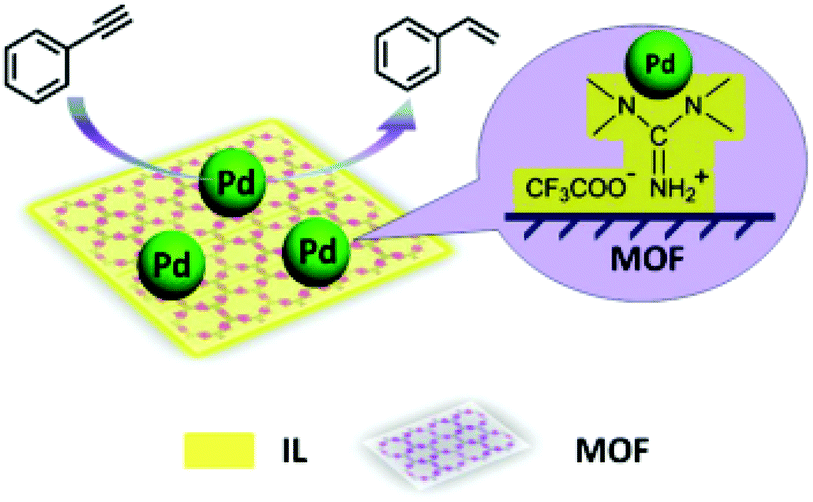 | ||
| Fig. 6 Synthesis of Pd/IL/Cu3(BTC)2 catalyst and its catalytic activity for the selective partial hydrogenation of phenylacetylene. Reproduced from ref. 52 with permission from the Royal Society of Chemistry. | ||
3.5 Hydrocarboxylation of cyclohexane
A Cu(II)-MOF was synthesised with [4-((4-([2,2′:6′,2′′-terpyridin]-4′-yl)benzyl)oxy)benzoate] (Fig. 7) as a flexible bifunctionalized terpyridine ligand.53 The catalytic activity of this terpyridinyl-containing Cu-MOF was examined in the low-temperature hydrocarboxylation of cyclohexane with carbon monoxide, water and potassium peroxodisulfate in water/acetonitrile or water/IL medium. The IL used was 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide, [BMPyr][NTf2] (Scheme 15), due to its stability towards CO. This reaction converts cyclohexane into the corresponding cyclohexanecarboxylic acid (Scheme 16). A noticeable advantage of this method is the use of an aqueous medium and an IL, under mild conditions, without the need for an acid, such as trifluoroacetic acid, that becomes a waste product in the reaction workup. In water/acetonitrile medium (3:3), the catalyst showed 33.4% yield of cyclohexanecarboxylic acid with a TON value of 44.5. On the other hand, the catalyst afforded 35.9% yield of the product with a TON value of 45.1 in water/[BMPyr][NTf2] medium (3:3), showing a slight improvement. Control experiments revealed that the solvent composition was an important parameter in the hydrocarboxylation of cycloalkanes, which does not proceed in pure water, in only acetonitrile, or in the absence of both (only in cyclohexane). Furthermore, the reaction did not also proceed in pure IL. The activity and selectivity of the catalyst was maintained for four consecutive cycles; however, the activity dropped by 16% and 35% in the 5th and 6th cycle, respectively. Powder XRD patterns of the four-times reused catalyst did not show any change in the structural integrity of the Cu-MOF compared to the fresh catalyst. No leaching of Cu was also detected under the present experimental conditions.
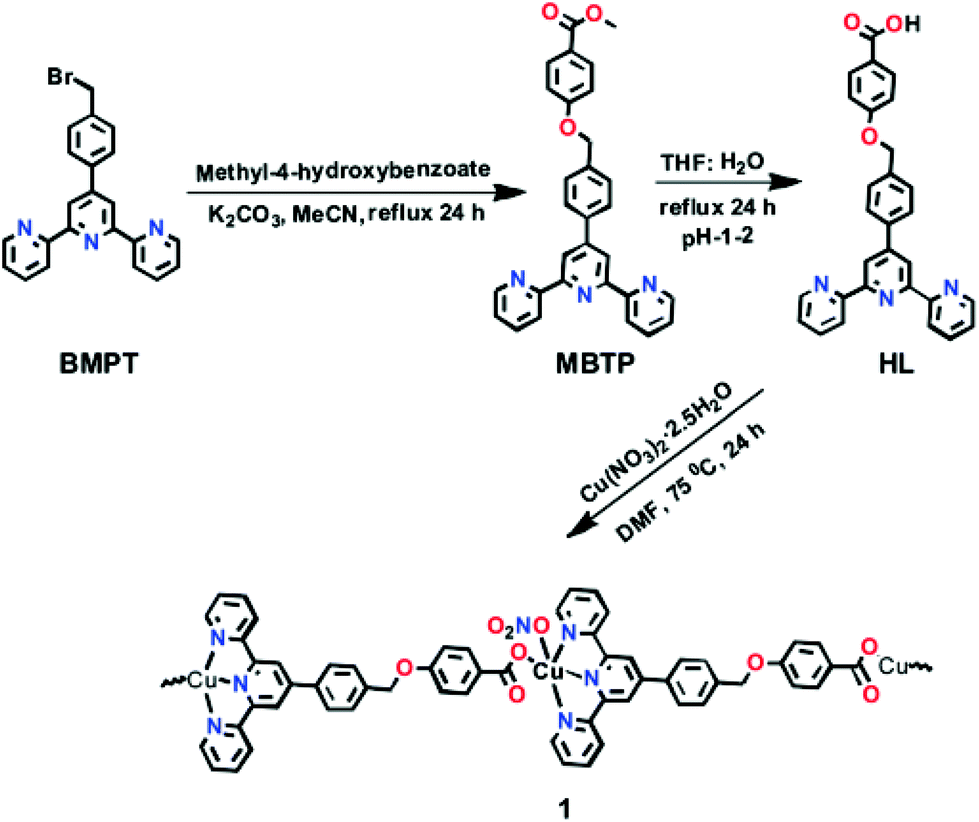 | ||
| Fig. 7 Synthesis of HL and formation of the corresponding MOF by reaction with copper nitrate. Reproduced from ref. 53 with permission from the Royal Society of Chemistry. | ||
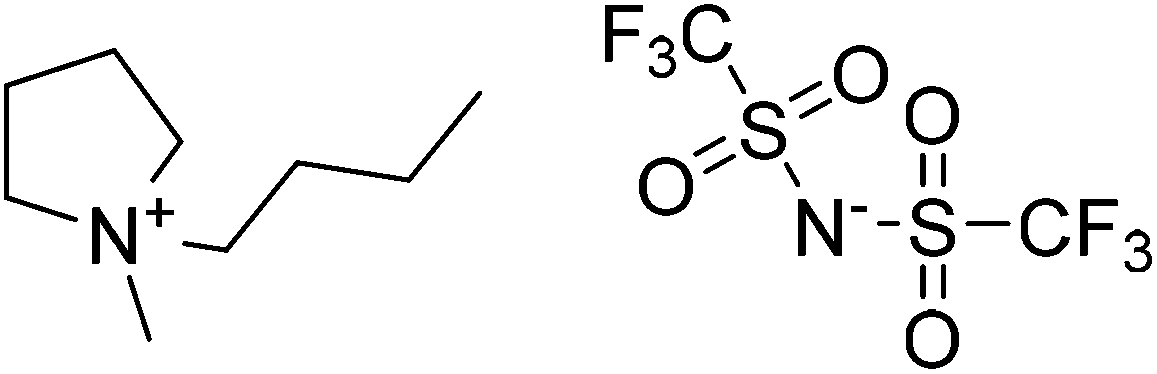 | ||
| Scheme 15 Structure of 1-butylmethylpyrrolidium bis(trifluoromethanesulfonyl)imide, [BMPyr][NTf2]. | ||
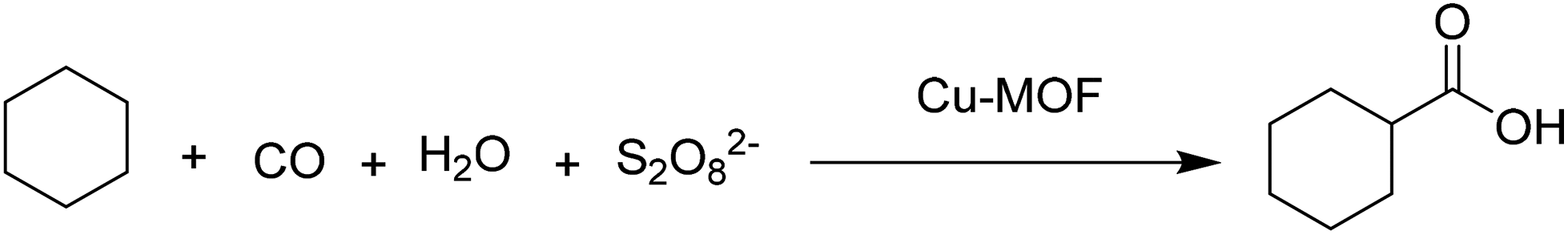 | ||
| Scheme 16 Hydrocarboxylation of cyclohexane to cyclohexanecarboxylic acid promoted by Cu-MOF. | ||
3.6 Azide–alkyne cycloaddition
Recently, CuI was loaded on UiO-67-type MOF, namely UiO-67-IM, to obtain CuI@UiO-67-IM. UiO-67-IM was already decorated with n-pentadecyl units attached to an imidazolium salt via direct ligand functionalization (Fig. 8) before the adsorption of CuI.54 Powder XRD patterns of UiO-67-IM and CuI@UiO-67-IM indicated that both samples were highly crystalline and their structure was identical to that of pristine UiO-67. Thus, the pre-embedded imidazolium salt with long alkyl linear chain and subsequent coincorporation of CuI did not affect the framework of UiO-67. The BET surface area of CuI@UiO-67-IM was 45.1 m2 g−1, which is significantly lower than that of UiO-67 (2113 m2 g−1). The activity of CuI@UiO-67-IM was examined in the Cu(I)-catalyzed azide–alkyne cycloaddition between phenylacetylene and benzyl azide to achieve 89% conversion with 99% selectivity of the expected 1,4-disubstituted triazole (Scheme 17) at 80 °C in water under air atmosphere for 2 h. Also, the reaction was heterogeneous in nature since there was no further conversion after removal of the solid catalyst from the reaction mixture. The recyclability of CuI@UiO-67-IM was studied in a one-pot, two-step domino reaction based on 4-nitrobenzyl bromide, NaN3 and phenylacetylene. The catalyst showed conversions of 94 to 90% with >99.9% selectivity within five catalytic runs. In addition, powder XRD patterns and SEM images of CuI@UiO-67-IM after five catalytic runs confirmed that the structural integrity and morphology of CuI@UiO-67-IM were retained after the reaction. Furthermore, no valence change was observed for the copper species as determined by XPS after five catalytic cycles. However, there was a leaching of CuI to the level of 10.7% of the initial CuI amount, as determined by ICP-AES.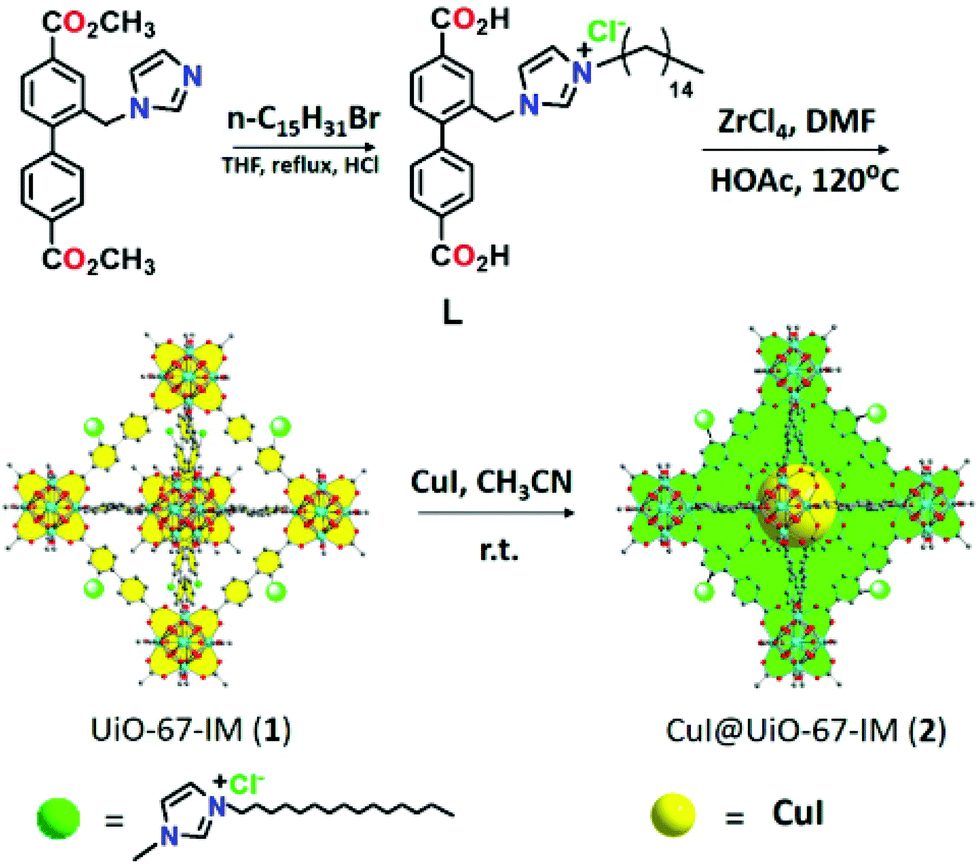 | ||
| Fig. 8 Synthesis of L, UiO-67-IM and CuI@UiO-67-IM. Reproduced from ref. 54 with permission from the American Chemical Society. | ||
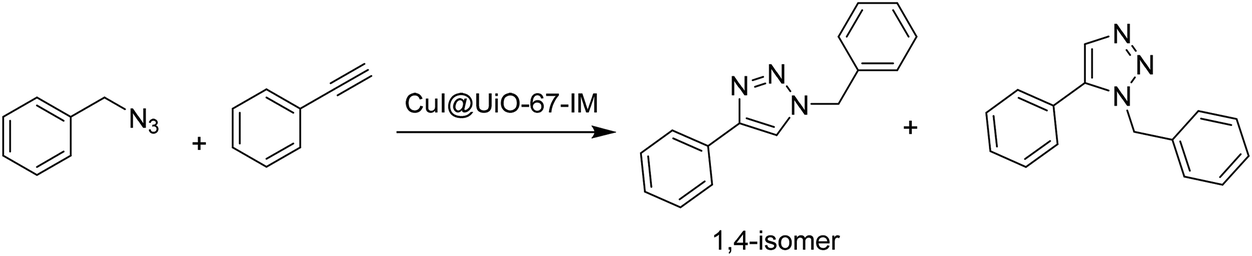 | ||
| Scheme 17 CuI@UiO-67-IM catalyzed cycloaddition reaction between benzyl azide and phenylacetylene. | ||
3.7 Coupling of CO2 with epoxide
ILs have been one of the preferred solvents for CO2 reactions, including CO2 insertion into epoxides. The advantages of IL in these reactions derive not only from the solubility that CO2 shows in these liquids, but also from halides as counter anions of the ILs acting as a nucleophile activating the C atom of CO2, favouring the distortion of this reagent from the linearity. Not surprisingly, ILs embedded within MOFs have also been used as catalysts for this reaction type.A strategy was developed to combine together coordinatively unsaturated metal sites based MOFs with functional IL to form heterogeneous bifunctional catalysts (Fig. 9) and its activity was tested in the cycloaddition of CO2 with epoxides under mild conditions in the absence of a co-catalyst. In this context, two bifunctional heterogeneous catalysts incorporating IL, MIL-101(Cr)-X(n-Bu)3Br (X: N or P) were synthesised with CUS as Lewis acid centres and embedding ILs as additional active sites.55 Characterization data by powder XRD and IR spectroscopy indicated the formation of MIL-101-NH2 and its modification by ILs. MIL-101-N(n-Bu)3Br and MIL-101-P(n-Bu)3Br converted CO2 and propylene oxide into propylene carbonate (PC, Scheme 18) with 99.1 (110.1 TON) and 98.6% (109.6 TON) yields, respectively. Control experiments using mixtures of pure components of MIL-101-NH2, (nBu)4NBr or (n-Bu)4PBr showed a comparatively lower yield of PC under the same conditions, which can be rationalized due to the existence of only monofunctional catalytic sites in these materials. Furthermore, although the physical mixtures of MIL-101-NH2 with (n-Bu)4NBr and MIL-101-NH2 with (n-Bu)4PBr exhibited 98.4 and 97.9% yields, respectively, the values of which are similar to those achieved by MIL-101-N(n-Bu)3Br and MIL-101-P(n-Bu)3Br, the advantage of the IL-functionalized MOFs was that there was no decline in activity even after three cycles, while the catalysts consisting of the physical mixture of IL and MOF showed a dramatic decrease in yield in the second cycle. In one case, ICP analysis revealed the presence of 0.012 mg of phosphorus in solution. IR spectroscopy and powder XRD patterns for the fresh and recycled MIL-101(Cr)-X-(n-Bu)3Br catalysts did not show appreciable changes, thus, supporting the stability of the material under the experimental reaction conditions. Furthermore, the advantage of these bifunctional MOF catalysts was confirmed by comparing their activity with some benchmark MOFs153–157 under the same reaction conditions. As shown in Fig. 10, MIL-101-X-(n-Bu)3Br showed the highest yield to PC (>98% conversion). In contrast, other MOFs exhibit only a very low conversion, with 2.5, 5.2, 5.4, 20.9 and 23.2% for MOF-5, IRMOF-3, HKUST-1, MIL-101 and Mg-MOF-74, respectively. This enhanced catalytic activity of MIL-101(Cr)-X-(n-Bu)3Br was believed to be due to the synergetic catalytic roles of Lewis acid sites and the Br− ion of ILs as a nucleophile. These experiments clearly demonstrate that bifunctional catalysts offer enhanced activity compared to monofunctional catalysts.
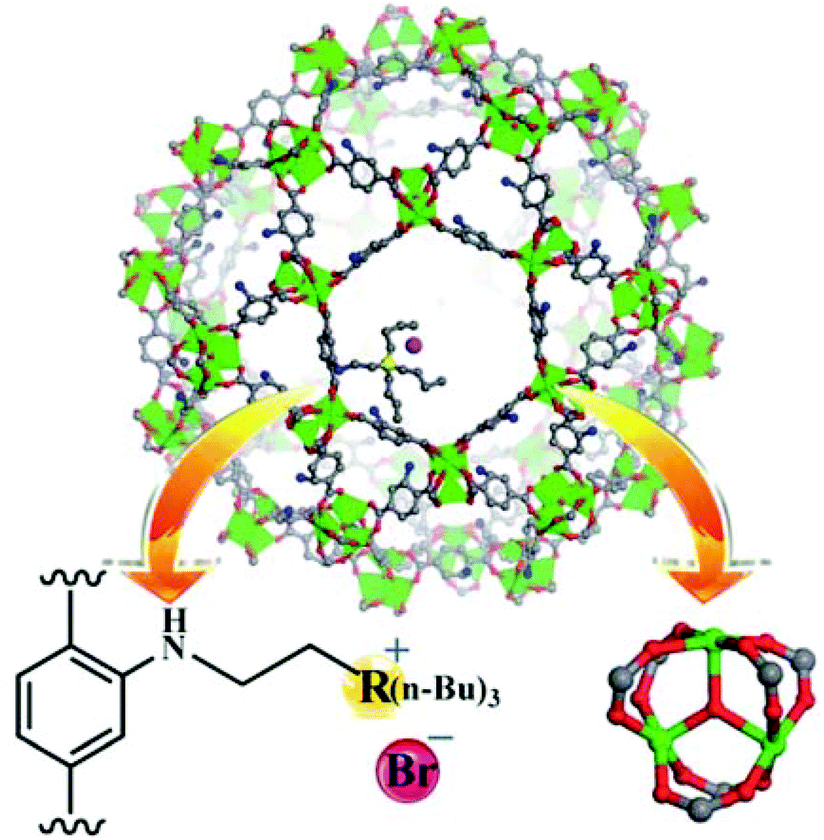 | ||
| Fig. 9 Structure of MIL-101(Cr)-X-(n-Bu)3Br (Cr: green, C: gray, O: red, N: blue, Br: amaranth, and R = N or P). Reproduced from ref. 55 with permission from the Royal Society of Chemistry. | ||
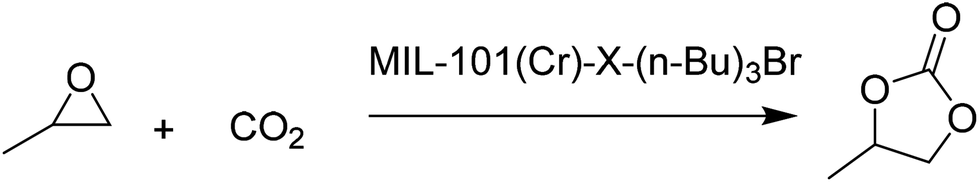 | ||
| Scheme 18 Reaction of propylene oxide with CO2 catalyzed by MIL-101(Cr)-X-(n-Bu)3Br. | ||
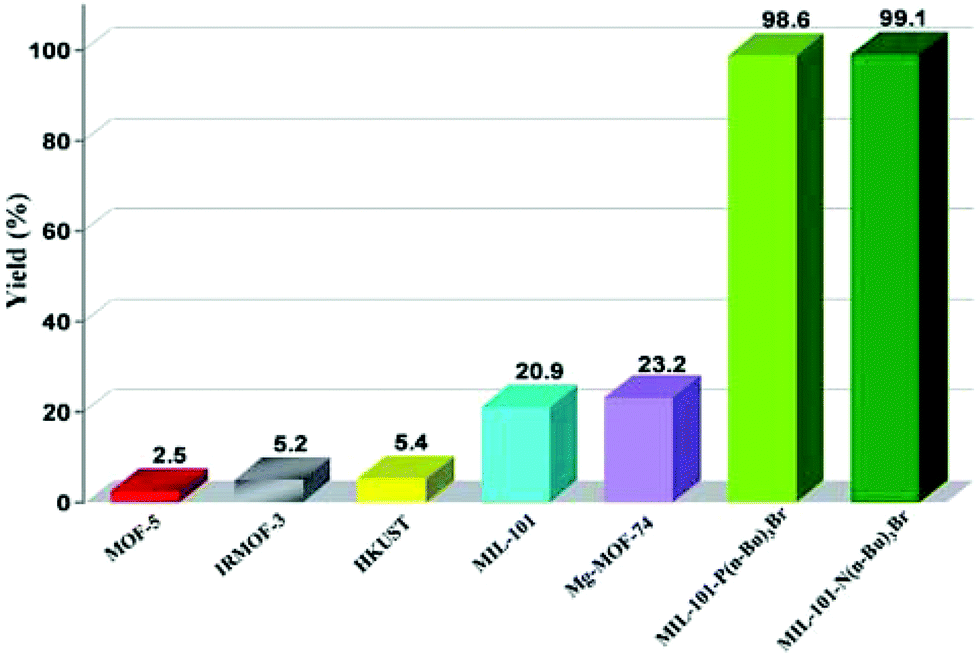 | ||
| Fig. 10 Comparison of PC yield in the cycloaddition CO2 to propylene oxide and catalyzed by bifunctional MIL-101(Cr)-X-(n-Bu)3Br compared to other MOFs. Reproduced from ref. 55 with permission from the Royal Society of Chemistry. | ||
A new imidazolium-based UiO-67 type MOF (UiO-67-IL) was synthesised using ligand functionalization (Fig. 11).56 The catalytic activity of UiO-67-IL was studied in the cycloaddition of CO2 to epichlorohydrin (Scheme 19) with or without TBAB as co-catalyst under 1 atm of CO2. The powder XRD pattern indicates that UiO-67-IL is highly crystalline and its structure is similar to that of pristine UiO-67. The use of UiO-67-IL afforded 99% yield (45.08 h−1 TOF) of the desired product with TBAB as a co-catalyst at 90 °C, while the same catalyst required, under identical conditions, a much longer time to reach 88% yield (13.66 h−1 TOF) without TBAB. On the other hand, no catalytic activity was observed with UiO-67 in the absence of IL even at 90 °C (<3% yield); however, the carbonate yield increased to 8% yield for longer time. In contrast, UiO-67-IL with imidazolium moieties afforded 95% yield under similar conditions. On the other hand, the UiO-67-TBAB catalytic system showed 98% yield, whereas UiO-67-IL gave 99% yield in less than half the time for epichlorohydrin CO2 cycloaddition. These data clearly illustrate the pivotal role played by the embedded imidazolium group in UiO-67-IL catalyzing this reaction. No leaching of active sites was detected under this experimental condition. Also, the catalyst was reused for five catalytic runs. Powder XRD patterns of the fresh and five-times reused catalyst showed that the structural integrity is maintained.
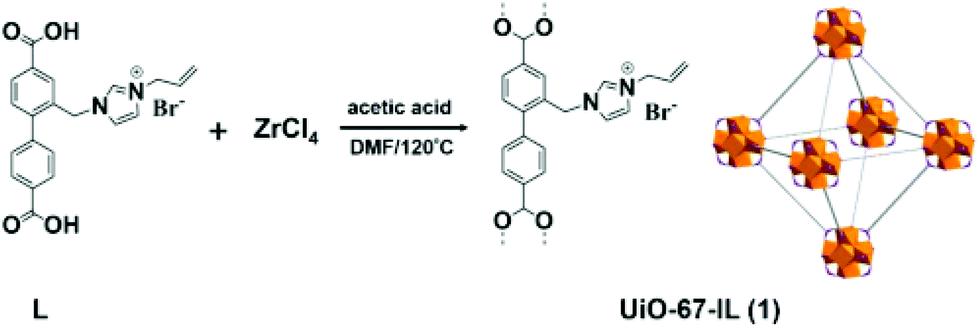 | ||
| Fig. 11 Synthesis of UiO-67-IL by using an IL-functionalized biphenyldicarboxylic acid as linker. Reproduced from ref. 56 with permission from the American Chemical Society. | ||
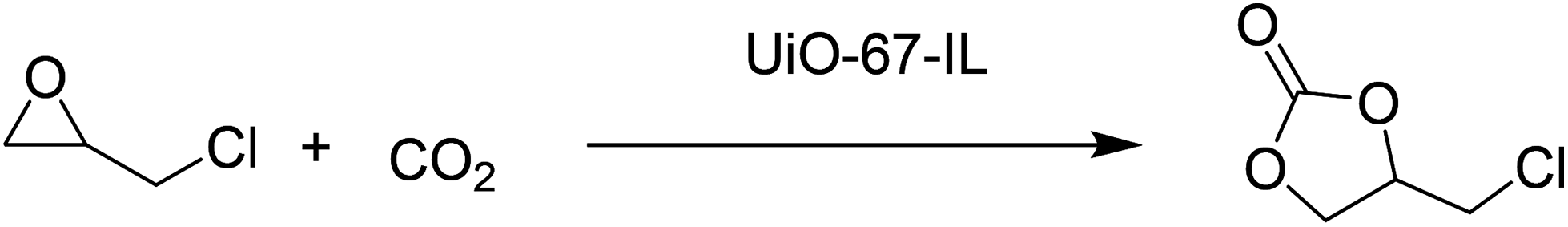 | ||
| Scheme 19 Cycloaddition of CO2 to epichlorohydrin catalyzed by UiO-67-IL. | ||
Recently, the covalent post-functionalization of zeolitic imidazolate framework-90 (ZIF-90) with a pyridinium based IL was performed to obtain IL-supported ZIF-90 (IL-ZIF-90) and its activity was examined in the solvent-free cycloaddition of epoxides with CO2.57 2-Imidazolecarboxaldehyde (ICA) showed 19% conversion while aminopyridinium iodide (AMPyI) afforded 44% conversion with 100% selectivity, whereas the combined physical mixture of all the starting materials (zinc nitrate, ICA and AmPyI) employed for the synthesis of IL-ZIF-90 resulted in slightly higher conversion of 55% under identical conditions. On the other hand, the use of ZIF-90 as a catalyst resulted in 51% conversion which is slightly lower than that of the combination of ZIF-90 and AmPyI (65% conversion). Interestingly, IL-ZIF-90 exhibited 97% conversion with 98% selectivity under identical conditions. These data clearly demonstrate the advantage of post-functionalization in MOFs, enhancing the activity of ILs within the frameworks of MOFs. Among the various epoxides screened, propylene oxide afforded a maximum conversion of 97%, whereas cyclohexene oxide showed 9% conversion under identical conditions. This lower activity was due to the steric hindrance caused by the cyclohexene ring. IL-ZIF-90 was reused in four successive runs without any significant decline in its activity. Powder XRD patterns of the fresh catalyst did not show any noticeable changes in the four-times reused catalyst.
4. Hierarchical porous MOFs for catalysis
In recent years, the development of hierarchically porous MOFs with controlled micro-/mesoporosity has received considerable attention due to their better performance in practical industrial processes such as adsorption, catalysis and chemical sensing.158 Also, achieving hierarchical structures in heterogeneous solids is of great interest as they feature high pore volumes, large surface areas and larger pore sizes. Very recently, a new method, namely “linker labilization”, has been developed to increase the MOF porosity and pore size, thus leading to hierarchical-pore architectures.159 In this method, microporous MOFs were initially synthesized using robust metal nodes and pro-labile linkers. Then, the mesopores were subsequently created as crystal defects through the splitting of a pro-labile-linker followed by the removal of the linker fragments by acid treatment. It was facilely shown that the linker labilization method can create controllable hierarchical porous structures in stable MOFs, which allows the diffusion and adsorption of cytochrome c in an efficient manner.Recently, a facile route has been reported for the preparation of carbon nitride (CN) foams as structural templates with micrometer-sized pores with the nitrogen content of 25.6 wt% by the fast carbonization of melamine foam (Fig. 12). The nitrogen functionalities of CN foam provide the feasibility of chemical anchoring and growth of ZIF-8 crystals, thus leading to the development of hierarchical porous MOFs.160 Furthermore, the growth of ZIF-8 crystals also changes the nature of the CN foam from hydrophilic to highly hydrophobic, thus creating an effective shield against water for the MOF crystals. As a result, the introduction of ZIF-8 crystals onto the CN foam provides selective absorption of oils up to 58 wt% from water/oil mixtures and also facilitates the highly efficient conversion of CO2 to 3-chloropropene carbonate in a quantitative yield with selectivity. In contrast, the use of ZIF-8-based catalysts for this reaction resulted in low product selectivity to 3-chloropropene carbonate (52%) and also produced by-products including diols (23.7%) and a dimer (24.3%).161 Also, ZIF-8 catalysts showed a significant loss in their catalytic activity after the first cycle whereas ZIF-8/CN foam retained high selectivity and conversion up to the fourth cycle. Hence, this approach could be extended to those MOFs that have poor stability in water.
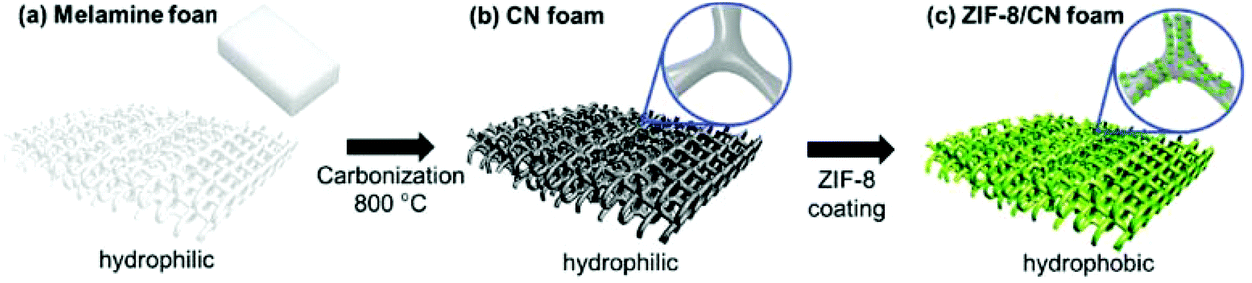 | ||
| Fig. 12 Schematic representation of the synthesis of ZIF-8/CN foam with hierarchical porosity. Reproduced from ref. 160 with permission from Wiley. | ||
The ability of MOFs to gelate under specific experimental conditions provides further opportunities in the preparation and shaping of hierarchically porous MOF monoliths, which could be used in catalysis and adsorption applications. Recently, xero- or aerogel monoliths consisting solely of nanoparticles of several prototypical Zr4+-based MOFs such as UiO-66-X (X = H, NH2, NO2, (OH)2), UiO-67, MOF-801, MOF-808 and NU-1000, have been reported.162 Furthermore, UiO-66 gels were shaped into monolithic spheres of 600 mm diameter using an oil drop method, thus creating different materials which could find applications in packed-bed catalytic or adsorptive applications, where hierarchical pore systems can greatly weaken mass transfer limitations.
A pyridyl-decorated MOF-505 analogue, [Cu2(L)(H2O)2]·Gx (H4L = 5,5′-(pyridine-2,5-diyl)-diisophthalic acid, G: solvent molecule), has been reported with rare hierarchical meso- and microporosity with exposed unsaturated CuII sites and Lewis basic pyridyl sites. The catalytic activity of this catalyst was reported in the cyanosilylation reaction under solvent-free conditions.163 The dehydrated catalyst showed 99% conversion in the cyanosilylation of benzaldehyde at 40 °C. The catalytic activity of this hierarchical MOF is comparable to that of other Cu or Zr-based MOFs72,164,165 in the cyanosilylation of benzaldehyde but is considerably higher than the catalytic acitvity of other catalysts like Cu3(BTC)2 (50–60%),129 Cd-MOF (77%),166 and Ce-MOF (94%, 2 h).167 This enhanced activity is due to the presence of a high density of the exposed metal sites and the improved molecular accessibility of the hierarchical meso- and microporosity. Catalysis was heterogeneous in nature and MOF-505 was recycled up to five times with only a minor loss of activity.
In another precedent, a continuous phase transformation processing strategy has been applied for fabricating and shaping MOFs into shaped bodies and MOF foams that exhibit reversible transformation among these states. In this context, a cup-shaped reactor based on a Cu-MOF composite and hierarchically porous MOF foam were prepared and their activities were compared in the oxidation of diphenylmethane with TBHP as an oxidant.168 The use of a cup-shaped reactor afforded 76% conversion with 93% selectivity, while using porous foam enhances the conversion to 92% with 97% selectivity under identical conditions. Furthermore, quenched solid-state density functional theory calculation revealed that the foam possesses hierarchically distributed porosity with the pore size distribution consisting of both micro- and mesopores expanding up to 40 nm. The hierarchically porous MOF foam was reused for three cycles with no significant decline in its activity.
Recently, a coordination replication strategy has been applied to construct hierarchical Cu-based MOF nanoarrays and their activity has been examined in the reduction of 4-nitrophenol to 4-aminophenol using sodium borohydride as a reducing agent.169 In this approach, Cu(OH)2 nanorods were the source for Cu that coordinated with a series of organic ligands to form different MOF crystals, and they also served as a 3 D substrate to support the growth of the Cu-based MOF (Fig. 13). Hierarchical MOF nanoarrays exhibited enhanced activity in the reduction of 4-nitrophenol compared with those on MOF-2 nanoparticles and Cu(OH)2 nanorod arrays. This is due to the unique 3D hierarchical nanoarray architecture of the nanocomposite. There was no significant loss in activity for eight successive catalytic cycles thus showing the long term stability of hierarchical MOFs.
 | ||
| Fig. 13 Preparation of hierarchical Cu-based MOF nanoarrays by the coordination replication strategy. Reproduced from ref. 169 with permission from Wiley. | ||
Two hierarchically porous MOFs namely, NUS-6 (NUS: National University of Singapore) composed of either zirconium (Zr) or hafnium (Hf) clusters were synthesised using a modulated hydrothermal method with high stability and strong Brønsted acidity.170 These MOFs exhibit BET surface areas of 550 and 530 m2 g−1 for NUS-6(Zr) and NUS-6(Hf), respectively, and a hierarchically porous structure of coexisting micropores (∼0.5, ∼0.7, and ∼1.4 nm) and mesopores (∼4.0 nm) with dangling sulfonic acid groups. Structural analysis showed that the hierarchical porosity of NUS-6 derives from missing linkers and clusters of the parent UiO-66 framework. The activity of these MOFs was tested in the dehydration of fructose to 5-hydroxymethylfurfural (HMF), in which NUS-6(Hf) showed 98% yield which is superior than NUS-6(Zr) (84% yield) after 1 h at 100 °C. This enhanced activity is due to the stronger Brønsted acidity contribution from Hf-μ3-OH groups as well as the availability of smaller pore sizes to restrict undesirable reactions.
The above examples illustrate how the preparation of hierarchical MOFs with controlled micro-/mesoporosity and particle shapes can enhance the catalytic activity of these solids by facilitating the accessibility of substrates and reagents to the active sites. This particle shaping is of great importance in commercial catalysts used in industry, but is an aspect that is frequently ignored in most catalytic studies. Shaping MOF particles with an adequate morphology and ordering is a necessary step to be considered in any potential application of these materials as adsorbents and catalysts.
5. Summary and future outlook
It has been shown that MOFs are suitable solid catalysts also under conditions in which the presence of conventional organic solvents are avoided or when incorporating ILs that possess active sites. This includes not only solvent-free conditions using MOFs, but also other examples where MOFs have been modified by adsorbing ILs, either embedded or attached to the MOF lattice. In the last case these ILs act by not only modifying the polarity of the reaction, but also frequently by adding additional acid/basic sites that cooperate in the reaction mechanism, increasing the efficiency of the catalytic process. At present, the number of studies are still limited, although there are several reaction types that have been sufficiently documented that can occur under these favourable green conditions in the absence of solvents such as condensations, silylations, selective partial hydrogenation of alkynes to alkenes and CO2 insertions. It will be important to expand the use of MOFs under solvent-free conditions to other reaction types, particularly to oxidations and couplings that are processes highly sensitive to the nature of solvent and that can generate high amounts of wastes, trying to delineate when solvents can be avoided and gain understanding of the reasons.One point that has not been, in our opinion, sufficiently addressed is MOF stability as a catalyst under solvent-free conditions. It should be considered that the most robust MOFs have pore sizes of 1 nm or below and structure stability decreases as the pore size increases. Besides the lack of structural stability that is a general issue to be carefully addressed when using MOFs as catalysts, specific points that need to be investigated are how product adsorption affects catalytic activity and what are the causes of deactivation under solvent-free conditions. A general claim in most of the reports presented is the lack of the catalyst deactivation without paying attention to why product inhibition or byproduct formation do not seem to occur. In other words, all the solid catalysts should deactivate upon reuse and solvent-free conditions are specially favourable for poisoning due to the high concentration of products/byproducts and the absence of desorption by solvent molecules. It would be important to establish an in-depth study of the reasons why this deactivation process does not happen and the system always appears stable.
Another important aspect that has been frequently neglected is particle shaping and structuring resulting in the formation of hierarchical MOF catalysts. The presence of porosity combining micro- and mesoporosity is known to facilitate mass transport and this results in an enhanced activity using heterogeneous catalysts. Examples have been presented showing that the flexibility in MOF synthesis allows developing different strategies like linker labilization, gelation or synthesis on foams that have resulted in hierarchical MOFs.
Overall, the present review has shown that MOFs can contribute to the greenness of chemical processes not only by acting as efficient and selective solid catalysts, but also by avoiding the use of, in some cases, volatile solvents without the penalty of fast deactivation. Similarly, decoration or coating of the internal pores of MOFs by IL units has shown that it is possible to combine the effects of molecular ILs with that of a solid catalyst with the minimum amount of IL and with advantages in terms of recovery of IL and workup of the reaction mixture. It has also been shown that the preparation of MOFs exhibiting a hierarchical structuring enhances their catalytic activity by facilitating mass transport. Since MOF catalysis is under intense investigation, it can be predicted that additional efforts will be made to expand the use of MOFs as heterogeneous catalysts using the three strategies commented above to other types of reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AD thanks the University Grants Commission (UGC), New Delhi, for the award of an Assistant Professorship under its Faculty Recharge Programme. AD also thanks the Department of Science and Technology, India, for financial support through Extra Mural Research Funding (EMR/2016/006500). Financial support by the Spanish Ministry of Economy and Competitiveness (Severo Ochoa and CTQ2015-69153-CO2-1) is gratefully acknowledged.References
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- J. H. Clark, R. Luque and A. S. Matharu, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 183–207 CrossRef CAS PubMed.
- R. Cernansky, Nature, 2015, 519, 379–380 CrossRef PubMed.
- K. Sanderson, Nature, 2011, 469, 18–20 CrossRef CAS PubMed.
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed.
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabres i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922–1947 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed.
- L. Zhu, X.-Q. Liu, H. L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856–867 CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. – Eur. J., 2016, 22, 8012–8024 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Opanasenko, J. Cejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed.
- K. Fujie and H. Kitagawa, Coord. Chem. Rev., 2016, 307, 382–390 CrossRef CAS.
- A. Bhunia, S. Dey, J. M. Moreno, U. Diaz, P. Concepcion, K. V. Hecke, C. Janiak and P. Van Der Voort, Chem. Commun., 2016, 52, 1401–1404 RSC.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Ángeles Monge, CrystEngComm, 2013, 15, 9562–9571 RSC.
- L. M. Aguirre-Diaz, M. Iglesias, N. Snejko, E. Gutierrez-Puebla and M. Angeles Monge, RSC Adv., 2015, 5, 7058–7065 RSC.
- L.-J. Zhang, C.-Y. Han, Q.-Q. Dang, Y.-H. Wang and X.-M. Zhang, RSC Adv., 2015, 5, 24293–24298 RSC.
- R. F. D'Vries, M. Iglesias, N. Snejko, E. Gutierrez-Puebla and M. A. Monge, Inorg. Chem., 2012, 51, 11349–11355 CrossRef PubMed.
- W. Jiang, J. Yang, Y.-Y. Liu, S.-Y. Song and J.-F. Ma, Inorg. Chem., 2017, 56, 3036–3043 CrossRef CAS PubMed.
- F. Liu, Y. Xu, L. Zhao, L. Zhang, W. Guo, R. Wang and D. Sun, J. Mater. Chem. A, 2015, 3, 21545–21552 CAS.
- M. Thimmaiah, P. Li, S. Regati, B. L. Chen and J. C. G. Zhao, Tetrahedron Lett., 2012, 53, 4870–4872 CrossRef CAS PubMed.
- S. Rostamnia and A. Morsali, RSC Adv., 2014, 4, 10514–10518 RSC.
- S. Rostamnia and H. Xin, Appl. Organomet. Chem., 2014, 28, 359–363 CrossRef CAS.
- M. Saikia, D. Bhuyan and L. Saikia, Appl. Catal., A, 2015, 505, 501–506 CrossRef CAS.
- S. Beheshti and A. Morsali, RSC Adv., 2014, 4, 37036–37040 RSC.
- N. Anbu and A. Dhakshinamoorthy, J. Colloid Interface Sci., 2017, 494, 282–289 CrossRef PubMed.
- S. Beheshti and A. Morsali, RSC Adv., 2014, 4, 41825–41830 RSC.
- P. Li, S. Regati, H. Huang, H. D. Arman, J. C.-G. Zhao and B. Chen, Inorg. Chem. Front., 2015, 2, 42–46 RSC.
- P. Li, S. Regati, H.-C. Huang, H. D. Arman, B.-L. Chen and J. C.-G. Zhao, Chin. Chem. Lett., 2015, 26, 6–10 CrossRef CAS.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- X. Zhou, Y. Zhang, X. Yang, L. Zhao and G. Wang, J. Mol. Catal. A: Chem., 2012, 361–362, 12–16 CrossRef CAS.
- R. Babu, R. Roshan, A. Cherian Kathalikkattil, D. W. Kim and D.-W. Park, ACS Appl. Mater. Interfaces, 2016, 8, 33723–33731 CAS.
- Q. X. Luo, X.-D. Song, M. Ji, S.-E. Park, C. Hao and Y.-Q. Li, Appl. Catal., A, 2014, 478, 81–90 CrossRef CAS.
- Q.-X. Luo, M. Ji, S.-E. Park, C. Hao and Y.-Q. Li, RSC Adv., 2016, 6, 33048–33054 RSC.
- J. Wu, Y. Gao, W. Zhang, Y. Tan, A. Tang, Y. Men and B. Tang, Appl. Organomet. Chem., 2015, 29, 96–100 CrossRef CAS.
- S. Abednatanzi, K. Leus, P. G. Derakhshandeh, F. Nahra, K. De Keukeleere, K. Van Hecke, I. Van Driessche, A. Abbasi, S. P. Nolan and P. Van Der Voort, Catal. Sci. Technol., 2017, 7, 1478–1487 CAS.
- S. Abednatanzi, A. Abbasi and M. Masteri-Farahani, Catal. Commun., 2017, 96, 6–10 CrossRef CAS.
- Z. Wu, C. Chen, H. Wan, L. Wang, Z. Li, B. Li, Q. Guo and G. Guan, Energy Fuels, 2016, 30, 10739–10746 CrossRef CAS.
- H. Wan, C. Chen, Z. Wu, Y. Que, Y. Feng, W. Wang, L. Wang, G. Guan and X. Liu, ChemCatChem, 2015, 7, 441–449 CrossRef CAS.
- H. M. A. Hassan, M. A. Betiha, S. K. Mohamed, E. A. El-Sharkawy and E. A. Ahmed, J. Mol. Liq., 2017, 236, 385–394 CrossRef CAS.
- Q.-X. Luo, M. Ji, M.-H. Lu, C. Hao, J.-S. Qiu and Y.-Q. Li, J. Mater. Chem. A, 2013, 1, 6530–6534 CAS.
- L. Peng, J. Zhang, S. Yang, B. Han, X. Sang, C. Liu and G. Yang, Green Chem., 2015, 17, 4178–4182 RSC.
- A. Paul, A. P. C. Ribeiro, A. Karmakar, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2016, 12779–12789 RSC.
- Y.-H. Hu, J.-C. Wang, S. Yang, Y.-A. Li and Y.-B. Dong, Inorg. Chem., 2017, 56, 8341–8347 CrossRef CAS PubMed.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 CAS.
- L.-G. Ding, B.-J. Yao, W.-L. Jiang, J.-T. Li, Q.-J. Fu, Y.-A. Li, Z.-H. Liu, J.-P. Ma and Y.-B. Dong, Inorg. Chem., 2017, 56, 2337–2344 CrossRef CAS PubMed.
- J. Tharun, K.-M. Bhin, R. Roshan, D. W. Kim, A. C. Kathalikkattil, R. Babu, H. Y. Ahn, Y. S. Won and D.-W. Park, Green Chem., 2016, 18, 2479–2487 RSC.
- B. Y. Park, K. Y. Ryu, J. H. Park and S. G. Lee, Green Chem., 2009, 11, 946–948 RSC.
- Y. Ogasawara, S. Uchida, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2009, 15, 4343–4349 CrossRef CAS PubMed.
- M. North, D. L. Usanov and C. Young, Chem. Rev., 2008, 108, 5146–5226 CrossRef CAS PubMed.
- J.-M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed.
- D. A. Evans, L. K. Truesdale and G. L. Carroll, J. Chem. Soc., Chem. Commun., 1973, 55–56 RSC.
- R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS PubMed.
- V. Chechik, M. Conte, T. Dransfield, M. North and M. OmedesPujol, Chem. Commun., 2010, 46, 3372–3374 RSC.
- Y. N. Belokon, M. North and T. Parsons, Org. Lett., 2000, 2, 1617–1619 CrossRef CAS PubMed.
- W. Xi, Y. Liu, Q. Xia, Z. Li and Y. Cui, Chem. – Eur. J., 2015, 21, 12581–12585 CrossRef CAS PubMed.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321–14323 CrossRef CAS PubMed.
- S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854–5855 CrossRef CAS PubMed.
- R. F. D'Vries, V. A. de la Peña-O'Shea, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. A. Monge, J. Am. Chem. Soc., 2013, 135, 5782–5792 CrossRef PubMed.
- M. Gustafsson, A. Bartoszewicz, B. Martín-Matute, J. Sun, J. Grins, T. Zhao, Z. Li, G. Zhu and X. Zou, Chem. Mater., 2010, 22, 3316–3322 CrossRef CAS.
- F. Gandara, B. Gomez-Lor, M. Iglesias, N. Snejko, E. Gutierrez-Puebla and A. Monge, Chem. Commun., 2009, 2393–2395 RSC.
- Y. Zhu, Y. M. Wang, S. Y. Zhao, P. Liu, C. Wei, Y. L. Wu, C. K. Xia and J. M. Xie, Inorg. Chem., 2014, 53, 7692–7699 CrossRef CAS PubMed.
- H. F. Yao, Y. Yang, H. Liu, F. G. Xi and E. Q. Gao, J. Mol. Catal. A: Chem., 2014, 394, 57–65 CrossRef CAS.
- G. Rajagopal, S. Selvaraj and K. Dhahagani, Tetrahedron: Asymmetry, 2010, 21, 2265–2270 CrossRef CAS.
- S. A. Pourmousavi and H. Salahshornia, Bull. Korean Chem. Soc., 2011, 32, 1575–1578 CrossRef CAS.
- R. El Osta, A. Carlin-Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. de Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS.
- M. Maes, F. Vermoortele, L. Alaerts, S. Couck, C. E. A. Kirschhock, J. F. M. Denayer and D. E. De Vos, J. Am. Chem. Soc., 2010, 132, 15277–15285 CrossRef CAS PubMed.
- A. Alizadeh and S. Rostamnia, Synthesis, 2010, 4057–4060 CrossRef CAS.
- K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A. Hedberg and B. C. Reilly, J. Med. Chem., 1991, 34, 806–811 CrossRef CAS PubMed.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS PubMed.
- R. A. Janis and D. J. Triggle, J. Med. Chem., 1983, 26, 775–785 CrossRef CAS PubMed.
- L. Martins, K. M. Vieria, L. M. Rios and D. Cardoso, Catal. Today, 2008, 133–135, 706–710 CrossRef CAS.
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed.
- X. Zhang, Z. Zhang, J. Boissonnault and S. M. Cohen, Chem. Commun., 2016, 52, 8585–8588 RSC.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, p. 1 Search PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC.
- Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC.
- K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- S. Lober, P. Rodriguez-Loaiza and P. Gmeiner, Org. Lett., 2003, 5, 1753–1755 CrossRef PubMed.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- R. Alvarez, S. Velazque, F. San, S. Aquaro, C. De, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS PubMed.
- V. K. Y. Lo, Y. G. Liu, M. K. Wong and C. M. Che, Org. Lett., 2006, 8, 1529–1532 CrossRef CAS PubMed.
- I. Matsuda, J. Sakakibara and H. Nagashima, Tetrahedron Lett., 1991, 32, 7431–7434 CrossRef CAS.
- C. P. Sar, T. Kalai, J. Jeko and K. Hideg, ARKIVOC, 2012, 47–59 CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, J. Am. Chem. Soc., 2005, 127, 13118–13119 CrossRef CAS PubMed.
- D. C. Stoian, E. Taboada, J. Llorca, E. Molins, F. Medina and A. M. Segarra, Chem. Commun., 2013, 49, 5489–5491 RSC.
- T. Jiang, X. Ma, Y. Zhou, S. Liang, J. Zhang and B. Han, Green Chem., 2008, 10, 465–469 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 CAS.
- Y. J. Kim and R. S. Varma, J. Org. Chem., 2005, 70, 7882–7891 CrossRef CAS PubMed.
- V. Calo, A. Nacci, A. Monopoli, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef CAS PubMed.
- Y. Xie, T. T. Wang, R. X. Yang, N. Y. Huang, K. Zou and W. Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- C. G. Li, L. Xu, P. Wu, H. Wu and M. He, Chem. Commun., 2014, 50, 15764–15767 RSC.
- Y. Zhang, S. Yin, S. Luo and C. T. Au, Ind. Eng. Chem. Res., 2012, 51, 3951–3957 CrossRef CAS.
- J. Q. Wang, D. L. Long, J. Y. Chen, F. Cai and L. N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS.
- W. Kleist, F. Jutx, M. Maciejewski and A. Baiker, Eur. J. Inorg. Chem., 2009, 3552–3561 CrossRef CAS.
- T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129–1130 RSC.
- H. Yasuda, L.-N. He and T. Sakakura, J. Catal., 2002, 209, 547–550 CrossRef CAS.
- Y. B. Xiong, H. Wang, R. M. Wang, Y. F. Yan, B. Zheng and Y. P. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC.
- W. L. Dai, L. Chen, S. F. Yin, W. H. Li, Y. Y. Zhang, S. L. Luo and C. T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
- C. R. Qi, J. W. Ye, W. Zeng and H. F. Jiang, Adv. Synth. Catal., 2010, 352, 1925–1933 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40, 1005–1013 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Nature, 1999, 399, 28–29 CrossRef.
- S. Luo, X. Mi, L. Zhang, S. Liu, H. Xu and J.-P. Cheng, Angew. Chem., Int. Ed., 2006, 45, 3093–3097 CrossRef CAS PubMed.
- K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Green Chem., 2014, 16, 3508–3514 RSC.
- W. Wu, B. Han, H. Gao, Z. Liu, T. Jiang and J. Huang, Angew. Chem., Int. Ed., 2004, 43, 2415–2417 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- J. H. Clark and S. J. Tavener, Org. Process Res. Dev., 2007, 11, 149–155 CrossRef CAS.
- R. V. Hangarge, D. V. Jarikote and M. S. Shingare, Green Chem., 2002, 4, 266–268 RSC.
- D.-Z. Xu, Y. Liu, S. Shi and Y. Wang, Green Chem., 2010, 12, 514–517 RSC.
- A. Dhakshinamoorthy, M. Alvaro, P. Concepcion and H. Garcia, Catal. Commun., 2011, 12, 1018–1021 CrossRef CAS.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81–88 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- T. K. Shing, Y.-Y. Yeung and P. L. Su, Org. Lett., 2006, 8, 3149–3151 CrossRef CAS PubMed.
- D. H. Barton, H. Patin and F. Launay, New J. Chem., 1998, 22, 559–563 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- P. Yin, L. Chen, Z. Wang, R. J. Qu, X. G. Liu, Q. Xu and S. H. Ren, Fuel, 2012, 102, 499–505 CrossRef CAS.
- C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, J. L. D. Macedo, J. A. Dias, S. C. L. Dias and K. S. P. Alvim, Appl. Catal., A, 2010, 372, 153–161 CrossRef CAS.
- A. H. M. Fauzi, N. A. S. Amin and R. Mat, Appl. Energy, 2014, 114, 809–818 CrossRef.
- C. Chen, Z. W. Wu, Y. G. Que, B. X. Li, Q. R. Guo, Z. Li, L. Wang, H. Wan and G. F. Guan, RSC Adv., 2016, 6, 54119–54128 RSC.
- A. H. M. Fauzi and N. A. S. Amin, Energy Convers. Manage., 2013, 76, 818–827 CrossRef.
- D. Lu, J. W. Zhao, Y. Leng, P. P. Jiang and C. J. Zhang, Catal. Commun., 2016, 83, 27–30 CrossRef CAS.
- L. Zhang, Y. Cui, C. Zhang, L. Wang, H. Wan and G. Guan, Ind. Eng. Chem. Res., 2012, 51, 16590–16596 CrossRef CAS.
- Y. Jiang, J. Lu, K. Sun, L. Ma and J. Ding, Energy Convers. Manage., 2013, 76, 980–985 CrossRef CAS.
- Q. Wu, H. Wan, H. Li, H. Song and T. Chu, Catal. Today, 2013, 200, 74–79 CrossRef CAS.
- A. H. Mohammad Fauzi and A. N. A. Saidina, Energy Convers. Manage., 2013, 76, 818–827 CrossRef CAS.
- B. Zhen, H. Li, Q. Jiao, Y. Li, Q. Wu and Y. Zhang, Ind. Eng. Chem. Res., 2012, 51, 10374–10380 CrossRef CAS.
- H. Zhang, F. Xu, X. Zhou, G. Zhang and C. Wang, Green Chem., 2007, 9, 1208–1211 RSC.
- M. C. Ferreira, A. J. Meirelles and E. A. Batista, Ind. Eng. Chem. Res., 2013, 52, 2336–2351 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- L. Sun, J. Li, J. Park and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 126–129 CrossRef CAS PubMed.
- D. Deng, Y. Yang, Y. Gong, Y. Li, X. Xu and Y. Wang, Green Chem., 2013, 15, 2525–2531 RSC.
- Y. Yabe, T. Yamada, S. Nagata, Y. Sawama, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2012, 354, 1264–1268 CrossRef CAS.
- W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 CrossRef CAS.
- H. Sajiki, S. Mori, T. Ohkubo, T. Ikawa, A. Kume, T. Maegawa and Y. Monguchi, Chem. – Eur. J., 2008, 14, 5109–5111 CrossRef CAS PubMed.
- H. Li, M. O'Keeffe, O. M. Yaghi and M. Eddaoudi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- L. Alaerts, E. Seguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Ferey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- S. Gadipelli, J. Ford, W. Zhou, H. Wu, T. J. Udovicn and T. Yildirim, Chem. – Eur. J., 2011, 17, 6043–6047 CrossRef CAS PubMed.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- S. Yuan, L. Zou, J.-S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme, T. Cagin and H.-C. Zhou, Nat. Commun., 2017, 8, 15356–15365 CrossRef CAS PubMed.
- D. Kim, D. W. Kim, O. Buyukcakir, M.-K. Kim, K. Polychronopoulou and A. Coskun, Adv. Funct. Mater., 2017, 27, 1700706–1700713 CrossRef.
- C. M. Miralda, E. E. Macias, M. Q. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- B. Bueken, N. V. Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. M. Denayer, R. Ameloot, S. Bals, D. D. Vos and T. D. Bennett, Chem. Sci., 2017, 8, 3939–3948 RSC.
- Q.-Q. Dang, Y.-F. Zhan, L.-N. Duan and X.-M. Zhang, Dalton Trans., 2015, 20027–20031 RSC.
- J. Yang, X. Wang, F. Dai, L. Zhang, R. Wang and D. Sun, Inorg. Chem., 2014, 53, 10649–10653 CrossRef CAS PubMed.
- R. Wang, Z. Wang, Y. Xu, F. Dai, L. Zhang and D. Sun, Inorg. Chem., 2014, 53, 7086–7088 CrossRef CAS PubMed.
- M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS.
- A. Karmakar, G. M. D. M. Rúbio, A. Paul, M. F. C. Guedes da Silva, K. T. Mahmudov, F. I. Guseinov, S. A. C. Carabineiro and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 8649–8657 RSC.
- Y. Chen, X. Huang, S. Zhang, S. Li, S. Cao, X. Pei, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2016, 138, 10810–10813 CrossRef CAS PubMed.
- T. Zhang, W. Liu, G. Meng, Q. Yang, X. Sun and J. Liu, ChemCatChem, 2017, 9, 1771–1775 CrossRef CAS.
- Z. Hu, Y. Peng, Y. Gao, Y. Qian, S. Ying, D. Yuan, S. Horike, N. Ogiwara, R. Babarao, Y. Wang, N. Yan and D. Zhao, Chem. Mater., 2016, 28, 2659–2667 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |