
The reactions of dimethyl carbonate and its derivatives
P.
Tundo
*,
M.
Musolino
 and
F.
Aricò
and
F.
Aricò
Department of Environmental Science, Informatics and Statistics, Ca’ Foscari University, Scientific Campus Via Torino 155, 30172 Venezia Mestre, Italy. E-mail: tundop@unive.it
First published on 30th October 2017
Abstract
The worldwide urge to embrace a sustainable and bio-compatible chemistry has led industry and academia to develop of chlorine-free methodologies focused on the use of CO2 and CO2-based compounds as feedstocks, promoters and reaction media. In this scenario, dialkyl carbonates (DACs) and in particular dimethyl carbonate (DMC) occupy a privileged position due to their low toxicity, high biodegradability and peculiar reactivity. Nowadays, the large-scale production of DACs is carried out through clean processes (i.e., phosgene-free processes), which include the direct insertion of CO2 into epoxides, allowing – in principle – recycling of the carbon dioxide emitted during carbonate degradation. This groundbreaking achievement has definitely drawn attention toward the conception of procedures to activate the rather stable DACs with the aim of employing these compounds as green alternatives to their reactive chlorinated analogues. DACs are ambident electrophiles, which under appropriate conditions can undergo BAc2- or BAl2-nucleophilic substitution to give, respectively, alkoxycarbonylation and alkylation reactions. The many efforts devoted to improving the industrial suitability of organic carbonates have unveiled an intriguing and innovative chemistry as demonstrated by the numerous publications and patents published on these compounds over the last thirty years. This review reports on DACs as alkoxycarbonylating agents and their applications in industry and fine synthesis, as well as alkylating agents including allylic alkylation using palladium catalysts and the Pd/Ti bimetallic system and anchimerically driven alkylations via mustard carbonates. Moreover, the reactivity of organic carbonates toward several substrates and under different reaction conditions is described along with some distinctive DAC-mediated cyclization and transposition reactions. The synthesis of olefins and ethers under both liquid and gas phase conditions via thermal decarboxylation of organic carbonates is also reported.
 P. Tundo | Pietro R. Tundo is Professor of Organic Chemistry (Torino, Messina and Venice Universities), retired from 2016. He was a guest researcher at College Station (Texas, 1979–1981), Potsdam (New York, 1989–90) and Syracuse (New York, 1991–92). He is a member of the Bureau of IUPAC and Chair of the IUPAC Committee on Green Chemistry. He is dedicated to the introduction and dissemination of Green Chemistry. The author of about 350 scientific publications, 40 patents and many books, his basic organic scientific chemistry research interests include: selective methylations, continuous flow chemistry, chemical detoxification of contaminants, hydrodehalogenation under multiphase conditions, phase-transfer catalysis, supramolecular chemistry, safe alternatives to harmful chemicals and green chemistry for cultural heritage. |
 M. Musolino | M. Musolino. After graduating with a degree in organic chemistry at the University of Milan in 2006, Manuele Musolino worked as a medicinal chemist in both academia (University of Milan) and industry (Sanofi-Aventis Spa) for six years as a research assistant. In 2012, he was awarded a Scottish Universities Life Sciences Alliance (SULSA) 4 year PhD Fellowship in Medical Sciences. He moved to Scotland where, in 2017, he obtained his PhD at the University of Aberdeen. Since March 2017 he has been working as a research fellow in the Green Chemistry group of Professor Tundo and Professor Aricò at Ca’ Foscari University of Venice. |
 F. Aricò | Fabio Aricò obtained his master's degree in chemistry from the University of Messina in 1999. He then moved to the UK, where was awarded a Ph.D. at Reading University; his work focused on macrocycle–polymer interconversion. He then spent two years as a postdoctoral fellow in Sir Fraser Stoddart's group at the UCLA (USA) working on supramolecular systems. In 2005 he moved back to Italy, where he joined the Interuniversity Consortium of Chemistry for the Environment collaborating with the University Ca’ Foscari. Since 2016 he has been an associate professor at Ca’ Foscari University (Venezia); his main research interests are green chemistry and new synthetic pathways. |
1. Introduction
In the last few decades, governments have imposed increasingly stringent environmental regulations upon the chemical industry, aiming at reducing waste generation and energy requirement. Therefore, companies’ priority has become the conservation of high process performances by means of eco-compatible methodologies employing non-toxic and biodegradable chemicals and the chemistry of organic carbonates, at the time only marginally exploited, seemed, in this prospect, very appealing. In fact, dialkyl carbonates (DACs) represent a safe and efficient alternative to chlorine (halogen) reagents,1 inasmuch as they can replace toxic: (1) methyl and acyl halides – as well as dimethyl sulfate – in alkylation and carbonylation reactions;2 (2) phosgene in the synthesis of urethanes, carbamates and isocyanates;3 (3) dichloromethane and chloroform as solvents. Furthermore, both chlorine and carbonate moieties are sacrificial groups (i.e. leaving groups) in nucleophilic displacements; however, the latter do not produce salts and harmful wastes to be disposed of and display low toxicity and high biodegradability, rendering organic carbonates green solvents and reagents (Scheme 1).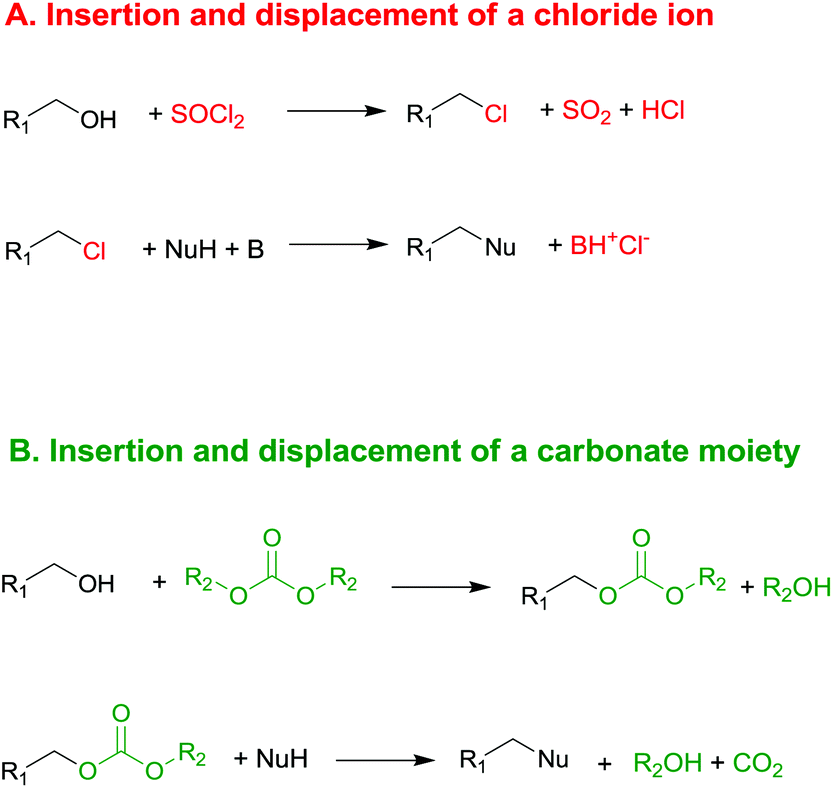 | ||
| Scheme 1 Examples of chlorine and carbonate moieties as sacrificial molecules: a similar synthetic pathway, and opposite environmental outcome. | ||
Currently, DACs and in particular dimethyl carbonate (DMC) are synthesised through clean processes (Scheme 2, eqn (2) and (3)); as a result these compounds have become particularly appealing for chemical companies as is shown by the increasing number of industrial applications (e.g. in the synthesis of herbicides and fungicides, pharmaceuticals, fragrances, varnishes, adhesives and additive for bio-fuels)4 and in turn of patents registered.5 It should be mentioned that DMC was initially produced by reaction of phosgene with methanol – acting as a nucleophile – under basic conditions (Scheme 2, eqn (1)),6 resulting in a considerable amount of NaCl dissolved in water to deal with. In the mid-80s, Enichem7 and UBE8 eventually developed a cleaner approach consisting of the catalytic oxidative carbonylation of methanol with oxygen (Scheme 2, eqn (2)), which de facto paved the way for a green industrial scale production of DMC. Nowadays DMC is mainly produced by insertion of CO2 into oxyrane to give ethylene carbonate, which generates DMC and ethylene glycol by reaction with methanol under basic conditions (Scheme 2, eqn (3)).9
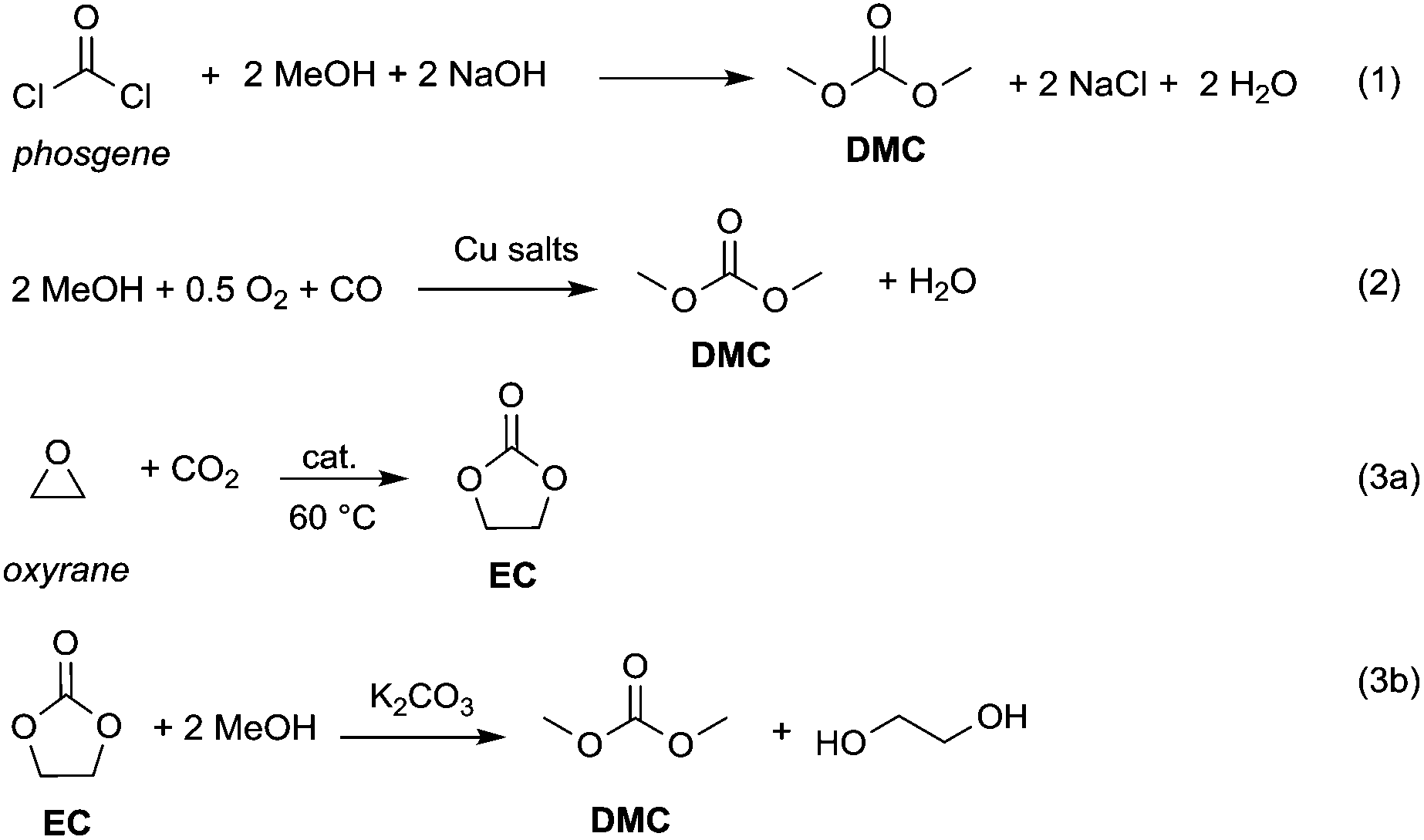 | ||
| Scheme 2 Syntheses of DMC. Via phosgene (eqn (1)); by catalytic oxidative carbonylation of methanol (eqn (2)); by CO2 insertion into oxyrane (eqn (3)).6–9 | ||
One major advantage of DMC is its peculiar selective behaviour that follows the Hard–Soft Acid–Base (HSBA) theory introduced by Pearson in 1963 and further developed by Mèndez.10 In fact, according to the reaction conditions and their nature, nucleophiles can discriminate between the electrophilic sites of DMC and undergo a methoxycarbonylation reaction via a BAc2 mechanism or a methylation reaction via a BAl2 mechanism (Fig. 1). The two methyl groups represent the softer electrophilic sites of the molecule as their saturated sp3-hybridised carbon atoms exhibit a weaker positive charge.
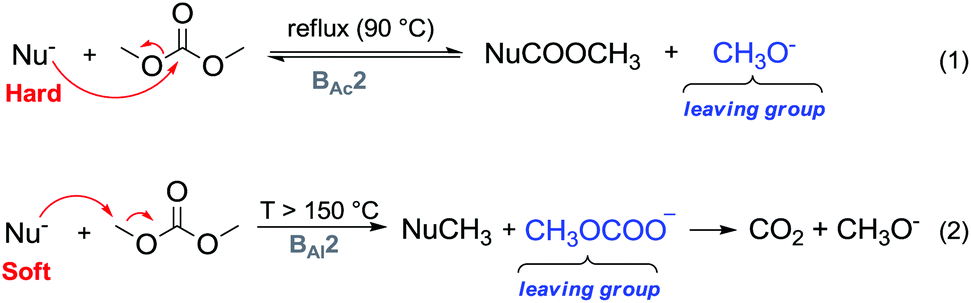 | ||
| Fig. 1 DMC reactivity according to the HSAB theory.10 | ||
Another appealing feature of DAC is that it does not produce inorganic salts in either acylation (Fig. 1, eqn (1)) or alkylation (Fig. 1, eqn (2)) reactions.
In a methylation reaction via DMC chemistry, the leaving group, i.e., the methyl carbonate – once formed – rapidly decomposes into methanol and methoxide anions, restoring the base that can then be used in truly catalytic amounts. The methyl carbonate anion is the conjugated base of methylcarbonic acid, which is a strong acid (pKa = 1.7) comparable to glycine (pKa = 1.99), maleic acid (pKa1 = 1.99) and HSO4− (pKa2 = 1.99).11 Its basicity renders it a better leaving group than the acetate anion (acetic acid pKa = 4.76) and as a consequence, DMC is a stronger electrophile than methyl acetate.
In this perspective, DMC-based chemistry allows also the utilization of continuous-flow (c-f) procedures, i.e., gas–liquid phase-transfer catalysis (GL-PTC), and a continuously stirred tank reactor (CSTR). As depicted in Fig. 1, methoxycarbonylation is an equilibrium reaction, while methylation is not; thus the product of the continuous process can be easily controlled by opportunely tuning the temperature. Moreover, it has been observed that also operating at 200–250 °C (mainly for methylation/alkylation reactions), decomposition and polymerization products or tars are not formed and usually clear reaction mixtures are obtained.
It should be pointed out that the HSAB theory is not exhaustive in explaining all the facets of DAC reactivity and further factors – such as the strong entropic effects occurring in DAC-assisted cyclisation reactions (see below) or the eventual anchimeric effect – (ref. 12) may drive the reaction mechanisms.
On the other hand, the major drawback of the use of DACs is the necessity to provide a high energy input to activate them toward both alkoxycarbonylation and alkylation, whereas chlorine reagents are very reactive and their inherent energy is directly transferred to intermediates, which easily leads to the desired compounds. Nevertheless, the challenge to activate DACs has revealed over the years their unexpected reactivity toward different substrates. The selectivity toward the mono-methylation of the acidic methylene group, the efficiency in promoting cyclisation and transposition reactions, the comparable reactivity of non-toxic carbonate analogues of mustards – both sulfur and nitrogen derivatives – and their parent halogenated compounds13 and the feasibility of producing new polymers from bio-feedstocks are only few examples of the versatility and effectiveness of DAC chemistry.
1.1 Rationale
This review is a comprehensive overview of all the main research fields of DAC chemistry including:1. Alkoxycarbonylation of α-carbons;
2. Alkylation reactions including peculiar cases such as the metal catalysed allylic alkylation and anchimerically driven alkylation reaction via mustard carbonate;
3. Cyclisation, which due to its two-step mechanism may be considered borderline between alkoxycarbonylation and alkylation;
4. Trasposition reactions;
5. Pyrolysis and decarboxylation.
O- and N-alkoxycarbonylation14 and the use of DACs for polymeric synthesis15 and as solvents are not addressed herein as they have already been extensively reported in the literature.
2. Alkoxycarbonylation of α-carbons
2.1 Reaction with ketones and aldehydes
Ketones and aldehydes – protected as acetals – react with DACs under basic conditions through a BAc2 alkoxycarbonylation. Strong bases such as NaH,16 BuLi,17 alkoxides18 and sodium amide19 are generally required to extract an acidic proton from the methylene at α position to the carbonyl function.Weaker bases, such as K2CO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and MgO,21 have also been reported to be suitable catalysts for the alkoxycarbonylation; however, they are efficient only at higher temperature (≥200 °C). Thus, the so formed enolate – acting as a nucleophile – can react with carbonates via Claisen condensation to give a β-ketoester and an alcohol (Scheme 3, eqn (4)).
20 and MgO,21 have also been reported to be suitable catalysts for the alkoxycarbonylation; however, they are efficient only at higher temperature (≥200 °C). Thus, the so formed enolate – acting as a nucleophile – can react with carbonates via Claisen condensation to give a β-ketoester and an alcohol (Scheme 3, eqn (4)).
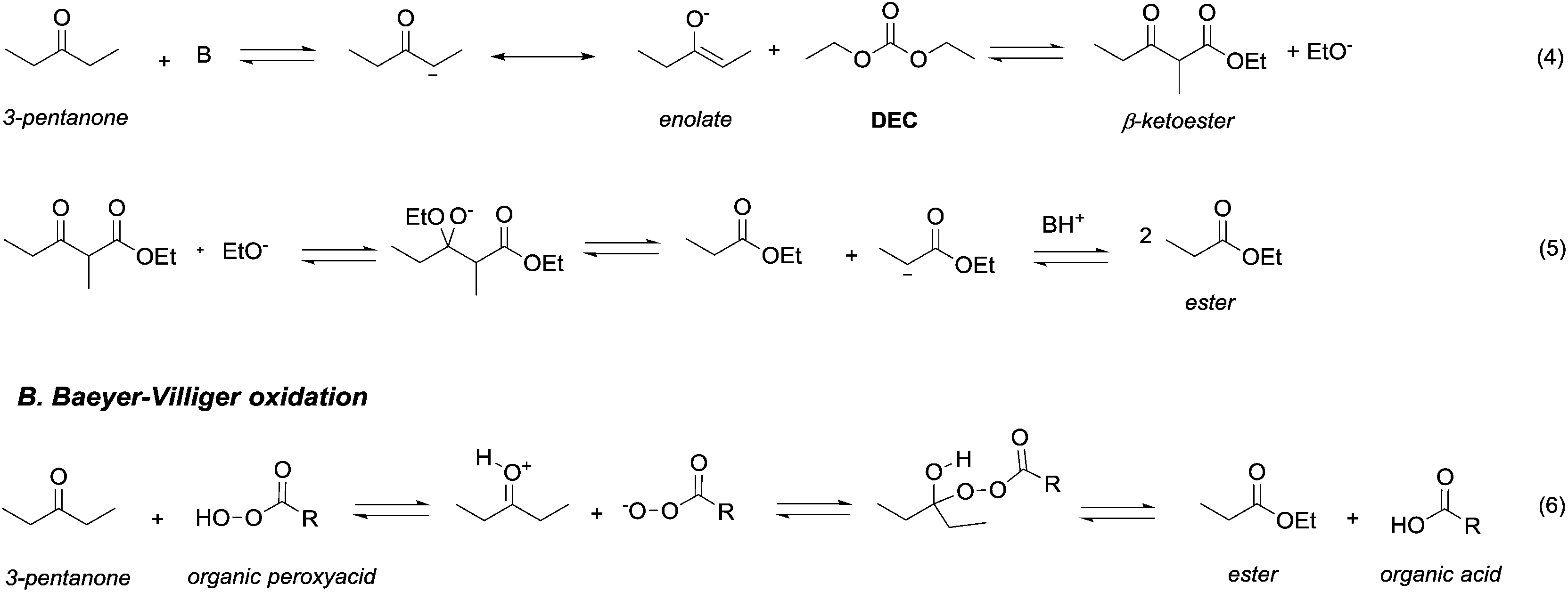 | ||
| Scheme 3 Oxidation of 2-propanone via retro-Claisen condensation with DEC (eqn (4) and (5); A) and via Baeyer–Villiger oxidation with a peroxyacid (eqn (6); B). | ||
Anhydrous reaction conditions, controlled reaction temperature (≤100 °C) and alcohol removal (e.g. by distillation) may increase the process yield of this step and limit the formation of by-products. In fact, in the presence of alcoholates in the reaction mixture, β-ketoester may undergo retro-Claisen carbon–carbon cleavage (Scheme 3, eqn (5))20 and produce two ester derivatives. This tandem Claisen and retro-Claisen condensation can be compared to the Baeyer–Villiger oxidation of ketones by means of organic peroxyacids (Scheme 3, eqn (6)). In this reaction, DACs may be considered as organic oxidants, inasmuch as a hydrogen replacement by a methoxycarbonyl group results in a carbon having a higher oxidation state.22 Tandem Claisen and retro-Claisen reactions have been investigated to produce methyl propionate (MP) starting from 3-pentanone and DMC. This ester is extensively used as solvent for paints and in fragrances and flavouring for its characteristic fruity odour, and due to its applications, the development of an efficient and green industrial synthesis may be rather attractive. The reaction was performed using different solid and homogeneous bases and heating to 260 °C; however, as shown in Table 1,21,23 it had poor to moderate conversion percentages and led to the formation of elimination and polycondensation by-products. The best results of this series were 41% conversion and 51% selectivity toward MP and only 11% toward the intermediate methyl 2-methyl-3-oxopentanoate (MO). The optimised reaction conditions were the use of MgO as a solid base, 6 equivalents of DMC and heating to 260 °C for 5 h.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | Cat. | T (°C) | t (h) | Conv. (%) | Product selectivityc (%) | ||
| MO | MP | Bpsd | |||||
| a 3-Pentanone/DMC 1.0/6.0 (molar ratio); catalyst wt% = 1.5%; reaction performed by heating in an autoclave. b The amount of catalyst equalled the amount of basic sites of MgO. c Determined by GC-MS analysis. d Bps: major by-products = 5-ethyl-4-methyl-4hepten-3-one, 2-methyl-3-pentanone, 3-methoxy-2-pentene.21,23 | |||||||
| 1a | MgO | 260 | 5 | 41 | 11 | 51 | 38 |
| 2b | NaOH | 260 | 5 | 46 | 17 | 43 | 40 |
| 3b | NaOCH3 | 260 | 5 | 48 | 24 | 38 | 28 |
| 4a | CaO | 200 | 5 | 30 | 0 | 45 | 55 |
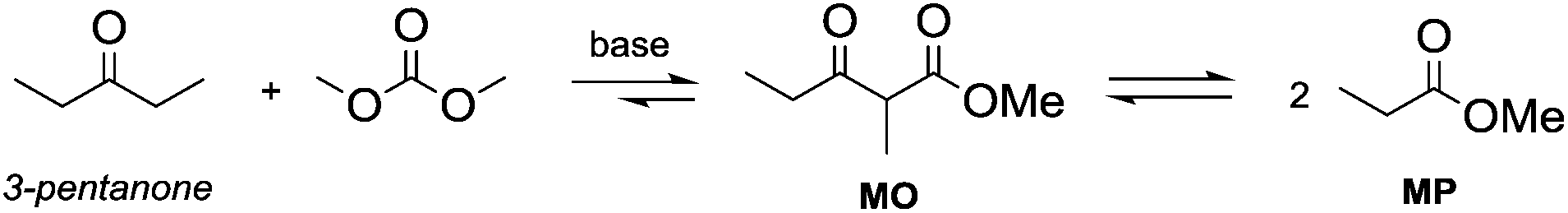
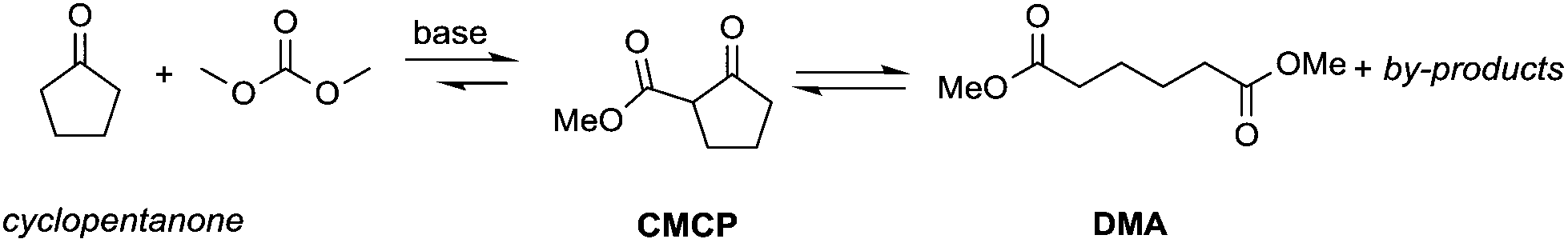
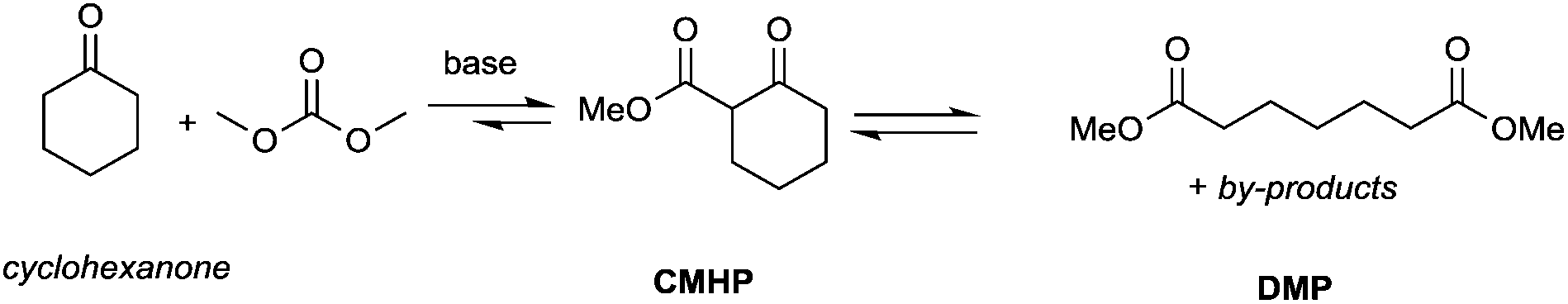
Particularly interesting is the methoxycarbonylation of cyclopentanone and cyclohexanone in the presence of weak bases. In fact, both cyclic aliphatic ketones yield linear diesters, namely adipic and pimelic methyl esters, by a reaction with DMC over K2CO3 and according to the retro-Claisen mechanism depicted in Scheme 4.20,24 The major industrial application of these esters may be their use as building blocks for an eco-friendly production of polyesters and polyamides like nylon 6,6 and nylon 7,7.25 In this perspective, retro-Claisen reaction conditions were optimised and the main results are shown in Tables 2 and 3.
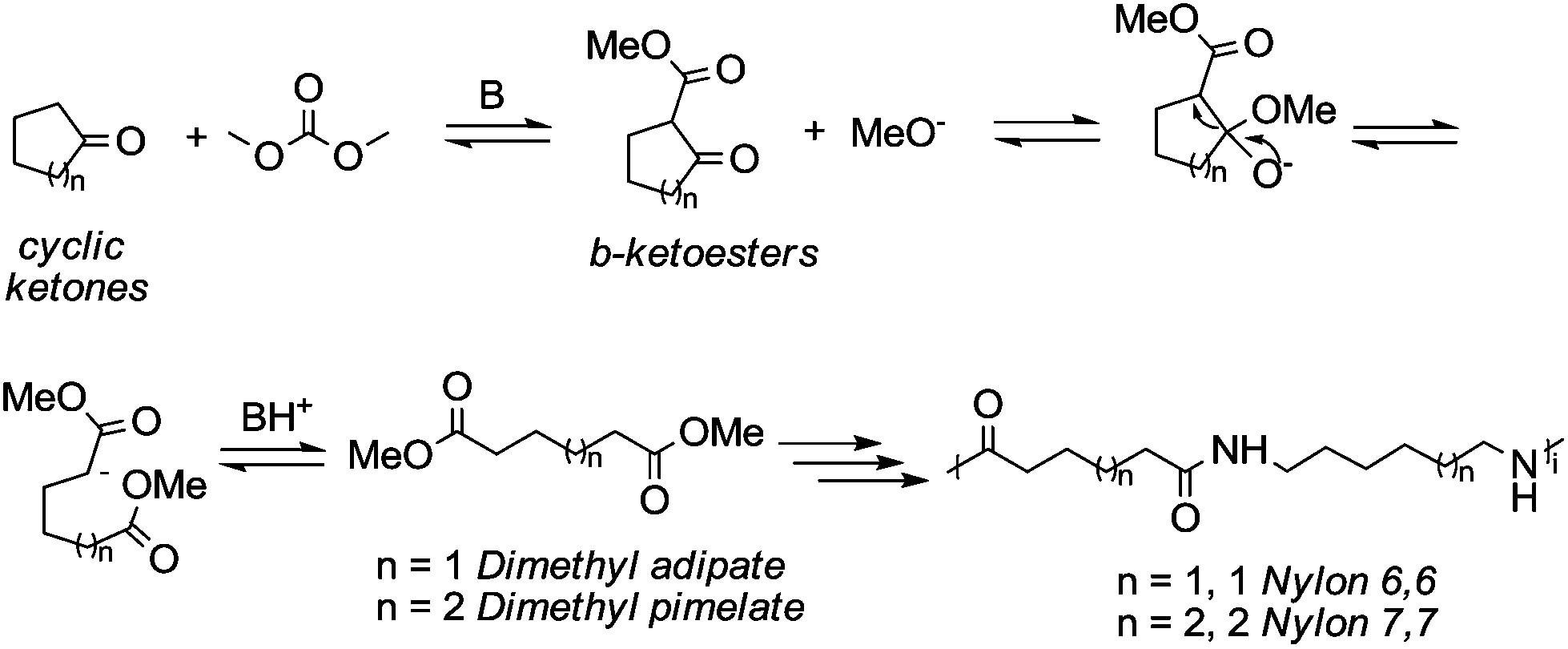 | ||
| Scheme 4 Ring opening mechanism via retro-Claisen reaction. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Cat. | MeOH (eq) | T (°C) | t (h) | Conv. (%) | Product selectivitye (%) | ||
| CMCPf | DMA | Bpsg | ||||||
| a Cyclopentanone/DMC 1.0/4.0 (molar ratio); catalyst wt% = 1.5%; reaction performed by heating in an autoclave. b Cyclopentanone/DMC/catalyst 1.0/20.0/2.0 (molar ratio); reaction performed by heating in an autoclave. c Cyclopentanone/DMC/catalyst 1.0/20.0/2.0 (molar ratio) and 0.4 eq. of PEG 250; reaction performed by heating in an autoclave. d Cyclopentanone/DMC 1.0/46.0 (molar ratio) and CeO2 nanorods; N2 (0.5 MPa); reaction performed via continuous flow. e Determined by GC-MS analysis. f CMCP: Carboxymethylcyclopentanone. g Bps: Major by-products = 2-methyldimethyladipate, 2,2′-methylenedicyclopentanone, 2-(cyclopentan-3-ylidene)cyclopentanone.24,26,27 | ||||||||
| 1a | MgO | — | 260 | 5 | 86 | 11 | 51 | 38 |
| 2a | CaO | — | 260 | 5 | 77 | 3 | 52 | 45 |
| 3b | K2CO3 | — | 260 | 11 | 33 | 11 | 48 | 41 |
| 4c | K2CO3 | 20 | 200 | 6 | 94 | 0 | 45 | 55 |
| 5c | Cs2CO3 | 20 | 200 | 6 | 96 | 0 | 46 | 54 |
| 6d | CeO2 | — | 260 | 5 | 96 | 0 | 85 | 15 |
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Cat. | MeOH (eq) | T (°C) | t (h) | Conv. (%) | Product selectivityc (%) | ||
| CMCHd | DMP | Bpse | ||||||
| a Cyclopentanone/DMC 1.0/4.0 (molar ratio); catalyst wt% = 1.5%; reaction performed by heating in an autoclave. b Cyclopentanone/DMC/catalyst 1.0/20.0/2.0 (molar ratio) and 0.4 eq. of PEG 250; reaction performed by heating in an autoclave. c Determined by GC-MS analysis. d CMCH: methoxycarbonylcyclohexanone. e Major by-products: 2-Methyl-dimethylpimelate, 2,2′-methylenedicyclohexanone, 2-methyl-2-methoxycarbonyl cyclohexanone, 2-(1-cyclohexenyl)cyclohexanone, 2-methylcyclohexanone.21,24 | ||||||||
| 1a | MgO | — | 260 | 5 | 84 | 6 | 52 | 42 |
| 2a | CaO | — | 260 | 5 | 65 | 2 | 52 | 46 |
| 3b | K2CO3 | — | 260 | 11 | 67 | 1 | 24 | 75 |
| 4b | K2CO3 | — | 200 | 6 | 82 | 0 | 20 | 80 |
| 5b | Cs2CO3 | 20 | 200 | 6 | 79 | 0 | 47 | 53 |
The reaction of cyclopentanone and DCM has been tested over solid and homogeneous bases and under different reaction conditions by heating in an autoclave (Table 2).24,26 Several experiments have also been carried out by adding polyethylene glycol (PEG) 250 and methanol to the reaction mixture using K2CO3 or Cs2CO3 as a base, a large excess of DMC (20 eq.) and heating to 200 °C for 6 h.
Although in almost all the experiments the conversion was quantitative, the selectivity toward adipic dimethylester (DMA) remained moderate due to the formation of several by-products. Recently, Zhang et al.27 reported the formation of DMA from cyclopentanone with high conversion and high selectivity by employing a continuous-flow system with a large excess of DMC (60 eq.) and using CeO2 nanorods as a catalyst. Similar results were accomplished by investigations of reaction of cyclohexanone with DMC to give pimelic dimethyl ester (DMP).21,24 In this case, the reaction proceeded with high conversion and moderate selectivity using MgO as a base, an excess of DMC (5 eq.) and Cs2CO3 upon adding PEG 250 and methanol to the reaction mixture and heating to 200 °C (Table 3).
In Scheme 5 are reported several examples of alkoxycarbonylation of less activated α-carbons via DAC chemistry in the presence of strong bases at temperatures below 100 °C. Pinacolone 1 was reacted with DMC at 85 °C for 5 h using NaH as a base to give methyl pivaloylacetate 2 in good yield (Scheme 5, eqn (7)).28 The same procedure was exploited to synthesise methyl pivaloylacetate-d64 in a moderate yield,29 which was used for the preparation of perdeuteriated 2,5-di-tert-butyl-3,4-di(methoxycarbonyl)pyrroloxyl 5, a stable nitroxide free radical (Scheme 5, eqn (8)).
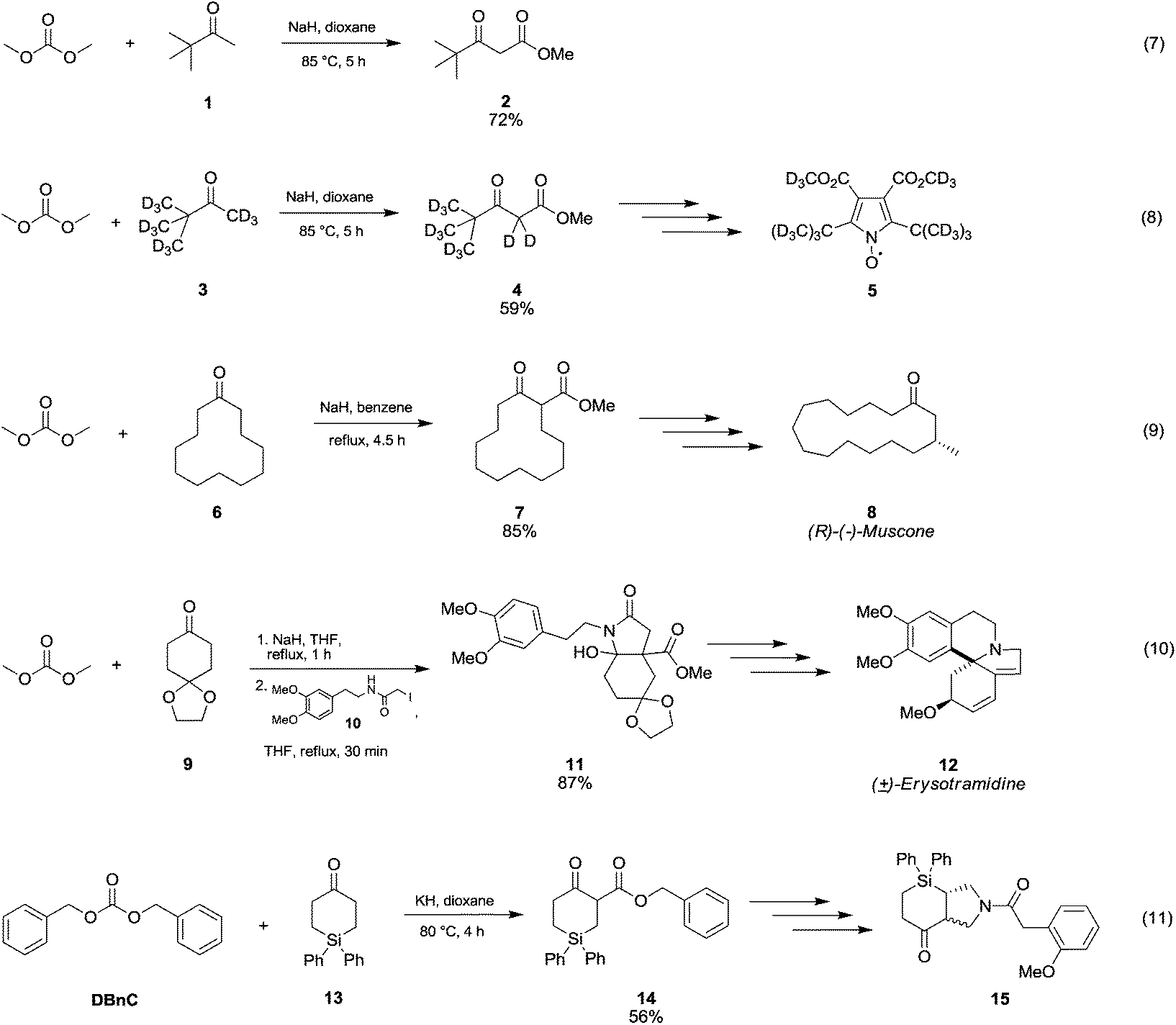 | ||
| Scheme 5 Synthesis of β-ketoesters via alkoxycarbonylation of non-activated α-carbons to a carbonyl group.28–32 | ||
The β-ketoester 7 (Scheme 5, eqn (9)) was obtained in good yield by reaction of cyclododecanone 6 and DMC;30 this compound (7) is an intermediate in the synthesis of (R)-(−)-Muscone 8, extensively used in perfumery.
In order to develop a general and efficient strategy to both aromatic-type and nonaromatic-type erythrinan and homoerythrinan alkaloids (Scheme 5, eqn (10), compound 12), Gao and co-workers31 used DMC for the synthesis of arylethylhydroindole 11 (87% yield) via methoxycarbonylation of ketone 9 with sodium hydride in refluxing THF followed by a reaction with compound 10.
Cycloaddition reactions of N-benzyl-azomethine ylide with 4-silacyclohex-2-en-1-ones provided 3-aza-1-silabicyclo[3.4.0] hexanes (Scheme 5, eqn (11), compound 15). According to the procedure described by Damour et al.,32 the reaction intermediate 14 was prepared starting from cyclic ketone 15 by reaction with dibenzylcarbonate (DBnC) and KH in dioxane at 80 °C for 4 h.
The benzylic/allylic α-carbon of a dissymmetrical ketone can be selectively methoxycarbonylated by reaction with DMC over an alkylic one. 5-Methoxy-2-tetralone 16 was reacted with DMC using sodium methoxide as a base and heating to 70 °C to give compound 17 in good yield (Scheme 6, eqn (12)).33 Instead, in the presence of two alkylic α-carbons, the methoxycarbonylation occurs preferentially on the less hindered one as confirmed by Taber and co-workers34 in their procedure to obtain the methoxycarbonylated compound 19, an intermediate in the synthesis of the phytotoxin coronatine 20 (Scheme 6, eqn (13)). Moreover, it has been observed that the vinyl protons of substituted cyclohexenones do not react with DMC also in the presence of a strong base, as a methoxycarbonylation reaction involves only the α-methylene moiety. As a matter of fact, the synthesis of the intermediate 6-carbomethoxy-4,4,6-triphenylcyclohex-2-en-1-one 22 was accomplished in 65% yield by methoxycarbonylation followed by insertion of a phenyl group in parent compound 21 (Scheme 6, eqn (14)).35 On the other hand, the sterically constrained 6,6-dimethylbicyclo-[3.1.1]hept-3-en-2-one derivative 23 (Scheme 6, eqn (15))36 underwent methoxycarbonylation exclusively on the vinylic position to give the unsaturated compound 24, which is an intermediate in the synthesis of chiral natural products containing the pinane skeleton (i.e. 6,6-dimethylbicyclo[3.1.1]heptane skeleton).
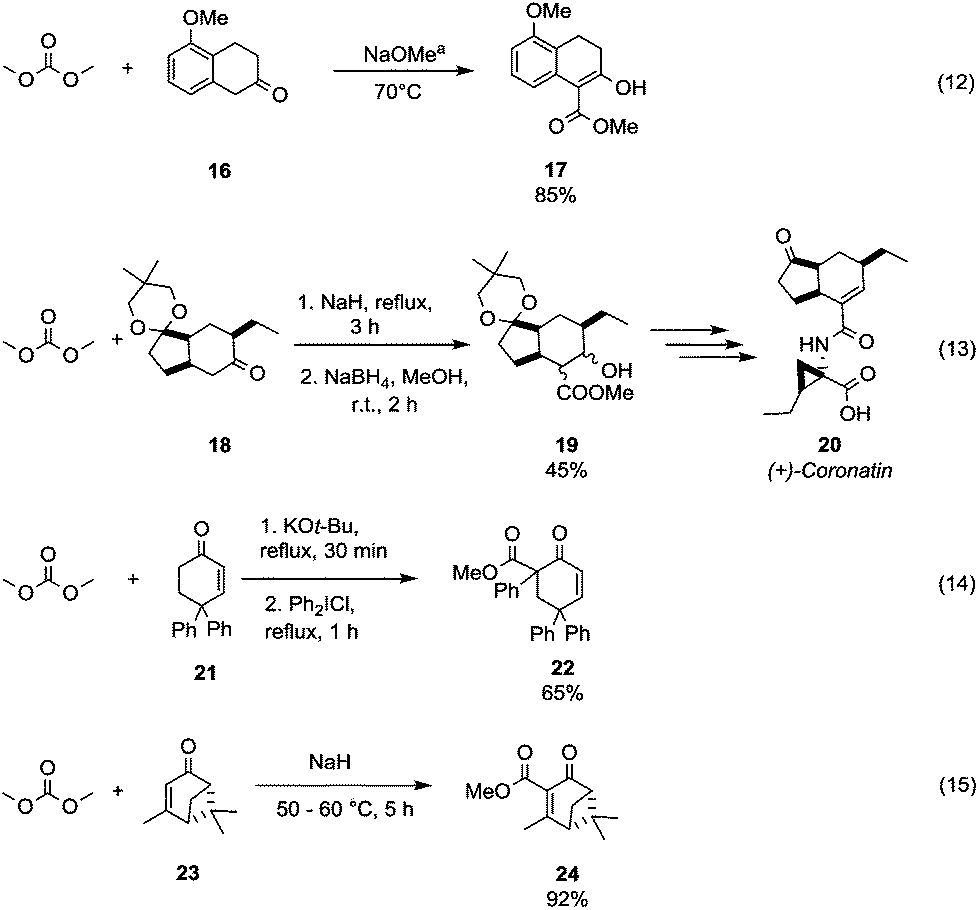 | ||
| Scheme 6 Examples of methoxycarbonylation selectivity toward different α-carbons to a carbonyl group. a Reaction conditions partially reported.33–36 | ||
The acidity of benzylic α hydrogens has been efficiently exploited to introduce a β-ketoester motif via DAC-chemistry into the scaffolds used for the organic synthesis of several bioactive compounds. Flavonoid scaffolds 1-methoxycarbonyl-3,4-substituted tetralin-2-one derivatives 26a–c (Scheme 7, eqn (16))37 were achieved in 88–93% yields by reaction of the corresponding ketones 25a–c with DMC and NaH in benzene.
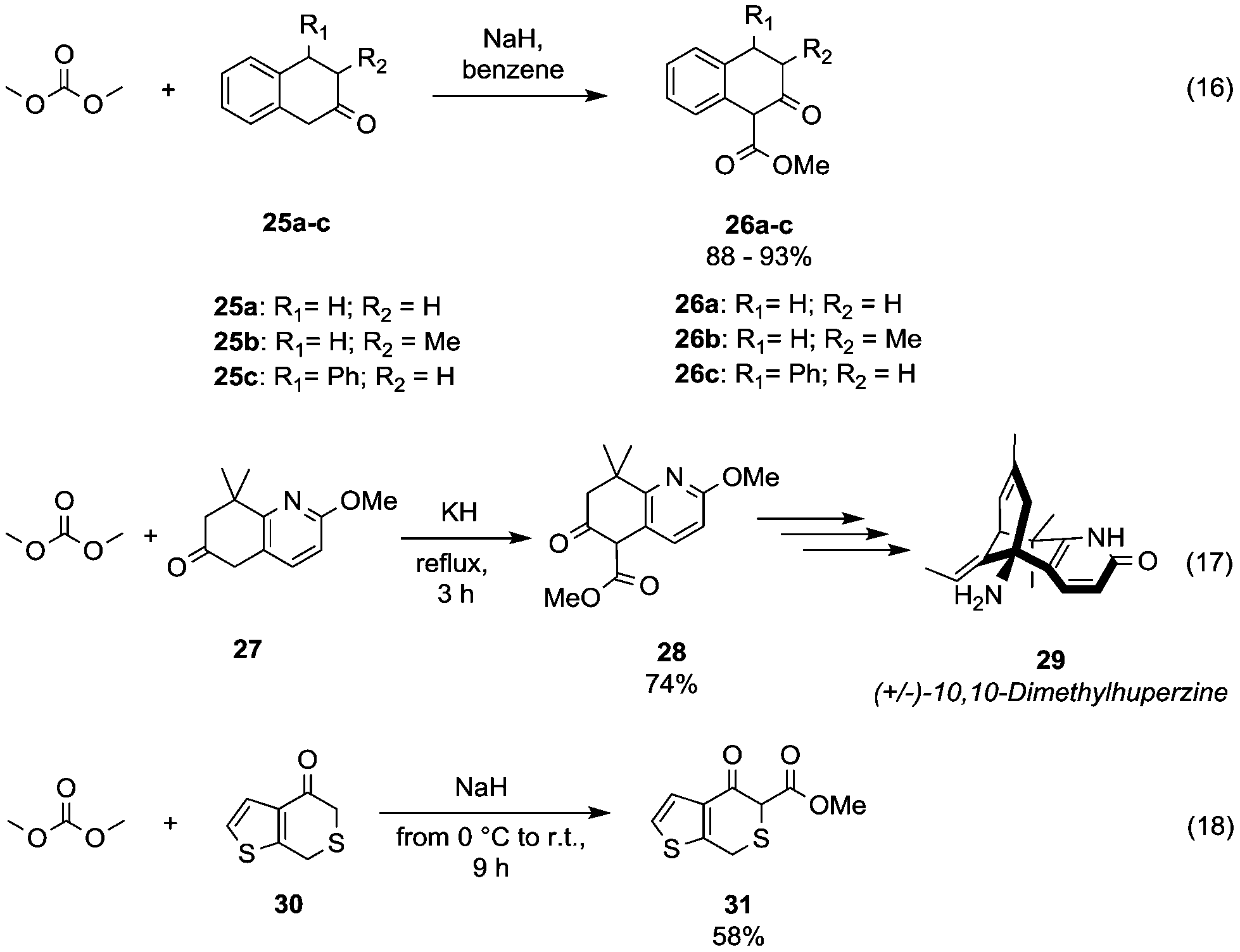 | ||
| Scheme 7 Synthesis of β-ketoesters via alkoxycarbonylation of activated α-carbons.38,39 | ||
Similarly, Kozikowski and co-workers,38 aiming at the synthesis of (+/–)-10,10-dimethylhuperzine 29 (Scheme 7, eqn (17); a drug used for the treatment of Alzheimer's disease), have prepared the key intermediate 28 by insertion of a methylester group at the benzylic position (5) of the 5,6,7,8-tetrahydro-6-oxo-quinoline derivative 27.
As an example of the methoxycarbonylation of highly activated carbon is reported the reaction of H-thieno[2,3-c]thiopyran-4(7H)-one 30 with NaH and DMC in benzene at 0 °C, which afforded compound 31 in 58% yield (Scheme 7, eqn (18)).39
Ring formation can be achieved straightforwardly via DAC chemistry by an alkoxycarbonylation reaction followed by intramolecular cyclisation of properly substituted ketones (Scheme 8). Cumarin derivatives 33a–c were obtained in rather good yield by a one-pot reaction of the corresponding 2-hydroxyacetophenones 32a–c with NaH and DEC in toluene, heating to reflux for 3 h (Scheme 8, eqn (19)).40
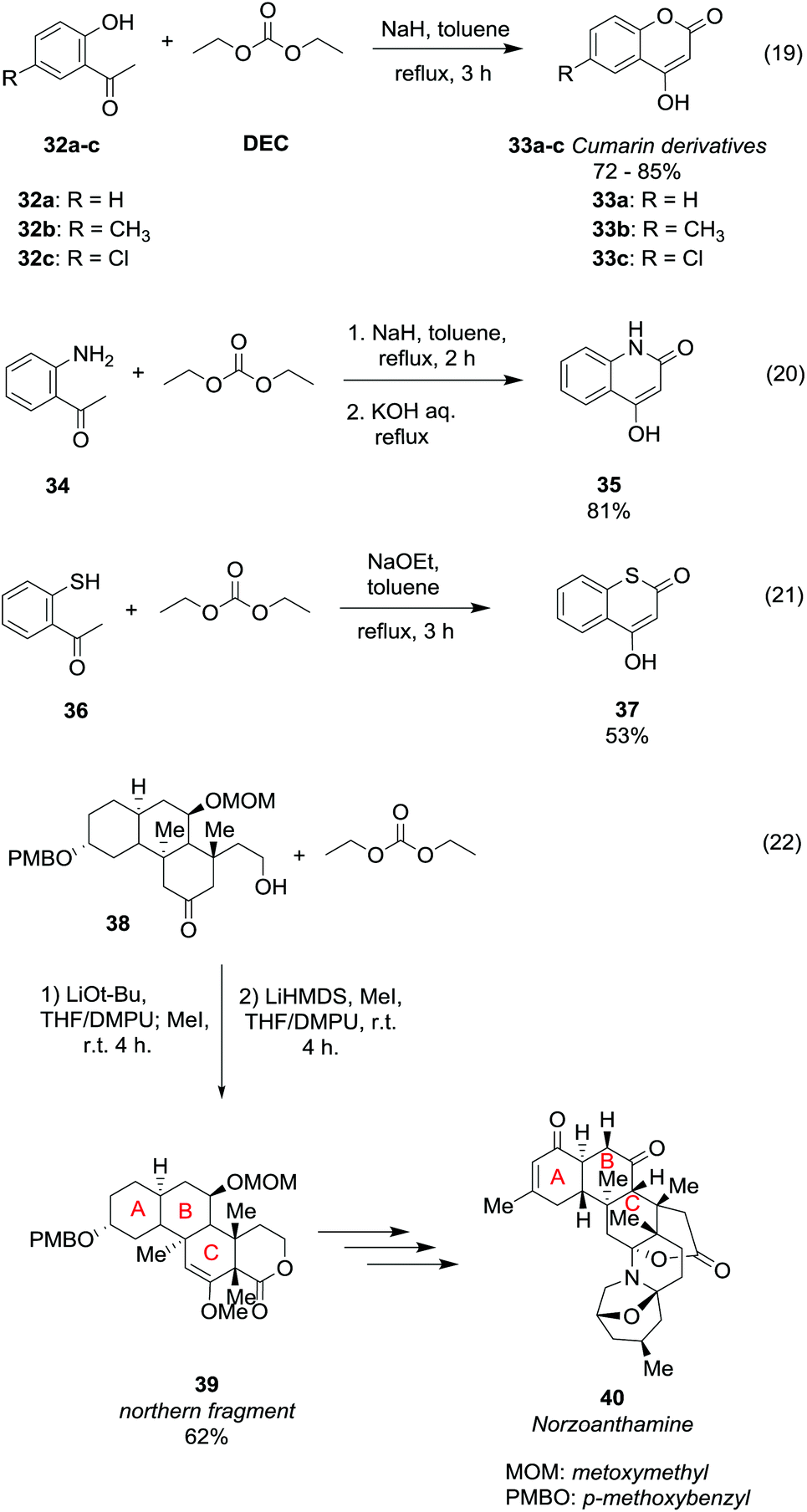 | ||
| Scheme 8 Ring formation via reaction of DEC with ketones.40–43 | ||
The cyclization proceeds via a double alkoxycarbonylation; in the first step the CH3 α to the carbonyl reacts with diethyl carbonate (DEC) via a BAc2 mechanism; thus the cyclic compounds are formed as a consequence of an intramolecular alkoxycarbonylation reaction. In this procedure DEC acts as a carbonylating agent as two moles of ethanol are released for each mole of cumarin 33 formed. Slightly modified reaction conditions were used to obtain 4-hydroxyquinolin-2(1H)-ones 35 in 81% yield, starting from 2-aminoacetophenone 34 (Scheme 8, eqn (20)).41 Besides, the reaction performed using 2-mercaptoacetophenone 36 as a substrate and sodium methoxide as a base afforded thiocumarin 37 in a moderate yield (Scheme 8, eqn (21)).41,42 This ring formation reaction was also employed for the enantioselective synthesis of the tetracyclic motif 39 (Scheme 8, eqn (22)),43 which is the northern fragment of norzoanthamine 40, a natural alkaloid extracted from a sea anemone species. Treatment of compound 38 with t-BuOLi as a base, iodomethane and DEC followed by a stereoselective methylation afforded methyl enol ether 39 in 65% yield.
Acetal carbanions, which can be considered synthetic equivalents of acyl anions, react with DACs, leading to alkoxycarbonylated products. This approach was applied to the synthesis of α-substituted alkoxy dienes from α,β-unsaturated acetals (Scheme 9).17 The reaction of (E)-1,1-diethoxybut-2-ene 41 with sec-butyllithium complexed with potassium tert-butoxide in THF by cooling to −95 °C resulted in the formation of 1-metalated 1-ethoxy 1,3-dienes. Subsequently, the addition of DMC led to the formation of the two main compounds 42 and 43, respectively mono- and bis-substitution products. Specifically, when an excess of DMC (5 eq.) was used, a mixture of products was obtained with 42 as a major component in 70% yield and 43 in 10%. On the other hand, when the reaction was repeated employing a molar ratio of acetal/DMC of 1/0.5 the bis-substituted product 43 was the solely isolated compound in 90% yield.
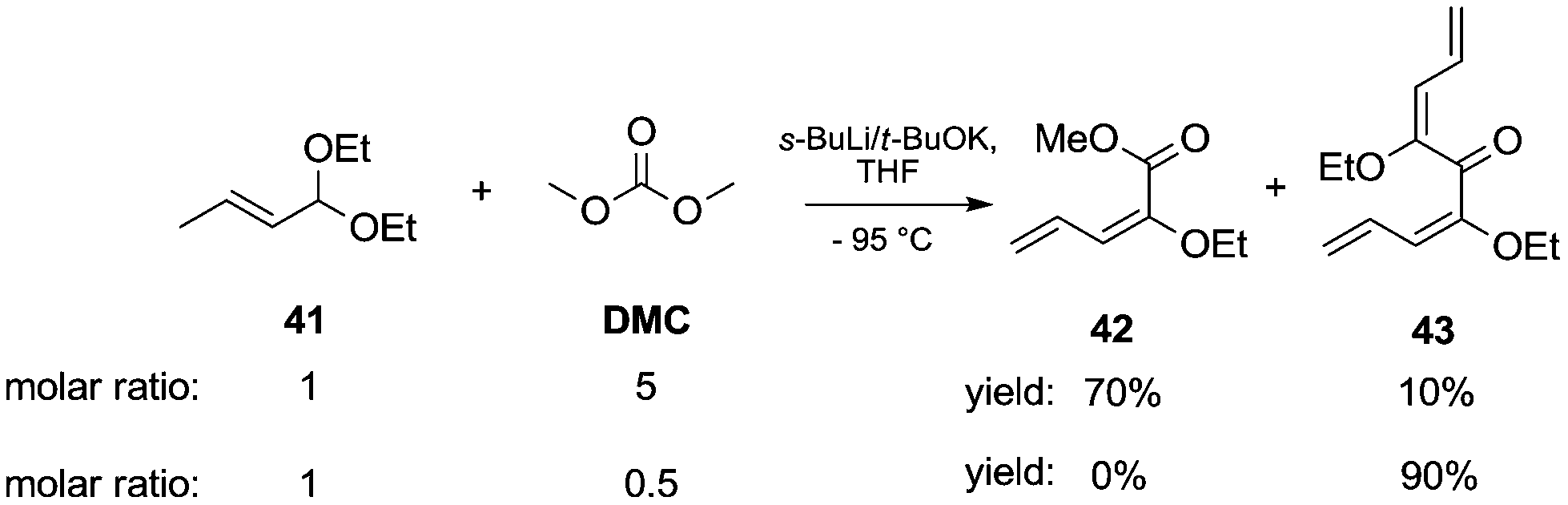 | ||
| Scheme 9 Synthesis of α-substituted alkoxy dienes from α,β-unsaturated acetals.17 | ||
2.2. Reaction with esters, lactones, lactams, nitriles and isonitriles
This section focuses on BAc2 methoxycarbonylation reactions of activated methylene of esters, lactones and lactams (Scheme 10) as well as of nitriles and isonitriles (Scheme 11). Reaction of DACs with esters under basic conditions leads to the formation of malonate derivatives (Scheme 10, eqn (23) and (24)) by insertion of an ester motif on the carbon at α position to the carboxylate group.44 Analogous to the formation of β-ketoester, the general procedure consists of the deprotonation of the α-methylene group by means of a strong base followed by a reaction with the proper dialkyl carbonate. The use of an excess of DAC in combination with alcohol removal from the reaction mixture may shift the equilibrium toward the product formation (Scheme 10, eqn (23)).45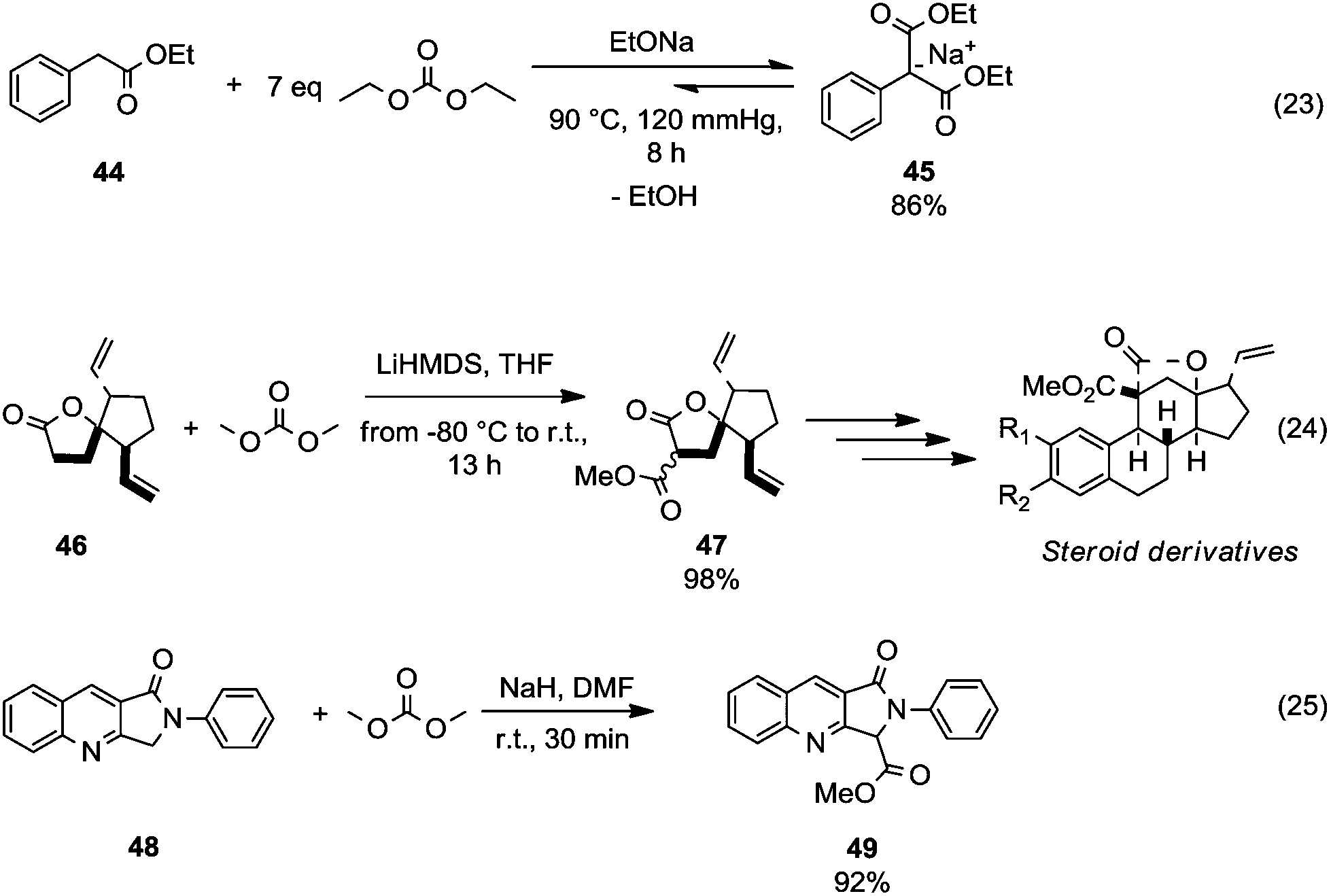 | ||
| Scheme 10 Alkoxycarbonylation of activate α-methylene of esters (eqn (23) and (24)) and cyclic amides (eqn (25)).45–47 | ||
 | ||
| Scheme 11 Alkoxycarbonylation of cyanide (eqn (26)) and isocyanide (eqn (27)) α-carbon.48,49 | ||
An alkoxycarbonylation reaction of substituted butyrolactones with DACs was used to prepare key intermediates in the synthesis of a variety of steroid-based compounds. Michellys et al.46 reported the synthesis of ester 47 (Scheme 10, eqn (24)) as a mixture of isomers by a reaction of butyrolactone 46 with lithium hexamethyldisilylamide (LiHMDS) and DMC carried out from −80 °C to room temperature. Similarly, N-protected lactams undergo alkoxycarbonylation on activated α-carbon.47 Compound 49 (Scheme 10, eqn (25)) – an intermediate for the synthesis of peripheral benzodiazepine receptor (PBR) ligands – was obtained in good yields, starting from lactam 48 by reaction with NaH and DMC in DMF at room temperature.
This procedure was also applied to alkylic and benzylic nitriles and isonitriles (Scheme 11). Takeuchi and co-workers48 investigated the reactivity of p-substituted phenylacetonitriles (50a–d) with diethyl, diallyl and dibenzyl (DBnC) carbonates in the presence of NaH and THF as a reaction medium (Scheme 11, eqn (26)). The corresponding esters (51a–d, 52a–d and 53a–d) were achieved in appreciable yields.
As shown in Scheme 11, eqn (27), the syntheses of α-isocianoacetates 55a and 55b49 were accomplished in moderate yields starting from benzyl isocyanide 54 and with DMC (70% yield) or DEC (63% yield) in the presence of NaH. Hydrolysis of 55 under acidic conditions gave phenylalanine 56. Therefore, this method represents a valuable approach to the synthesis of amino acids.
2.3 Reaction with Grignard-like and lithiated carbanions
Among the plethora of strategies reported in the literature, Grignard-like reactions and lithium promoted reactions are the principal methods to generate carbanions suitable to perform alkoxycarbonylation via DAC chemistry.In 1905 Chichibabin50 was the first to report the reaction of DEC and the ethyl ester of orthocarbonic acid (C(OC2H5)4) with various Grignard reagents, which led to the formation of esters in good yields along with small amounts of ketones and tertiary alcohols. Afterwards, it has been established that the use of specific carbanion/DAC molar ratios may direct the reaction to the formation of esters (1/1), ketones (2/1) or tertiary alcohols (3/1) (Scheme 12).
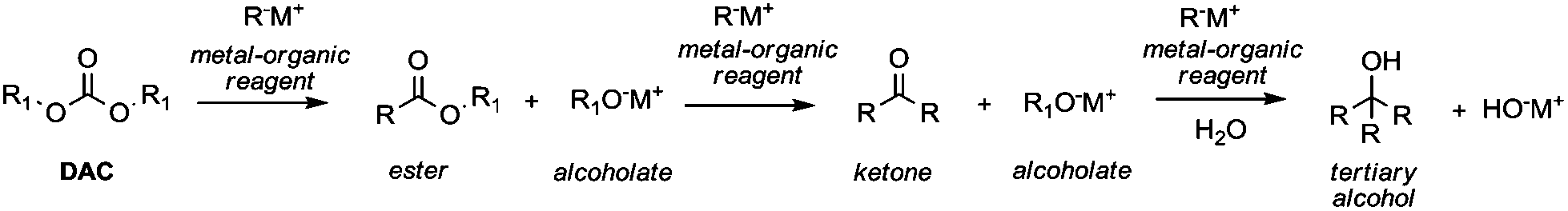 | ||
| Scheme 12 General synthetic pathway of the reaction of carbanions with a dialkylcarbonate. | ||
However, the selective synthesis of a predicted product – instead of a mixture of ester, ketone and tertiary alcohol – depends on several factors such as steric hindrance, electrophilicity of the DAC and nucleophilicity of the anion employed. In Scheme 13 gives a select series of examples of halogen–metal (Mg or Li) exchange reaction followed by insertion of an ester group by the addition of DAC. Terpstra et al.51 described the one pot synthesis of methyl 3-methylthiophene-2-carboxylate 59 (Scheme 13, eqn (28)) via the Grignard reaction as the intermediate step in their procedure to obtain substituted benzothiophenes.
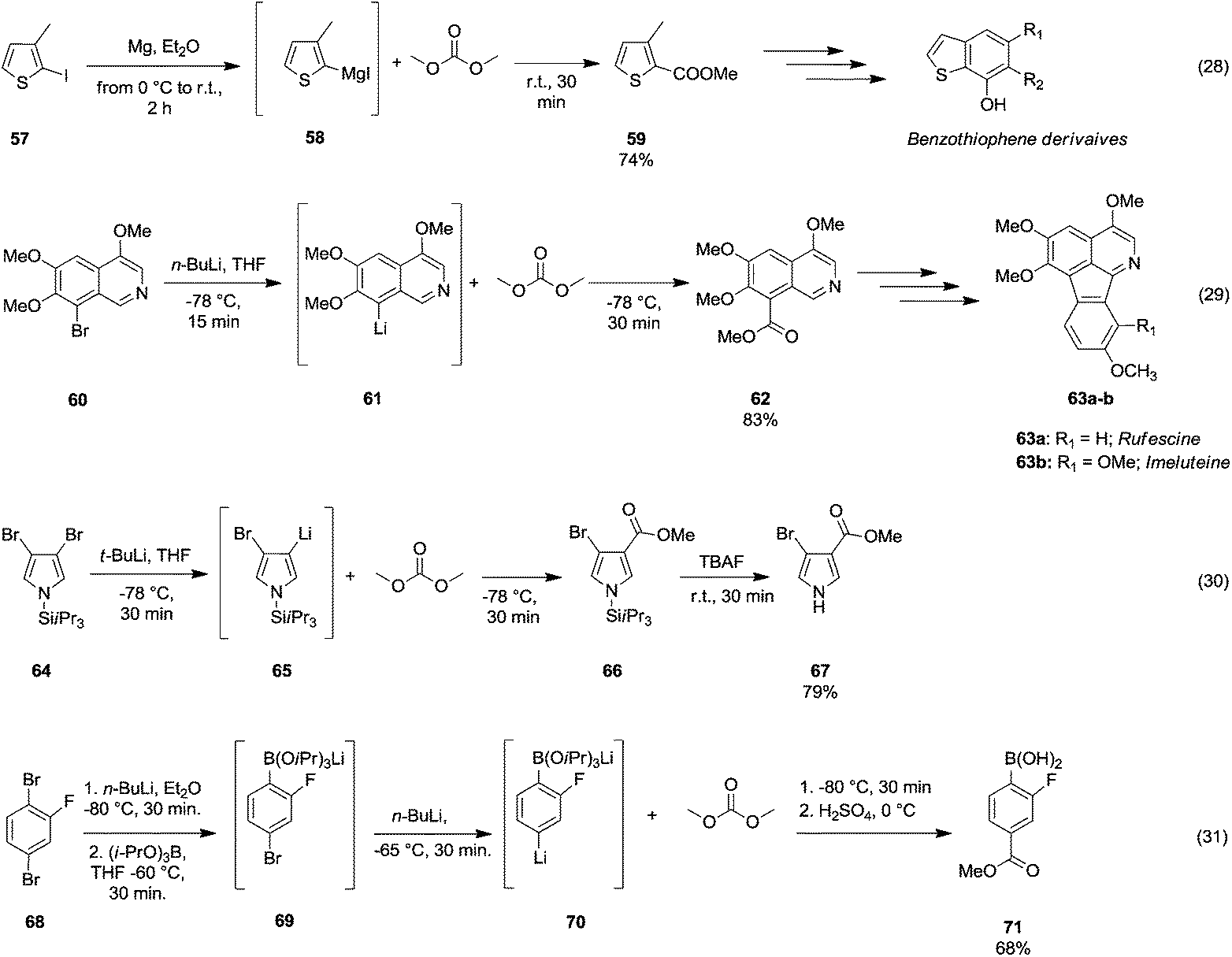 | ||
| Scheme 13 Examples of bromine–Mg or –Li exchange reaction followed by alkoxycarbonylation via DACs chemistry.51–54 | ||
The precursor 2-iodo-3-methylthiophene 57 was first treated with Mg in Et2O to generate the reactive Grignard reagent 58, which by reacting with DMC gave compound 59 in 74% yield. 8-Methoxycarbonyl-5,6,7-trimethoxyisoquinoline 62 (Scheme 13, eqn (29)) has been used as an intermediate of a total synthesis of rufescine 63a and imeluteine 63b,52 natural azafluoranthene alkaloids isolated from Abuta imene and Abuta rufescens. The ester 62 was obtained in 83% yield by metalation of 8-bromo-5,6,7-trimethoxyisoquinoline 60 with n-butyllithium followed by treatment of the resulting 8-lithio-5,6,7-trimethoxyisoquinoline 61 with DMC.
A similar synthetic approach was developed for the methoxycarbonylation of a substituted pyrrole (Scheme 13, eqn (30)).53 3,4-Dibromo-1-[tris(1-methylethyl)silyl]-1H-pyrrole 64 was treated with t-butyllithium (1 eq.) at −78 °C, resulting in a selective halogen–metal exchange to afford the mono-lithiated derivative 65, which gave compound 66 in good yield by reaction with DMC followed by TBAF (tetrabutylammonium fluoride)-assisted N-desilylation.
Arylboronic acids may be easily synthesized starting from 1,3- and 1,4-dibromobenzenes 68 (Scheme 13, eqn (31)).54 The general procedure consists of the bromine–lithium selective exchange using n-butyllithium, followed by a boronation reaction with a trialkyl borate to give the lithium(bromophenyl)trialkoxyborate derivative 69. Further addition of n-butyllithium promotes the second bromine–lithium exchange to give the reactive dilithiated intermediate 70, which upon treatment with DMC and quenching under acidic conditions affords the substituted aryl boronic acid derivative 71 in moderate to good yields. Unlike Grignard reagents, whose formation occurs almost exclusively by halogen–metal exchange, organolithium reagents can generate efficiently carbanions of a variety of substrates through hydrogen–metal exchange (Scheme 14). Optically active 2,2′-bis(oxazolyl)-1,1′-binaphthyls 74 (boxax) – incorporating various substituents at their C3 and C3′ positions – are employed as chiral ligands in the enantioselective palladium(II)-catalyzed Wacker-type cyclization of o-allylphenol derivatives. The diester derivative 74 (Scheme 14, eqn (32))55 was prepared in good yield via ortho-lithiation of compound 72 – promoted by the oxazolyl groups – by using sec-butyllithium and N,N′,N′-tetramethylethylenediamine (TMEDA) in THF at −78 °C for 2 h followed by the addition of DMC and warming the mixture to r.t. A series of lithiation–substitution reactions at the α- and β-positions of N-Boc N-alkyl cyclopropyl amines was reported by Park et al.56 The cyclopropylamine 75 was reacted with sec-butyllithium and TMEDA in Et2O at 78 °C to give selectively lithiation at position β (Scheme 14, eqn (33)). Subsequently, DMC was added to the reaction mixture and the diastereoisomer 77 was isolated after purification by flash chromatography (FC) in 61% yield. Phenylboronic acid 80 (Scheme 14, eqn (34)) was obtained in good yield by deprotonative o-lithiation of the electron poor phenyldioxazaborocane 78 by means of lithium diisopropylamide (LDA) in THF at −75 °C, followed by quenching the reaction mixture with DMC and acidification with 2 M H2SO4 aq. solution.57
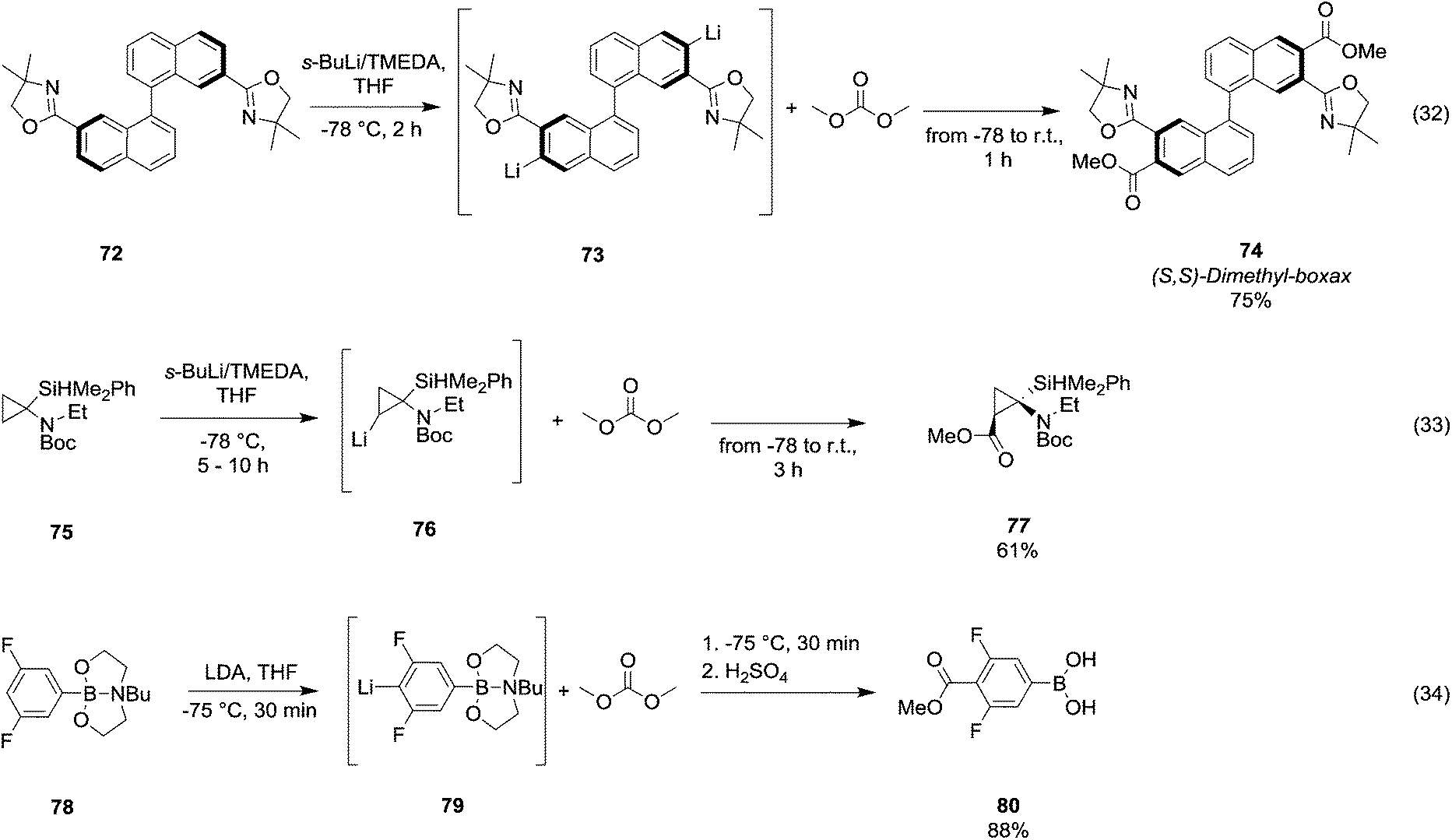 | ||
| Scheme 14 Examples of H/Li-exchange reaction followed by alkoxycarbonylation via DACs chemistry.55–57 | ||
Ketone synthesis by using metalation and DMC chemistry was employed in the total synthesis of the marine alkaloid Variolin B (Scheme 15, eqn (35) compound 83).
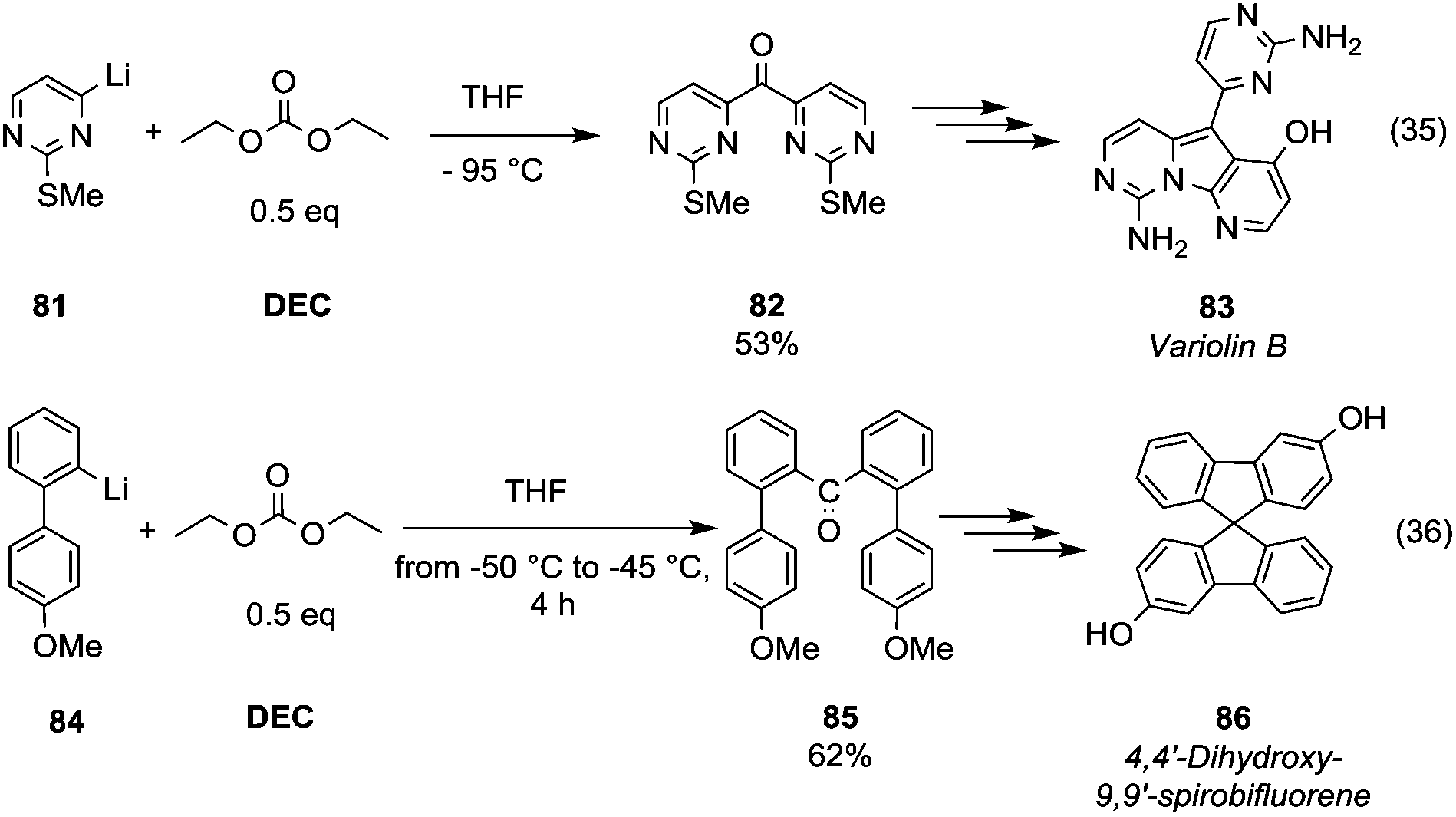 | ||
| Scheme 15 Ketones formation via DACs chemistry.58–60 | ||
The lithiated species 81 – generated by iodine–lithium exchange using n-butyllithium in THF at −95 °C – was reacted with DEC (0.5 eq.) at −95 °C to give the symmetric ketone 82 in 53% yield. DMC was used as a key reagent to prepare the ketone intermediate 85 (Scheme 15, eqn (36))59 in a synthetic route to achieve chiral dihydroxy-9,9′-spirobifluorenes, which belong to a class of compounds widely exploited in molecular electronics, light-emitting materials and enantioselective molecular recognition. Lithiated intermediate 84 – obtained by bromine–lithium exchange using n-butyllithium – was reacted with DMC (0.5 eq.) to obtain di[2-(methoxyphenyl)phenyl]ketones 85 in a moderate yield. Subsequently, ring closure induced by methanesulfonic acid and demethylation reaction gave dihydroxy-9,9′-spirobifluorene 86 in a racemic mixture resolvable by chiral agents or chiral HPLC.
In 1931, Moyer et al.60 published a synthetic procedure conceived to obtain a tertiary alcohol, i.e., triethyl carbinol 87 (Scheme 16, eqn (37)) by reaction of ethylbromide with Mg and DEC. However, over the years, the synthesis of tertiary alcohols by DAC chemistry has been principally developed along with the generation of carbanions by means of organolithium reagents. Thus, triaryltrityl alcohol 89 was obtained in 73% yield (Scheme 16, eqn (38))61 by reaction of acetonide 88 with n-BuLi (1.1 eq.) and tetramethylethylenediamine (TMEDA; 1.1. eq) in toluene increasing the temperature from −4 °C to r.t. for 2 h, followed by the addition of DMC (0.33 eq.) at −20 °C. A further methoxycarbonylation reaction (i.e. the insertion of three methylester groups) and radical generation gave the triaryltrityl radical 90 in 35% yield.
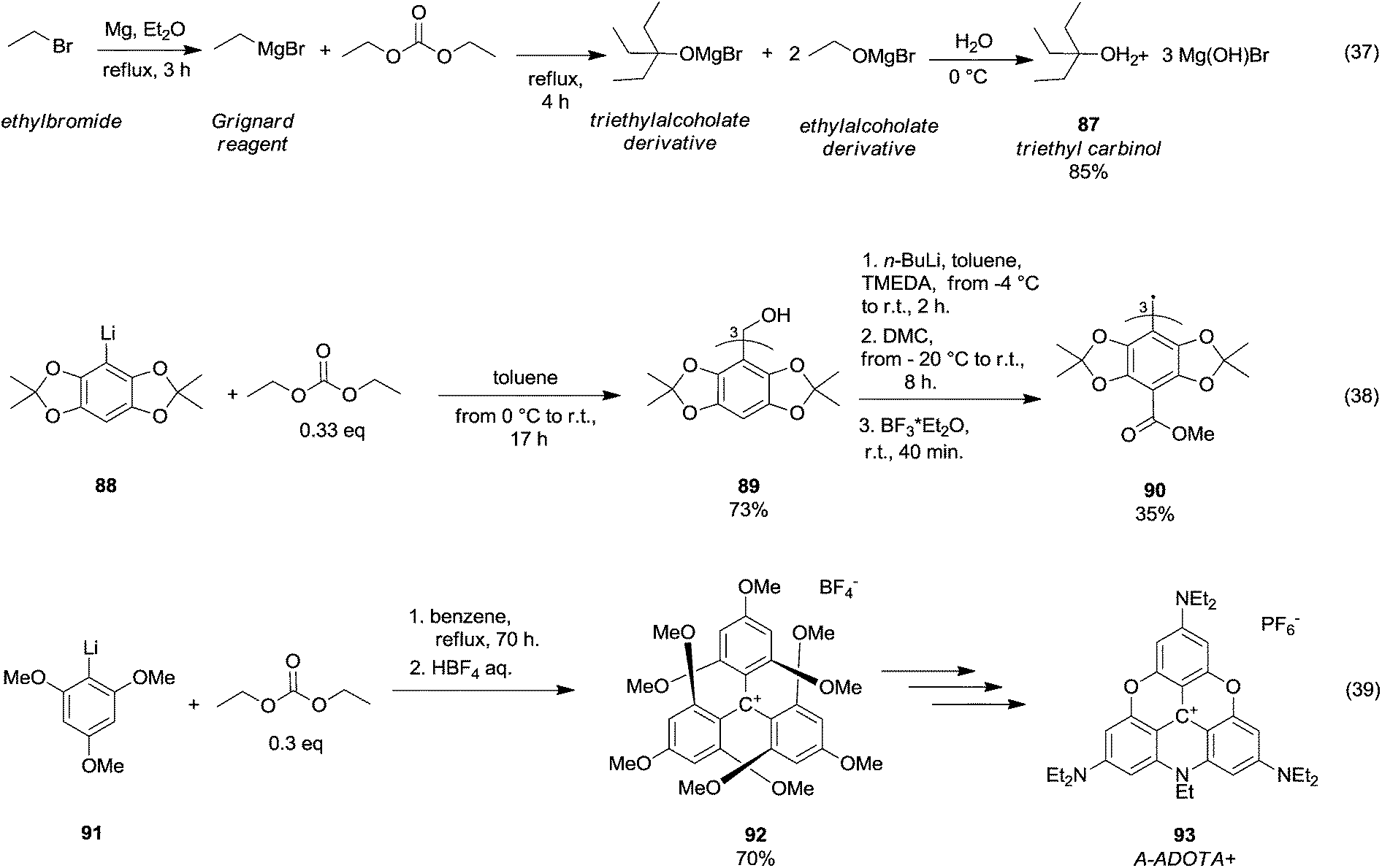 | ||
| Scheme 16 Tertiary alcohols formation via DACs chemistry.61,63 | ||
Tris(p-carboxyltetraoxoaryl)methyl radicals – as well as tris (p-carboxyltetrathiaaryl)methyl radicals achievable through a similar synthetic approach – are persistent radicals used as spin probes for Electron Paramagnetic Resonance (EPR) imaging of biological systems. DEC was also employed in the formation of triamino-substituted planar carbenium ions (Scheme 16, eqn (39)).62 This procedure was based on a straightforward and selective nucleophilic aromatic substitution on the tris(2,4,6-trimethoxyphenyl)carbenium ion 92 with amines and it permitted producing a wide variety of amino-substituted carbenium ions, such as tris(dialkylamino)-azadioxatriangulenium carbenium ion 93 (A-ADOTA+), i.e. the most stable triangulenium ion ever synthesised and characterised.63
In the literature, only a few examples of characterized nuleophilic boron reagents have been reported.64 In such a perspective, Monot et al.65 developed the synthesis and reactions of an unsubstituted N-heterocyclic carbene boryl anion (Scheme 17). Boryl iodide 95 was prepared in almost quantitative yield by in situ reaction of the precursor 94 with iodine (in a 2/1 molar ratio) in benzene. Among the several organolithium reagents screened, lithium 4,4-di-tert-butylbiphenylide (LDBB) in the presence of TMEDA produced – by iodine–lithium exchange – significant concentrations of the anion 96, which when quenched with an excess of DEC at −78 °C provided the stable ethoxycarbonylborane complex 97 isolated in 61% yield by FC purification.
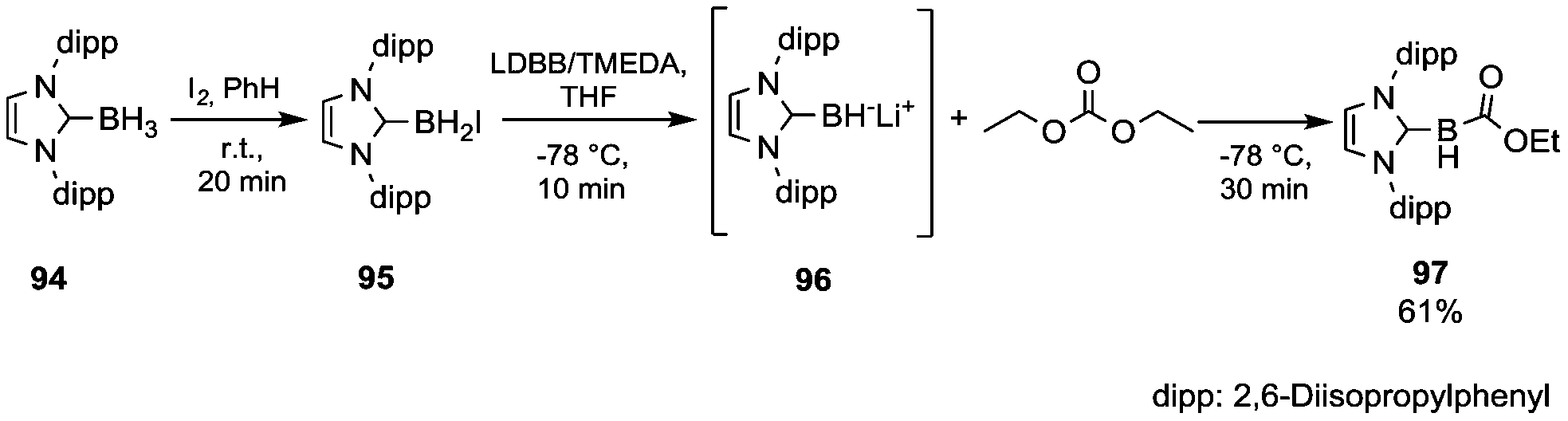 | ||
| Scheme 17 Reductive metalation of a stabilised borane and quenching with DEC.65 | ||
2.4. Special cases: magnesium- and potassium-methyl carbonates
Magnesium methyl carbonate 99a (MMC) or the Stiles reagent is a versatile reagent for methoxycarbonylation of active α-methylene groups.66 Finkbeiner et al.67 used this synthetic procedure on phenylhydantoin 98 (Scheme 18, eqn (40)) by reaction with MMC in DMC at 80 °C. Likely, the methoxycarbonylation reaction occurred via a six membered intermediate 100, in which magnesium acted as a chelator.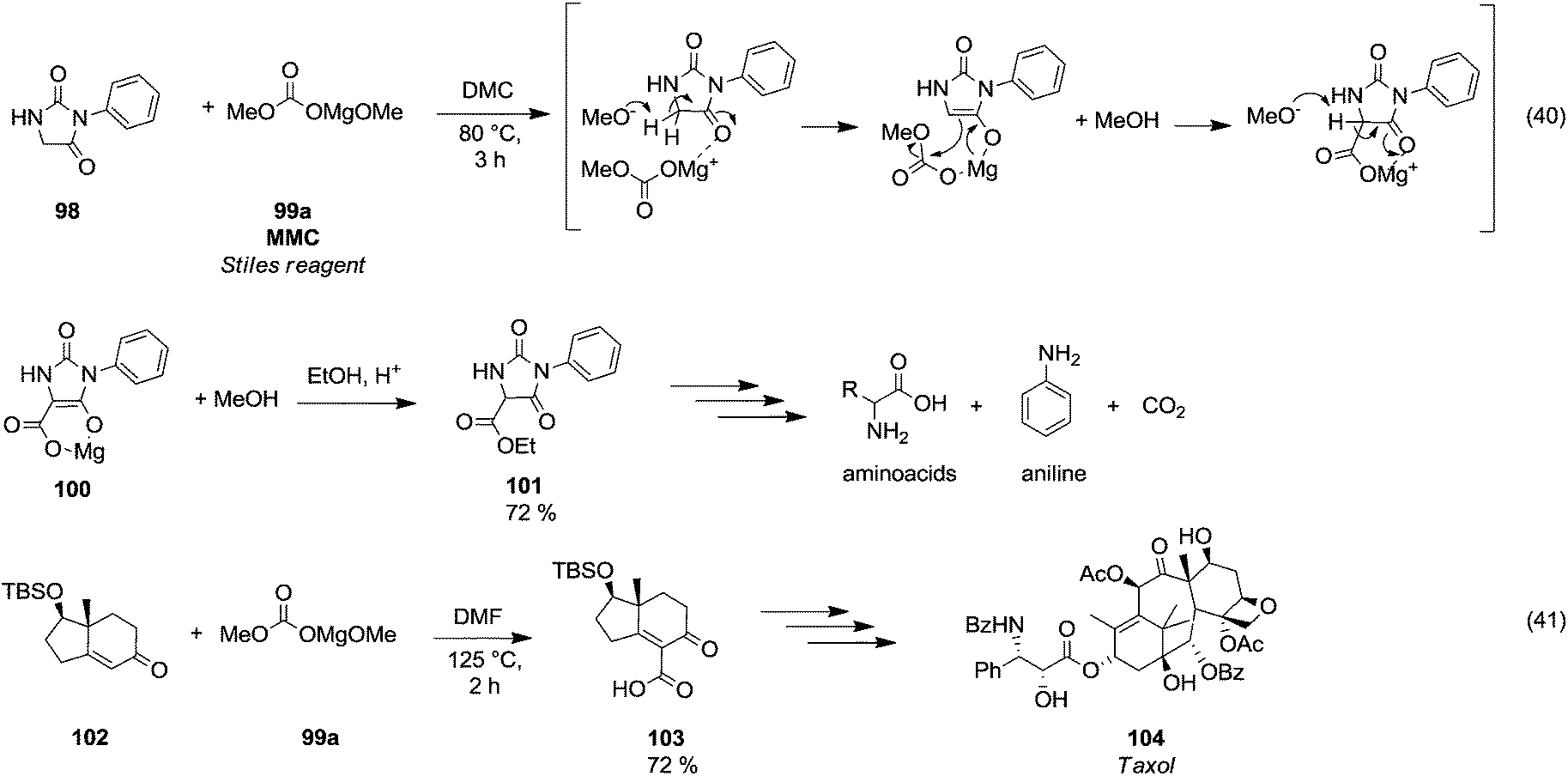 | ||
| Scheme 18 Magnesium assisted carboxylation reactions using MMC.67,68 | ||
This synthetic approach has been widely used to synthesise amino acids. In a total synthesis of the anticancer diterpenoid Taxol (104),68 magnesium methyl carbonate was used to introduce selectively a carboxylic moiety on the vinylic position of the enantiomerically pure intermediate 102 by reaction in DMF at 125 °C, giving compound 103 in 74% yield (Scheme 18, eqn (41)).
Potassium methyl carbonate 99b (PMC) was also proved to be an efficient methoxycarbonylating agent of allyl and arylboronic esters (Scheme 19) in the presence of a catalytic amount of chloro [1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]copper(I) (Cu(iPr)Cl).69
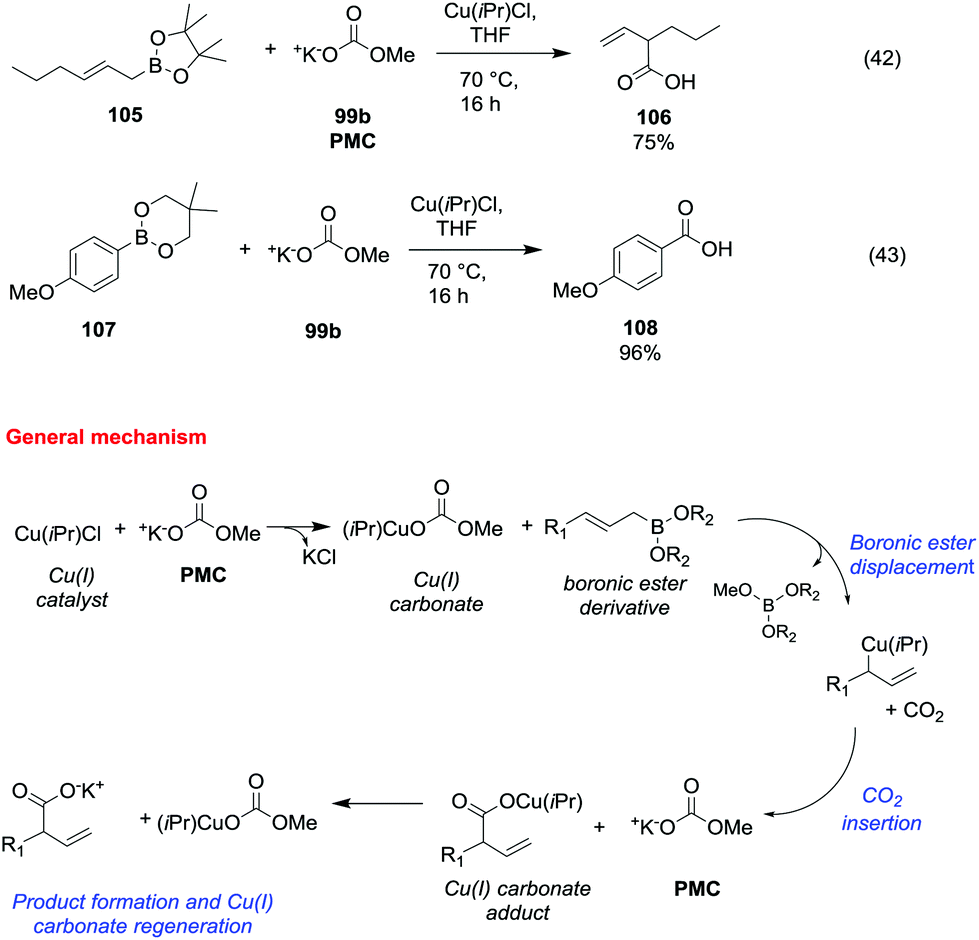 | ||
| Scheme 19 Copper(I) catalysed carboxylation reactions of allyl and arylboronic esters with PMC.69 | ||
The general synthetic procedure – carried out in a glove box under an argon atmosphere – consisted of the reaction of an allyl or arylboronic ester (105 and 107) with a catalytic amount of Cu(iPr)Cl and a stoichiometric amount of 99b, in THF heating to 70 °C for 16 h (Scheme 19, eqn (42) and (43)). The corresponding β,γ-unsaturated carboxylic acid 106 and benzoic acid 108 were obtained in moderate to good yields following the proposed mechanism depicted in Scheme 19.
3. Alkylation
Typical alkylating agents can be classified into three main categories according to the reaction conditions used, i.e., basic, acidic or neutral (Table 4).70| Basic conditions | Neutral conditions | Acid conditions |
|---|---|---|
| a Triazene. b Trimethylsilyldiazomethane. c Isourea. d N,N-Dimethylformamide dimethyl acetal. e Nowadays, DACs are also used under acidic and neutral conditions. | ||
| Me2SO4 | CH2N2 | MeOH/H+ |
| TsOMe/TsOMs | Ph–NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMea NMea |
MeOH/BF3 |
| AlkHal | TMSCHN2b |
|
| Me4NX | RNHC(OMe)NRc | |
| Me3O·BF4 | (MeO)2CHNMe2d |
|
| ROCOORe | (MeO)3CR | |
Regio- and chemoselectivity related to this chemical transformation may represent an issue, inasmuch as they might play a pivotal role for complex substrates that incorporate different reactive sites. Besides, most of the classic highly reactive alkylating agents are toxic and corrosive chemicals.71
Reactions conducted under acidic conditions require corrosive chemicals; meanwhile, alkylating agents used under neutral conditions may be carcinogenic, explosive (CH2N2; Ph–NH–NNMe), toxic (N,N-dimethylformamide, dimethyl acetal) or expensive (TMSCHN2).72
Alkyl iodides, mesylates, tosylates and dimethyl sulphate are alkylating agents commonly used under basic conditions. Most of these reagents are chlorine-based chemicals that encompass toxic/corrosive properties and are well known to be carcinogens.73 Besides, in order to perform an efficient alkylation, these chemicals require a stoichiometric amount of a base, producing in turn a stoichiometric amount of inorganic salts that need to be disposed of.
Non-volatile tetraalkylammonium salts are possible substitutes, even though they showed an inferior reactivity. As an example, Meerwein salts (Me3O·BF4) have been employed as an alternative to classic alkylating agents, although these compound rapidly hydrolysed and are difficult to handle.
In this perspective, dialkylcarbonates (DACs) are nowadays extensively investigated as alkylating agents. Dimethyl carbonate (DMC) – the most exploited organic carbonate – has been shown to be a safe, environmentally friendly, green substitute for metal halides and dimethylsulphate.2,74
It should be, however, mentioned that DACs are clearly less reactive than toxic alkylating compounds such as alkyl halides and dimethyl sulphates. This is not necessarily inconvenient since the high Gibbs energy required to activate DACs is the key for regio- and chemoselective processes such as the mono-methylation of the acidic methylene group or aniline by DMC.
In this section is reported a comprehensive overview on applications, mechanisms and eventual limitations on alkylation reactions via DAC chemistry. The reactivity of oxygen, nitrogen, sulfur and acidic CH2-substrates is described focusing on new synthetic pathways, different reaction conditions and activated substrates. The peculiar case of metal catalysed allylic alkylation is also reported. Finally, the novel anchimerically driven alkylation reaction via mustard carbonate and its future applications in the realm of DAC chemistry are also discussed in detail.
It is also noteworthy that more recently DACs have demonstrated their great versatility as alkylating agents able to react efficiently under basic, acidic and neutral conditions as is shown in the following specific examples.
3.1 O, N, S and C alkylation reactions
However, primary alcohols can be converted into methyl ethers through DMC chemistry in a two step reaction: first a BAc2 transesterification, followed by the decarboxylation of the resulting methylcarbonate (Scheme 20).76
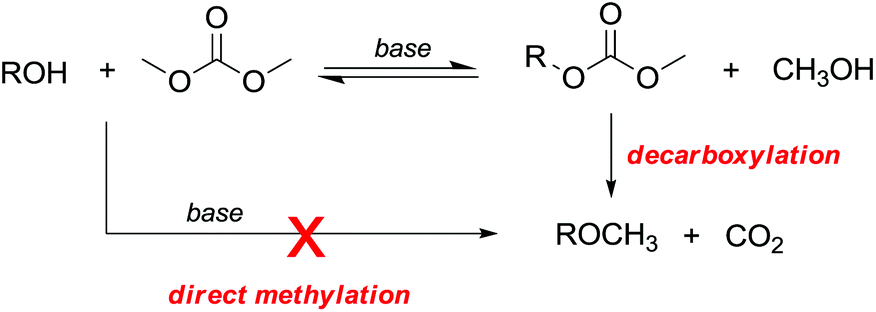 | ||
| Scheme 20 Synthesis of ethers via DAC chemistry. | ||
A few but significant exceptions involving direct methylation of hard alkoxides via DMC chemistry (Scheme 21) are related to: (a) substrates incorporating strategically located ether functional groups or electron pairs and (b) benzyl alcohols.
 | ||
| Scheme 21 Direct synthesis of ethers from alcohol via DMC chemistry. | ||
In both cases, methylations were ascribed to a stabilizing effect on the transition state of the BAl2 mechanism, due to the involvement of a secondary base effect or through the intramolecular assistance of the leaving group.
A striking example is the unexpected methylation of isosorbide. In Table 5, the influence of the molecule backbone in the reaction of selected secondary alcohols in BAl2 reactions with DMC is shown. The selected substrates have a similar structure and their methylation was studied under identical reaction conditions. In a typical procedure the alcohol was reacted with an excess of DMC at its reflux temperature (90 °C) in the presence of a strong base, i.e., NaOMe.12
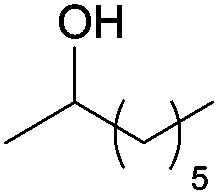
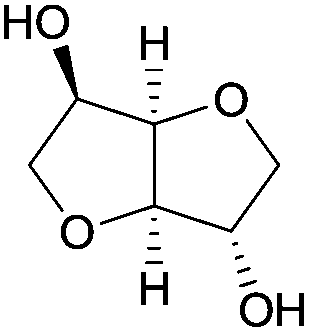
As reported in Table 5, there is a relevant difference in the reactivity of the selected substrates. 2-Octanol gave only the carboxymethyl derivative, while the other secondary alcohols, i.e., n-propyl 2-hydroxypropyl ether and 3-hydroxy-tetrahydrofuran formed methyl derivates in low to modest yields, 9 and 14% yields, respectively. On the other hand, isosorbide showed by far the higher affinity towards methylation, resulting in the quantitative formation of dimethyl isosorbide (DMI). Competitive reactions among isosorbide and secondary alcohols showed the same trend as well. For instance, when 2-octanol and isosorbide were reacted in the same batch reaction, 2-octanol underwent only carboxymethylation reaction, while isosorbide was fully converted into DMI.
The easy methylation of isosorbide has been ascribed to the neighbouring effect of four oxygen atoms all at β position to each other, which enhanced the nucleophilicity of the hydroxyl groups.
Similar assistance of neighbouring ether groups has also been invoked in the case of the methylation, ethylation and benzylation of glycerol acetals, namely formal and solketal. At T ≥ 200 °C, in the presence of K2CO3, glycerol formal (GlyF) and (2,2-dimethyl-1,3-dioxolan-4-yl)methanol (solketal) were alkylated by DMC to produce the corresponding O-methyl ethers with selectivity up to 99% and excellent yields (86–99%).77 The reaction profile of solketal with DMC is reported in Fig. 2.
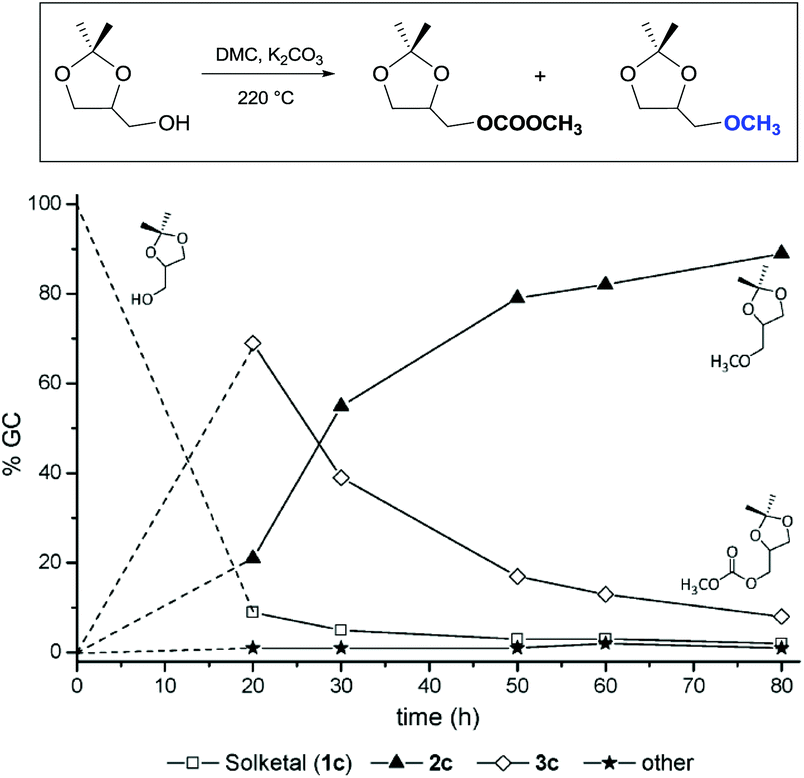 | ||
| Fig. 2 Reaction of solketal with DMC carried out at 220 °C. The stainless steel autoclave was charged with a mixture of solketal (7.7 mmol), DMC (molar ratio of solketal:DMC 1:30) and K2CO3 (molar ratio of substrate:K2CO3 1.0:1.2). | ||
These results corroborated the results previously observed for isosorbide and underlined the importance of the intramolecular base centre in the mechanistic pathway of a BAl2 reaction.
The K2CO3-catalysed etherification of solketal with other carbonates – such as dibenzyl and diethyl carbonate (DBnC and DEC, respectively) – proceeded with the same excellent selectivity observed for DMC. However, at 220 °C, the solketal conversion did not exceed 81%.
In the investigation of isosorbide reactivity, it was also observed that its secondary hydroxyl groups were efficiently methylated by DMC in the presence of a range of bases, without any racemisation (Scheme 22), affording solely DMI as a pure product. It should be mentioned that this molecule (b.p. 235 °C) is industrially appealing as a potential substitute of high-boiling solvents (DMSO and DMF).12c
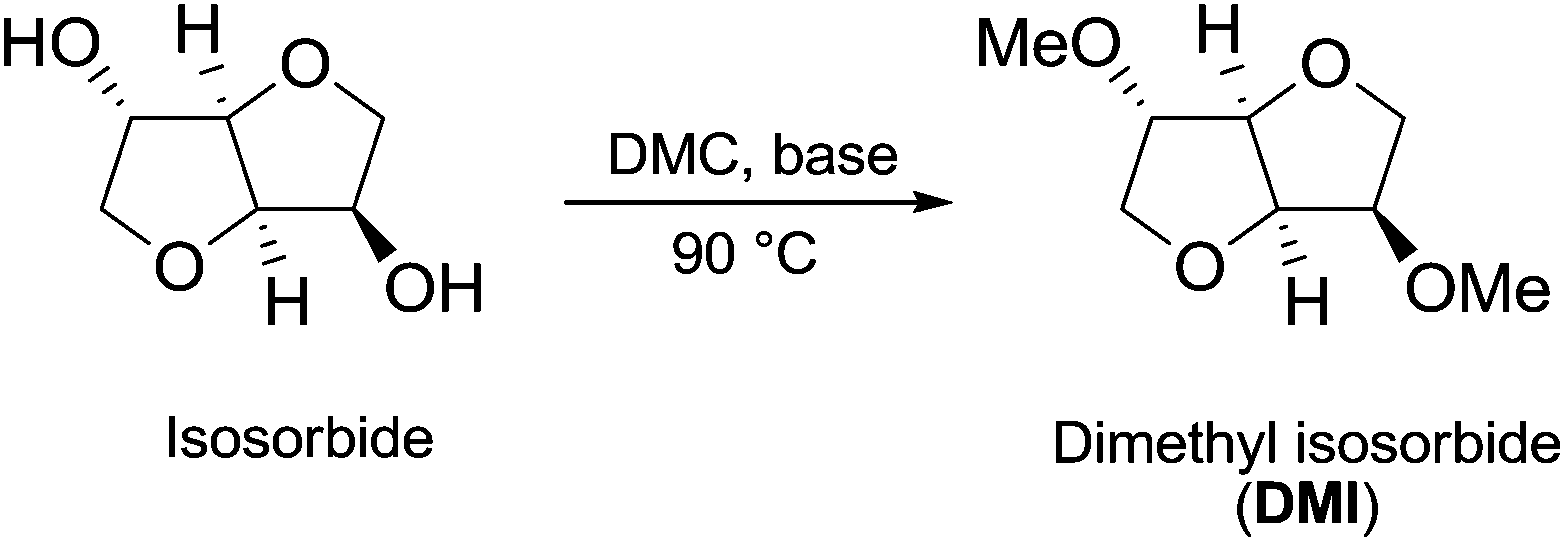 | ||
| Scheme 22 Synthesis of DMI via DMC chemistry.77 | ||
Many trials were carried out aiming at speculating on the reaction mechanism. Methylation, carboxymethylation and decarboxylation reactions are, respectively, depicted in pathways a, b and c in Scheme 23. All these chemical transformations do not involve any chiral centres, which remain unaffected.
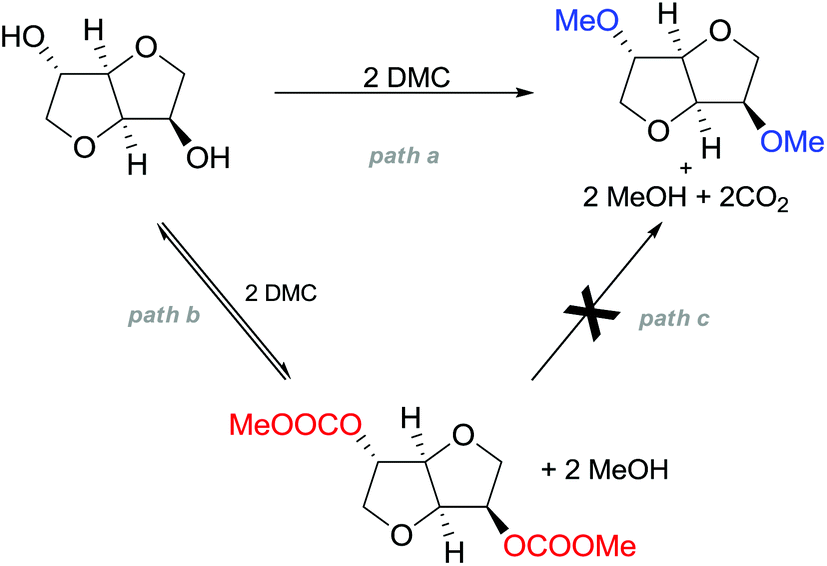 | ||
| Scheme 23 Possible reaction pathways for the methylation of isosorbide. Path a is not an equilibrium reaction; path b is an equilibrium reaction; evidence of path c has never been observed.77 | ||
Path a represents a direct SN2 nucleophilic replacement. Paths (b + c) describe the formation of one intermediate which could generate DMI by a decarboxylation reaction.
In order to test the latter synthetic pathway, dicarboxymethyl isosorbide (DC) was isolated pure and was reacted with NaOMe using either DMF or diglyme as a solvent at 90 °C; however, the formation of DMI did not occur at all, showing that a methylation reaction does not proceed via a decarboxylation reaction (Scheme 23, path c). Thus, according to these results, the mechanism seems to be a distinct BAl2 substitution on DMC.
The remarkable reactivity of isosorbide towards methylation with DMC could be ascribed to its unique structure. In fact, an isosorbide “V”-like configuration is formed by two cis-connected tetrahydrofuran rings with an opening angle of 120°. Besides, as mentioned above, each hydroxyl group is at β-position to both furanic oxygens78 and this vicinity might direct its reactivity towards DMC (Fig. 3).
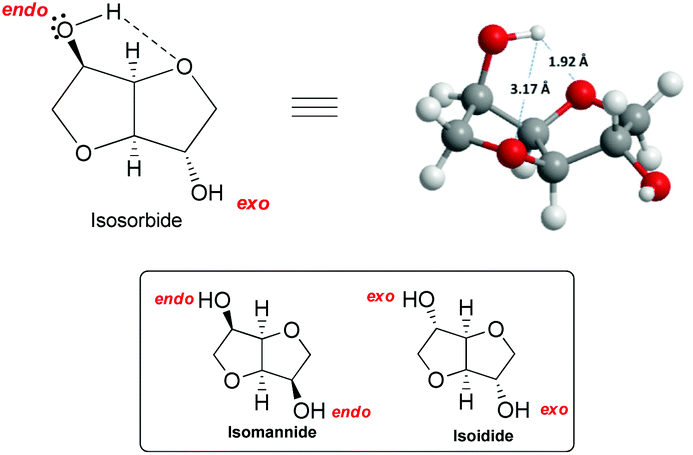 | ||
| Fig. 3 (a) View of isosorbide molecule. (b) The reactivity of the endo hydroxyl group of isosorbide. Hydrogen bond is depicted in the structure (––). | ||
Isosorbide incorporates two secondary hydroxyl moieties, one at the 2-position directed toward the V-shaped cavity labelled as endo, and the second at the 5-position pointing outside of the sugar cavity indicated as exo (Fig. 3). The hydrogen of the endo hydroxyl group can form an intramolecular hydrogen bond, while the exo-OH can be solvated by the ethereal oxygen; the solvation of the latter is necessarily weaker.79
The configuration of the two hydroxyl groups has been shown to influence their reactivity. In fact, isosorbide epimers, isoidide and isomannide – which incorporate, respectively, only exo or endo hydroxyl groups – have different physical/chemical properties and in turn diverse reactivity.
In this view, Table 6 reports the methylation of isomannide and isoidide by DMC conducted under similar reaction conditions to those employed for isosorbide.
| No. | Substrate | Time | Dimethyl etherb | Other productsb |
|---|---|---|---|---|
|
a Reaction conditions: Isosorbide:DMC:K2CO3 1.0:30.0:0.1 eq.; temperature 90 °C. All the reactions have been conducted under anhydrous conditions. Selectivity and conversion have been calculated by GC-MS analysis.
b A mixture of the products reported in Scheme 6.77,78
|
||||
| 1 | Isosorbide | 24 | 98 | 2 |
| 2 | Isomanide | 5 | 93 | 7 |
| 3 | Isoidide | 24 | 61 | 39 |
Isomannide underwent faster methylation reaction possibly as a consequence of the two OH-groups that are both endo and thus involved in H-bonding able to promote the methylation reaction.78
In contrast, when the groups were at exo position as in isoidide, the reaction rate was lower.
A few patents reported on the synthesis of DMI from isosorbide using either zeolite or catalysts such as 4-(N,N-dimethlyamino)pyridine (DMAP). In particular, Archer Daniels Midland Howard Company disclosed that, reacting isosorbide with DMAP as a catalyst and DMC as a solvent, in a microwave at 150 °C for 15 min resulted mainly in the formation of DMI. Monomethyl isosorbide was also detected.80
It should be noted that the synthesis of isosorbide and its subsequent alkylation have always been carried out in two steps under different reaction conditions and catalysts.
However, recently, there was reported a one-pot procedure to produce DMI from D-sorbitol via DMC chemistry employing a single catalyst able to promote both the preparation and derivatization of isosorbide (Scheme 24).81
 | ||
| Scheme 24 One-pot synthesis of DMI.81 | ||
Under the best found reaction conditions, a mixture of D-sorbitol, DMC and the selected catalyst was charged into the autoclave. The reaction was first heated at 90 °C (DMC boiling point) and then the temperature was increased up to 200 °C. Table 7 summarises the results obtained in the presence of different catalysts, i.e., Et3N, 1,5,7-triazabicyclo-[4.4.0]dec-5-ene (TBD), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and hydrotalcite. The best result – 69% conversion – was achieved when TBD was used as a catalyst (Table 7, entry 2), although also Et3N gave DMI in good yield. A small amount of DMI epimers was also detected, i.e., dimethyl isomannide (DMIm) and dimethyl isoidide (DMIi).
| No. | Base | P (bar) | DMI (%) | DMIm (%) | DMIi (%) |
|---|---|---|---|---|---|
|
a Reaction conditions: D-Sorbitol:DMC:catalyst 1.:50:1 eq. in an autoclave at 90 °C for 48 h and then at 200 °C for 24 h; yields were calculated via GC-MS.
b In this experiment, the presence of monomethyl derivatives MMI1 and MMI2 (16% and 11%, respectively) and methyl carboxymethyl derivatives MCE1 and MCE2 (32%) (Scheme 6) was also observed.
c (1/1 w/w).81
|
|||||
| 1 | Et3N | 85 | 60 | 14 | 26 |
| 2 | TDB | 26 | 69 | 14 | 17 |
| 3 | DBU | 58 | 59 | 19 | 22 |
| 4b | KW2000c | 50 | 41 | 0 | 0 |
This high yielding preparation of DMI was quite surprising considering the complexity of the reaction pathway. In fact in order to achieve DMI, D-sorbitol had to undergo a double carboxymethylation (BAc2) followed by a double intramolecular alkylation (BAl2) where the carboxymethyl group acting as the leaving group prompted the formation of isosorbide (Scheme 25).
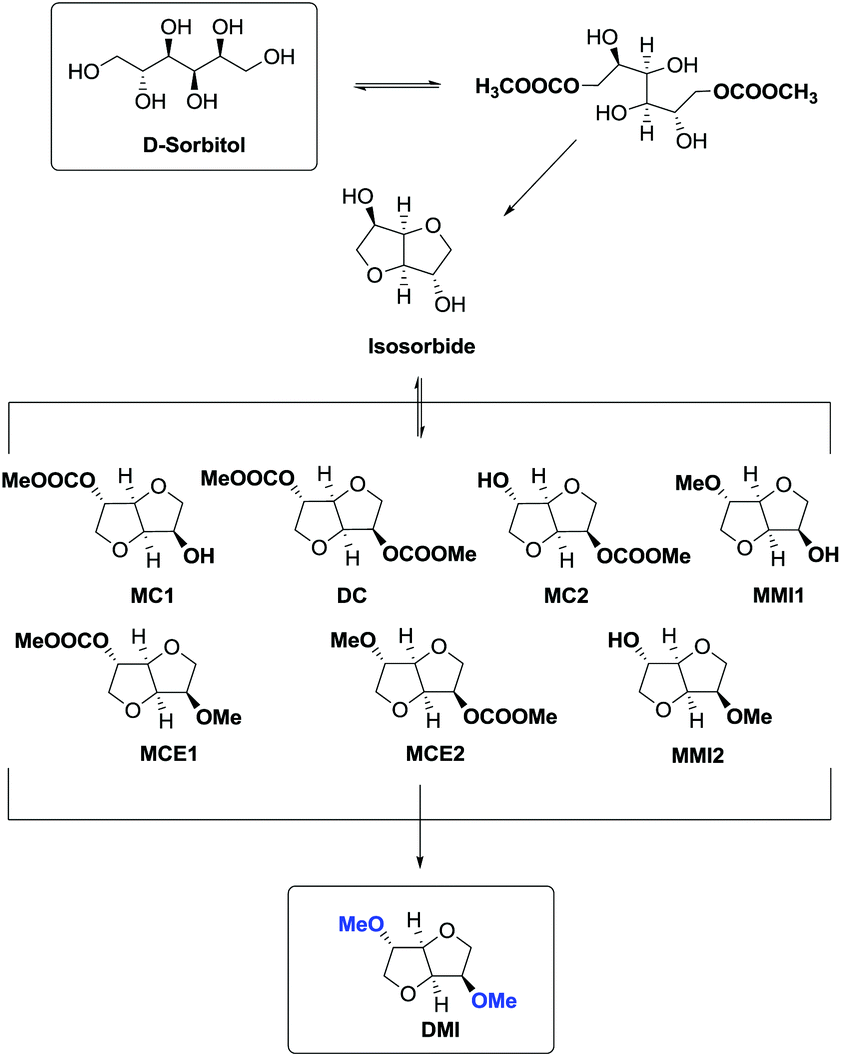 | ||
| Scheme 25 Synthesis of DMI starting from D-sorbitol via DMC chemistry. Reaction intermediates include monomethyl derivatives (MMI1, MMI2), carboxymethyl derivatives (MC1, MC2, DC) and methyl carboxymethyl derivatives (MCE1, MCE2). All the reactions are equilibrium reactions except the last methylations leading to DMI. | ||
The consequential methylation of isosorbide via DMC was complicated by the presence by several by-products as a result of numerous simultaneous reactions.
Simplifying the overall process, the one-pot synthesis of DMI takes place through at least six subsequent reactions.
In this procedure DMC was used to its full extent acting as a carboxymethylating, leaving group in the cyclization reaction and a methylating agent. Furthermore, DMC was also the reaction medium.
In this view, an equally unexpected methylation via DMC chemistry concerns a sugar-based material, i.e. cassava, a woody shrub native to South America of the spurge family, Euphorbiaceae. Cassava is extensively cultivated as an annual crop in tropical and subtropical regions for its edible starchy tuberous root that represents a major source of carbohydrates. Methylation of cassava starch was performed efficiently to a high degree of substitution of 0.6 within 4 min by combining thiourea as a base catalyst and DMC as a solvent and a methylating reagent and applying microwave irradiation as an energy source.82
The improved reactivity of DMC was ascribed to two reasons: the first is the enhanced nucleophilicity of –OH functions due to the internal hydrogen bonds with the cellulose skeleton and the second is the equilibrium reaction among DMC and thiourea, which may create a more reactive intermediate. This intermediate most probably encompasses a methyl group that is more reactive toward nucleophilic displacement (SN2) due to the release of a good leaving group (CH3O− H3N+CSNHCOO−). The suggested reaction mechanism is reported in Scheme 26.
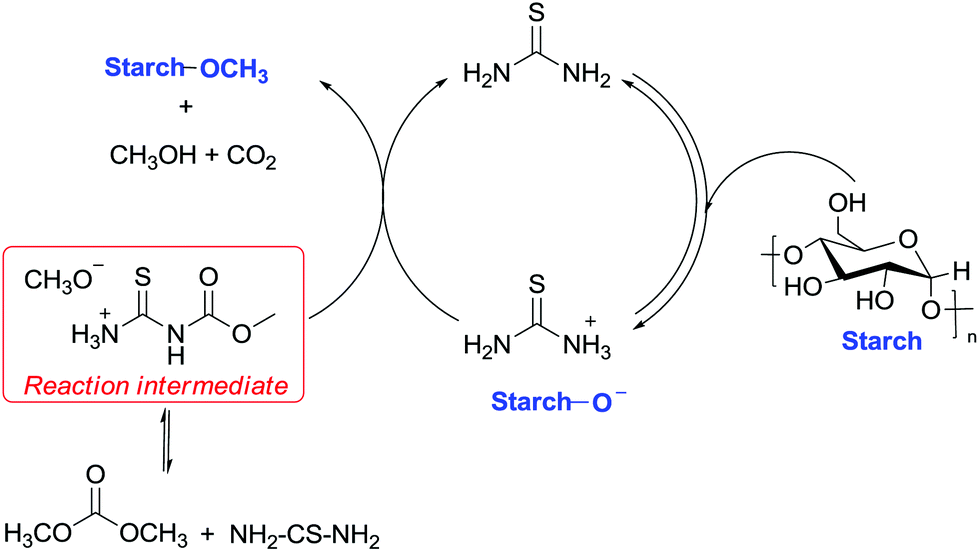 | ||
| Scheme 26 Catalytic cycle for the enhanced methylation of cellulose by DMC catalysed by thiourea. | ||
Another example of direct methylation of hard alkoxides by DMC concerns benzyl alcohols. It was reported that reaction of DMC with several benzyl alcohol derivatives (o- and p-methoxy-, p-hydroxyl-, diphenylmethyl-, and triphenylmethyl-) in the presence of sodium-exchanged faujasites (NaX or NaY) as catalysts led to the formation of the corresponding methyl ethers in yields up to 98%.17 On the other hand, reactions conducted under similar conditions and in the presence of potassium carbonate resulted in the formation of a methoxycarbonyl adduct and dibezyl carbonate (Scheme 27).
 | ||
| Scheme 27 Reaction of DMC with benzyl alcohol. Reactions were carried out using a solution of benzyl alcohol, DMC and faujasites X and Y. The weight ratio of substrate:catalyst:DMC was 1.0:0.2:60.0.17 | ||
Primary, secondary, and tertiary benzyl-type alcohols also gave good yields of the related methylated products by reaction with DMC at 165–200 °C, in the presence of both faujasites NaY and NaX (Scheme 28).83
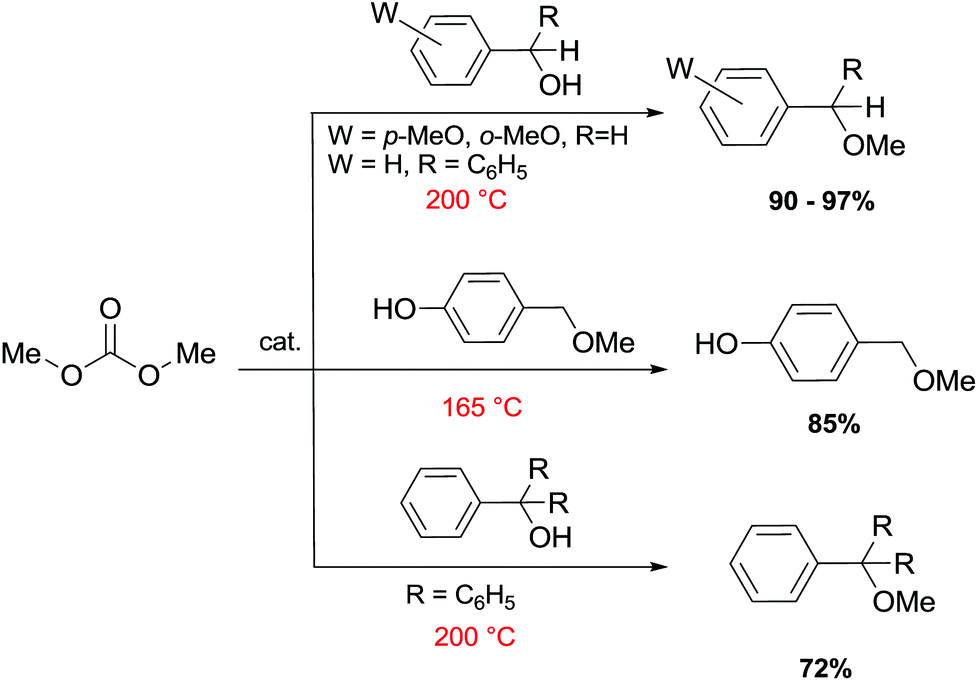 | ||
| Scheme 28 Reaction of DMC with several benzyl alcohol derivatives. General reaction conditions: benzyl alcohol (1.9 mmol), DMC (30 mL) and faujasites X and Y (1.5–3.0 weight ratio).17,75,83 | ||
In the case of hydroxybenzyl alcohols, the reaction was also highly chemoselective: for instance, at 200 °C, methyl p-hydroxybenzyl ether was obtained in 85% yield. Owing to the acidity of the zeolite, the aromatic OH-substituent was fully preserved from methylation and methoxycarbonylation side-reactions (Scheme 28).
However, in the case of substrates with mobile protons at the β-position (i.e. 1-phenylethanol and 1,1-diphenylethanol), the dehydration reaction to olefins was the major, if not the exclusive, process.
The authors stated that as the conditions are quite similar to those already reported by Tundo et al.,75 it might be plausible that the reaction mechanism involves the formation of the unsymmetrical carbonate followed by the decarboxylation reaction. In fact, DMC was employed in a large excess as the reaction medium; thus only methyl benzyl carbonate was eventually present as an intermediate. However, in the meantime, decomposition of DMC into CO2 and dimethyl ether should have also been observed; as no details in that sense were reported, the mechanistic behaviour remains indeterminate.
This reaction was also investigated by Chong et al., who studied the methylation and carboxymethylation reactions of benzyl alcohol over a 3 T cluster of a faujasite zeolite and a 36 T extended framework by density functional theory (DFT).84 The authors proposed that the methylation reactions occur via two transition states, i.e., the formation of the alkoxide (TS1), immediately followed by its attack on the unhindered primary alkyl group of DMC (TS2) leading to the CO bond cleavage. According to the data reported, the authors suggested that methylation via the two-TS pathway was found to be more kinetically stable than a single TS pathway, i.e., direct attack of the benzyl alcohol on DMC to produce the benzyl methyl ether.
 | ||
| Scheme 29 ArOH methylation via DMC chemistry.85 | ||
Unlike the alkylation with alkyl halides, where a stoichiometric amount of base is needed for neutralizing the formed acid, in the case of DACs the base is not consumed and may be used just in catalytic amounts.85
Similarly, the O-methylation of phenol with the less reactive esters takes place under harsh conditions, while the O-methylation of phenols with DACs generally requires temperatures of 100–200 °C depending on the reaction conditions and might be carried out also by using microwave irradiation.86
Among methylating reagents, DMC seems to be the most promising for industrial use as well.2,87 In fact, compared to MeOH, DMC proved to be a more efficient methylating agent under basic conditions, since the reaction can be carried out at lower temperatures and with improved selectivity.88
As an example, ArOEts were obtained in quantitative yields by the reaction of the corresponding ArOHs with DEC in the presence of N,N-dimethylacetamide used as a polar aprotic cosolvent and sodium ethoxide as the base. The reactions were carried out under batch conditions under atmospheric pressure and at a temperature of 137 °C.89
Aryl methyl ethers via DMC chemistry were also synthesised operating under anion activation (phase-transfer catalysed reactions) with excellent yields (>99%) at 100% selectivity for O-methylation. The formation of C-methylation products was not observed.90
Lissel et al.91 were the first to report the methylation of phenols with DMC using phase-transfer (PT) catalysts. The reaction was performed at 373 K for 8 h under atmospheric pressure in an efficient batch system. The reactions of DMC with phenol, p-methylphenol and p-tert-butylphenol in the presence of K2CO3 and 18-crown-6 for 8 h at 373 K gave the corresponding methylated products in 40%, 23% and 36% yields, respectively. 2-Naphthol was also methylated to give 2-methoxy naphthalene in 82% yield.
Ouk and co-workers reported that the PT catalyst obtained combining K2CO3/Bu4N+Br− was the most effective one for the O-methylation of 2,4-dihydroxybenzophenone (2,4-DHB) by DMC.92 Aryl methyl ethers were also obtained in good yields, also when starting from highly hindered phenols, such as 4′-hydroxyacetophenone and 2,6-di-tert-butyl-4-methylphenol.
Interestingly, when 2,4-dihydroxy benzophenone 109 was selected as a substrate in the presence of Bu4N+Br− (5 mol% with respect to the diol), the reaction was regioselective as 2-hydroxy-4-methoxybenzophenone 110 was the only product obtained (Scheme 30). The reaction was carried out at 90–100 °C under atmospheric pressure with DMC as a reagent and a solvent.
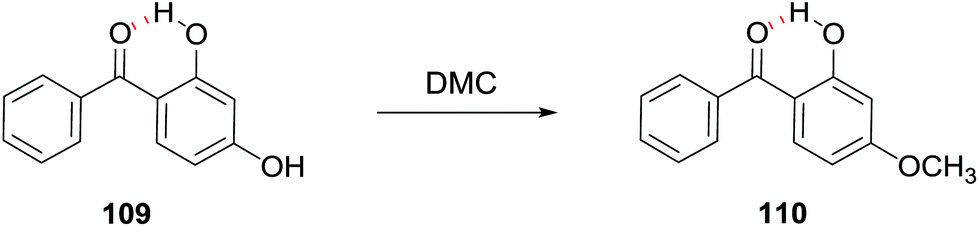 | ||
| Scheme 30 Regioselective synthesis of 2-hydroxy-4-methoxybenzophenone 110.92 | ||
Prior to this study, 2-hydroxy-4-methoxybenzophenone could be synthesised only via selective mono-methylation in an autoclave at the temperature range between 140 and 180 °C in the presence of an alkaline base.93
Aryl methyl ethers obtained from phenols via DMC chemistry can also be synthesised in the presence of many quaternary onium salts, often called Ionic Liquids (ILs).94 Several ammonium salts including 1-n-butyl-3-methylimidazolium chloride ([BMIm]Cl), 1-n-butyl-3-methylimidazolium bromide ([BMIm]Br), 1-n-butyl-3-methylimidazolium tetrafluoroborate ([BMIm]BF4), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([BMIm]PF6) and 1-ethyl-3-methylimidazolium bromide ([EMIm]Br) were synthesised and used as solvents at this scope.95
Aryl methyl ether syntheses were conducted at 120 °C under atmospheric pressure. This procedure has the advantage that sterically hindered phenols can be efficiently O-methylated. However, the amount of onium salt, namely ILs, used was 50% with respect to the substrate to be alkylated.95
Likewise, Ouk et al. reported that O-methylation of phenols with DMC was achieved in good yield in the presence of Bu4N+Br− at 130 °C in a semi-continuous process.96 The reaction was carried out in the presence of Bu4N+Br− (in a 1:1 molar ratio with phenols) at atmospheric pressure.
O-Methylation of mono-, di- and tri-hydroxy benzenes, i.e., phenol, catechol and pyrogallol, with DMC as a solvent in the presence of a phosphonium ionic liquid as a catalyst has also been reported.97 Trihexyl (tetradecyl) phosphonium bromide (PB) was found to be the best catalyst. The maximum conversion and selectivity were found at 200 °C, in a 1:6 molar ratio of the reactant to DMC and at a catalyst concentration of 0.88 × 10−5 mol cm−3.
Recently, it has been shown that SN2 displacement reactions on DMC are enhanced by guanidinium-like compounds used in stoichiometric or catalytic amounts. In particular DMAP and pentaguanidinium salts (i.e. pentamethyl guanidine) were found to be superior catalysts for the preparation of aryl and aralkyl ethers. Usually, the reactions required only a catalytic amount of these compounds (5 mol%).98
Several mono- and dimethylated flavonoids were also synthesised by using DMC as a reagent and a solvent at 90 °C in the presence of a stoichiometric amount of DBU.99
O-Methylation of flavonoids is a common xenobiotic transformation occurring in plants. The products were isolated in high yields and with a high degree of purity (yields >98% with conversions >98%).
Another interesting application of DMC as a methylating agent concerns the O-methylation of aryl alcohols contained in bio-oil. This represents a potential method for upgrading bio-oil100 as the phenolic component is one of the factors leading to the thermal instability of biomass pyrolysis oils.
It was reported that bio oils – obtained from the fast pyrolysis of rice husk (reaction carried out at about 450–550 °C) – were O-methylated in [BMIm]Cl at 130 °C with DMC.101 The phenolic compounds present were completely converted to the corresponding aryl methyl ethers and carboxylic acids were esterified to the corresponding methyl esters. As a result, the modification of phenolic bio-oil has enhanced the heating value by 30%.
Cyclic carbonates for phenol alkylation. Cyclic carbonates have been efficiently employed for the hydroxyalkylation of phenols in the presence of alkali loaded large pore zeolites as catalysts. This study showed that KL type zeolite (a crystalline mesoporous nanomaterial) was selective in producing mono-ethylene glycol phenyl ethers.
In the case of PhOH, 98.5% yield of 2-phenoxyethanol 111 was obtained with a 1.0 ratio of ethylene carbonate (EC/PhOH). A ratio of 2.0 resulted in the formation of an appreciable amount of the diether derivative 2-(2-phenoxyethoxy) ethanol 112 (Scheme 31).102
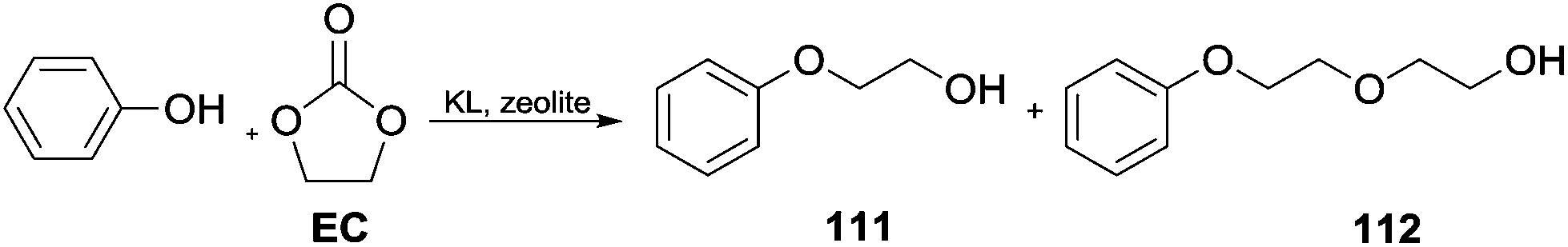 | ||
| Scheme 31 Hydroxyalkylation of phenols by cyclic carbonates catalysed by alkali loaded large pore zeolites.102 | ||
The yield of mono-ethylene glycol phenyl ether was controlled both by the intermediate basicity and the channel architecture of the zeolite structure.
Employing this synthetic approach, various substituted simple phenols were converted into mono-ethylene glycol ether of phenols with >90% yields.
Di-hydroxyl aromatics – such as hydroquinone, 4-4′-biphenol and bisphenol-A (2,2-bis(4-hydroxyphenyl)propane) – also produced the corresponding mono- and di-ethylene glycol ethers. The ratio of ethylene carbonate to phenols was found to be critical for the selective formation of their monoethylene glycol phenyl ether.
When propylene carbonate was used for the hydroxyalkylation of phenol it was found that it has a lower reactivity compared to EC. As expected, the ring opening at the less hindered methylene carbon of 1,2-propylene carbonate prevailed, giving rise to selective formation of β-hydroxy phenyl ethers.
In another example, in situ generated glycerol carbonate (GC) was used for the synthesis of aryloxypropanediols. This synthetic approach is based on the idea of preparing valuable chemicals from renewable sources.103
The protocol was carried out under solvent-free conditions and allowed the utilization of glycerin and DEC instead of GC as a reagent (Scheme 32). Interestingly, the reactivity of the CH2 moiety of DEC in BAl2 reactions is lower than the one in GC, rendering this one-pot synthesis possible.
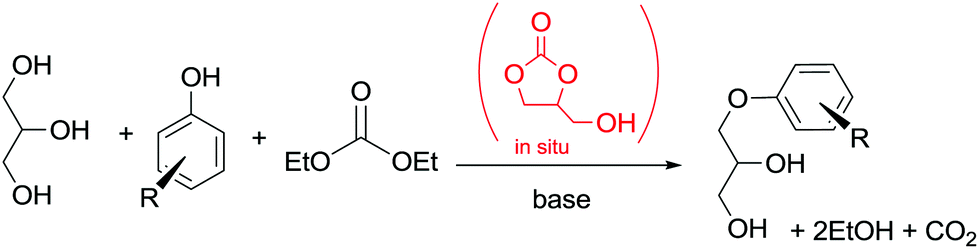 | ||
| Scheme 32 One-pot synthesis of aryloxypropanediols. Yields: R = H, 99%, o-OCH3 99%, p-OCH3 98%, o-CH3 97%; p-Cl 95%; p-isopropyl 99%; α-naphthyl 99%.102 | ||
The method represents an easy route to aryloxypropanediols, which are used as intermediates in biological products and in polymer chemistry. The catalyst and the unreacted reagent could be recycled.
Scheme 33 depicts the reaction conducted in the presence of phenol and the possible by-products that can be formed. In this perspective, the effect of the amounts of DEC and glycerol on the reaction selectivity was investigated (Table 8). The best result was achieved using 1.4 eq. of DEC, and an excess (3 eq.) of glycerol in the presence of a catalytic amount of K2CO3.
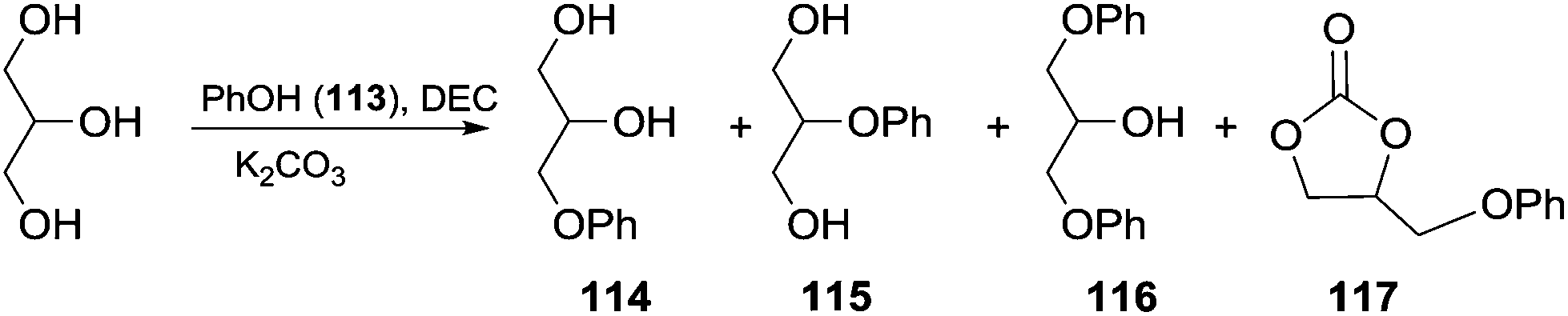 | ||
| Scheme 33 One-pot synthesis of aryloxypropanediol 114via in situ generated GC.103 | ||
| No. | Glycerol (equiv.) | DEC (equiv.) | t (h) | 113 (%) | 114 (%) | 115 (%) | 116 (%) | 117 (%) | GC (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 0.1 equiv. of K2CO3, 105–110 °C; the yields of 114–117 were referenced to phenol 113, and the yields of GC were referenced to DEC and evaluated by NMR analysis after work-up. b This reaction was conducted in the presence of glycerol carbonate; unconverted GC (glycerol found in variable amounts in the crude).103 | |||||||||
| 1 | 1 | 1 | 8 | 19 | 51 | 2 | 19 | 1 | — |
| 2b | GC (1) | — | 8 | 20 | 44 | 2 | 21 | 2 | 1b |
| 3 | 3 | 1 | 12 | 26 | 60 | 2 | 3 | — | — |
| 4 | 3 | 1.4 | 12 | 9 | 71 | 3 | 8 | 3 | 9 |
| 5 | 3 | 1.4 | 18 | 1 | 82 | 4 | 8 | — | — |
Alkylation of phenols by DMC in continuous-flow apparatus. The alkylation reactions are of particular relevance when they can be carried out under continuous-flow (c.-f.) conditions.
Over the years, Gas–Liquid Phase-Transfer Catalysis (GL-PTC) has been applied to many processes and is particularly relevant for DAC chemistry.104
Under GL-PTC conditions, a gaseous stream of reagent and DMC flows over a catalytic bed usually composed of an inert support of a porous inorganic material, usually α-alumina in the form of a spherical extrudate. The latter acts as a support for both an inorganic base (an alkaline carbonate) and a phase-transfer (PT) agent.
Accordingly, the methylation of 2-naphthol with DMC was performed at 180 °C at atmospheric pressure using a continuous-flow gas-phase reaction (Scheme 34). The product, 2-methoxynaphthalene, is an important fragrance (orange scent) and a pharmaceutical intermediate.105
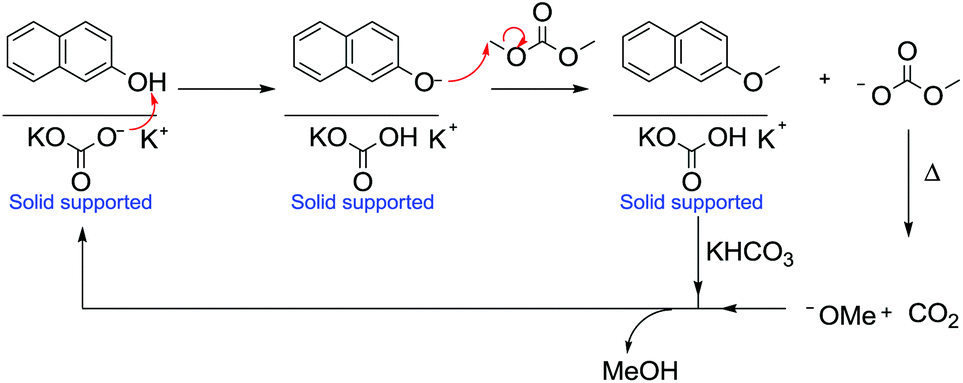 | ||
| Scheme 34 Methylation of 2-naphthol under GL-PTC conditions and the base catalytic cycle.104 | ||
For simplicity, this synthetic approach has been also reported in the form of a students’ laboratory exercise in order to introduce them to the concepts of continuous-flow systems.
A semi-continuous apparatus (Fig. 4) was used for the synthesis of other aryl alkyl ethers (ArOR) in good yields by reacting phenols with DMC or DEC in the presence of potassium carbonate as a catalyst.
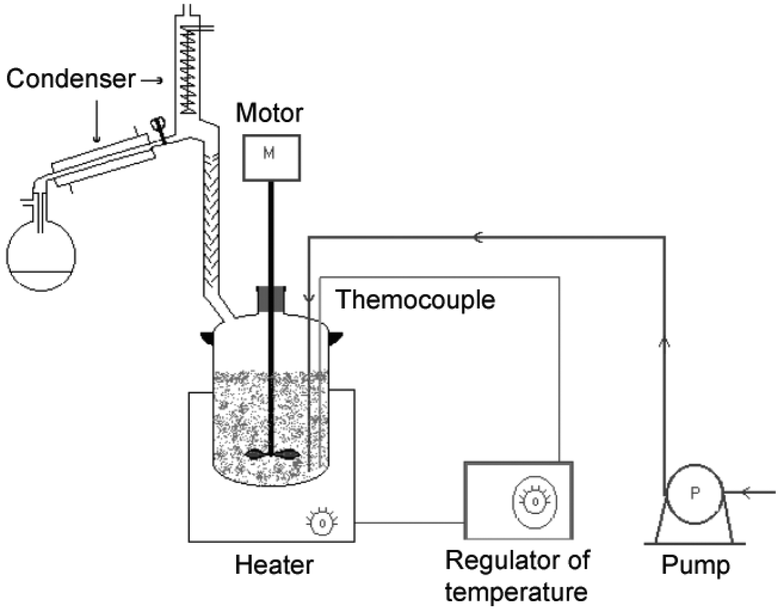 | ||
| Fig. 4 Schematic plot of the reactor used in O-alkylation of phenols with DMC. | ||
In one example the reaction was carried out at 160 °C, in a semi-continuous procedure in which DMC was progressively fed into the preheated reactor already containing p-cresol and K2CO3 under atmospheric pressure, in a homogenous process.
The p-cresol/K2CO3 molar ratio was 120, and after 30 h of reaction, the substrate was totally converted into 4-methylanisole. Other phenols were tested using the same procedure and the reaction was, in any case, totally O-selective except in the case of catechol in which a mixture of by-products was also observed.
O-Methylation of phenol with DMC can also be achieved in vapor phase using alkali–metal ion-exchanged faujasite-type zeolites as the catalysts at 553 K.106 Reactions were carried out in a c.-f. reactor at atmospheric pressure. The catalysts were packed in a reactor of silica tubing placed in a vertical furnace and the mixture of phenol and DMC was fed into the reactor by a syringe.
Alkali exchanged zeolites showed a high catalytic activity for anisole formation, while the zeolites containing alkaline earth metal cations (CaX, MgY) gave a low yield of the product. The latter catalysts gave methyl tolyl ether and cresols as by-products due to their acidic properties.
Among alkali-exchanged zeolites, NaX (X-type zeolite containing Na+ as the cation) gave a high and stable activity: the selectivity for anisole did not change with time on stream, although the activities decayed except for NaX. The highest selectivity was observed for KX (95%), although also NaX gave very good result.
In order to understand better the effect of the catalyst structure on the reactivity of DMC, the interaction of phenol and DMC with acid–base sites in zeolite NaX has been studied using FT-IR, UV and mass spectroscopy.107 At room temperature, phenol is predominantly adsorbed by hydrogen bonding to basic oxygen atoms of NaX. At higher temperature, phenol is partially deprotonated over basic sites to form phenolate anions. The latter undergo H-bonding to zeolitic hydroxy groups and Na+ ions. Meanwhile, DMC forms a chelating complex with Na+ ions at room temperature (Fig. 5, eqn (3)), which decomposes at 150 °C into dimethyl ether and CO2 (Fig. 5, eqn (4)).
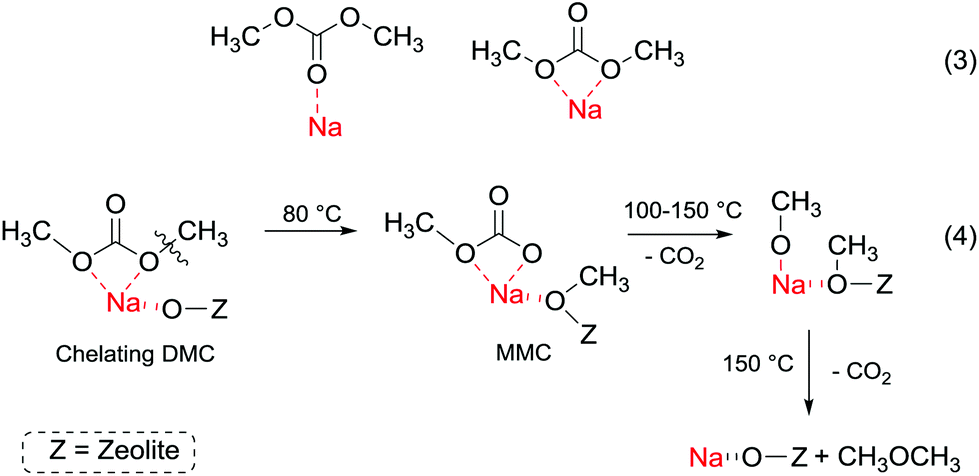 | ||
| Fig. 5 (a) DMC with acid–base sites in zeolite NaX at different temperatures;107 (b) DMC with acid–base sites in zeolite NaX at different temperature. | ||
FT-IR experiments of co-adsorbed reactants showed that, in the presence of phenol, DMC is mainly bonded by its carbonyl oxygen to Lewis acid sites. The formation of anisole occurs at 150 °C and proceeds most likely according to Fig. 6.
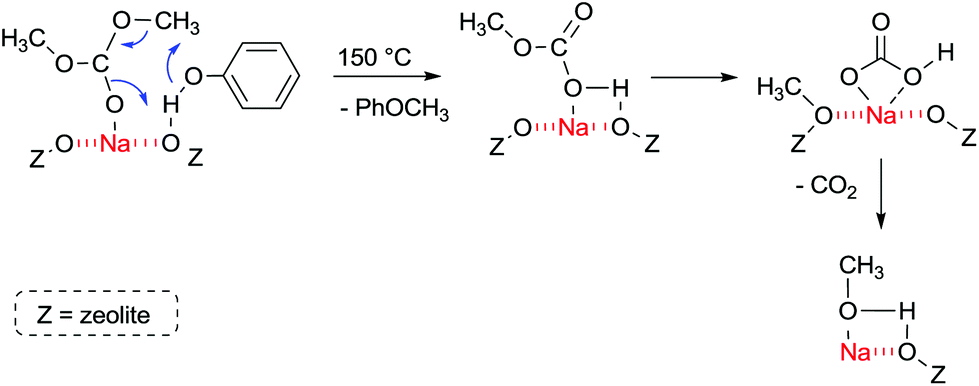 | ||
| Fig. 6 Methylation of phenol with acid–base sites in zeolite NaX at 150 °C.107 | ||
Xue and co-workers reported on the use of newly prepared KNO3/NaY solid base catalysts in the O-ethylation of phenol with DEC.108 The solid catalyst was prepared by supporting KNO3 on NaY zeolites and then calcinated. The performance of the solid base catalyst was investigated for the O-ethylation of phenol with DEC. High selectivity for phenetole along with high conversion of phenol was obtained in the vapour phase.
Base-catalysed methylation of phenols by DMC – in the presence of a small amount of DMF – was also conducted under c.-f. conditions employing microwave-heated reactors.109 The reaction was efficient for different types of phenols, giving a clean reaction in high yields. The transfer of the microwave procedure to the c.-f. method proved to work very well; the reaction could be further optimised to a catalytic procedure with 10 mol% of DBU and DMC (3 eq.).110
The reaction was also of general application to different types of phenols, giving a clean reaction in high yields.
The methylation reaction using 2,3,4,5-tetrahydrothiophene-1,1-dioxide, namely sulfolane, as a solvent instead of DMF was then reported in the presence of 10% DBU.111 Conversion at 220 °C for 10 min was complete and the selectivity toward the corresponding ether was >95%.
Alkylation reactions of dihydroxybenzenes. Selective methylation of ortho-dihydroxybenzene to give the mono and the di derivatives has also been widely investigated under c.-f. conditions.
Ono et al. reported the reaction of catechol with DMC on alumina to give as the main product guaiacol with a selectivity of ca. 70% (Scheme 35).112 The reaction pathway included both guaiacol and catechol carbonate as intermediates. The conversion of catechol was 68% at time-on-stream of 1 h at 553 K, although it decreased to 35% after 5 h. The addition of carbon dioxide to the feed considerably decreased the catechol conversion, indicating that basic sites on alumina were the catalytic centers for methylation.
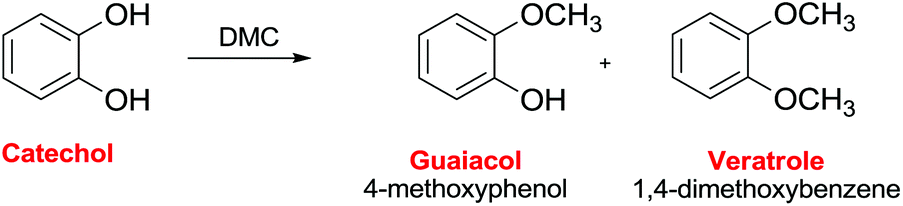 | ||
| Scheme 35 Selective methylation of cathecol.112 | ||
Working with alumina loaded with LiOH (5 mmol g−1), the guaiacol selectivity was 84% at 100% conversion of catechol at 583 K.113 The authors also reported that DMC is a much more efficient methylating agent than methanol for the O-methylation over LiOH/alumina.
Furthermore, alumina loaded with potassium nitrate showed a high catalytic activity for the vapor-phase methylation of catechol with DMC to afford veratrole.114 In this case, a veratrole yield of 97% was obtained at a catechol conversion of 99% at 583 K with a DMC/catechol ratio of 4.0 (Scheme 35).
Calcined Mg–Al hydrotalcite has also been reported as a catalyst in the reaction of catechol with DMC at 250–350 °C.115 Guaiacol and veratrole were obtained as the major products, along with small amounts of catechol carbonate and C-alkylated products. At 573 K, the total O-alkylation (guaiacol and veratrole) was 96.1% with a guaiacol selectivity of 84%. At higher DMC/catechol molar ratios and longer contact times, guaiacol selectivity clearly decreased with a consequent increase of veratrole. An optimum value – of the strength of acid–base pair sites involved in the adsorption of DMC and catechol – renders hydrotalcite-based oxides better catalysts compared to alumina and basic MgO.
Tundo et al. reported the methylation of hydroquinone in a c.-f. reaction over 10 wt% KOH on α-alumina at 553 K with a DMC/hydroquinone ratio of 3.0.116
The formation of p-dimethoxybenzene was 90% at a contact time of 21 s. By working at a lower molar ratio of 1.5 and a contact time of 13 s, conversion of 98.2% and 77.9% yields in the monomethylether was observed, respectively.
Such selectivity in the formation of the monomethylether is higher than those reported for the phase-transfer catalysis system117 (using toluene, aqueous NaOH and CH3I as methylating agents) as well as homogeneous alkylation conditions (using NaOH and KI).118
This result was explained in terms of different partition ratios of hydroquinone and its monomethyl derivative between the two-liquid phase (in the case of LL-PTC) and between a liquid and a gaseous phase (in the case of GL-PTC). Owing to the fact that compound partition arises from different factors, different results should be expected. In fact, in LL-PTC, hydroquinone was less soluble in the organic phase than its monomethyl derivative so the latter can react far more efficiently to yield the dimethyl derivative. When GL-PTC was used instead, partition between liquid and gaseous phases is mainly determined by the boiling points of the compounds involved: hydroquinone (b.p. = 285–288 °C) remained longer in liquid phase than its monomethyl derivative (b.p. = 244 °C); as a result, the latter was more plentiful in the gaseous phase and so it reacted with DMC to a lesser extent than hydroquinone; thus the methylation was more selective.
Some reactions of DMC with phenol and pyrocatechol have been industrially carried out in the pilot plant described in Fig. 4.38
A comprehensive study of the O-methylation of dihydroxybenzenes (DHBs) catechol, resorcinol, and hydroquinone with DMC was carried out on MgO and alkali metal ion (Li+, K+, and Cs+)-loaded MgO under vapor-phase conditions.119 Selective O-methylation of DHBs to methoxyphenols (MPs) was carried out with DMC in the temperature range of 523 and 603 K.
When pure MgO was employed, O-methylation activity declined rapidly with time on stream. On the other hand, alkali metal ion-loaded MgO showed better activity with all substrates. Among the catalyst systems screened, K+-MgO showed the best selectivity for mono O-methylation.
Catalytic activity and product selectivity varied with respect to DHB substrates. Selectivity for O-methylated products increased with increasing basicity of alkali ions. K+-MgO showed high and stable activity toward MPs. Selectivity for the MPs obtained from three substrates increased in the following order: hydroquinone > resorcinol > catechol. The mode of interaction of substrates on the catalyst surface influenced the reactivity and product selectivity.
One of the most commonly used methods for the preparation of asymmetric sulfides is the alkylation of thiols with alkyl halides. In 1975 Tamura et al. reported that the alkylation of thiols with DACs offered an alternative method for the synthesis of sulfides.120
As an example, sodium benzenethiolate was treated with DEC in refluxing ethanol for 15 h to give ethyl phenyl sulfide in a 70% yield (Scheme 36).
 | ||
| Scheme 36 Synthesis of ethyl phenyl sulphide via DEC. | ||
Methylation of thiols has been then described also via phase-transfer catalysis using a system composed of K2CO3 and 18-crown-6. The reaction was carried out at 373 K for 8 h in very good yield (Scheme 37).121
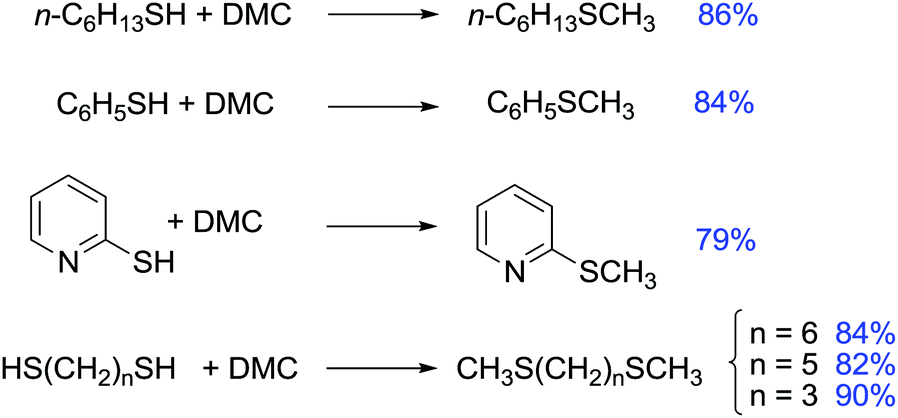 | ||
| Scheme 37 Examples of methylation of thiols by DMC in phase-transfer catalysis.120,121 | ||
Benzenethiol was also converted into the corresponding thioether under gas–liquid phase-transfer conditions. Similarly, the reaction of n-octylmercaptan with DMC gave the corresponding thioether at 453 K.122
According to Scheme 38, when a mixture of gaseous DMC and mercaptans flows through a catalytic bed of K2CO3/PEGs, thioethers are produced.
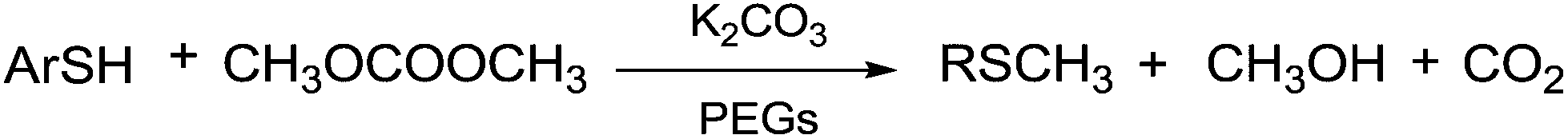 | ||
| Scheme 38 Methylation of mercaptans by DMC under c.-f. conditions.122 | ||
An efficient S-methylation of mercaptans or benzenethiols with DMC at room temperature in the ionic liquid [Bmim]Cl as a solvent has also been reported.123
Similarly, the reaction of HS-containing heterocycles with DMC in the presence of K2CO3 and Bu4N+Br− gave as major products the heteroaryl methyl thioethers in 44–93% yields.124 This method provided a useful synthetic method for the preparation of various heteroaryl methyl thioethers without the use of methylic halides or dimethyl sulfate.
NaY faujasite has also been investigated as an efficient catalyst for the reactions of DMC with several S-ambident nucleophiles such as o- and p-mercaptophenols.125
Reactions were performed at 150 °C showing high chemoselectivity as the substrates undergo only an S-methylation reaction, without affecting –OH or the –CO2H moieties. Typical selectivities are in the range of 90–98% (Scheme 39).
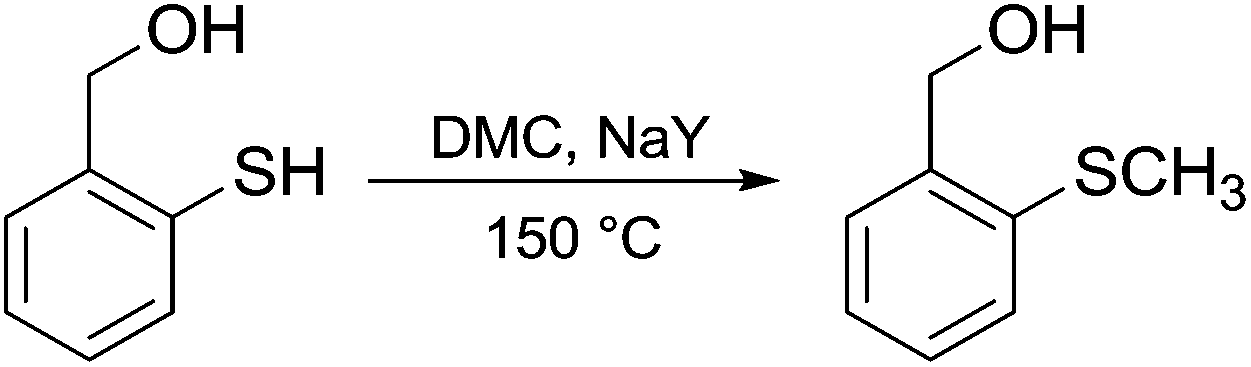 | ||
| Scheme 39 Methylation of mercaptophenols by DMC in the presence of NaY faujasite.125,126 | ||
The comparison between the methylation via DMC in the presence of the K2CO3 base-catalyst or NaY faujasite showed that the zeolite was always less active than the base. However, NaY was by far the best catalyst in terms of chemoselectivity, i.e., at 150 °C, mercaptobenzoic acids undergo S-methylation reactions without affecting the acid groups.
In contrast, in the presence of the K2CO3 catalyst, competitive reactions of O- and S-methylation, esterification, and O-methoxycarbonylation occurred simultaneously. K2CO3 and NaY allowed a comparable selectivity solely in the case of mercaptophenols, where hydroxythioanisoles were the only products in >90% yields.
Methylation of CH2-acidic compounds. Methylation and alkylation reactions on CH2-acidic compounds – carried out under batch and c.-f. conditions with DACs – were performed on a number of different substrates.126
Arylacetonitriles and methyl arylacetates (ArCH2X; X = CN, CO2Me, respectively) react with DMC (20 molar excess) at 180–200 °C in the presence of K2CO3 to produce monomethylated 2-arylpropionitriles and methyl 2-arylpropionates, respectively, with a selectivity >99.5% (Scheme 40).127
 | ||
| Scheme 40 Methylation of methyl aryloxyacetates and aryloxyacetonitriles by DMC chemistry.127 | ||
The reaction, with wide application, proceeds by DMC acting as a methoxycarbonylating agent towards the ArCH–X anion (X = CN, CO2Me) and as a methylating agent to the ArC–(CO2Me)X anion. DMC also proved to be the best solvent for such reactions.
This mechanism mirrors the allylic alkylation of Tsuji and Trost (see section 3.2) where the base is continuously regenerated by the decomposition of the methoxycarbonate anion into CO2 and the methoxide anion.
The unusual mono-C-methylation selectivity (>99%) of arylacetic acid derivatives with DMC is due to a reaction pathway that involves consecutive methoxycarbonylation, methylation, and demethoxycarbonylation steps.
In this synthetic procedure, the methoxycarbonyl group plays a double role, i.e., (i) it enhances the acidity of ArCH(COOCH3)X, favouring the formation of the corresponding anion, and (ii) it acts as protecting group which prevents further methylation. The key step is the attack of the ArC−(COOCH3)X anion on a second DMC molecule. These results are in perfect agreement with the Hard–Soft Acid–Base (HSAB) theory.10c The combination of the dual electrophilic character of DMC with its reaction products allows two consecutive steps to occur in a selective way: first the hard–hard reaction occurs leading to the formation of a soft anion; then a soft–soft nucleophilic displacement leads to the final product. Since hard–soft and soft–hard interactions are inhibited, neither double methylation nor double carboxymethylation occurs (see also Fig. 1).
In order to achieve the related mono-methyl derivatives 2-aroxypropionitriles and methyl 2-aroxypropionates in quantitative yield, the methylation of both arylacetonitriles and arylacetoesters was carried out under batch conditions, at 180–200 °C, in the presence of K2CO3 or t-BuOK as the catalyst. Also in the presence of 30 molar excess with respect to the substrate, no dialkylated by-products were formed.
In general, the reaction occurred faster on nitriles than on esters, which required higher temperatures and longer reaction times for the conversion to be completed.
It is noteworthy that in the case of methyl sulfones (ArSO2Me), the reaction proceeded with the homologation of the methyl to an isopropyl group (ArSO2CH3 to ArSO2CH(CH3)2).
Immobilised catalysts were also used as solid base for the methylation of acetonitrile. These catalysts were prepared by ion exchange of zeolites and Na–Al-MCM-41 and by organosilane grafting over mesoporous materials.128 The reaction was carried out in a stainless-steel Parr autoclave equipped with a mechanical stirrer. Operating at 180 °C, 90% selectivity towards the mono-methyl derivative was observed at 82% conversion.
Alkylation of CH2 acidic compounds by DACs. Although the alkylation of methylene active compounds by alkyl halides in the presence of K2CO3 and DMF has been extensively reported, a high monoalkyl selectivity is elusive when reactive halides such as benzyl halide are involved.129 In contrast, dibenzyl carbonate (DBnC) is also an efficient benzylating agent of both methylene-active compounds and phenol at high temperature (150 °C–180 °C).
The high boiling point of DBnC allows benzylation to be performed batchwise at atmospheric pressure. In a typical reaction a mixture of the substrates DBnC and K2CO3 in a 1.0:1.1–1.5:2.0 molar ratio reacted in a refluxing high boiling solvent, the best solvents being N,N-dimethyl- and N,N-diethyl-formamide (DMF and DEF, respectively).
Also, O-benzylation of phenol by DBnC gave benzyl phenyl ether in a good yield. Specific examples are reported in Table 9.
| No. | Substrate ArCH2CN | Solv. | DACs | Temp. (°C) | Time (h) | Productb | Yieldc (%) |
|---|---|---|---|---|---|---|---|
|
a All reactions were carried out in an autoclave using substrate, DAC and base (entries 1–4: 1.0:18.0:2.0; entry 5: 1.0:1.1:2.0; entry 6: 1.0:1.5:2).
b Conversions were determined by gas chromatography.
c All yields are based on distilled products.
d Yields are based on isolated product.129
|
|||||||
| 1 | PhCH2CN | — | DMC | 180 | 3.75 | PhCH(Me)CN | 90c |
| 2b | PhCH2CO2Me | — | DMC | 220 | 8 | PhCH(Me)COOMe | 80c |
| 3 | PhCH2CN | — | DEC | 180 | 47.5 | PhCH(Et)CO2Me | 97c |
| 4 | PhCH2CO2Et | — | DEC | 220 | 14.3 | PhCH(Et)CO2Et | 12c |
| 5 | PhCH2CN | DMF | DBnC | 155 | 6.0 | PhCH(Bz)CN | 82d |
| 6 | PhCH2CO2CH2Ph | DEF | DBnC | 177 | 4.5 | PhCH(Bn)CO2CH2Ph | 83d |
Methylation of lactones. DMC has been also employed for the selective mono-methylation at the α-position of lactones, i.e., γ-butyrolactone (Scheme 41, eqn (44)).128 When a 1
:10 (molar ratio) mixture of γ-butyrolactone and DMC was passed over a catalytic bed (300 g of α-alumina coated with 5 wt% of PEG 6000 and 5 wt% of K2CO3) at 180 °C and a flow rate of 6.0 mL h−1, 61% conversion to the only mono-methyl derivative was observed. This process allows the easy one-pot production of high-cost compounds.
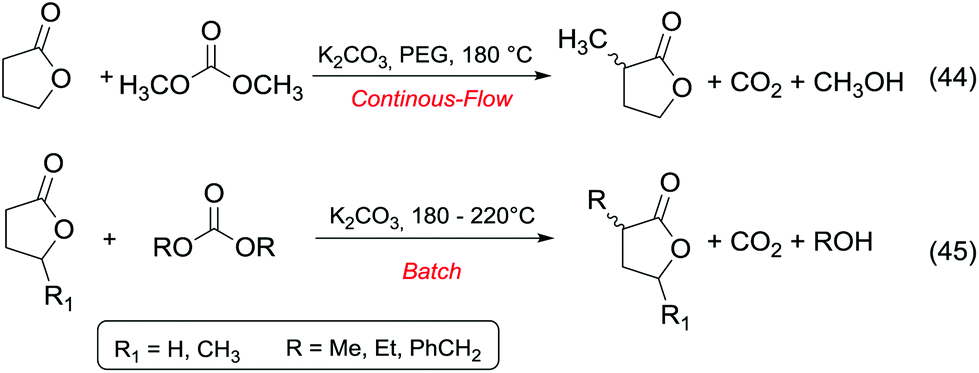 | ||
| Scheme 41 Selective monomethylation of lactones by DMC.128 | ||
Recently, a similar procedure has been used for the alkylation of γ-butyrolactone and δ-valerolactone under batch conditions.5a In a typical procedure, the selected lactone was reacted with DMC, DEC or DBnC in the presence of a stoichiometric amount of K2CO3 at 200–220 °C in an autoclave for 24 hours. α-Alkyl derivatives formed as major products in 45–82% yields. The proposed reaction mechanism is similar to the one discussed above for the selective monomethylation of CH2-acidic compounds.
Methylation of alkenes. Although not strictly related to the CH2 acidic compounds, an interesting industrial application of DMC is its use as a methyating agent of 2,3-dimethylbut-2-ene to give triptene.
Triptene, namely 2,3,3-trimethylbut-1-ene, is part of the triptyls, the general term for a mixture containing 2,2,3-trimethylbutane (commonly known as triptane) and triptene. Triptyls are liquid antiknock additives used in aviation fuels. Triptane has the molecular formula C7H16 and is the most compact and heavily branched of the heptane isomers, the only one with a butane (C4) backbone.
Triptane (Scheme 42) has a research octane number, RON, of 113 and can be blended into gasoline to increase its octane number and it is produced by reacting methanol over ZnI2 or InI3130 or by reacting dimethyl ether over solid acids.131 Triptene has a slightly lower RON of 108; however, it can be easily hydrogenated to triptane. A research goal is the development of a process for the production of octane-rich fuel additives from lighter olefins.
 | ||
| Scheme 42 Selective monomethylation of lactones by DMC.132 | ||
In this perspective, a series of methylating reagents: methanol, dimethyl ether and DMC, have been evaluated for their ability to methylate 2,3-dimethylbut-2-ene to yield triptyls. Zeolite Y, zeolites FER, Beta, ZSM-5 and MOR were used as catalysts.
It was demonstrated that the methylation of 2,3-dimethylbut-2-ene (Scheme 42) can be achieved in much higher yield using DMC as opposed to MeOH or DME reactants over an H-Y catalyst, which has been shown to be the best catalyst among a series of zeolites tested.132
Furthermore, it has been shown that the double bond isomerisation of 2,3-dimethylbut-2-ene to 2,3-dimethylbut-1-ene did not impact the selectivity of the methylation reaction towards 2,3,3-trimethylbut-1-ene.
Alkylation of aliphatic amines. DAcs have been used as alkylating agents of primary, secondary and tertiary amines, as well as of ammonia halide salts (NH4+Hal−). In these reactions DACs were confirmed to be a versatile family of ambiphilic electrophiles, which generated an unusual double reactivity perfectly explained in terms of Pearson's HSAB theory.10 Accordingly to this theory, it was possible to prepare urethanes and N-methyl derivatives starting from aliphatic and aromatic amines.133 However, as DACs are less reactive than the normally used chlorine-based alkylating agents, they need to be activated either by a higher reaction temperature or by employing catalysts. In particular, depending on the catalyst selected a different selectivity in the products has been observed.
Generally, DACs could be activated by bases, by anion activation (phase-transfer catalysis) and by acidic conditions like Lewis acids134 and zeolite. All these approaches have been used for either aliphatic or aromatic amines, although to different extents, as the latter are notoriously scarcely active as nucleophiles.
Under neutral conditions (without considering the basicity of the substrate), aliphatic and aromatic amines do not react selectively with DMC, as both BAl2 and BAc2 mechanisms occur concurrently (Scheme 43). However, in the presence of strong bases, the BAc2 mechanism accelerates, so that urethanes formed in a time span of a few minutes.
 | ||
| Scheme 43 Reaction of benzylamine with DMC in the absence of a base. | ||
Strong bases – such as potassium tert-butoxide or sodium methoxide – catalyse the reaction of aliphatic and aromatic amines to give the corresponding carbamates quantitatively at 90 °C. Since aliphatic amines are harder nucleophiles than aromatic ones, they react faster with DMC (Table 10).
| No. | Amine | Time (min) | Base | Carbamate (%) |
|---|---|---|---|---|
| Reaction conditions: Reflux temperature. 90 °C; amine:base:DMC molar ratio: 1.0:1.2:40.0.a After 18 h 73% of N-methyl carbamate.b After 18 h 87% of N-methyl carbamate.c After 0.5 h 70% of N-methyl carbamate.d After 6 h, 78% of N-methyl carbamate.133 |
||||
| 1 | p-Anisidinea | 25 | C4H9OK | 77 |
| 2 | p-Chloroanilineb | 20 | C4H9OK | 86 |
| 3 | Benzylaminec | 1 | C4H9OK | 100 |
| 4 | n-Decylamined | 3 | CH3ONa | 100 |
| 5 | 1,6-Hexanediamine | 10 | CH3ONa | 100 |
| 6 | n-Octylamine | 5 | CH3ONa | 100 |
| 7 | Phenethylamine | 5 | CH3ONa | 100 |
Under the conditions of entry 4 (Table 10), n-decylamine reacted with DMC under a nitrogen atmosphere to give, after 3 min, 100% of the carbamate. By protracting the reaction, the initially formed carbamates underwent a further reaction with DMC and gave the corresponding N-methylurethanes in 99% yield refluxing for 21 hours (20% after 2 h, and 46% after 5 h).
It should be pointed out that once formed, the urethanes needed the presence of a base to further react with DMC. Under these conditions, the related RN–COOCH3 anions are softer nucleophiles than the starting amines due to higher polarizability and underwent selectively BAl2 reactions. Double methoxycarbonylation products (RN(COOCH3)2) were never observed.
It is, in fact, well known that bases significantly accelerate aminolysis and transamination reactions.135 Thus, also in this case, the base removed H+ from the protonated nitrogen during or after the attack, thereby increasing the negative charge on the nitrogen atom.136
The reactivity of amines with DMC can be explained according to the HSAB theory. As the DMC molecule presents ambident electrophilic character, it reacts with hard nucleophiles (amines in the presence of strong bases) according to a BAc2 mechanism, forming the related carbamates. The anions of the so-produced urethanes, being softer in character, can then react via a BAl2 mechanism with the softer part of the molecule, the methyl group, and give the N-methyl derivatives.
In conclusion, the dual electrophilic character of DMC allows two consecutive steps to occur both in a selective way. As previously stated, since hard–soft and soft–hard interactions are inhibited, double methylation and double methoxycarbonylation do not occur.
Alkylation of amines via phase-transfer catalysts. Dibenzyl carbonate (DBnC) in the presence of catalytic amounts of tetraalkylphosphonium salts allowed primary aliphatic amines (RNH2; R = PhCH2, Ph(CH2)2, n-decyl, and 1-naphthylmethyl) to be efficiently N-benzylated to the corresponding RN(CH2Ph)2 under solventless conditions (Table 11)137 In comparison with the reaction carried out without quaternary salts, where the competitive formation of the benzyl carbamate is favoured, the phosphonium salt promoted selectivity toward the benzylated amine, showing also an increase in the reaction rate.
However, in a single case, i.e., 4-(aminomethyl)benzoic acid, both N,N-dibenzylation and esterification of the acid group were observed.
Analysis of the IR vibrational modes of benzylamine in the presence of tetrabutylphosphonium bromide supported the hypothesis that the enhanced selectivity, observed for this synthetic approach, may be due to an acid–base interaction between the salt and the amine, which increases the steric bulk of the amine and favors the attack of the nucleophile on the less hindered alkyl terminus of DBnC.
Synthesis of quaternary ammonium salts. DACs have been also used for the synthesis of quaternary ammonium salts from simple ammonium halides (NH4+Hal−) over an ionic liquid catalyst, 1-ethyl-3-methylimidazolium bromide (EMImBr).138 The latter was prepared according to procedures described in the literature.139
In a typical reaction, the ammonium halide, DAC, and EMImBr were transferred into a stainless steel autoclave and heated to 443 K for 8 hours. Quaternary ammonium salt yields depend on both the ammonium salt and the alkyl group on DACs. When DMC was reacted with different ammonium salts, the quaternary ammonium salt yield followed the anion order of Br– ∼ NO3− ∼ Cl− > F− > SO42− > C2O42− ∼ CO32− ∼ CH3COO− (Table 12). The results reported in Table 12 demonstrate that for the same ammonium salt anion, DMC was a much better alkylation reagent than DEC. Dibenzyl carbonate was also a good alkylation reagent, showing a similar alkylation efficiency to that of DMC.
| No. | Ammonium salt | DAC | Product | Yield (%) |
|---|---|---|---|---|
|
a In each reaction, 2.0 mmol of AM was employed. The molar ratio of hydrogen or nitrogen of the ammonium ion to DAC is 1:1 and the amount of EMImBr is 10.0 mol% based on DAC.138
|
||||
| 1 | NH4F | DMC | [N(CH3)4]F | 82 |
| 2 | NH4Cl | DMC | [N(CH3)4]Cl | 96 |
| 3 | NH4Cl | DEC | [N(CH3)4]Cl | 0 |
| 4 | NH4Br | DMC | [N(CH3)4]Cl | 96 |
| 5 | (NH4)2SO4 | DMC | [N(CH3)4]2SO4 | 70 |
| 6 | (NH4)2C2O4 | DMC | [N(CH3)4]2C2O4 | 48 |
| 7 | (NH4)2CO3 | DEC | [N(CH3)4]2CO3 | 2 |
With only a few exceptions, i.e., ammonium carbonate (with DMC), ammonium oxalate (with DMC), and ammonium salts (with DEC), quaternary ammonium salt were achieved in yields above 80%. Moreover, it was found that the electron-donating property of the alkyl moieties of ammonium cations, the electrophilic nature of the alkyl group of the carbonate, the acidity of the acid related to the ammonium salt, the steric hindrance of the ammonium salts and the DAC used are all key factors that influence the yields of quaternary ammonium salts.140
For ammonium salts bearing bulky alkyl groups on the nitrogen atoms, such as cyclohexyl and n-C12H25, high yields of quaternary ammonium salts were achieved as well.
In contrast with the reported methods141 which proved to be efficient solely in the case of tertiary amines, this synthesis of quaternary ammonium salts worked for almost all kinds of ammonium salts, primary and secondary aliphatic ammines. Tertiary ammines have already been reported to give the methyl trialkyl ammonium methyl carbonate in quantitative yield.141
An interesting example of the synthesis of quaternary ammonium salts is the synthesis of pentaalkylmethylguanidinium methylcarbonates from the reaction of pentaalkylguanidines and DMC (Scheme 44).142
 | ||
| Scheme 44 Reaction of pentaalkylguanidines with DMC and subsequent anion exchange via acid–base reaction. (i) 120 °C, 3 d; (ii) 50 °C, 6 h. R′ = Et, Bu, iOct; R2 = Et, Bu.142 | ||
Most of the title compounds are room temperature ionic liquids which provide convenient access to halide-free guanidinium-based ILs via acid–base reactions and subsequent decarboxylation, similarly to industrially important imidazolium methylcarbonates.
Methylation of aliphatic amines employing acidic catalysts. MY faujasites are efficient catalysts for the N-methylation of aliphatic amines via DMC chemistry.143 The high chemoselectivity of the reaction (methylation versus carbamation) was reached through the tuning of the acid–base properties of the zeolites and by continual removal of CO2 from the reactant mixture.
N-Methylations of aliphatic amines [XC6H4(CH2)nNH2; n = 1, X = H, o-MeO, p-MeO; n = 2, X = H, o-MeO; PhCH(Me)NH2] with DMC led to N-methyl- and N,N-dimethyl-amines (RNHMe and RNMe2) in good overall yields (70–90%).144 Conversely, when the reaction was performed in the presence of CO2, the related carbamates (RNHCO2Me) were competitively formed to a large extent.145
This methylation reaction probably proceeds through a BAl2 displacement of the amine on DMC (Scheme 45). Importantly, the procedure has valuable potential for the synthesis of tertiary amines as the mono-N- versus N,N-dimethylation selectivity cannot be controlled.
 | ||
| Scheme 45 Mono-methylation selectivity of RNH2 with the DMC competition mechanism. | ||
Alkylation of aromatic amines. Aromatic amines are softer in character than aliphatic ones, and thus they showed a remarkable selectivity toward mono-alkylation reactions.
In particular, there has been reported a straightforward and selective N-mono-methylation of anilines incorporating a variety of functional groups, which remained unaffected albeit susceptible to undergo methylation reactions themselves.146 This chemoselective methylation was catalysed by NaY faujasite, and DMC was used as a solvent and a reagent. Several functionalised anilines – such as aminophenols, aminobenzyl alcohols, aminobenzoic acids and aminobenzamides – have been investigated as substrates. Scheme 46 reports a comparison of the selectivity in methylation reaction via DMC either in the presence of NaY or in the presence of a base.
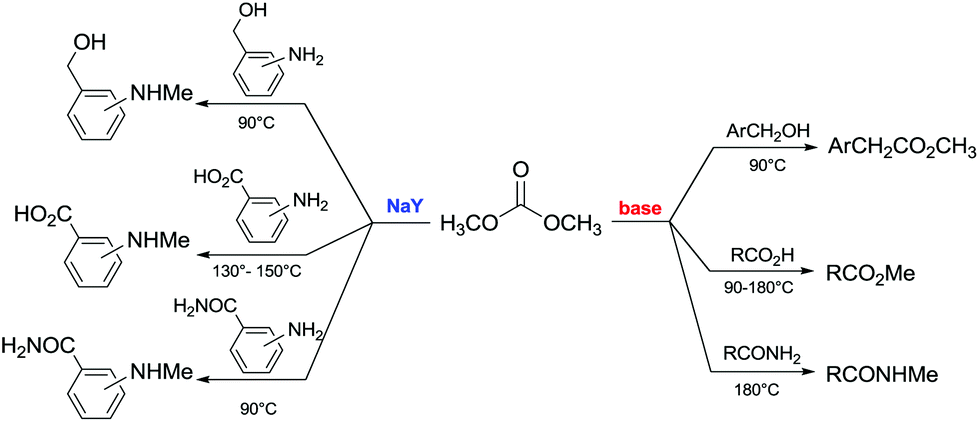 | ||
| Scheme 46 Selectivity in methylation reaction with DMC in the presence of NaY and in the presence of a base.143–145 | ||
In the presence of zeolites, the reaction proceeded with the exclusive formation of N-methylanilines without any concurrent O-methylation or N-/O-methoxy carbonylation side processes. In particular, solely mono-N-methyl derivatives [XC6H4NHMe, X = o-, m-, and p-OH; o- and p-CH2OH; o- and p-CO2H; o- and p-CONH2] were obtained with a selectivity of up to 99% and isolated yields of 74–99%.
Aminobenzoic acid is the only compound of this series to require a higher reaction temperature (130 °C).
Particularly notable is the reaction of p-aminophenol reported in Scheme 47. In this case the chemoselectivity was particularly evident. O-Aminophenol behaved similarly, thus it led to N-methyl o-hydroxyaniline in 91% yield.
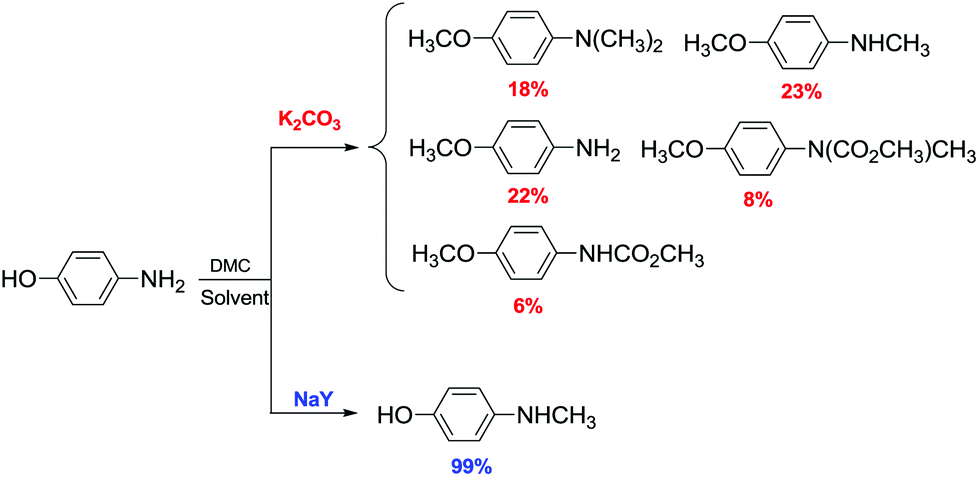 | ||
| Scheme 47 Selective N-methylation of p-aminophenol with DMC in the presence of faujasites.147 | ||
For these reactions, the nature of the co-solvent is critical: glycol-derived dimethyl ethers such as triglyme and glyme give the best results while the methylation is hindered when carried out in DMF and MeCN.
In order to understand better the reaction mechanism, a study of the modification of the IR properties of DMC, induced by the interaction with NaY has been performed and the experimental results obtained were compared with ab initio simulations.147
It was observed that the enhanced activity and selectivity was due to the electric fields present in the confined spaces of the supercages. The interaction of DMC with Na+ ions present in the NaY structure is the key factor in promoting the methylation and carboxymethylation activity of DMC.
Also, arylenediamines can be mono-N-alkylated by DACs in the presence of the NaY zeolite catalyst in a regioselective process (Scheme 48) without any evidence of the formation of N,N′-dialkylated ones.148
 | ||
| Scheme 48 Mono-N-alkylation of arylenediamines.148 | ||
Many N-alkylated arylenediamines are commercially important building blocks in dyestuff applications,149 and are components of pharmaceuticals.150
Table 13 reports the results for a series of arylenediamines reacted with DMC or DEC at reflux, or EC at 130 °C. All the selected amines showed a high selectivity for mono-N-alkylation. For the simplest diamines, selectivities are higher for DEC than for DMC, and, surprisingly, o-phenylenediamine (OPD) and m-phenylenediamine (MPD) showed comparable yields and high selectivities.
| No. | Substrate | DAC | Product,b isolated yield (%) | Impurities (%) |
|---|---|---|---|---|
| a Reactions in DMC and DEC carried out at reflux; reaction with ethylene carbonate (EC) at 130 °C; OPD = o-phenylenediamine; MPD = m-phenylenediamine. b All products were characterised either by spectroscopic and chromatographic comparison with authentic samples, or by NMR, mass spectroscopy and microanalysis. c OPD. d SM = Starting material. | ||||
| 1 | o-Phenylenediamine | DMC | N-Me-OPD, 75 | 6c |
| 2 | o-Phenylenediamine | DEC | N-Et-OPD, 78 | — |
| 3 | m-Phenylenediamine | DMC | N-Me-MPD, 90 | 10d |
| 4 | m-Phenylenediamine | DEC | N-Et-MPD, 99 | — |
| 5 | m-Phenylenediamine | EC | N-2-OH-ethyllMPD | 80 |
The reaction of OPD with DMC at 160 °C performed in the absence of the zeolite gave only 30% conversion comprising a mixture of N-methyl-OPD, N,N-dimethyl-OPD and 2-benzimidazolone in a 75:20:5 ratio. This result highlighted that the catalyst was clearly essential for both the reactivity and selectivity.
Selective N-alkylation of 2,4-diaminotoluene with DMC (Scheme 49) has been also reported in the presence of two series of catalysts, either zeolites exchanged with transition metal ions (Zn2+, Pb2+ and Sc3+) or mesoporous aluminosilicates containing Lewis acid sites introduced by ion exchange (Zn2+) or forming part of the framework (SnIV, TiIV and ZrIV) (Table 14).151
 | ||
| Scheme 49 Reaction of 2,4-DAT and DMC in the presence of solid catalysts. | ||
The predominant products were those arising from N-methylation, although hard Lewis acid sites (Sc3+ in zeolite Y) and mesoporous catalysts also led to N-carbamoylation to a significant extent (over 50% for Sc in zeolite Y or Zr in the MCM-41 framework).
These results have been explained by considering the influence of the hardness/softness and the dimensions of the catalyst cavities on the selectivity. The enhanced efficiency of transition metal containing aluminosilicates was due to the presence of stronger acid sites compared to Na+.
On the other hand, larger channel dimensions of MCM-41 were more favorable for N-methoxycarbonylation than smaller pore size of zeolites (Table 14).
Besides zeolites, a variety of supports have been investigated for the selective N-methylation of aniline with DMC. In particular, Zn1−xCoxFe2O4 (x = 0, 0.2, 0.5, 0.8 and 1.0) type systems – prepared via the coprecipitation route – proved to be quite efficient catalysts.151 It was observed that systems possessing low x values are highly selective and active for the synthesis of N-methyl aniline. Higher activity was observed in the temperature range of 200–250 °C.152
Recently, also organocatalysts have shown great selectivity in the methylation of aniline by DMC.153
Finally, it should be mentioned that dialkylated aromatic amines can be achieved by anion activation via phase-transfer catalysts.154 In this view, unsymmetrical methyl alkyl carbonates with general formula ROCO2Me gave interesting results in the methylation reactions of anilines. In particular, the rapid and selective N,N-dimethylation reaction of aromatic amines using onium salts, especially phosphonium (e.g., Ph3Pet+I− and n-Bu4P+Br−), as catalysts was reported.
In a typical reaction the selected primary aromatic amines (XC6H4NH2, X = p-OMe, p-Me, H, p-Cl, p-CO2Me, o-Et, and 2,3-Me2C6H3NH2) were reacted with methyl alkyl carbonates (ROCO2Me, R = MeO(CH2)2[O(CH2)2]n; n = 2–0, respectively) in the presence of onium salts (0.1 molar equiv. amount with respect to the amine) and under solvent-free conditions, at 140–170 °C. The corresponding N,N-dimethyl derivatives (ArNMe2) were obtained with high selectivity (up to 96%) and good isolated yields (78–95%).
N-Methylation of heterocycles. Methylation of various nitrogen-containing heterocycles was first reported by Lissel and coworkers.155
Several heterocycles were methylated using K2CO3 and 18-crown-6, including substituted imidazoles, benzimidazole, pyrimidine bases, xanthine, theobromine and theophylline; a few examples are reported in Scheme 50.
 | ||
| Scheme 50 Methylation of various nitrogen-containing heterocycles by a phase-transfer system using K2CO3 and 18-crown-6.155 | ||
DMC was also used as a methylating agent for N-heterocyclic compounds without any catalyst or additional solvents at the temperature range of 110–170 °C under atmospheric pressure.156
Several substrates—including imidazole, pyrazole, pyrrole, morpholine, and piperazine—were employed to afford N-methylated products.
Blacklock et al. reported that catalytic amounts of 1,4-diazabicyclo[2.2.2]octane (DABCO) or DBU promoted the alkylation of nitrogen, oxygen, or sulfur atoms with DBnC. DABCO could effectively accelerate this benzylation, which worked efficiently (within hours) with several soft nucleophiles.157
The general applicability of this synthetic approach was tested on a variety of heterocycles. For the DABCO- and DBU-catalysed benzylation reaction, the results are reported in Table 15.
The same authors reported that ionic liquids can effectively accelerate slow N-benzylation reactions on benzoimidazole.158 Furthermore, it was proved that the reaction rate was enhanced from hours to minutes by applying microwave irradiation. A method for the synthesis of dialkylimidazolium ionic liquids was also developed by accomplishing alkylation with DACs in an autoclave with the corresponding acidic ionic liquid precursors.159
N-Methylation of indoles has also been carried out by using DMC and the effect of various functional groups on the substrate has been investigated.160 As an example, N-methylation of 6-nitroindole was conducted by reacting 6-nitroindole with DMC in the presence of K2CO3 in DMF at a temperature of 126 °C. The reaction was complete within 2 h, in 96% yield and a high purity suitable for large-scale production.
In another example, N-methylation of NH-containing heterocycles has also been achieved using TMEDA as an active organocatalyst.161 Accordingly, DMC methylated imides, indoles, benzimidazoles, and piperazines in high conversion.
The nitrogen bicyclic base DBU was also employed as an efficient catalyst in the methylation reaction of indoles, and benzimidazoles with DMC under mild conditions.110 Additional rate enhancement was accomplished by applying microwave irradiation in the presence of Bu4N+I−. By combining both these acceleration strategies, chemical transformations could be performed efficiently in high yield within minutes.
As an example, N-methylation of benzimidazole with DMC was obtained in a shorter reaction time and at low temperature using DBU (1.0 eq.) as a catalyst. The resulting N-methyl derivative was obtained in 2 h at 90 °C (98% yield) and in 6 min under microwave irradiation (76% yield).162
DMC has been also employed in the N-methylation of electron-deficient pyrroles in the presence of DMF and a catalytic amount of DABCO.163 This alkylation methodology proved to be useful for the alkylation of a variety of pyrroles in 72–98% yields. Some examples are reported in Table 16.
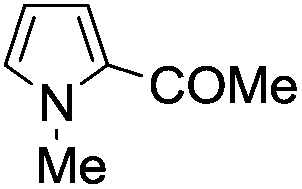
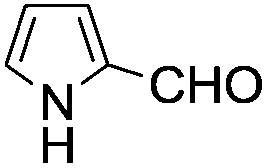
Quaranta and co-workers reported the reaction of pyrrole with DMC under phosphazene catalysis and evaluated the N-methoxycarbonylation versus the N-methylation reaction.164
Phosphazene (t-butylimino-tris(dimethylamino)phosphorane, tert-butylimino-tris(pyrrolidino)-phosphorane (BTPP)) and amidine (e.g. DBU) superbases have been investigated as catalysts.
At 423 K, in the presence of 10 mol% of BTPP, pyrrole was quantitatively methylated to 1-methylpyrrole within 3 h. The main reaction pathway involved the decarboxylation of the intermediate 1-methoxycarbonyl pyrrole and led to the formation of 1-methylpyrrole.
N-Methylation of amides. It was reported that propionamide and benzamide can be N-methylated with DMC in the presence of a quaternary ammonium salt cetyl trimethyl ammonium bromide (CTAB) at 493 K.165 Only mono-methylated products were obtained.
Lactams such as 2-pyrrolidone, 8-valerolactam, and ε-caprolactam were also N-methylated in high yields in the presence of CTAB (Scheme 51).
 | ||
| Scheme 51 N-Methylation with DMC in the presence of CTBA.165 | ||
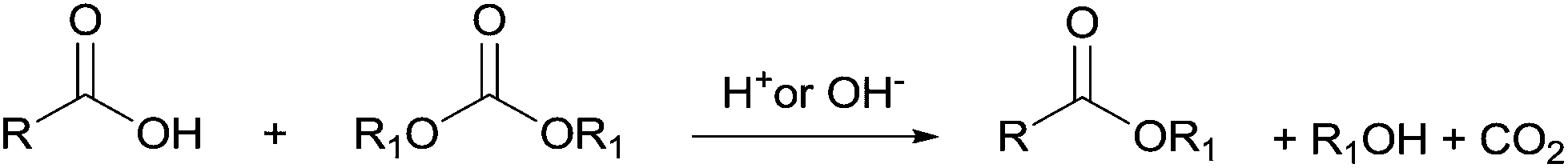 | ||
| Scheme 52 Esterification of carboxylic acids using DMC. | ||
Synthesis of esters under acidic conditions. The Fischer esterification of carboxylic acid with DMC (or DEC) proceeds in a high yield via solvent-free acidic catalysis.166 In a typical synthetic procedure a carboxylic acid and DMC were reacted in neat at 80–85 °C (at 110–120 °C using the less reactive DEC) in the presence of conc. H2SO4 or p-toluenesulfonic acid (PTSA) as a catalyst (0.1 eq.) to achieve straightforwardly the corresponding ester in high yield (up to 99%) and high purity (95–99%). This method may be efficiently used with different substrates such as aliphatic, aromatic, cinnamic, nicotinic and aminobenzoic acids and no significant differences in reactivity were observed between aromatic and aliphatic carboxylic acids.
Mild reaction conditions were employed in the mesoporous sulfated zirconia (MSZ)-catalysed methylation of 4-methoxyphenylacetic acid with DMC (Scheme 53).167 MSZ is an insoluble catalyst which exhibits superior features to those of conventional sulfated zirconia (CSZ) catalysts, such as a larger surface area, more regularly shaped and uniformly distributed mesopores and higher acidity. Therefore, the reaction carried out in an autoclave at 160 °C in the presence of MSZ afforded methyl-4-methoxyphenylacetate in both high conversion and selectivity not accessible by means of CSZ-catalysed methylation.
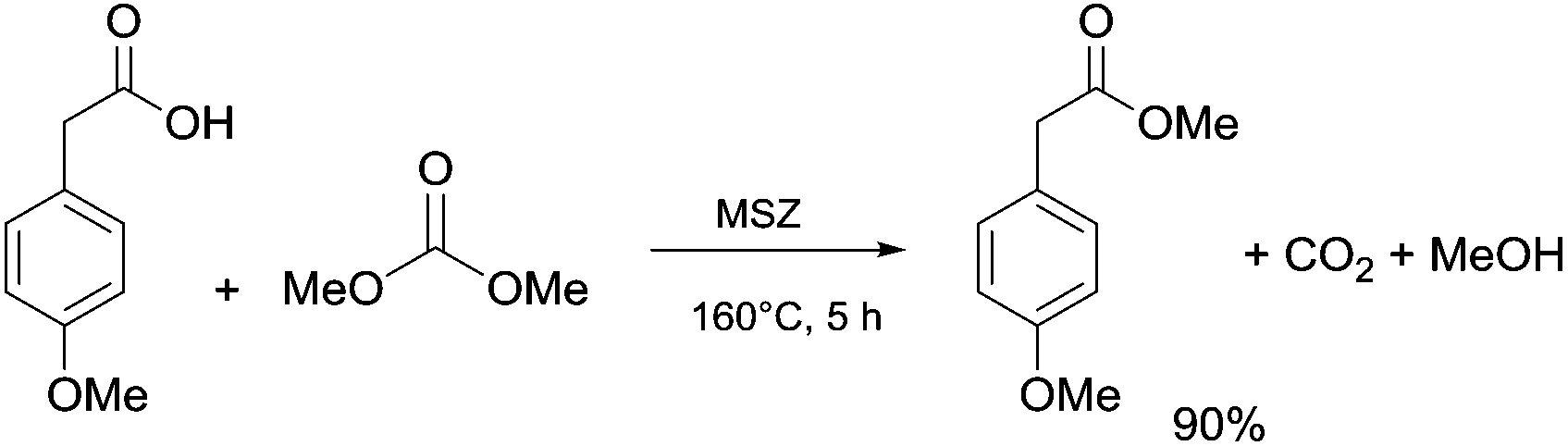 | ||
| Scheme 53 Esterification of 4-methoxyphenylacetic acid by DMC chemistry in the presence of MSZ.167 | ||
In addition, the Fischer esterification was performed by reaction of DACs (pyrocarbonates) with rather activated carboxylic acid under mild conditions and in the presence of a catalytic amount of a Lewis acid (Scheme 54).168
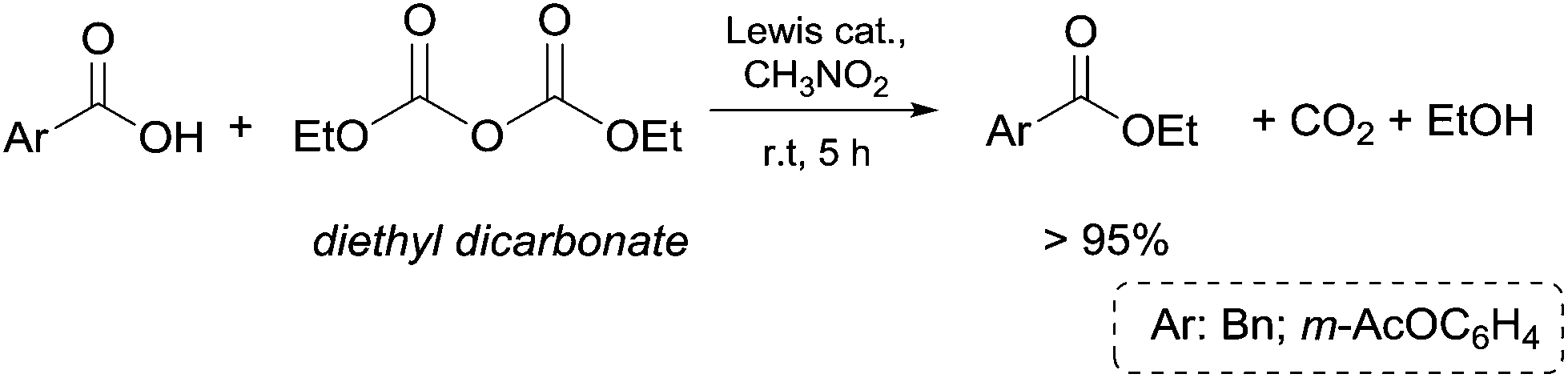 | ||
| Scheme 54 Lewis acid-catalysed esterification via diethyldicarbonate.168 | ||
Diethyl dicarbonate was reacted with benzylic or 3-acetoxybenzoic acid and different Lewis acids – such as Mg(ClO4)2 and Cu(OTf)2 – to give the corresponding esters with high conversion and stereoselectivity.
In another synthetic procedure, DMC was reacted with several ambident nucleophiles, such as o- and p-mercaptophenols, o- and p-mercaptobenzoic acids, o- and p-hydroxybenzoic acids, mandelic and phenyllactic acids, in the presence of NaY faujasite and under batch conditions at 150–165 °C (Scheme 55).169
 | ||
| Scheme 55 Reactions of DMC with several ambident nucleophiles in the presence of NaY faujasite.169 | ||
Under these conditions mercaptophenols and mercaptobenzoic acids underwent chemoselectively S-methylation reactions, without affecting OH and CO2H groups, respectively. In contrast, at 165 °C, hydroxybenzoic, mandelic and phenyllactic acids formed the corresponding methyl esters, while both their aromatic and aliphatic hydoxyl substituents were preserved from methylation and/or transesterification processes. Typical selectivities were f 90–98% and products (S-methyl derivatives and methyl esters, respectively) were isolated in 85–96% yields.
A comparative study between NaY faujasite and K2CO3 as esterification catalysts was performed. Although potassium carbonate showed a superior reactivity to the zeolite, it led to a loss of chemoselectivity, inasmuch as o- and p-mercaptobenzoic acids underwent simultaneous S-methylation and esterification reactions, and hydroxybenzoic, mandelic and phenyllactic yielded complex mixtures of products of O-methylation, O-methoxycarbonylation, and esterification of their hydroxyl and carboxyl groups, respectively.
Esterification of benzoic acids was carried out efficiently over zeolite Hβ and HZSM-5 in an autoclave using DMC as a methylating agent.170 The reaction proceeded both inside and outside the pores of zeolites and thus the pore size along with catalyst acidity affected the conversion of bulky acids to the corresponding esters.
As an example, the methylation of salicylic acid (SA) by DMC was reported to occur over zeolites Hβ, HZSM-5 and HY at 370–425 K in an autoclave under autogenous pressure (Scheme 56).
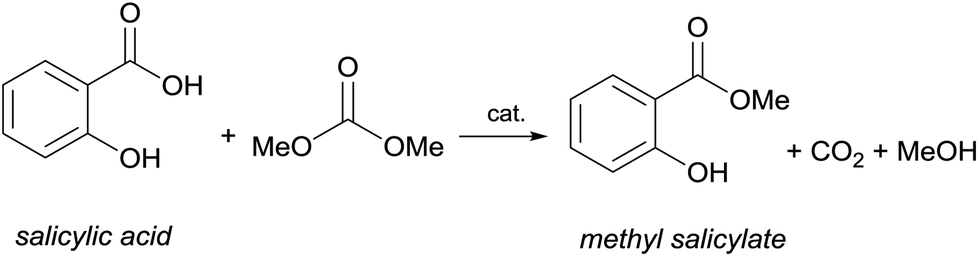 | ||
| Scheme 56 Esterification of salicylic acid with DMC in the presence of a catalyst such as zeolites Hβ and HZSM-5 or a mesoporous material such as AlSBA-15-SO3H. | ||
The SA conversion was over 90% for both Hβ and HZSM-5 at 423 K. As expected, the use of the less acidic HY led to a poor reaction yield, albeit the selectivity was greater than 95%. Besides, kinetics studies were performed and the zeolite-catalysed esterification of SA was shown to obey the first order kinetics. A comparative study of the esterifying ability of DMC and methanol has been also reported.
Esterification of salicylic acid with DMC has been also carried out by one-pot synthesis of mesostructured AlSBA-15-SO3H with various Si/Al ratios.171 Mesoporous materials functionalised with sulfonic groups were prepared under acidic conditions by oxidative co-condensation of tetraethoxysilane and 3-mercaptopropyltrimethoxysilane (MPTMS) in the presence of tri-block copolymers (such as poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol)), Al2O3 as an aluminium source and hydrogen peroxide. This methodology was exploited to prepare functionalised mesopourous solids containing other functional groups such as arensulfonic, carboxylate and peroxycarboxilic acid. AlSBA-15-SO3H showed high activity in the esterification of salicylic acid with DMC, and the yield of methyl salicylate was correlated with the Brønsted acidity of the catalysts.
Synthesis of esters under basic conditions. DBU was reported to be an effective nucleophilic catalyst for carboxylic acid esterification with DMC.110,158,172O-Methylation of benzoic acid with DMC conducted using DBU (1 eq.) was completed within 3 h, faster than those carried out using other bases, i.e., tributylamine, DABCO, DMAP, NH4OH and N-methylmorpholine. In particular, the widely used acylation catalyst DMAP gave only 2.5% conversion to benzoate ester in 2.5 h, whereas the reaction performed using DBU afforded 97% yield at the same time point.
In order to elucidate the reaction mechanism, kinetics studies were performed. A direct BAl2 displacement was excluded by measurements and the proposed pathway was reported to involve an initial N-acylation of DBU with DMC to form a carbamate intermediate (see section 4, Cyclisation, Scheme 81).173 Subsequently, carboxylate reacts with this intermediate, generating the corresponding methyl ester via O-alkylation. The amount of DBU employed seemed not to influence linearly the rate, as the transition state may involve DBU itself. However, reaction rates dramatically decreased when using a substoichiometric amount of DBU (from 0.2 to 0.5 eq.). Therefore, it was postulated that 1.0 equivalent of DBU was almost entirely consumed in the diffusion-controlled deprotonation of carboxylic acid and only a slight amount of base was available via equilibrium to react with DMC to form the active intermediate. This DBU-assisted esterification was efficiently used with sterically hindered and poorly activated aromatic acids, amino acids, carbohydrate acids, as well as carboxylic acids. Shieh and coworkers174 reported a large scale synthesis – up to 100 g – of a series of esters via microwave irradiation at 160 °C. This approach increased 20–80 fold the reaction rate obtained under batch conditions.
Aromatic esters have been prepared in high to moderate yields by reaction of carboxylic acids with diphenyl carbonate dissolved in pyridine at room temperature.175p-Toluic acid, p-anisic acid and cinnamic acid were isolated in almost quantitative yield after four days at 20 °C.
Chau and coworkers176 developed a phase-transfer (PT) catalytic system (i.e. K2CO3/tetrabutylammonium chloride, also known as TBAC) to methylate carboxylic acids under mild conditions and atmospheric pressure through an SN2 mechanism. The PT catalyst TBAC was used not only to increase the interaction between the base and substrates, but also as a reaction medium to ensure high reaction temperatures (150 °C) necessary for an effective methylation via DMC.
O-Alkylation of sulfonic acids. Sulfonic acids – unreactive toward esterification by alcohols – were directly O-alkylated by DACs (Scheme 57).177 In a typical procedure a solution of TsOH hydrate dissolved in toluene was refluxed through a Dean–Stark trap – until no more water was collected – followed by the addition of DEC. Then the reaction mixture was refluxed for 24 h to afford ethyl-p-toluenesulfonate in 64% yield.
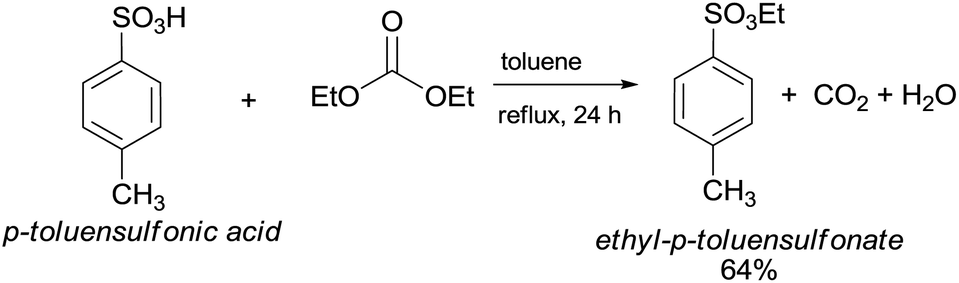 | ||
| Scheme 57 Esterification of p-toluenesulfonic acid by DEC.177 | ||
Albeit the esterification of p-toluenesulfonic acid was conducted with different esters, such as β-ketoesters and malonates, DEC proved to be the more efficient alkylating agent.
3.2 Synthesis of silicium, titanium and germanium ortho-esters
Suzuki et al.178 developed a facile and quantitative gas-solid synthesis of tetramethoxysilane employing silica gel supporting alkali hydroxides (5 wt%) and gaseous DMC at 500–600 K (Scheme 58, eqn (50)). | ||
| Scheme 58 Synthesis of tetramethoxysilane by DMC.178 | ||
Silica gel – loaded with the catalyst – was packed in a fixed-bed flow reactor and heated to the reaction temperature. Then, the reaction of DMC with the catalyst generated an alkoxide that promoted Si–O–Si bond cleavage and O-methylation (Scheme 58, eqn (51)). The continuous gas flow of DMC alkylated the silicate and regenerated the base, permitting the reaction to continue until the complete conversion of silica gel to its ortho-ester (Scheme 58, eqn (52)).
The reaction was completed in about 40 min by using KOH, RbOH, or CsOH as a catalyst. Kinetics studies showed that the activation of the silica surface occurs at the first stage of the reaction leading to a rapid increase of the reaction rate followed by a gradual decrease to zero, due to the complete conversion of silica gel into tetramethoxysilane.
Alkali halides such as cesium fluoride and sodium chloride turned out to be effective catalysts in the reaction of silica gel and DEC at 700 K to give tetraethoxysilane.178
Several silicon dioxide sources—such as commercially available diatomaceous earth materials, silicate rocks and minerals—were investigated as reagents in the base-mediated reaction with DMC to synthesize tetramethoxysilane.179
Rice hull ash (92% SiO2 purity), when loaded with 5 wt% of KOH, reacted with DMC at 625 K to give almost quantitatively tetramethoxysilane. At higher temperature the decomposition of DMC into dimethyl ether and carbon dioxide has been reported as a significant side reaction.
The ash was also reacted with DEC to give an 80% yield of tetraethoxysilane at 725 K.180
Okamoto et al.181 reported the complete depolymerisation of a series of polydimethylsiloxanes terminated by a methyl group by reaction of DMC, methanol and alkali metal halides to form dimethoxydimethylsilane (DMDMS) and methoxytrimethylsilane (MTMS) monomers. The various alkali metal halides tested as catalysts gave the same monomer yield and the observed depolymerization rate depended solely on the alcohol employed.
Depolymerization of poly(methylphenylsiloxane-random-dimethylsiloxane) and poly(methylvinylsiloxane-random-dimethylsiloxane) with DMC in methanol using potassium fluoride as a catalyst led to dimethoxymethylphenylsilane and dimethoxymethylvinylsilane, respectively, along with dimethoxydimethylsilane and methoxytrimethylsilane from polydimethylsiloxane parts (Table 17).
| Products | Yields | |||
|---|---|---|---|---|
| DMC + MeOH | DEC + EtOH | DEC + MeOH | DMC + EtOH | |
| a Reaction conditions: Polydimethylsiloxane: 1.5 g (containing 20 mmol of silicon atom); DAC: 30 mmol; ROH: 150 mmol; KF: 0.5 mmol; T: 453 K; reaction time: 5 h.181 | ||||
| Me3SiOMe | 5 | — | 5 | 3 |
| Me3SiOEt | — | 4 | — | 2 |
| Me2Si(OMe)2 | 80 | — | 45 | 5 |
| Me2Si(OMe)(OEt) | — | — | 36 | 27 |
| Me2Si(OEt)2 | — | 43 | 1 | 31 |
Base-catalysed depolymerisation with DMC represents a useful approach to recycle silicone rubber: dimethoxydimethylsilane was the major product isolated along with a small amount of tetramethoxysilane formed from the silica additive.
It has been reported that a simple route to produce titanium tetraalkoxides free from chloride impurities is provided by the reaction of titania (TiO2) with DACs.182 Titanium tetraalkoxide is formed by cleavage of Ti–O–Ti bonds in hydrous titanium dioxide by reaction with DACs with the formation of Ti–OR bonds and release of carbon dioxide (Scheme 59).
 | ||
| Scheme 59 Synthesis of Ti(OR)4 by reaction with DACs.182 | ||
In a typical procedure, hydrous titanium dioxide (TiO2·nH2O with n = 0.3–1.5) was reacted with DEC in an autoclave at 493–533 K for 16 h to give Ti(OC2H5)4 – isolated by vacuum distillation – in 95% yield. The use of a catalyst, such as sodium hydroxide, remarkably reduced the time required to complete the reaction.
Reaction of the catalyst with hydrous titanium dioxide likely occurs by cleavage of the Ti–O–Ti bond and the simultaneous formation of a Ti–OEt bond. The catalyst may facilitate the formation of ethoxide anions from a DEC molecule, which may attack the Ti–O–Ti bond to form the Ti–OEt bond. Similarly, Ti(OC3H7)4 was obtained in a 92% yield from TiO2·0.7H2O and dipropyl carbonate at 493 K.
Germanium dioxide was also modified with DMC in the presence of 5% KOH at 250 °C to give (MeO)4Ge exhibiting a reactivity similar to that of SiO2 (Scheme 60).
 | ||
| Scheme 60 Synthesis of tetramethoxygermanium by DMC.182 | ||
Therefore, GeO2 and SiO2 were reacted with DMC in a competition experiment. As a result, it was observed that the rate of the base-catalysed reaction of germania with DMC was approximately an order of magnitude higher than that of silica, albeit the surface area of silica was two orders of magnitude higher.
A mixture of Ge(MeO)4 and MeGe(OMe)3 was achieved in a 7:3 ratio by performing the reaction of GeO2 and DMC with 5% KOH at 350 °C. Their chemical identity was confirmed by 1H and 13C NMR spectroscopy and GC-MS.
3.3 Allylic alkylation
The development of facile procedures to introduce allyl groups into a carbon backbone has always been considered rather attractive, inasmuch as the double bond may be conveniently converted into other functional groups. In order to present a complete work on DACs, allylation reactions should be obviously included. The green aspect of this reaction is mainly related to the possibility of recycling the base (if used) and the catalyst. However, compared to other DAC based reactions the greenness is somewhat more limited.In 1940, Carrol183 discovered that allyl β-keto carboxylates – synthesised by a base-catalysed reaction between an allyl alcohol and a β-ketoester – could rearrange into γ,δ-unsaturated ketones by thermal [3,3]sigmatropic rearrangement as depicted in Scheme 61, eqn (53). Successively, Tsuji et al.184 adapted the palladium-catalysed allylation – known as the Tsuji allylation and further developed by Trost185 by means of the introduction of phosphine ligands – for allylic esters of acetoacetic acid, obtaining under mild conditions the same ketones thermally produced via the Carrol rearrangement (Scheme 61, eqn (54)).
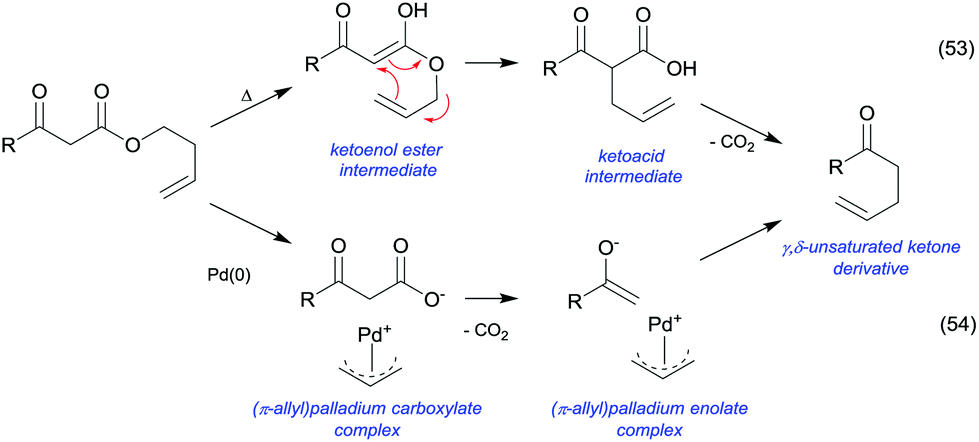 | ||
| Scheme 61 The Carroll rearrangement mechanisms: [3,3] sigmatropic rearrangement (eqn (53)) and Pd(0)-assisted (eqn (54)). | ||
Consequently, the same author carried out the formation of carbon–carbon bonds via allylic alkylation employing allyl carbonate derivatives.186 The peculiar reactivity of allyl carbonates – relative to that of traditional allylation reagents such as allyl halides – combined with Pd-catalysed chemistry allows performing allylic alkylation of activated ketones under mild conditions (generally, T < 70 °C) and without the employment of any additional base. In fact, allyl carbonates act either as nucleophiles towards palladium catalysts or as electrophiles (i.e. as alkylating agents) towards the substrate (e.g. primary alcohols, phenols, enols, β-ketoesters, amines, azides, imides, sulphonamides, and sulfones), which may be deprotonated by the alkoxides generated in situ through the decarboxylation of the carbonate moiety (Scheme 62).
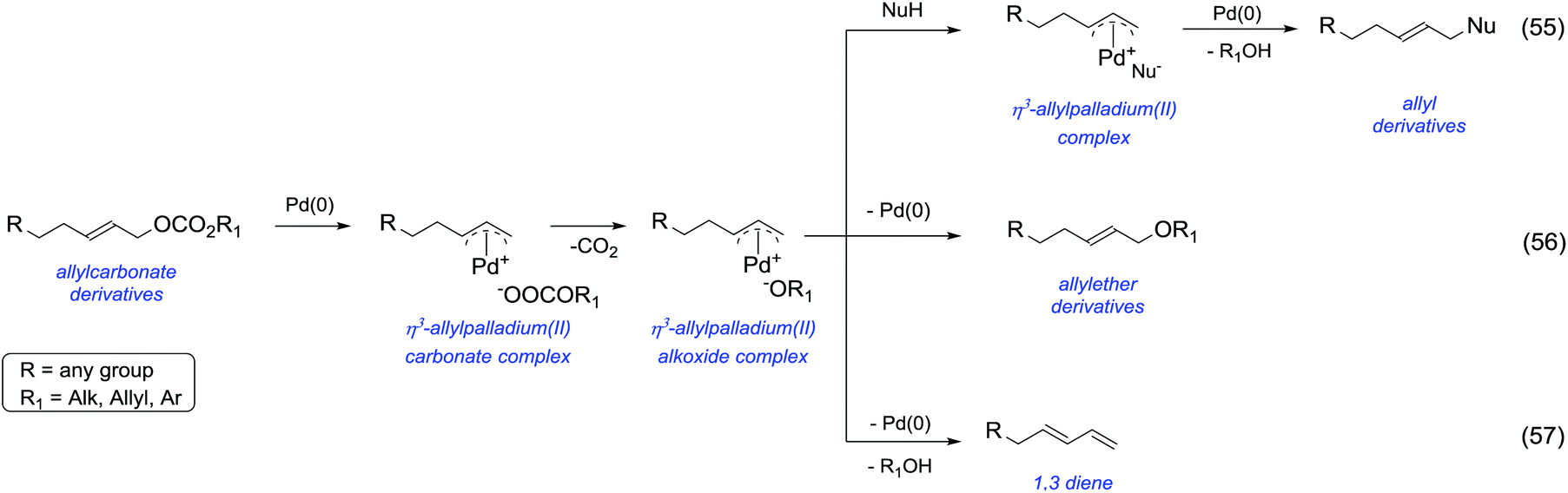 | ||
| Scheme 62 Reactions pathways of allyl alkyl carbonates in the presence of a Pd catalyst. | ||
The oxidative addition of an allyl carbonate to a Pd(0) species complexed with phosphine ligands generates a (π-allyl)Pd carbonate complex (i.e. a η3-allylpalladium complex), which then produces the key (π-allyl)Pd alkoxide complex upon CO2 release. This intermediate may undergo SN2 (BAl2): (1) by addition of a nucleophile to give the corresponding allyl derivatives (Scheme 62, eqn (55)); (2) by being attacked by the alkoxide formed in situ to give an allyl ether derivative (Scheme 62, eqn (56)). Moreover, β-elimination of Pd–H from the (π-allyl)Pd complex and in turn alkoxide protonation may lead to the formation of a 1,3-diene derivative(Scheme 62, eqn (57)).
Transition-metal-catalysed allylation represents a versatile method to form carbon–carbon and carbon–heteroatom bonds that is extensively used in organic synthesis and widely reported in the literature.187 The development of chiral ligands further improved the processes’ reactivity and stereoselectivity and broadened the range of applications of this synthetic approach in both total syntheses of natural products188 and pharmaceuticals.189 A select list of examples of allylation of various substrates employing allylcarbonates as alkylating agents and carried out under different reaction conditions is given below.
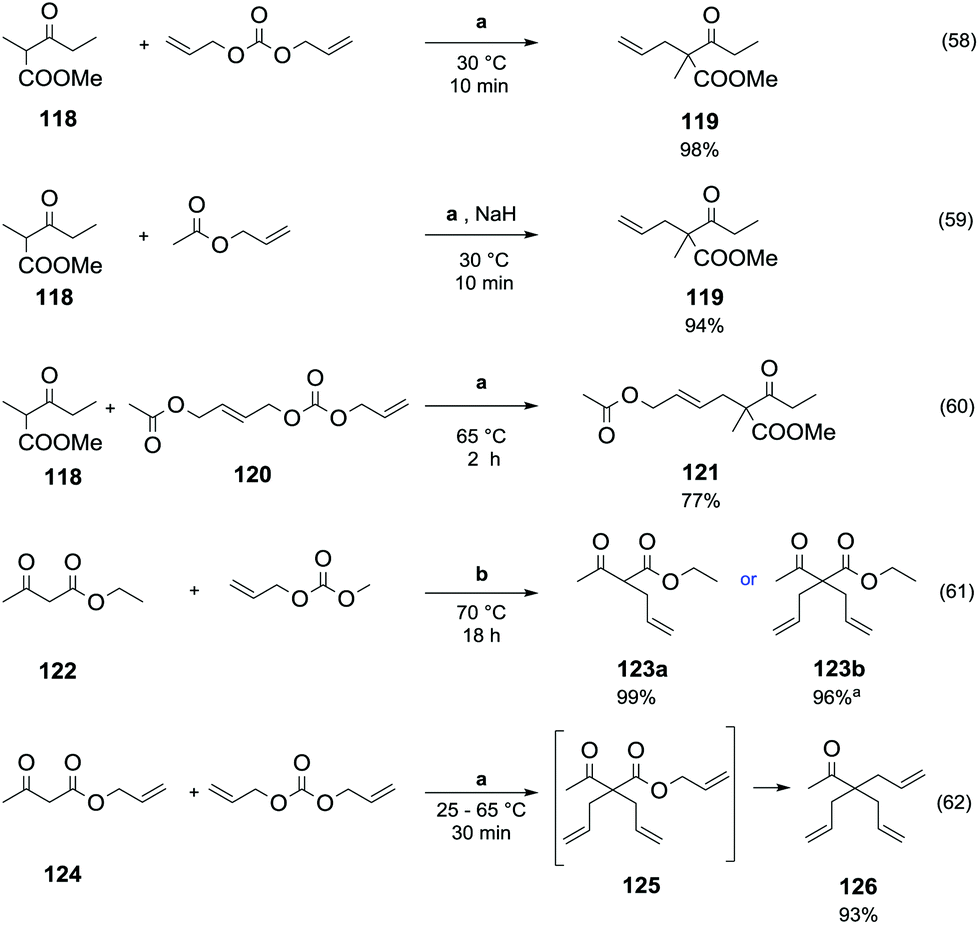 | ||
| Scheme 63 Examples of palladium catalysed Tsuji allylation. Reaction conditions a: Pd2(dba)3·CHCl3, PPh3, THF; reaction conditions b: MSN catalyst, K2CO3, THF. aCalculated by GC-MS analysis.190–193 | ||
Tsuji and co-workers190 reported the allylic alkylation of 2-methyl-3-oxopentanoate 118 using both diallyl carbonate (Scheme 63, eqn (58)) and allylacetate (Scheme 63, eqn (59)).
This study demonstrated the higher efficiency of the reactions carried out with allyl carbonates compared to those with acetates, which required the addition of a strong base (1.5 eq. of NaH) to the reaction mixture to achieve the desired product (methyl 2-allyl-2-methyl-3-oxopentanoate 119) in good yield.
Further evidence of the superior reactivity of the allyl carbonate is shown by the chemoselectivity of the reaction of 118 with the 4-acetoxy-2-butenyl methyl carbonate 120, inasmuch as the allylation involved solely the allylic position binding the carbonate moiety (Scheme 63, eqn (60)).
In order to selectively achieve the mono versus the double allylation (generally favoured), Dickschat et al.191 developed a series of immobilised and cooperative catalysts i.e. bifunctional mesoporous silica nanoparticles (MSNs) incorporating Pd-complexes and additional basic sites.
Thus, mono allyl derivative 123a and diallyl derivative 123b (Scheme 63, eqn (61)) were obtained pure and in quantitative yield by reaction of ethyl acetoacetate 122 with methyl allyl carbonate using a properly functionalised catalyst, along with the addition of K2CO3 (1 eq.) and controlling the reaction temperature. Interestingly, a Pd-catalysed reaction between allyl acetoacetate 124 – incorporating two acidic protons – and diallyl carbonate (Scheme 63, eqn (62))192 gave α,α,α-triallylacetone 126 in 93% yield, via decarboxylation/alkylation of diallyl carbonate intermediate 125.
Simple ketones, esters, nitriles, and sulfones are unreactive under the conditions generally used for allylic alkylation of malonates, β-keto esters, β,γ-unsaturated ketones, esters, nitriles and sulfones. In fact, the alkoxides formed during the reaction are not sufficiently basic to efficiently deprotonate those species with pKa higher than 20 (pKa values of alkoxide conjugated acids are in the range of 16–18).
Enol silyl ethers are stable enolates and thus ketone equivalents, which can undergo the Tsuji–Trost allylation by employing slightly modified reaction conditions (Scheme 64).193 Cyclohexanone 128 (Scheme 64, eqn (63)) and aldehyde 130 (Scheme 64, eqn (64)) were obtained in high yield by refluxing a THF solution of silyl enol ethers 127 and 129, respectively, with diallylcarbonate, using Pd2(dba)3·CHCl3 as a catalyst. Moreover, 1,2-bis(diphenylphosphino)ethane (dppe) was used as a Pd(0) bidental ligand (in place of PPh3), as it was shown to contribute to forming a (π-allyl)palladium(I) complex more reactive toward nucleophilic attack.
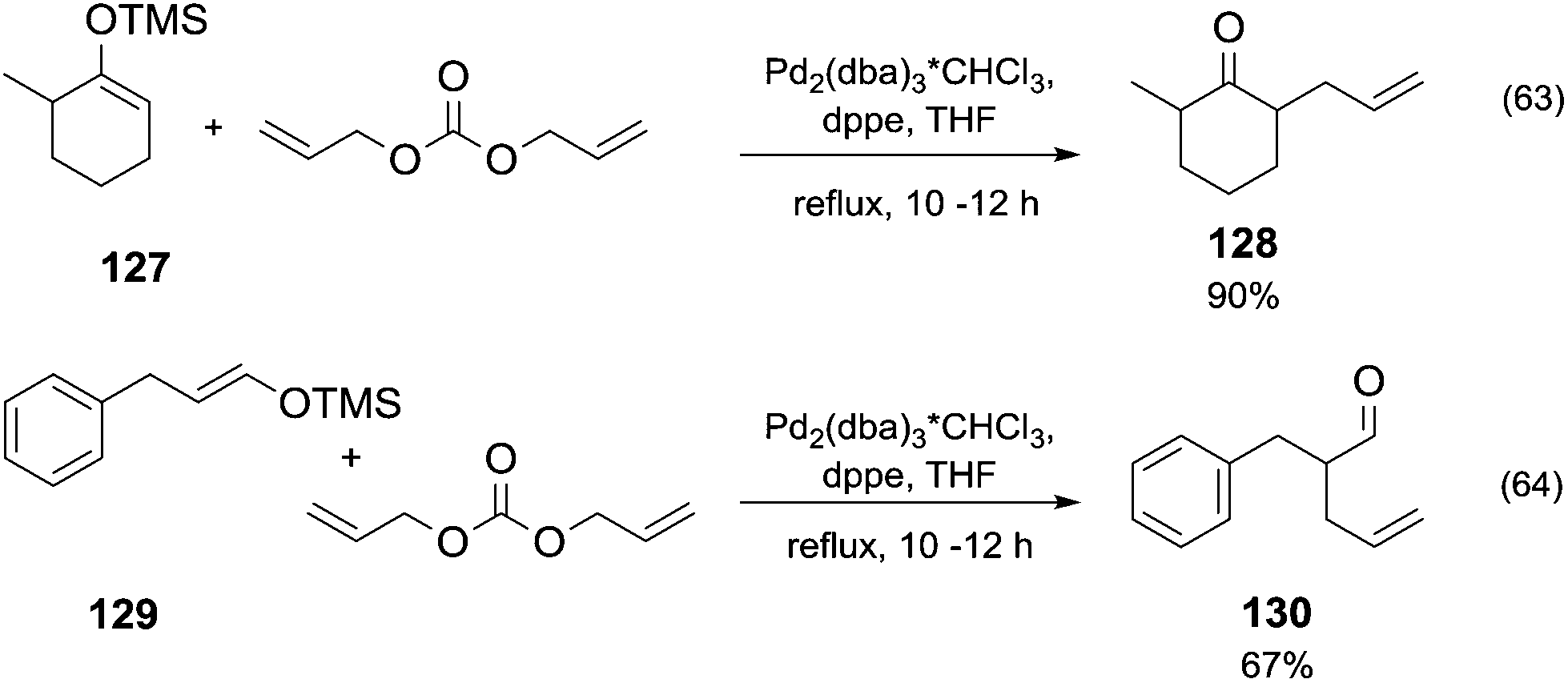 | ||
| Scheme 64 Tsuji allylation of silyl enol ethers.193 | ||
Asymmetric alkyl allylation. Quaternary stereocenters at a prochiral nucleophile can be achieved through decarboxylative Asymmetric Alkyl Allylation (AAA), which represents an enantioselective version of decarboxylative allylation.
AAAs are performed using either palladium or molybdenum catalysts and the stereoselectivity may be modulated according to the features of both the ligand and the nucleophile employed in the reaction.
Ligands may affect steric hindrance and electronic density of catalysts and induce chirality of the final products. On the other hand, nucleophiles can be divided into stabilised or “soft” nucleophiles and unstabilised or “hard” nucleophiles.194 Usually, the former have a pKa of conjugate acids <25 and they were described to attack directly the carbon of the allyl moiety complexed with the catalyst from the face opposite the metal and the chiral ligand, leading to a product exhibiting the same configuration of the ligand (outer sphere mechanism). The latter – with a pKa of conjugate acids >25 – instead were reported to pre-coordinate the metal center, and the product with the opposite stereochemistry – relative to the ligand – was isolated after reductive elimination (inner sphere mechanism).195
The development of AAA synthetic procedures has focused on the use of soft nucleophiles rather than the hard ones, scarcely investigated and exploited (to date no efficient synthesis involving both hard nucleophiles and allyl carbonates has been reported in the literature).196
In 2004, Stoltz and co-workers197 firstly described the enantioselective decarboxylative allylation of a series of enolcarbonates, testing a variety of (S)-alkyl-PHOX chiral ligands (Scheme 65, eqn (65)).
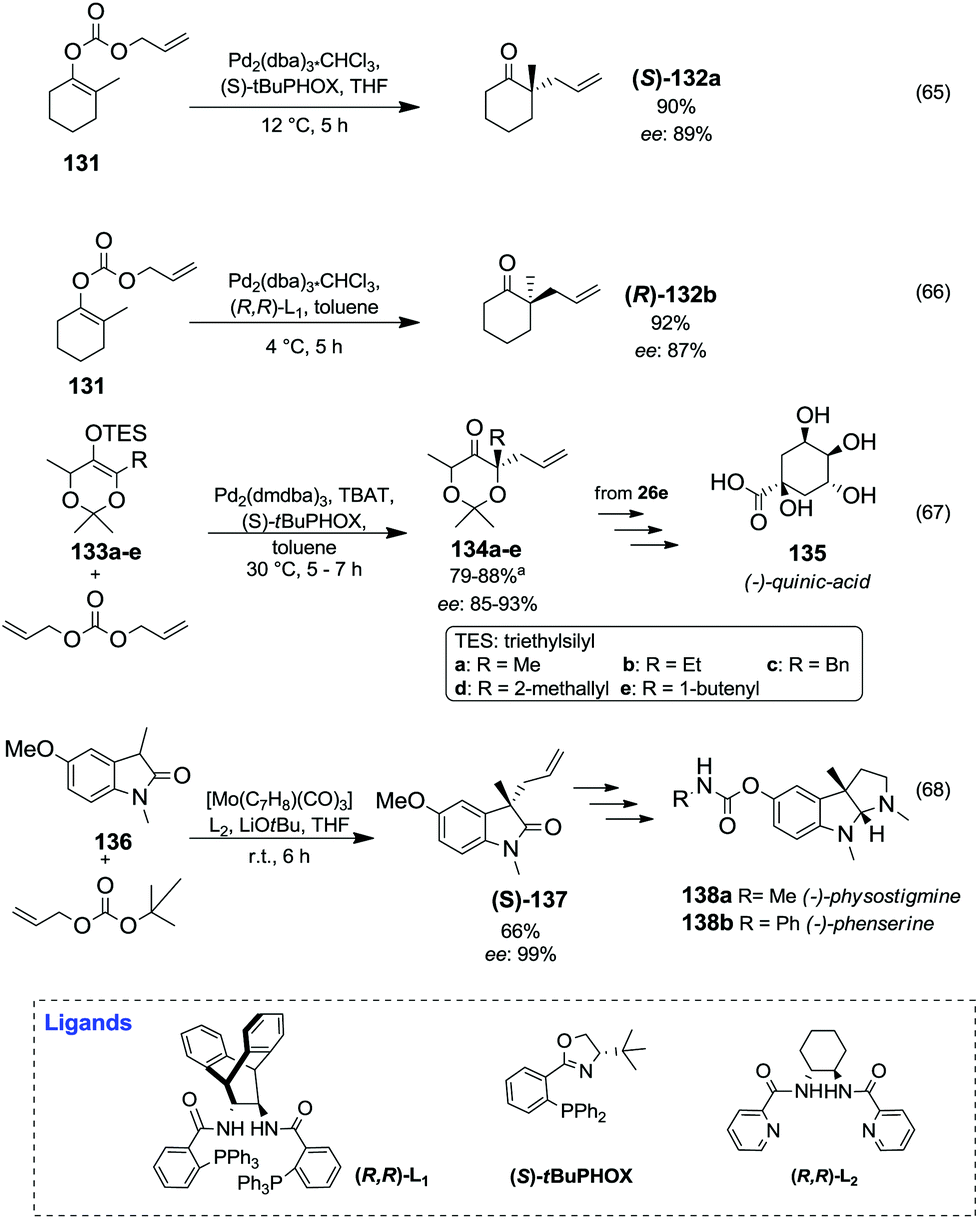 | ||
| Scheme 65 Examples of Pd and Mo-catalysed AAA synthesis. aIsolated yield measured by gas chromatography or HPLC analysis.197–200 | ||
(S)-2-Allyl-2-methylcyclohexan-1-one 132a was obtained in 90% yield and with an ee of 89% by reaction of enol carbonate 131 with Pd2(dba)3·CHCl3, in THF at 12 °C using (S)-tBu-PHOX as a chiral ligand. Trost et al.198 achieved similar high conversion and enantioselectivity (92% yield and 87% ee) in their attempt to synthesise the enantiomer (R)-2-allyl-2-methylcyclohexan-1-one 132b using slightly modified reaction conditions and (R,R)-L1 as a chiral ligand (Scheme 65, eqn (66)).
Successively, Stoltz's group199 further investigated the efficiency of (S)-tBu-PHOX in the reaction of enolsilyl derivatives 133a–e with diallylcarbonate in toluene using bis(3,5-dimethoxybenzylidene)acetone-palladium(0) (Pd2(dmbda)2) as a catalyst and Bu4NPh3SiF2 (TBAT) as a cleavage agent of the triethylsilyl group (Scheme 65, eqn (67)). It is noteworthy that (−)quinic acid 135 – a scaffold for the synthesis of pharmaceuticals and usually extracted from coffee beans – was synthesised in rather good yield starting from the silylenol ether 134e.
Owing to the poor enantiospecificity achieved by a Pd-catalysed AAA reaction – involving the outer sphere mechanism – of alkyloxyindoles, Trost et al.200 performed this decarboxylative allylation via Mo-catalysed AAA – known to proceed via the inner sphere mechanism. Thus 3-alkyloxyindole 136 was reacted with allyl tert-butyl carbonate in THF using [Mo(C7H8)(CO)3] as a catalyst and (R,R)-L2 as a chiral ligand and adding a strong base to the reaction mixture (LiOtBu) to assist the nucleophile deprotonation (Scheme 65, eqn (68)).
As expected, the reaction afforded the (S)-allylated oxindole derivative 137, which was isolated enantiomerically pure and in moderate yield after crystallisation.
Compound 137 represents a key intermediate for the synthesis of the natural alkaloid (−)-physiostigmine 138a, a potent and reversible cholinesterase inhibitor, as well as of its synthetic analogue (−)-phenserine 138b.
Besides, basic pronucleophiles with pKa ranging far above 25 may act as soft nucleophiles by addition of a base to the reaction mixture, which can facilitate the deprotonation step. In this perspective, the dibenzomethane 139 with pKa = 32 was reacted with the chiral allyl carbonate derivative 140 in glyme using dichloro(1,5-cyclooctadiene)palladium(II)201 as a catalyst and Xanphos as a phosphine ligand and adding KHMDS as a strong base to the reaction mixture to give compound 141 as pure diastereoisomer and in 88% yield (Scheme 66).
 | ||
| Scheme 66 Example of an AAA reaction using basic pronucleophiles.201 | ||
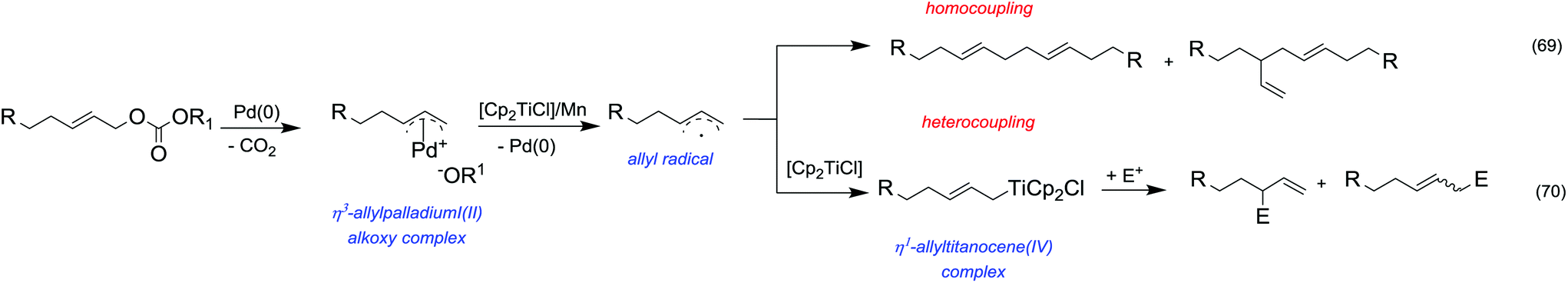 | ||
| Scheme 67 Proposed reaction mechanism for Pd/Ti-catalysed allylation. | ||
The addition of bis(cyclopentadienyl)titanium(III) chloride (Cp2TiCl) and Mn dust promotes the single electron reduction of the η3-allylpalladium(II)complex to yield an allyl radical that may react with: (1) a second allyl radical to give a homocoupling reaction (Scheme 67, eqn (69)); (2) Cp2TiCl to generate a nucleophilic η1-allyltitanocene(IV)complex, which in turn may react with an electrophile (Scheme 67, eqn (70)).
Standalone allylcarbonates undergo dimerization reaction – Wurtz-type reaction203 – by a Pd/Ti-catalysed reaction under mild conditions to give a mixture of α,α- and γ,α-coupling products. Typical reaction conditions were used to synthesise a 3:2 mixture of compounds (α,α)-143a and (γ,α)-143b in 70% yield (Scheme 68, eqn (71)).
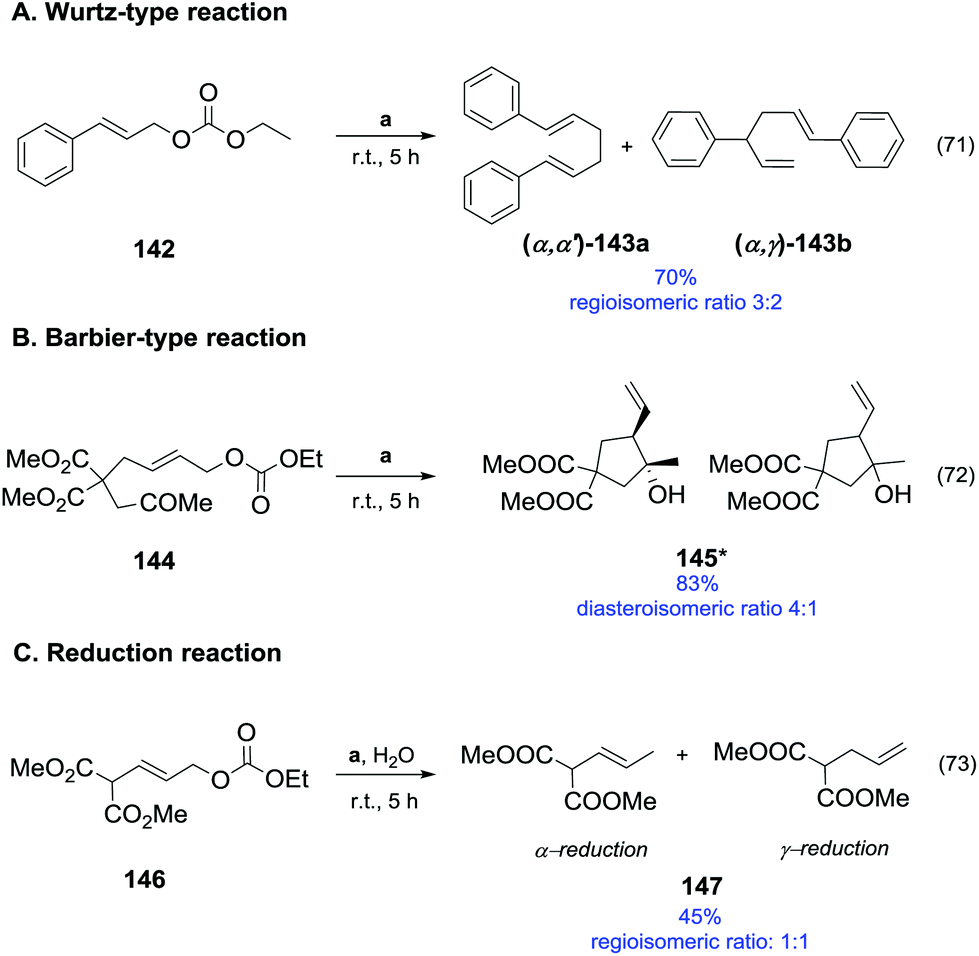 | ||
| Scheme 68 Examples of Pd/Ti-catalysed allylations: Wurtz-type reaction (A), Barbier-type reaction; * the exact configuration of the second isomer is not reported (B) and reduction reaction (C). Reaction conditions a: [Cp2TiCl], PdCl2/2PPh3, Mn dust, TMSCl, 2,4,6-collidine, THF.203,204 | ||
Allyl carbonate 142 was reacted with PdCl2 and Ph3P to give a η3-allylpalladium complex, which was converted into the allyl radical by [Cp2TiCl]-mediated single electron transfer. The rate of this step was increased by adding Mn dust into the reaction medium.
The allyl α-radical may attack either the α- or γ-position of another allyl radical to give the corresponding products (143a-b). The Ti(III) catalyst was usually regenerated in situ by the presence of trimethylsilylchloride (TMSCl) and 2,4,6-collidine in the reaction mixture. The stereospecificity may further increase on introducing a substituent at γ-position.
A Barbier-type204 reaction usually involves the formation of the nucleophilic η1-allyltitanocene(IV) complex (Scheme 67, eqn (70)) and is used to selectively homoallylate, crotylate and prenylate aldhehydes and ketones. In Scheme 68, eqn (72), the cyclisation of β,γ-ene-η-ketoethylcarbonate 144 was reported as efficient intramolecular allylation of a carbonyl group. This reaction was carried out using similar conditions to those employed in Wurtz-type reactions except for the concentration of Cp2TiCl that was used in a stoichiometric excess (2 eq.), instead of substoichiometric (0.2 eq.), in order to prompt its coordination to the allyl radical and give the η1-allyltitanocene(IV) complex.
Target compound 145 was then accomplished in 83% yield as a mixture of trans/cis diastereoisomers – relative to the configuration of the hydroxyl and vinyl groups – by a nucleophilic attack of the Ti(IV) complex on the carbonyl group at η position. trans Diastereoisomer is the major product in a five-membered ring cyclisation – and the minor one in a six-membered ring cyclisation – and cis isomer formation seemed to be due to the direct attack of an allyl radical on the carbonyl moiety, rather than the nucleophilic attack of the Ti(IV)-complex.
Once the η1-allyltitanocene(IV) complex formed, it can react with water to cause reduction of the allylic substrate. Albeit the protonolysis of the Ti(IV) complex should favour the reduction, a mixture of α- and γ-products (147) in an almost equimolar ratio was commonly obtained (Scheme 68, eqn (73)).
Allylic alkylation of nucleophiles: C-heteroatom bond formation. Palladium-catalysed allylation of hydroxy and amino groups has been extensively treated in the literature.205 However, methodologies involving a Pd catalyst predominantly provide regioisomers resulting from the nucleophilic attack at the less hindered carbon – mainly for the amination reaction – and with low stereoselectivity.
Interesting synthetic procedures that employ iron, iridium and rhodium complexes have been developed in order to overcome the poor regio- and stereoselectivity of Pd-catalysed allylation of N compounds.
Plietker206 proposed an efficient iron(II)-catalysed decarboxylative amination of primary and secondary allylcarbonates. A primary aromatic amine, such as aniline, was reacted with chiral allylcarbonate 148 using Bu4N[(FeCO)3(NO)] as a catalyst, and PPh3 as a ligand in the presence of piperidinium chloride buffer in order to preserve the catalyst efficiency (Scheme 69, eqn (74)).
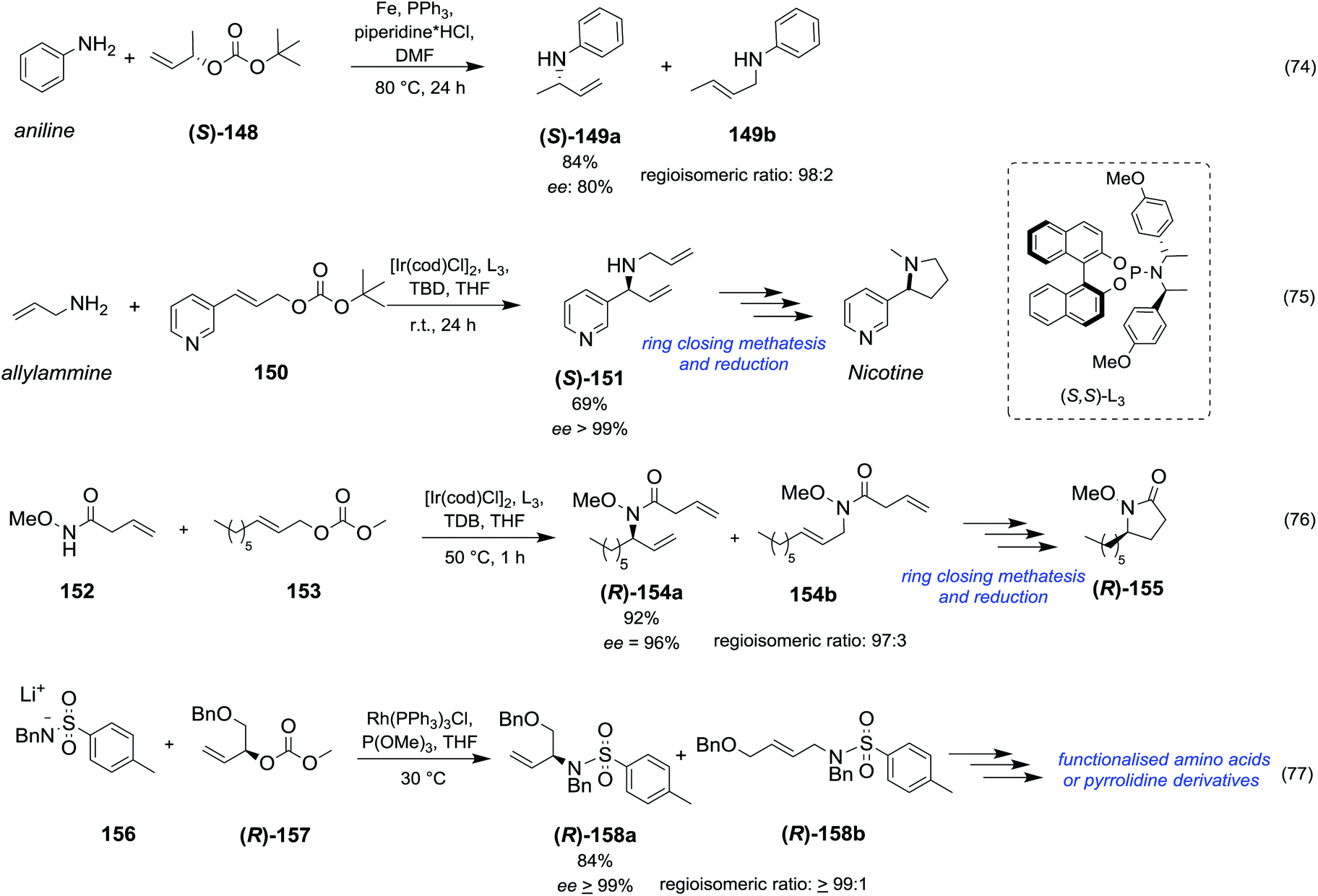 | ||
| Scheme 69 Fe-, Ir- and Rh-catalysed allyl alkylation of nitrogen.206–209 | ||
Compound 149a was achieved in good yield with high regioselectivity by heating the reaction mixture to 80 °C for 24 h.
Asymmetric Ir-catalysed allylation has been rather exploited as a valid alternative to Pd-catalysed allylation and a number of strategies involving amine and hydroxammic acid derivatives have been developed.
Enantiopure nicotine was synthesised by Grubb (II)-catalysed ring closing metathesis of compound 151 followed by racemisation-free double bond reduction (Scheme 69, eqn (75)).207 Allyl derivative 151 was obtained in moderate yield and with an ee > 99% by reaction of allylamine with carbonate 150 in the presence of bis(1,5-cyclooctadiene)diiridium dichloride [Ir(cod)Cl]2 as a catalyst, L3 as a ligand and TBD as a base.
A similar approach was used in the synthesis of pyrrolidinones (155) (Scheme 69, eqn (76))208via Ir-catalysed decarboxylative amination of carbonate 153 by hydroxyamic acid derivative 152 to give 154a with high regio- and enantioselectivity.
To date, only a few examples of enantiospecific asymmetric Rh-catalysed allylation of amine have been reported. Evans et al.209 developed a useful method employing Wilkinson's catalyst (Rh(PPh3)3Cl), P(OMe)3 as a ligand and lithium anions of differently substituted secondary amines protected by sulfonyl groups (156) as nucleophiles (Scheme 69, eqn (77)).
The enantioenriched sulfonylamminoallyl derivatives (158), obtained in good yield through this synthesis, may be used as synthons for the preparation of functionalised α-amino acids and nitrogen containing heterocycles.
Far less investigated is the transition metal-catalysed dercaboxylative allylation of thiol groups, which involves exclusively Pd and iridium-complex procedures.
As a representative example, [Ir(cod)Cl]2-catalysed reaction of 2-aminobenzene sodiumthiolate 159 with (E)-cinnamyl methyl carbonate 160 is reported in Scheme 70.210 The reaction led to 161a in good yield and excellent regio- and enantioselectivity. Traces of N,S-bisallylated derivative 161c were detected by NMR spectroscopy and GC-MS.
 | ||
| Scheme 70 Ir-Catalysed allyl alkylation of sulfur.210 | ||
Reductive allylation of alkyl halides. Qian et al.211 recently reported the direct allylation of various alkyl halides with allyl carboxylates and carbonates through C(sp3)alk–C(sp3)all coupling catalysed by the Co/Mn system.
3-Bromopropanenitrile was reacted with allylcarbonate 162 at 80 °C in acetonitrile using the CoBr2/Mn system as a catalyst, trifluoroacetic acid (TFA) as a catalyst-system activator and pyridine to stabilise the low-valent Co intermediate formed during the reaction (Scheme 71). The allylated product 163a was achieved in 93% yield and in an excellent isomeric ratio.
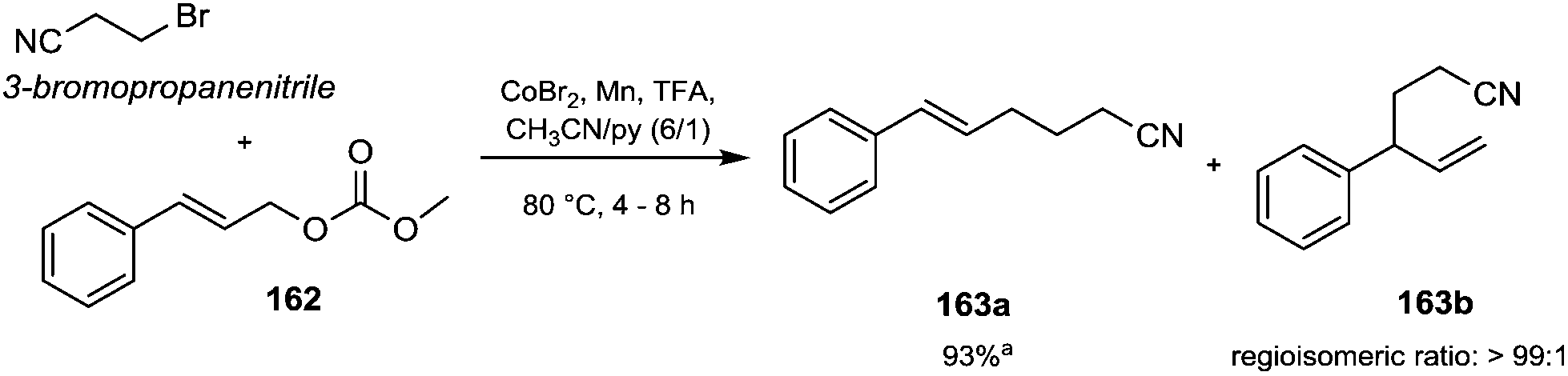 | ||
| Scheme 71 Example of reductive allylation of a functionalysed alkyl bromide catalysed by the Co/Mn system. aCalculated by GC-MS analysis.211 | ||
Allylation of styrenes via a Heck-type reaction. The [Ir(cod)Cl]2 catalyst has been employed to efficiently catalyse the cross-coupling reaction of 2-vinylanilines and allyl carbonates leading to the formation of skipped Z,E-dienes.212 This method provides the cis isomer and thus it exhibits the opposite regioselectivity relative to the Heck reaction, which instead affords the trans isomer.
O-Vinylaniline 164 was reacted with 2-furyl-substituted allyl carbonate 165 in THF using L2 as a ligand to give diene 166 in 92% yield (Scheme 72). Moreover, various aliphatic carbonate substrates – derived from γ-alkylallyl alcohols – and different aryl allylic carbonates incorporating either electron-releasing groups or electron-withdrawing groups were well tolerated and afforded the corresponding cross-coupling products in an excellent yield.
 | ||
| Scheme 72 Iridium-catalysed allylation of vinyls.212 | ||
Direct allylation of arenes. Arenes can be allylated directly via allyl carbonate by rhodium(III)-catalysed intermolecular allylation under mild reaction conditions, with a high γ-selectivity and isomeric ratio and good compatibility with a variety of functional groups.213
N,N-Diisopropylacetamide 167 allylation was catalysed by a permethylated cyclopentadienyl ruthenium complex ([Cp*RhCl2]2) adding AgSbF6 as an activator and pivalic acid (PivOH) in toluene at 30 °C (Scheme 73). The allylated compound 168 was achieved in 86% yield and a 50:1 regioisomeric ratio.
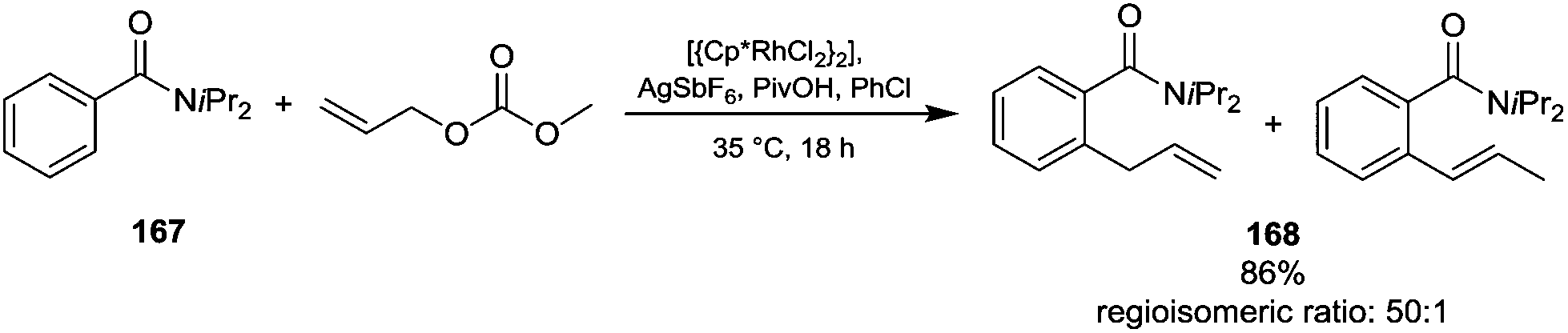 | ||
| Scheme 73 Rhodium-catalysed direct C–H allylation of arenes.213 | ||
Alder-ene reaction. Recently, Trost and co-workers214 reported a regio- and geometrical selective ruthenium complex catalysed strategy to produce allylate alkynes The diene 170 was obtained in 74% yield as a (Z/Z)
:(Z/E) mixture in a 1:3 ratio by a reaction of methyl 4-cyclohexylbut-2-ynoate 169 and allyl t-butyl carbonate using Cp*Ru(CH3CN)3+PF6− as a catalyst (Scheme 74). The nature of the allylic oxygen substituent may direct the isomer ratio.
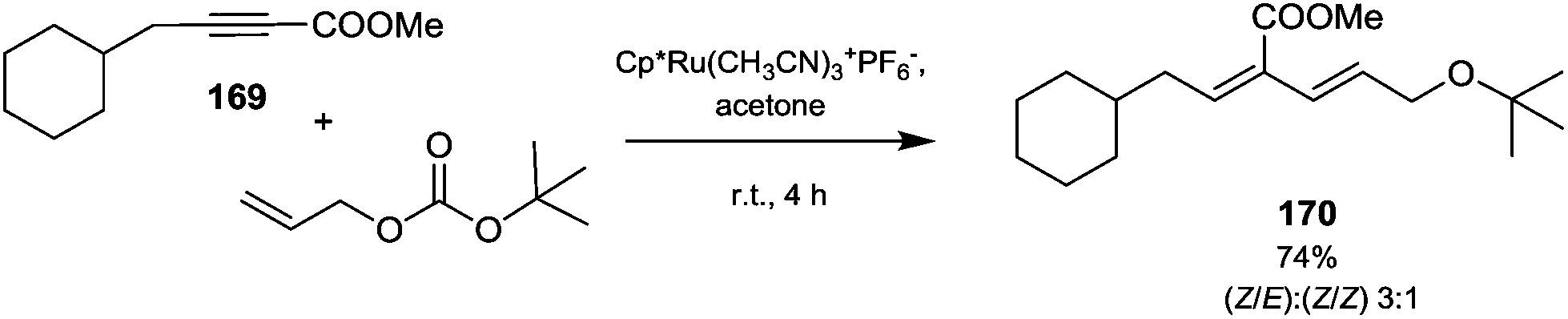 | ||
| Scheme 74 Ruthenium-catalysed Alder-ene reaction.214 | ||
Ni-Catalysed allylation. The synthesis of quaternary homoallyl alcohol 172 was accomplished in an excellent yield via reductive allylation by a Ni2I/Zn catalytic system using 2,6-bis[4′-(S)-isopropyloxazolin-2′-yl]pyridine ((S)-iPr-Pybox) as a chiral ligand – albeit no enantioselectivity was reported – (Scheme 75).215
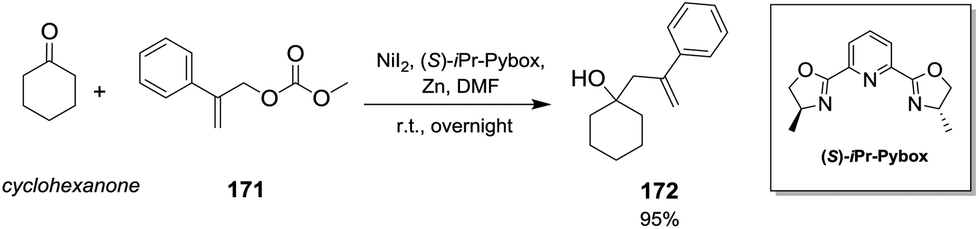 | ||
| Scheme 75 Ni-Catalysed reductive allylation.215 | ||
3.4 Nitrogen and sulfur-mustard carbonate analogues
Bis(2-chloroethyl) sulfide (Fig. 7) is sadly known as mustard gas, inasmuch as it was released in several chemical warfare mainly for its vesicant effects.216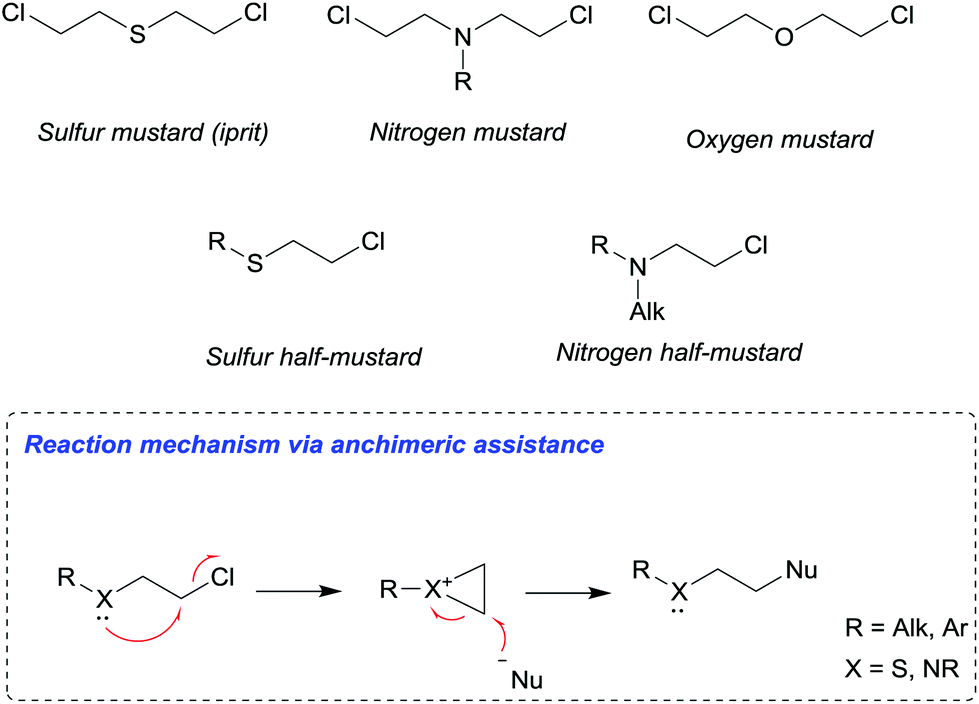 | ||
| Fig. 7 Chemical structure and reactivity of O, S, N mustards and half-mustards. | ||
Monochloro derivatives i.e. sulfur half-mustard, as well as mono- and dichloro oxygen/nitrogen analogues (i.e. oxygen/nitrogen half-mustards and mustards), exhibit high toxicity to humans and the environment.217
Despite their harmfulness, sulfur and nitrogen mustards have been extensively used in inorganic218 and organic synthesis,219 especially in the development of anticancer pharmaceuticals,220 for their peculiar reactivity. In fact, a chlorine displacement via anchimerically assisted – by sulfur and nitrogen – intramolecular nucleophilic substitution generates a highly electrophilic three-membered cyclic episulfonium/aziridinium ion, which easily undergoes nucleophilic attack (Fig. 7).221
In this perspective, mustard carbonate analogues may represent an intriguing green alternative to chlorine mustards as they are safe to handle, in principle non- or low toxic and many facets of their chemistry still need to be explored and disclosed.
O, S, and N mustard and the corresponding half-mustard carbonates were straightforwardly synthesized in good yield by reacting the related commercially available alcohols 173, 175a-d, 177, 179a–c and 181 with DMC or DEC via a BAc2 mechanism, using potassium carbonate as a base at refluxing temperature (Scheme 76, eqn (78)–(82)).222
 | ||
| Scheme 76 Syntheses of O-, S-, and N-half-mustard and mustard carbonates.222 | ||
The only exception was the reaction of 4-(N,N-dimethylamino)-1-butanol 179c with DEC (Scheme 76, eqn (81)), which failed to yield 180d and instead it gave quantitatively the stable quaternary N,N-dimethyl pirrolidinium salt 180e (Scheme 76, eqn (81) and Scheme 77, eqn (90)). The formation of compound 180e was described as a two-step cyclization reaction promoted by DEC via methoxycarbonylation of the hydroxyl group through a BAc2 mechanism and followed by intramolecular alkylation through a BAl2 mechanism.223
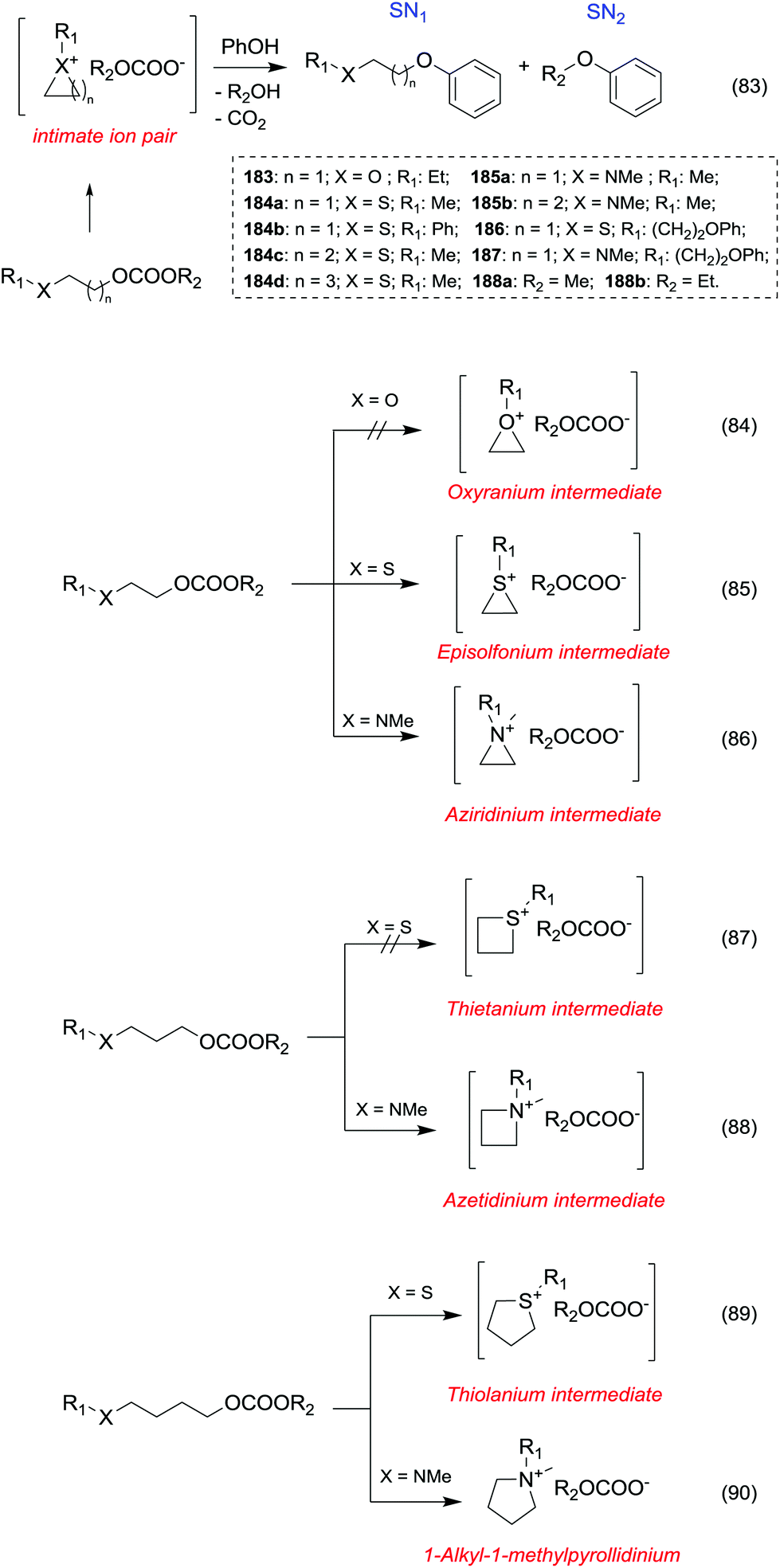 | ||
| Scheme 77 Syntheses of N-, S-, and O-half-mustard and mustard carbonates. | ||
In order to investigate the chemical behaviour of mustard carbonate as a neutral alkylating agent,224 a series of reactions were performed using phenol as a nucleophile and by testing different reaction conditions under both autoclave225 and neat conditions.226 The main results have been summarised in Table 18 and they provided clear evidence about the reaction mechanism (Scheme 77).
| No. | MC | N | X | R1 | R2 | Base (eq.) | Solvent | T (°C) | t (h) | Conv. (%) | Product selectivityf (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1X(CH2)nOPh | PhOR2 | |||||||||||
| Reaction of O-, S-, and N-half-mustard carbonate and mustard carbonate analogues with phenol.a Carbonate:phenol 1.0:3.0 (molar ratio) reaction performed in an autoclave.b Carbonate:phenol:base 1.0:3.0:1.0 (molar ratio) reaction performed in an autoclave.c Carbonate:phenol 1.0:2.0 (molar ratio) reaction performed in batch at 1.0 atm.d Carbonate:phenol 1.0:1.0 (molar ratio) reaction performed in an autoclave.e Carbonate:phenol 1.0:2.0 (molar ratio) reaction performed in an autoclave.f Calculated by GC-MS analysis.224–226 |
||||||||||||
| 1 | 174b | 1 | O | Me | Et | None | CH3CN | 180 | 24 | 0 | 0 | 0 |
| 2 | 174b | 1 | O | Me | Et | K2CO3 (1.0) | CH3CN | 180 | 24 | 97 | 28 (183) | 72 (188b) |
| 3 | 174a | 1 | O | Me | Me | K2CO3 (0.2) | none | 150 | 5 | 100 | 19 (183) | 81 (188a) |
| 4 | 176a | 1 | S | Me | Me | None | CH3CN | 180 | 24 | 100 | 100 (184a) | 0 |
| 5 | 176a | 1 | S | Me | Me | None | None | 150 | 5 | 56 | 86 (184a) | 6 (188a) |
| 6 | 176a | 1 | S | Me | Me | K2CO3 (0.2) | None | 150 | 5 | 100 | 90 (184a) | 0 |
| 7 | 176a | 1 | S | Me | Me | DBU (0.2) | None | 150 | 5 | 75 | 92 (184a) | 6 (188a) |
| 8 | 176b | 1 | S | Me | Et | None | CH3CN | 180 | 24 | 81 | 100 (184a) | 0 |
| 9 | 176b | 1 | S | Me | Et | K2CO3 (1.0) | CH3CN | 180 | 24 | 100 | 58 (184a) | 41 (188b) |
| 10 | 176c | 1 | S | Ph | Me | None | CH3CN | 180 | 24 | 36 | 45 (184b) | 0 |
| 11 | 176d | 1 | S | Ph | Et | None | CH3CN | 180 | 24 | 50 | 16 (184b) | 0 |
| 12 | 176e | 2 | S | Me | Me | None | CH3CN | 180 | 24 | 0 | 0 (184c) | 0 |
| 13 | 176f | 3 | S | Me | Me | None | CH3CN | 180 | 8 | 90 | 85 (184d) | 13 (188a) |
| 14 | 180a | 1 | NMe | Me | Me | None | CH3CN | 180 | 5 | 100 | 100 (185a) | 0 |
| 15 | 180b | 1 | NMe | Me | Et | None | CH3CN | 180 | 5 | 100 | 100 (185a) | 0 |
| 16 | 180c | 2 | NMe | Me | Me | None | CH3CN | 180 | 6 | 80 | 94 (185b) | 0 |
| 17 | 178 | 1 | S | (CH2)2OCO2R2 | Me | None | CH3CN | 180 | 24 | 0 | 0 (186) | n/a |
| 18 | 182 | 1 | NMe | (CH2)2OCO2R2 | Me | None | CH3CN | 180 | 5 | 100 | 90 (187) | n/a |
| 19 | 182 | 1 | NMe | (CH2)2OCO2R2 | Et | None | CH3CN | 180 | 6 | 100 | 90(187) | n/a |
Oxygen carbonate half-mustards 174a and 174b needed the presence of a base to react with phenol both in an autoclave using acetonitrile as a solvent and in neat at atmospheric pressure (no. 1–3, Table 18). Albeit quantitative conversions were achieved, the selectivity registered was modest as the formation of the oxyranium intermediate (Scheme 77, eqn (84)) via the anchimeric effect followed by the SN1 attack by phenol is rather unfeasible due to the instability of the cyclic cation. As a consequence, anisole 188a (or ethoxybenzene 188b) was detected by GC-MS as the major product.
On the other hand, sulfur half-carbonates 176a and 176b both reacted with phenol in an autoclave for 24 h at 180 °C with 100% and 81% conversions, respectively, and showed quantitative selectivity toward the desired alkylated product 184a (no. 4 and 8, Table 18), confirming the generation of an episolfonium intermediate (Scheme 77, eqn (84)).
From a mechanistic point of view, in this type of reaction, the episolfonium ion is trapped in a molecular cage in an intimate ion pair (Scheme 77) where diffusion phenomena limit and influence the reaction rate. The trapped cyclic intermediate is in equilibrium with the starting carbonate 176. Once the episulfonium intermediate is freed from the solvent cage it can then either react with the nucleophile or, in the absence of CH3OCOO− anions and nucleophiles, decompose into other products.
The same alkylation reaction was then performed adding 1 eq. of K2CO3 (#9, Table 18) to a mixture of phenol and carbonate 176b and it led to 100% conversion, but drastically increased the formation of ethyl phenyl ether 188b (41%). This result showed that in an autoclave the presence of a base activates the nucleophile toward the BAl2 mechanism (SN2), which was competitive with the anchimeric effect (SN1).
The reaction of 176a gave quantitative conversions and 90% selectivity toward the alkylated product 184a without detection of anisole by GC-MS analysis when it was conducted solventless in batch at 150 °C for 5 h and adding a substoichiometric >amount (0.2 eq.) of potassium carbonate (no. 6, Table 18).
A slightly higher selectivity (92%) was achieved using DBU as a base; however, the conversion decreased to 75% and 6% anisole was produced (no. 7, Table 18).
Owing to a lower sulfur nucleophilicity, the reaction in an autoclave of 2-(phenylthio)ethyl alkyl carbonates 176c and 176d with phenol led to products 184b in modest conversion and selectivity (no. 10 and 11, Table 18).
Compound 176e incorporating a propyl chain as a spacer between the methyl thio group and the carbonate moiety failed to react with phenol in the autoclave (no. 12, Table 18), whereas compound 176f with a butyl chain as a spacer gave the desired product 184d in 90% conversion and 94% selectivity (no. 13, Table 18). Most likely, the formation of the sterically constrained thietanium intermediate, via anchimeric effect, is not as favoured as in the case of homologues three- and five-membered (i.e. thiolanium) cyclic cation intermediates (Scheme 77, eqn (87) and (89)).12b
N,N-Dimethyl-2-phenoxyethylamine 185a was achieved in excellent yields by reaction of nitrogen half-mustard carbonates 180a and 180b with phenol in an autoclave through an aziridinium intermediate (Scheme 77, eqn (86)) in only five hours (no. 14 and 15, Table 18). Further evidence of the superior stability of cyclic nitrogen cation intermediates was provided by the reaction of 2-(dimethylamino)propyl methylcarbonate 180c, which proceeded with 80% conversion and 94% selectivity (no. 16, Table 18), suggesting the formation of a four-membered azetidinium intermediate (Scheme 77, eqn (88)).
The symmetric bis-(2-methylcarbonate)ethyl sulphide 187 failed to react with the nucleophile even at higher temperatures i.e. 200 °C (no. 17, Table 18). This result may be ascribed to its steric hindrance, which was predominant over the anchimeric effect of the sulfur under these reaction conditions. As expected, the reaction of bis-N,N-[(2-alkylcarbonate)ethyl]methylamines 182a–b with phenol resulted in a double alkylation to afford 187 in quantitative conversions and high isolated yields (no. 18 and 19, Table 18).
From these data it is also evident that nitrogen mustard carbonates are more reactive in autoclave conditions, while sulfur analogues are more reactive under neat conditions.
Moreover, kinetics studies were conducted on both sulfur and nitrogen half-mustard analogues.225 In Fig. 8, the reaction kinetics of (dimethylamino)ethyl ethyl carbonate 180b and phenol in an autoclave has been reported. This studies clearly highlighted that alkylation followed the first order kinetics (in black in Fig. 8).
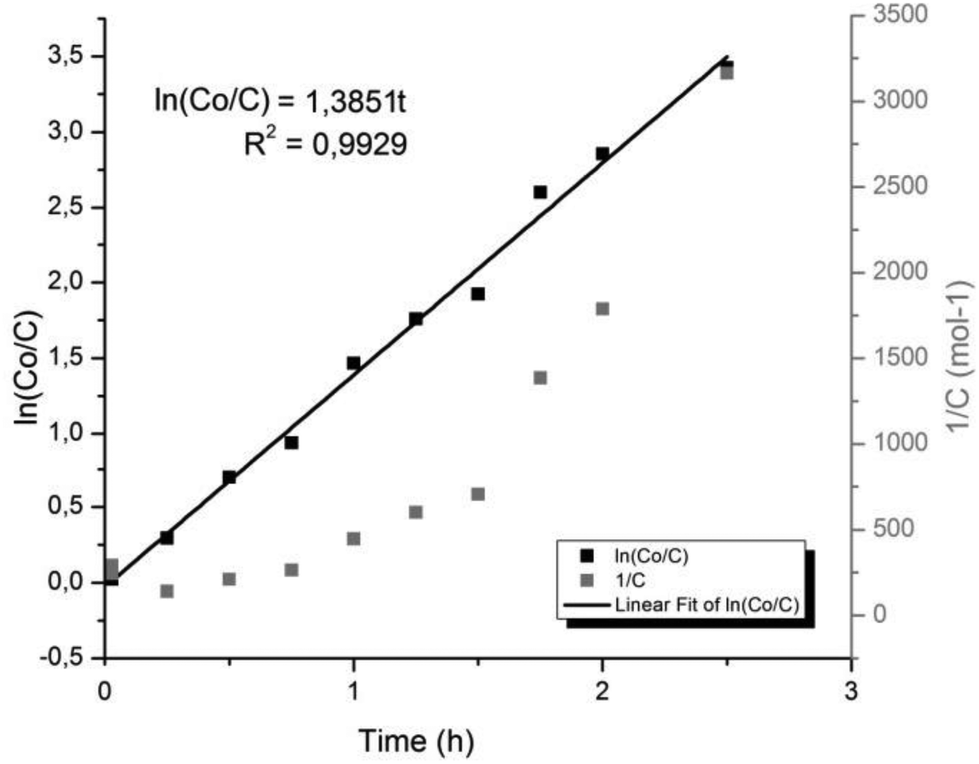 | ||
| Fig. 8 A comparison between first (black) and second (grey) order kinetics plotting of the reaction of carbonate 180b with phenol (1:1 molar ratio heating in an autoclave at 180 °C) where C is the concentration of the carbonate (mol l−1); p-xylene was used as internal reference. The data support that the reaction kinetic is SN1-like. | ||
A variety of nucleophiles, such as aromatic alcohols and diols and compounds incorporating acidic CH2, were tested under batch conditions with or without the addition of a base, aiming to further study the reactivity of sulphur mustard analogues.227
Among these nucleophiles, p-cyanophenol turned out to be particularly reactive due to the electron-withdrawing nitrile moiety, which prompted the alkylation reaction in neat at 150 °C and unde neutral conditions (Scheme 78). The effectiveness of p-cyanophenol as an activate nucleophile was highlighted by its reaction with several sulfur mustard carbonates. The resulting anchimerically driven alkylated products were achieved in good to excellent yield (54–99%).
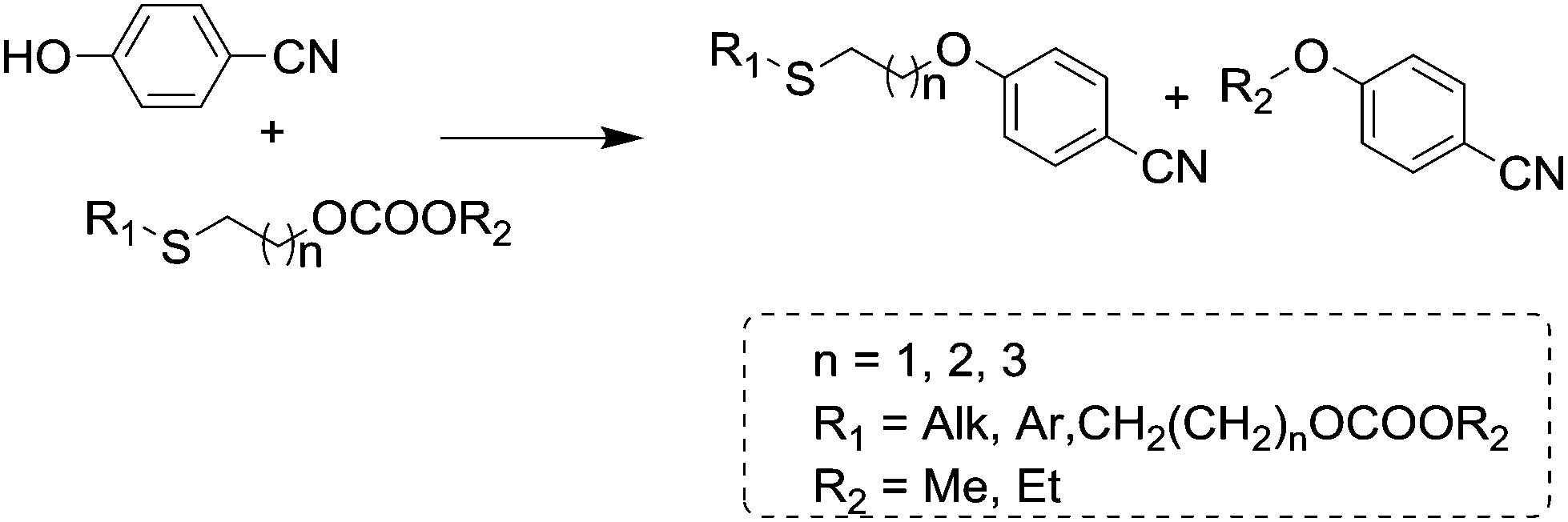 | ||
| Scheme 78 Reaction of sulphur half-mustard carbonate and mustard carbonate analogues with p-cyanophenol under batch conditions at 150 °C.227 | ||
Nitrogen mustard carbonate analogues were also reacted with a wide range of nucleophiles in an autoclave under neutral conditions (Table 19).
|
|
|||||
|---|---|---|---|---|---|
| MC | X | R1 | R2 | Nu | Product selectivityc (%) |
|
a Carbonate:nucleophile 1.0:3.0 (molar ratio) reaction performed at 180 °C in an autoclave for 5 h and using ACN as solvent. Conversion of the nucleophile was 100% in all the reactions.
b Carbonate:nucleophile 1.0:1.0 (molar ratio) reaction performed at 180 °C in an autoclave for 5 h and using ACN as solvent.
c Calculated by GC-MS analysis.
d 60% Isolated yield.225
|
|||||
| 180b | NMe | Me | Et | KCN |
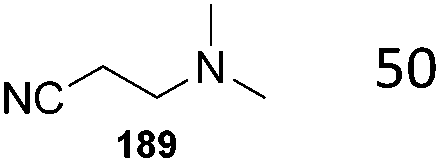
|
| 180b | NMe | Me | Et |
|
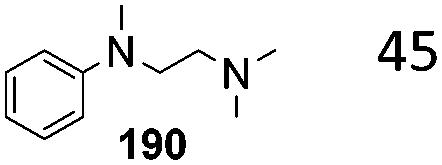
|
| 180b | NMe | Me | Et |
|

|
| 182a | N | (CH2)2OCO2R2 | Me |
|
|
2-(N,N-Dimethylamino)ethyl cyanide 189 was achieved in 50% yield by reaction of 2-(N,N-dimethylamino)ethyl ethyl carbonate 180b with potassium cyanide. A similar result was obtained by reaction of the same carbonate 180b with N-methylaniline to give compound 190.
Particularly interesting was the reaction involving the acidic methylene of phenylsulphonyl acetonitrile. Half-mustard 180b was selectively alkylated to give the expected compound 191 in 94% yield. Instead, the reaction between symmetrical mustard 182a and phenylsulphonyl acetonitrile led to the piperidine derivative 192 – isolated in 60% yield – via an intramolecular cyclisation mechanism (i.e. a double alkylation reaction).
Dimethyl ((methylazanediyl)bis(ethane-2,1-diyl)) dicarbonate 182a has been successfully used for the synthesis of a novel series of azacrowns (Fig. 9).228 Carbonate 182a polycondensated with biphenyl-2,2-diol and 1,1′-bis-2-naphthol under dilution conditions and in the presence of potassium nitrate to give the corresponding azacrown and diazacrown isolated by column chromatography on silica gel.
 | ||
| Fig. 9 Example of azacrown synthesis from nitrogen mustard carbonate 182a and related X-ray structures. | ||
Diazacrown structural elucidation was accomplished by X-ray diffraction analysis (Fig. 9).229
This approach was also employed for the synthesis of dibenzo 18-, 20- and 22-diazocrown-6 starting from, respectively, catechol, resorcinol and hydroquinone with mustard carbonate 182a without the addition of any base. Cyclic dimers were obtained pure after gravimetric chromatography on silica gel and they were fully characterised, including by single crystal X-ray diffraction analysis (Fig. 9).
All the azacrowns were achieved in good yields. It should be pointed out that previously these macrocycles could be achieved only using a chlorine-based chemistry or by multistep reactions.
4. Cyclization
Five- and six-membered nitrogen- and oxygen-based heterocycles, such as substituted benzofuran, benzodioxines, indolines and indoles, are key structural elements in biologically active natural products and pharmaceuticals.The 2,3-dihydrobenzofuran motif, for instance, is present in natural neolignans and lithospermic acids230 and is incorporated into several drugs exhibiting antimicrobial,231 antioxidant,232 antimitotic, antiangiogenis233 or neuritogenic activities.234 Benzodioxane derivatives are mainly employed for their effects on the nervous system (such as antidepressant,235 neurotrophic236 and α-adrenergic blocking agents237) and, moreover, a simple 2,3-dihydro-1,4-benzodioxine is present in isovanillyl sweetening agents. Likewise, indolines are present as structural subunits in vitamins, hormones and alkaloids and their synthesis is industrially relevant for the development of pharmaceuticals, herbicides, pesticides, dyes, etc.238 From this perspective, the reaction of DMC with aliphatic and aromatic 1,4- and 1,5-bifunctional compounds represents a green and valid alternative to cycloaddition and/or cyclization reactions involving transition-metals or halogens – as well as tosylate and mesylate – chemistry, which still are the most common approaches to synthesise five- and six-membered N- and O-based heterocycles.
4.1 Synthesis of O-based heterocycles
Oxolane and oxane derivatives (alkyl- and benzo-) are obtained by reaction of DAC with proper 1,4- and 1,5-diol derivatives under basic conditions.5e,12a,239 First attempts at this reaction were carried out by using a stoichiometric excess of a strong base, such as NaOMe and t-BuOK, in acetonitrile and heating to reflux.12aThe related five- and six-membered rings were achieved in quantitative yield and preliminary computational studies demonstrated that after methoxycarbonylation of one hydroxyl group (generally following the reactivity order: primary alcohol > secondary alcohol > tertiary alcohol > phenyl alcohol) the formation of cyclic ethers was the most energetically favored pathway. This in silico evidence confirmed the proposed two-step mechanism for the DMC-assisted cyclization (Scheme 79) that involved a methoxycarbonylation of the less hindered alcohol via BAc2 followed by intramolecular alkylation reaction via BAl2. Definitely, the former step occurs according to the HSAB theory, whereas the latter is most likely promoted by strong entropic effects, which lead to ring closure.
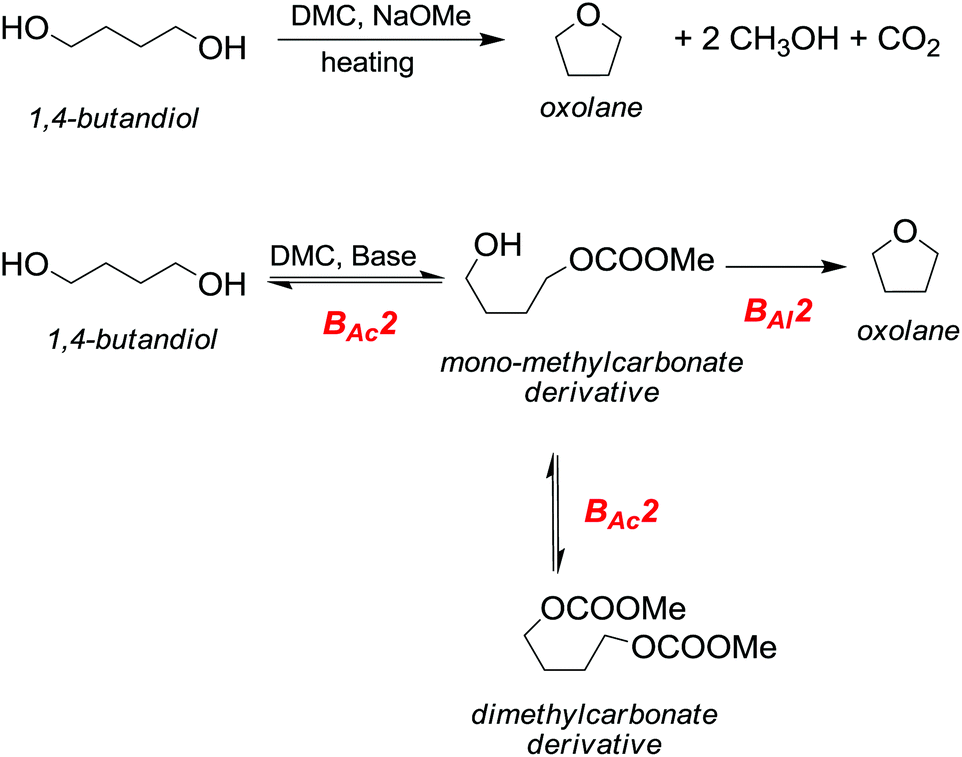 | ||
| Scheme 79 General mechanism for the synthesis of O-based heterocycles from 1,4-bifunctional compounds via DMC chemistry. | ||
The cyclization of Amberlyn diol 193 to Norlabdane oxide 194 – known with the trade name Ambroxan – is a poignant example of the DMC chemistry efficiency (Scheme 80, eqn (91)). Ambroxan is used in perfumery for its characteristic ambergris-type odour and it is industrially synthesised under acidic conditions in ca. 60% yield, due to a mixture of by-products formed as result of competing elimination reactions. On the other hand, the reaction of Amberlyn diol with DMC and t-BuOK at reflux temperature (90 °C) gives the desired product 194 in almost quantitative yield. Aiming to further investigating the reaction conditions, DMC-assisted cyclisation was tested upon different aromatic 1,4- and 1,5-diol derivatives in neat by using Lewis bases such as DABCO, TBD, and DBU 1,5-diazabiciclo[5.4.0]undec-5-ene (DBU) in stoichiometric (Scheme 80, eqn (93) and (94)), substoichiometric (Scheme 80, eqn (92)) or catalytic amounts (Scheme 82, eqn (98) and (99)).
 | ||
| Scheme 80 Synthesis of O-heterocycles 194, 196, 198, 199 and 201.5e,12a,239 | ||
These bicyclic amines display, indeed, a double function in methoxycarbonylation reactions by operating as base and as nucleophiles for the activation of DAC (Scheme 81).171 It has been reported that the reaction of a catalytic amount of a bicyclic base with DMC most probably generates an N-methoxycarbonyl amidinium adduct via a BAc2 mechanism and thus promote the methoxycarbonylation of the substrate hydroxyl group. The carbonate derivative obtained may undergo a further BAc2 by reacting with the bicyclic amine and giving a second N-alkoxycarbonyl amdinium adduct. The incorporation of this adduct into 1,4- and 1,5-bifunctional systems may activate the ring formation via intramolecular BAl2.
 | ||
| Scheme 81 Proposed mechanism for the cyclization of 1,4-diol with DMC in the presence of DBU. | ||
The N-carboxy amidinium group displacement and its decarboxylation will then regenerate the Lewis base. The synthesis of 1,3-dihydrobenzofuran 198 (Scheme 80, eqn (93)) from 2-bis(hydroxymethyl)benzene 197 was the only one reported within this series of cyclisations to occur in moderate yield and to give as a major by-product the dicarbonate derivative 199.
This fact highlights that in addition to the activation of DMC and basicity of nitrogen bicyclic bases (TBD > DBU ≫ DABCO), the cyclization may be affected by other effects (e.g. steric and entropic) strictly related to the affinity between substrate and base, which may hinder the ring closure.
A further application of base-catalysed DAC-assisted cyclization of 1,4-diols is the synthesis of isosorbide and its epimer isommanide (Scheme 82, eqn (98) and (99)), belonging to the family of anhydro sugar alcohols extensively employed in the food and pharmaceutical industries, as well as monomers in the formation of polymers and co-polymers.5c Isosorbide and its derivatives are used as diuretics,240 and in the treatment of cardiac failure and angina pectoris241 and oesophageal spasms242 and they are obtained industrially by acid-catalysed dehydration of their parent alcohols. The reactions of D-sorbitol and D-mannitol with DMC have been reported first using an excess of NaOMe (4 eq.) and MeOH as solvent and then in neat with a catalytic amount of DBU heating to 90 °C.5e,12
 | ||
| Scheme 82 Synthesis of anhydro sugar isosorbide and isomannide. aDetermined by GC-MS.5e,12 | ||
Both the procedures gave the desired anhydro sugar in very good yield through a stereoselective double cyclisation. Moreover, the dimethoxycarbonyl derivative of isosorbide can undergo a third five membered ring closure (Scheme 82, eqn (98), compound 202) by reaction with KW2000 and heating to 240 °C in continuous flow.
Roy et al.243 reported an efficient two-step large-scale synthesis of 2′-O-alkyl pyrimidine ribonucleosides (Scheme 83), commonly introduced into oligonucleotides to confer them with stability and binding capability toward proper gene targets.
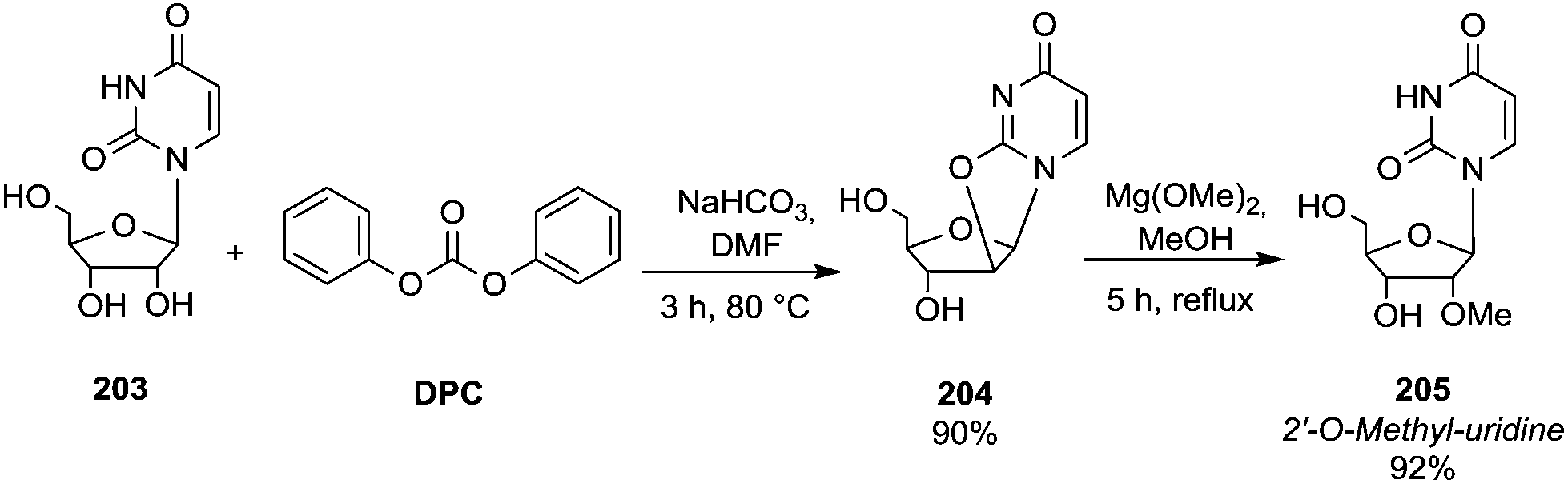 | ||
| Scheme 83 Synthesis of 2′-OMe-uridine 205via a cyclic intermediate.243 | ||
The synthesis of 2,2′-anhydrouridine 204 was carried out selectively and in 90% yield by reaction of uridine 203 with diphenyl carbonate (DPC) in DMF, using sodium bicarbonate in a catalytic amount and heating to 80 °C for 3 h. Treatment of compound 204 with magnesium methoxide in MeOH at reflux afforded 2′-O-methyl-uridine 205 in 92% yield.
4.2. Synthesis of the cyclic carbamates 1,3-oxazinan-2-ones
A series of six-membered cyclic carbamates 1,3-oxazinan-2-ones 208a–f (Scheme 84, eqn (100))244 were synthesized in a short time span and in the absence of a solvent via reaction of primary amines or hydrazines 206a–c with dimethoxycarbonyl derivatives 207a–b – obtained by reaction of DMC with proper 1,3-diols. The one-pot reaction proceeds via a two-step mechanism: an intermolecular substitution to give a linear carbamate intermediate (BAc2) and an intramolecular cyclization (BAl2), which leads to the cyclic carbamate in moderate yields by carbonate moiety displacement. Further procedure developments245 provided carbamates 212a–e in good yield (except 212a) through a double BAC2 mechanism by reaction of substituted aminopropanols 211a–e with EC using a substoichiometric amount of TBD (Scheme 84, eqn (102)). The general reaction pathway consisted of the alkoxycarbonylation of the hydroxyl group (the first BAC2) to obtain – by EC ring opening – a carbonate intermediate, which gave ring closure by intramolecular attachment of the amino group (the second BAC2) and consequent displacement of ethylene glycol.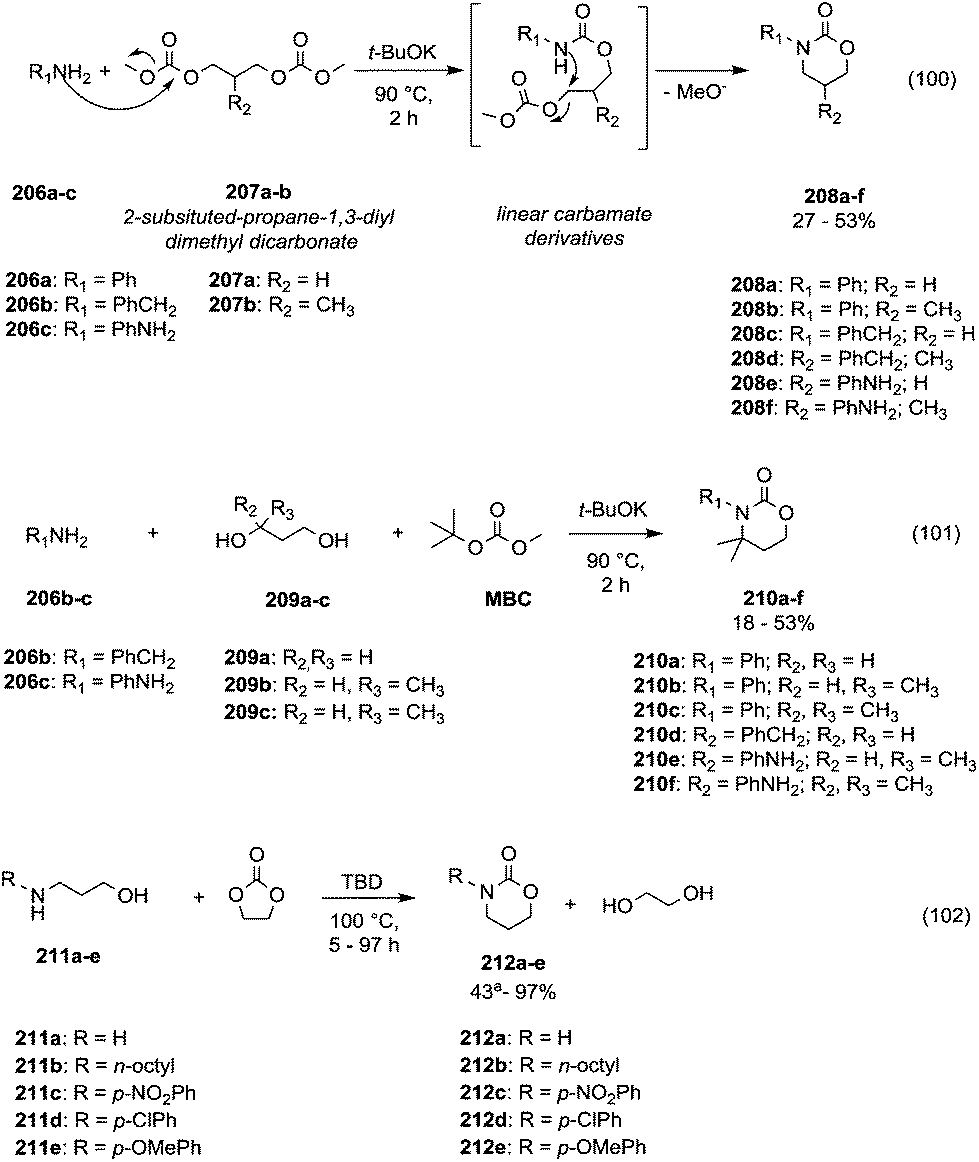 | ||
| Scheme 84 Synthesis of 1,3-oxazinan-2-ones via cyclisation. a Isolated yields: 212a = 43%, 212b–e > 93%.244,245 | ||
4.3. Synthesis of N-based heterocycles
The one-pot cyclisation of alkyl- or aryl-amino alcohol bifunctional compounds may proceed similarly to that of diols through methoxycarbonylation of both hydroxyl and amino groups via a BAc2 mechanism, followed by entropically favoured ring closure via a BAl2 mechanism to give N-methoxycarbonylated final compounds (Scheme 85).246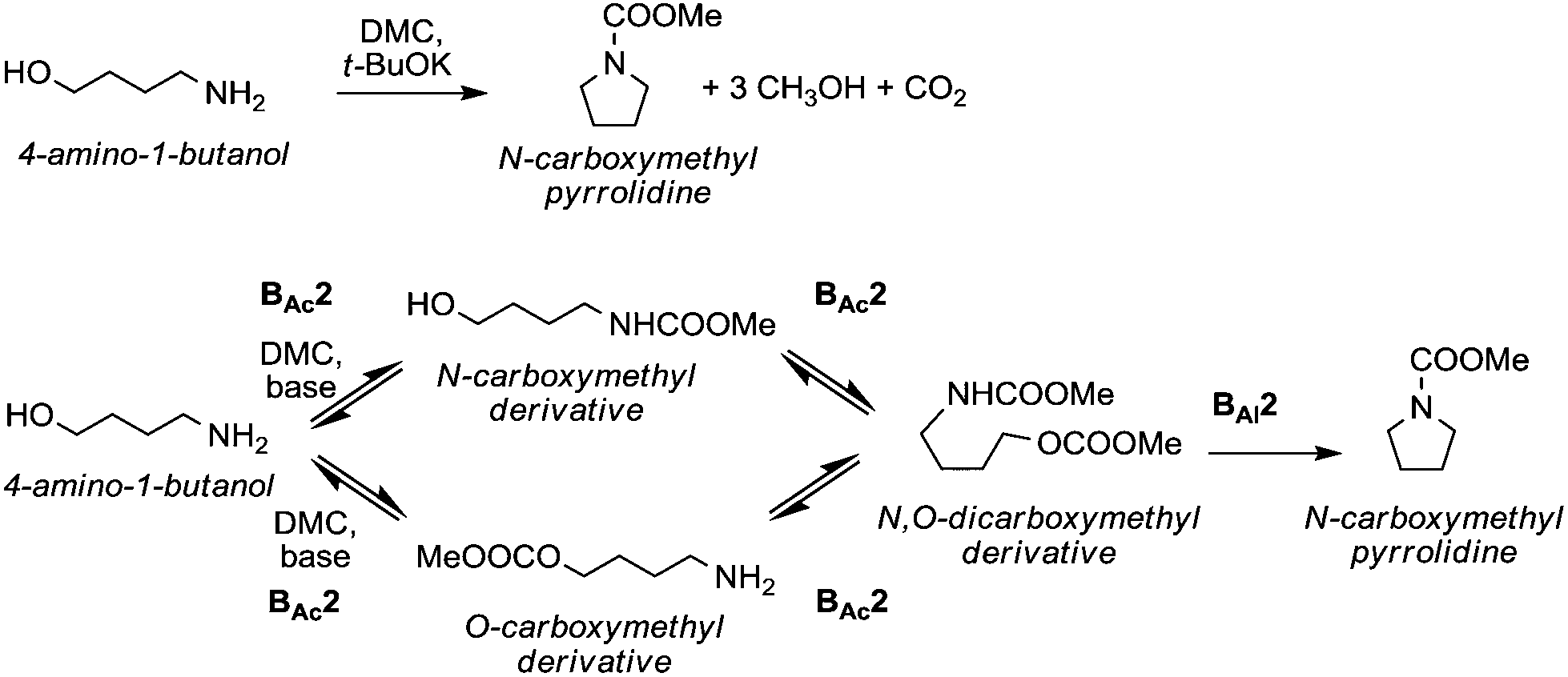 | ||
| Scheme 85 General mechanism for the synthesis of N-based heterocycles from 1-O-4-N-bifunctional compounds via DMC chemistry. | ||
The desired pyrrolidine and indoline derivatives were achieved in good yield by reaction with DMC and a stoichiometric excess of strong base (Scheme 86, eqn (103), compound 214) or with a substoichiometric amount (0.5 eq.) of TBD (Scheme 86, eqn (104), compound 216) in neat and heating to reflux.
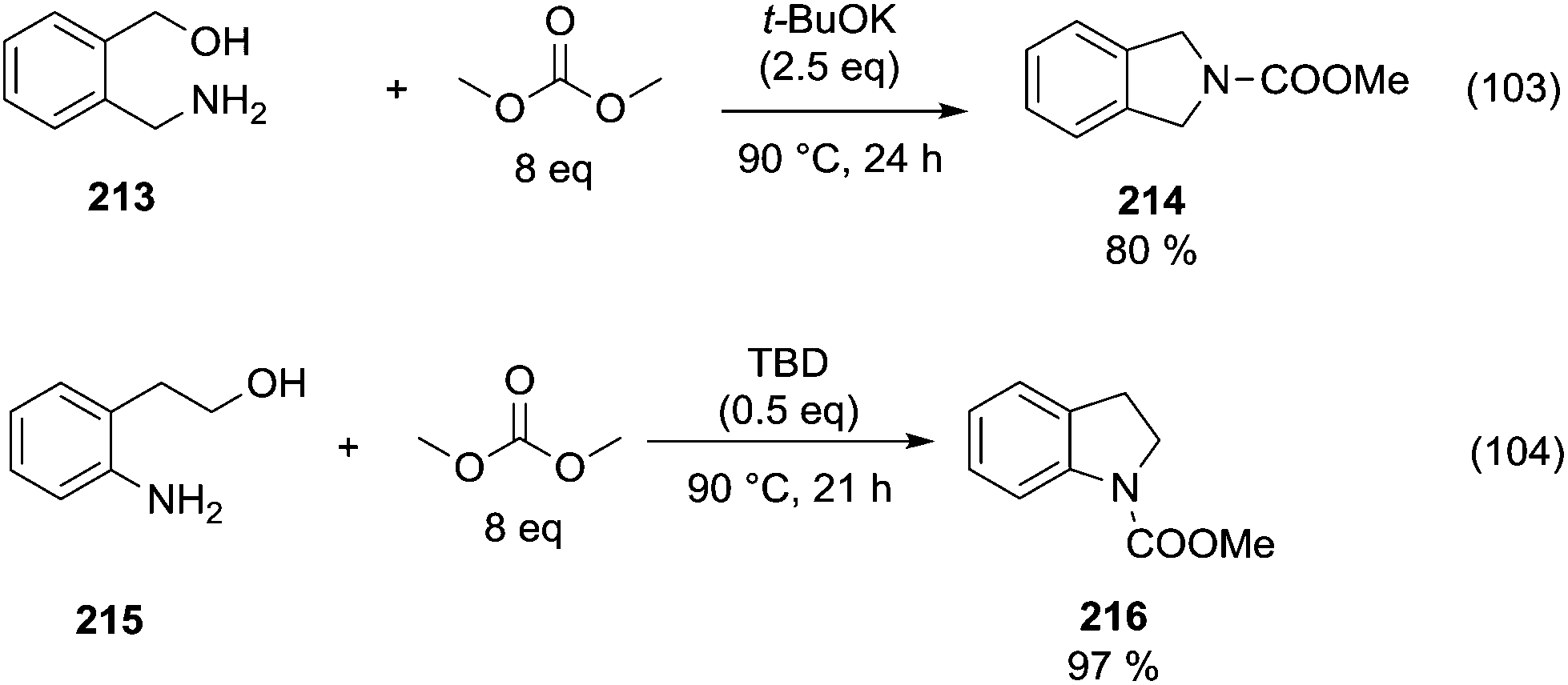 | ||
| Scheme 86 Synthesis of N-heterocycles 214 and 216.246 | ||
5. Transposition
DACs have been reported to promote rearrangements by reaction under basic conditions with alkyl and aryl hydroxamic acids (the Lossen rearrangement), substituted 1,3-oxaz-5-ol (the Steglich rearrangement) and indol-2-ol derivatives (carboxyl migration of indol esters), and alkyl and benzyl oximes ([3,3] sigmatropic rearrangement). A brief overview of DAC-assisted-transposition is given below.5.1. The Lossen rearrangement
In 1872 W. Lossen247 demonstrated that activated benzoylated benzhydroxamic acid underwent facile rearrangement under basic conditions into an isocyanate intermediate, which degraded into aniline upon aqueous treatment. Subsequently the same reaction was carried out under anhydrous conditions to obtain carbamates and ureas upon addition to the reaction medium of, respectively, alcohols or amines.Recently, Kreye et al.248 have reported an efficient alternative to the Lossen rearrangement by using a substoichiometric amount of tertiary base (e.g. TDB, DABCO and triethylamine (TEA)), different DACs as activators, and the corresponding alcohol as initiators and heating the mixture to reflux (Schemes 87 and 88).
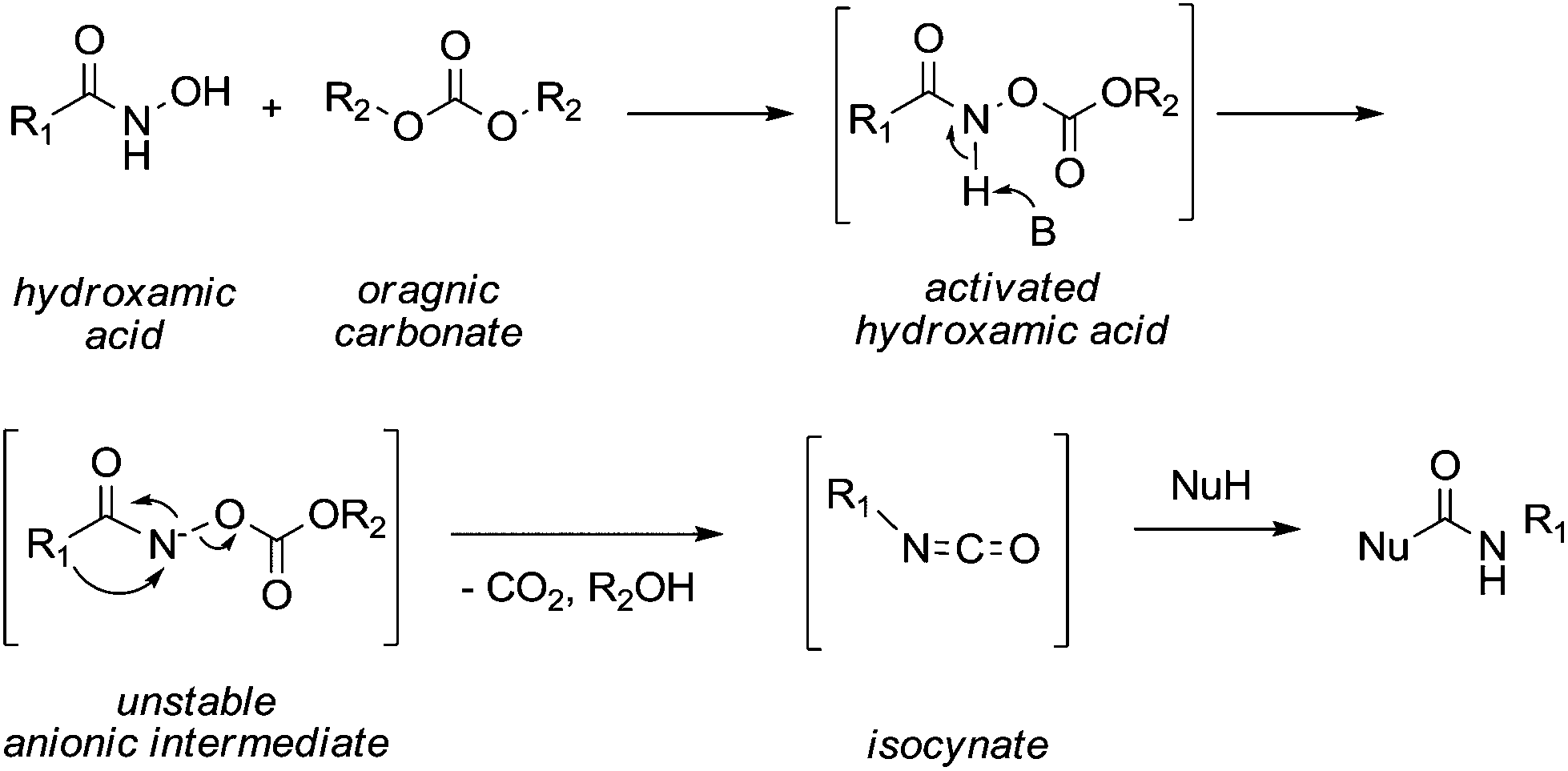 | ||
| Scheme 87 The Lossen rearrangement mechanism in the presence of organic carbonates. | ||
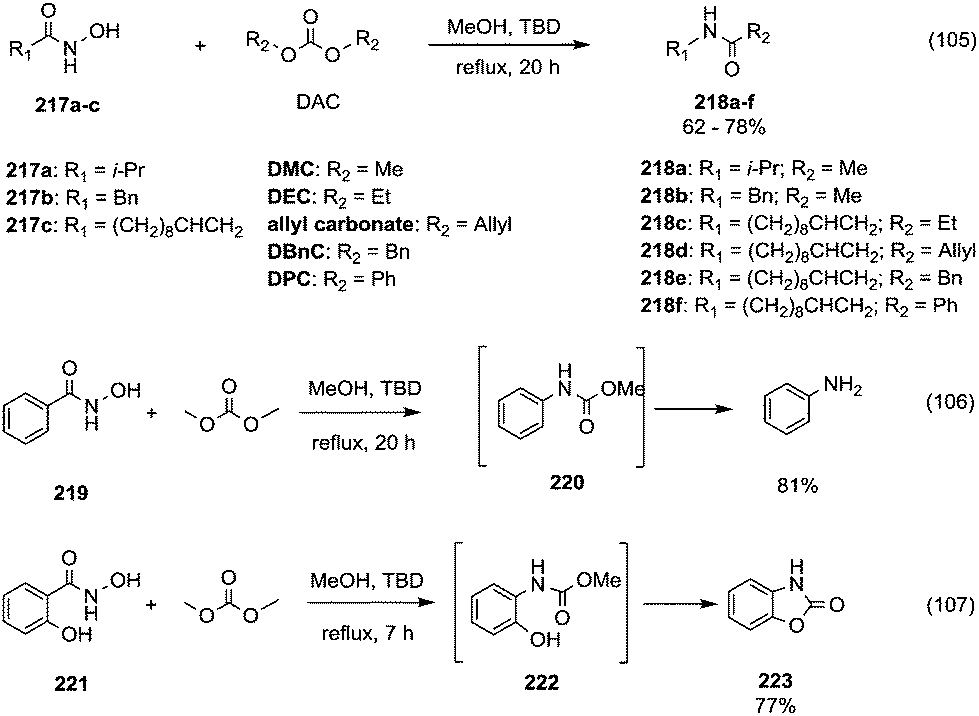 | ||
| Scheme 88 The Lossen rearrangement of different hydroxamic acids by reaction with DACs and a substoichiometric amount of base (0.2–0.4% eq.).248 | ||
In this procedure, the hydroxyl group of a hydroxamic acid derivative is activated by methoxycarbonylation (BAc2 mechanism) under basic conditions to give the unstable anionic intermediate (Scheme 87), which by decarboxylation prompts the transposition of the alkyl group attached to the carbonyl moiety to the nitrogen, generating a highly reactive isocyanate intermediate, alcohol and CO2. This procedure was tested on several alkylic, allylic, and benzylic derivatives 217a–c (Scheme 88, eqn (105)) giving the desired carbamates 218a–f in moderate to good yields. The reaction of N-hydroxybenzamide 219 with DMC afforded aniline in good yield (Scheme 88, eqn (106)), whereas N-2-dihydroxybenzamide 221 gave the oxazol-2-one 223 in 77% yield by intramolecular cyclization of the carbamate intermediate 224 (Scheme 88, eqn (107)).
5.2. The Steglich rearrangement
The Steglich [1,3] rearrangement249 is the conversion of an oxazol-5-yl carbonate to the corresponding 4-carboxyl azlactone via base-catalysed transposition (Scheme 89).250 Lewis bases such as 4-dimethylaminopyridine (DMAP) derivatives and achiral N-heterocyclic carbenes (NHCs) were shown to be excellent catalysts for O- to C-carboxyl rearrangement of a number of heterocyclic carbonate derivatives.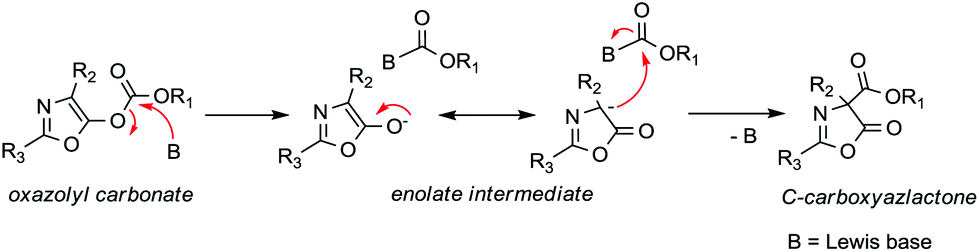 | ||
| Scheme 89 The Steglich rearrangement mechanism of oxazolyl carbonate. | ||
Shaw et al.251 reported that both R- and S-enantiomers of the hindered [3-(2,2,2-triphenyl-1-acetoxyethyl)-DMAP (TADMAP) in t-amyl alcohol at 0 °C catalysed the enantioselective Steglich rearrangement of oxazolyl enol carbonates, leading to the formation of an asymmetric quaternary carbon (Scheme 90, eqn (108) and (109)).
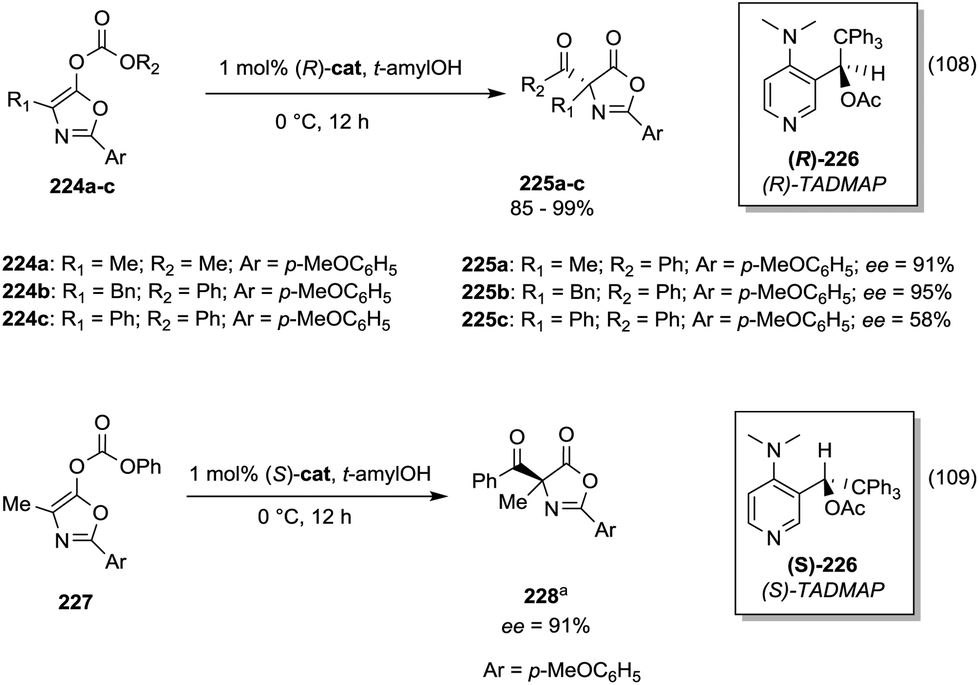 | ||
| Scheme 90 Enantioselective Steglich rearrangements catalysed by chiral TADMAPs. aReaction yield not reported.251 | ||
Although differently substituted carboxylated azlactones (225a–c) were achieved in good yields, the enantioselectivity efficiency of the chiral base seemed to be negatively affected by steric constraints as confirmed by the ee of compound 225c (Scheme 90, eqn (108)).
A similar rearrangement mechanism is involved in the conversion of benzofuran-2yl-carbonate and N-protected indo-2yl-carbonate derivatives into 2,3-dihydrobenzofuran-3-carboxylate 230 and N-protected 2-oxoindoline-3-carboxylate derivative 233, respectively (Scheme 91, eqn (110) and (111)).252 Enantiomerically enriched compounds 230 and 233 were obtained in very good yield by treatment of the corresponding enol carbonates 229 and 232 with only 1% of a chiral base 231.
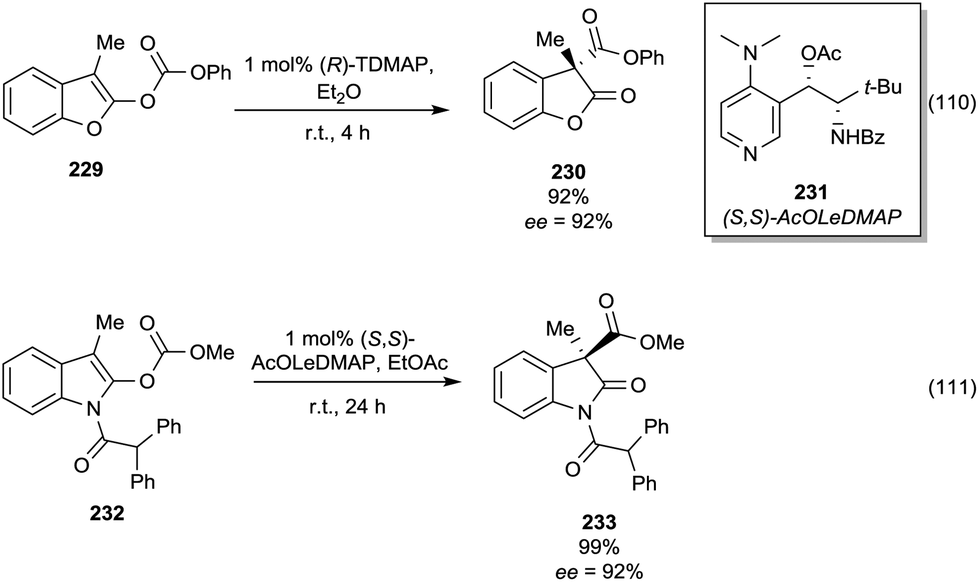 | ||
| Scheme 91 Enantioselective rearrangements of benzofuran enol carbonate (eqn (110)) and indole enol carbonate (eqn (111)) catalysed by chiral DMAP derivatives.252 | ||
5.3. [3,3] Sigmatropic rearrangement
Oximes react with DMC in the presence of a stoichiometric amount of base at 180–190 °C in an autoclave to give 4-oxazolin-2-one derivatives in moderate yield through a one-step mechanism. The [3,3] Sigmatropic rearrangement depicted in Scheme 92 was proposed by Marques et al.253 to elucidate the reaction pathway. The methoxycarbonylation of the oxime hydroxyl group (BAc2) is followed by an N-methylation reaction (BAl2) to give the quaternary ammonium intermediate. Subsequently, the base-assisted proton extraction from the methylene at α position to the CN bond prompts the formation of an enamine intermediate. The presence of a methylene group is thus essential for the [3,3] sigmatropic rearrangement (or 1-aza-1′,3′-dioxa-Cope rearrangement),254 which provides first an imine derivative which in turn leads to the oxazolinone derivative through cyclisation.
 | ||
| Scheme 92 4-Oxazolin-2-one formation via [3,3] sigmatropic rearrangement by reaction of oxime and DMC. | ||
Albeit this reaction can be applied to both aliphatic and aromatic oximes, the ring closure occurs regioselectively on benzylic methylene rather than on the alkylic one (Scheme 93, eqn (112)).
 | ||
| Scheme 93 Reaction of oximes in the presence of DMC.253,254 | ||
Cyclohexiloxazolin-2-ones 237 (Scheme 93, eqn (113)) and cycloheptyloxazolin-2-ones 239 (Scheme 93, eqn (114)) were achieved as pure compounds in moderate yields starting, respectively, from cyclohexiloxime 236 and cycloheptyloxime 238 and using similar reaction conditions.
Conversely, the attempts to synthesise the cyclopentyloxazolinone failed likely due to the steric strain of cyclopentenyl (i.e. enamine) intermediates formed during the reaction, which may hinder the achievement of the highly ordered transition state that is characteristic of this type of rearrangement.
5.4 Phenonium ion based reactions
Barontini and co-workers255 developed an efficient one-pot synthesis for selective etherification of 2-aryl-ethylalcohols 240a–d by using Amberlyst-15 as an acid catalyst and DMC as a reagent and a solvent (Scheme 94). | ||
| Scheme 94 Methylation reaction via phenonium ion formation. aAmberlyst-15 is a polystyrene based ion exchange resin with a strongly acidic sulfonic group.255 | ||
The reaction involved the methoxycarbonylation of primary alcohols to give carbonate derivatives 241a–d. Transient spiro[5.2]oct-5,7-diene-4yl carbocation (phenonium anion) was generated by the release of CO2 and MeOH; the latter may act as a nucleophile and form ether derivatives 242a–d. Reaction yields were affected by the substituents on the aromatic ring. In fact, electron releasing groups at ortho and para positions led to the formation of ether derivatives (e.g.242b and 242c) as major products by stabilisation of the phenonium ion. Conversely, electron withdrawing groups limited the production of ethers due to their destabilising effect (e.g.242d). Evidence of this transposition mechanism was provided by reactions performed on branched substrates (e.g. 2-phenyl-3-pentanol and 3-phenyl-3-pentanol),256 by using different nucleophiles255 and by computational studies.257
6. Pyrolysis and decarboxylation
DACs undergo thermal degradation essentially through two different pathways depending on the reaction conditions (Scheme 95):• Pyrolysis: Non-catalyst-assisted degradation, which may lead to the formation of an alkene, an alcohol and CO2 (Scheme 95, eqn (115)). The pyrolitic decomposition of dialkyl carbonates is far less investigated and used as a synthetic procedure to produce alkenes than the pyrolysis of the corresponding xanthates and esters (Scheme 96).
• Decarboxylation: Catalyst-assisted degradation, which promotes the formation of an ether and CO2 (Scheme 95, eqn (115)). This reaction is distinctive of organic carbonates as xanthates and esters exhibit different reactivities under similar experimental conditions.
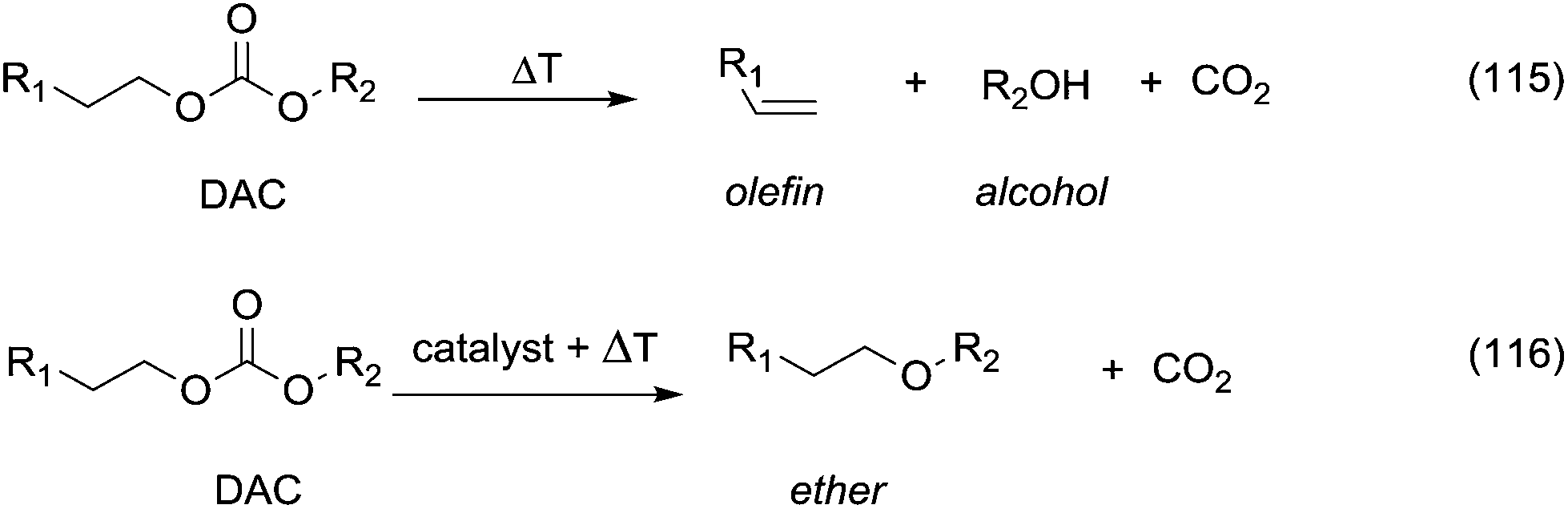 | ||
| Scheme 95 Thermal degradations of DACs: pyrolysis (eqn (115)) and decarboxylation (eqn (116)). | ||
 | ||
| Scheme 96 Proposed mechanism for the pyrolysis of esters, carbonates and xanthate. | ||
6.1 Pyrolysis
Pyrolysis of carbonates is a concerted mechanism occurring at T > 300 °C,258 which follows a first-order homogeneous kinetics with negative entropy of activation. The investigation of the pyrolysis of diethyl carbonate and methylethyl carbonate (MEC)259 at 300–375 °C highlighted that the degradation may be substantially an intramolecular mechanism.Clear evidence of this hypothesis is provided by DEC and MEC degradation products, which are solely ethylene and CO2 in 1/1 ratio and the corresponding alcohols. Besides, DEC and MEC exhibit inferior stability relative to DMC (T of decomposition >390 °C), inasmuch as the energy of activation for the abstraction of a proton through a cyclic state from DEC (Scheme 96, eqn (117)) is lower than the energy of activation for the abstraction of a hydrogen from DMC (∼7.4 kcal mol−1 against 8.9 kcal mol−1). Thus, the pyrolysis may represent a two-step mechanism, of which the first one proceeds through a six-membered cyclic transition state, which in turn may favour a cyclic cis-elimination (Scheme 96, eqn (117)).260 This rearrangement also occurs during the thermal decomposition of esters261 and xanthates262 – known as the Tschugaeff reaction and extensively used for the synthesis of alkene from alcohol – and it is largely accepted as a crucial mechanism step. In addition to olefin derivatives, carboxylic acids, hydrogen carbonodithioate, and hydrogen carbonate derivatives were produced by thermal rearrangement of, respectively, xanthates, carbonate and esters. As shown in Scheme 96 carbonodithioate derivatives further degrade to give carbonyl sulfide and the corresponding mercaptans (eqn (118)) and hydrogen carbonate derivatives may follow a similar second step to give CO2 and related alcohols (eqn (119)) as supported by the work of Notario et al.263 by means of theoretical studies on DEC and MEC thermolysis in the gas phase.
The decomposition rate follows the order xanthates > carbonates > esters and in the literature, comparative investigations mainly involve esters and carbonates,264 as the pyrolysis of xanthates typically occurs at lower temperatures (<200 °C). According to several studies conducted over a number of differently substituted alkyl or aryl esters and carbonates, the rates of pyrolysis—at T = 400–500 °C—of these compounds are similarly affected by the effect of their chemical structures. However, in their comprehensive research about methylcarbonate and acetate, Bigley and co-workers264a,265 showed that the pyrolysis of carbonates is generally ca. 22–24 times faster than their corresponding esters, due to lower energies and entropies of activation (Ea and ΔS* values).
6.2. Decarboxylation
In 1961, Wijnen76a discovered that thermal decarboxylation of DMC on quartz resulted in the formation of dimethyl ether (DME), CO2 and occasionally methanol due to the presence of water or acidic hydrogen on the support.Subsequently, different reaction conditions have been investigated76b–e with the aim of optimising and thus standardising the conversion of DACs into ethers either by heating in an autoclave or under continuous flow. Essentially this optimisation involves the modification of parameters, such as the type of catalyst, reaction temperature, reaction media, the type and amount of DACs employed and the type and amount of alcohols employed to obtain dissymmetrical ethers. Fu et al.76d reported the decarboxylation of DMC by heating from 100 °C to 350 °C and using the catalyst MgO as a strong solid base, H-ZSM as a strong solid acid, γ-Al2O3 as amphoteric oxide with surface acidity stronger than basicity, ZnO as amphoteric oxide with stronger surface basicity and weaker surface acidity than γ-Al2O3, and SiO2 as neutral oxide. The main results of this work have been summarised in Table 20.
|
|
|||
|---|---|---|---|
| Catalyst | T (° C) | Conv. (%) | Product selectivity (%)b DME (+MeOH and CO2) |
| a ZSM-5, zeolite Socony Mobil-5 is an aluminosilicate zeolite belonging to the pentasil family of zeolites; its chemical formula is NanAlnSi96–nO192·16H2O (0 < n < 27). b Determined by using gas chromatography. Catalyst amount and reaction time not reported. c Methane and unidentified by-products were also present.76d–e | |||
| MgO | 250 | 96 | 99 |
| H-ZSM-5a | 150 | 98 | 98 |
| γ-Al2O3 | 150 | 98 | 87c |
| SiO2 | 300 | 9 | 72 |
| ZnO | 350 | 3 | Mixture |
Excellent conversions and selectivities toward DME were achieved by using H-ZMS and γ-Al2O3 as catalysts at 150 °C and MgO at 250 °C.
An efficient one-pot synthetic procedure to produce methylethers by reaction of alcohols and DMC in the presence of Al2O3 or hydrotalcite has been developed by Tundo et al.76e This etherification involves a two-step mechanism as shown in Scheme 97. An alcohol reacting with DMC gives transesterification, resulting in a methylcarbonate intermediate (Scheme 97, eqn (120)), which upon decarboxylation produces a methylether and CO2 (Scheme 97, eqn (121)).
 | ||
| Scheme 97 General synthesis of dissymmetrical ethers by DAC chemistry. | ||
In an example, 1-octanol was converted quantitatively into the corresponding methyloctyl ether (MOE) by reaction with DMC using basic alumina and hydrotalcite KW2000 and heating to 200 °C in an autoclave (Table 21), whereas a reaction performed employing K2CO3 or MgO gave methyloctyl carbonate (MOC) as a major product. Under these conditions, benzyl alcohol was converted into the corresponding methyl ether as well.
|
|
|||||
|---|---|---|---|---|---|
| Catalyst | T (°C) | t (h) | Conv. (%) | Product selectivitya (%) | |
| MOE | MOC | ||||
| a Determined by using gas chromatography. b Hydrotalcite KW2000 is a synthetic layered double hydroxide composed of a brucite-like positively charged layer due to the partial substitution of Mg2+ and Al3+, which is compensated for by negatively charged interlayers containing anions and water. Its chemical formula is Mg6Al2(CO3)(OH)16·4H2O.76f,g | |||||
| K2CO3 | 200 | 2.5 | 99 | 0 | 100 |
| MgO | 200 | 2 | 78 | 2 | 98 |
| Acidic Al2O3 | 200 | 1 | 98 | 24 | 76 |
| Basic Al2O3 | 200 | 1 | 100 | 100 | 0 |
| KW2000b | 200 | 1 | 100 | 97 | 3 |
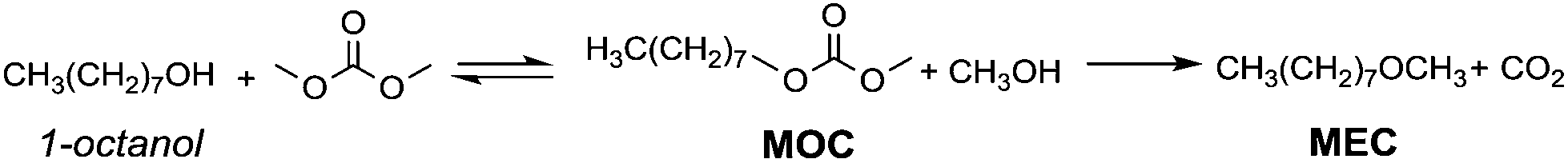
This method showed its limitations when applied to secondary alcohols, such as 2-octanol and 2-phenyl 2-propanol and tertiary alcohols, such as 2-phenyl-2-octanol by giving as by-products octenes and styrene. Moreover, continuous-flow methylation of 1-pentanol was carried out using a solid base of basic alumina and after 24 h no loss of activity was detected (i.e. no catalyst deactivation) and methylpentylether was constantly produced at 180 °C. Under liquid phase conditions, dismutation of alkyl carbonates represents one of the main drawbacks of the decarboxylation of dissymmetrical DACs performed to produce the corresponding ethers. The presence of acidic hydrogens on the surface of the catalyst (principally hydrotalcite KW2000) – such as water, alcohols and surface hydroxyls – may promote the formation of symmetrical DACs and dialkyl ethers as by-products. In order to increase the formation of dissymmetrical ethers, decarboxylation reactions are performed at high temperature (>200 °C) for short reaction time (1 h) as decarboxylation is favoured over transesterification.
Solvent polarity has also been observed to affect reaction yields. Tundo and coworkers76g investigated the decarboxylation reaction of n-decylmethylcarbonate in non-polar alkanes such as n-dodecane (b.p. 216 °C), n-tetradecane (b.p. 250 °C) and trans-decaline (b.p. 190 °C) and in polar aprotic solvents such as polyethyleneglycol dimethyl ether (b.p. > 250) and triethylene glycol (b.p. 216 °C). The reaction conducted in non-polar solvents gave the desired dissymmetrical ether in moderate to good yields (68–80%) based on the reaction temperature as decarboxylation requires higher activation energy than transesterification. On the other hand, glycols seemed to inhibit the product formation even at high reaction temperature, likely because polar solvents may prevent the absorption of polar compounds on the catalyst.
Cyclic ethers (number of carbons = 3–6) were prepared by decarboxylation of cyclic carbonate esters having at least one hydroxyl group in the molecule (Scheme 98).266 The general procedure consisted of the reaction of a triol derivative (243a–d) with EC or DEC heating to 180–230 °C and removing ethylene glycol (or ethanol) by distillation to obtain first a cyclic carbonate derivative (244a–d), which upon decarboxylation gave a cyclic ether (245a–d). Besides, it was observed that cyclic carbonates containing no hydroxyl groups are much more stable than carbonate ester containing a hydroxyl group as their degradation occurred at T > 230 °C.
 | ||
| Scheme 98 Synthesis of cyclic ethers by decarboxylation of cyclic carbonate ester. aCompound 243c was reacted with DEC to give 245c in 34% yield.266 | ||
7. Conclusions
Over the last thirty years, since the first green process to DMC has been patented by Enichem, organic carbonates have been extensively investigated. As a result, more than five thousand papers and patents have been published where DACs are employed as a green substitute of chlorine-based reagents, solvents or polymers.For this reason, a comprehensive scientific review on the reactivity of DACs is not feasible considering the complexity and versatility of these compounds. However, herein we have focused our attention on specific facets of organic carbonates that have never or only marginally been reported so far. In particular, this work deals with the alkoxycarbonylation of α-carbon to an acyl moiety, as well as of N, O, S, and C alkylation reactions. The use of DACs in the substitution of halogen-based compounds has been reported. The similarities and differences between DACs and chlorine parent compounds have been highlighted in several examples.
Halogen-based molecules are undoubtedly more reactive than organic carbonates; however, as a consequence they usually do not allow selective or chemeoselective reactions. Conversely, the less reactivity DACs often lead to alkylation with high selectivity. The high Gibbs energy required to activate DACs is, along with their affinity for the HSAB theory, the driving forces for regio- and chemoselective processes. In this view pivotal examples are the mono-methylations of numerous CH2 and NH2 compounds with selectivity often higher than 95%.
Another appealing feature of DACs is that they do not produce inorganic salts. In fact, both Pd-catalysed reactions and nucleophilic displacement (via a BAl2 mechanism) release alkyl carbonates as the leaving group that, once formed, rapidly decomposes into CO2 and an alkoxide anion. The latter restores the base, allowing its use in a catalytic amount. As a result, DAC-based chemistry can be employed in continuous-flow procedures such as gas–liquid phase-transfer catalysis and a continuously stirred tank reactor. As alkoxycarbonylation is an equilibrium reaction, while alkylation is not, the product of the continuous process can be easily controlled using the temperature as the key factor.
It is also noteworthy that, nowadays, thanks to the extensive exploitation of DACs as alkylating agents and to the versatility of these compounds they can be used under basic conditions (i.e., O, S and C alkylation), under acidic conditions (i.e., N-alkylation via zeolites) and under neutral conditions (anchimerically driven alkylation via mustard carbonates).
Furthermore, an overview on less known DAC-mediated transposition, cyclisation, pyrolysis and decarboxylation reactions has also been addressed. What is evident from the examples herein discussed is that behind the apparent simplicity of organic carbonates hides a non-immediate and multifaceted chemistry because it combines the reactivity of an sp2 (hard electrophilic site) and sp3 (soft electrophilic site) carbon. Clear examples are the selective alkoxycarbonylation on differently activated α-carbons or the DAC-based transposition reactions.
It is also noteworthy that organic carbonate chemistry has an intrinsic elegance; DACs are both green reagents and (good) solvents, they produce only alcohols and CO2 as by-products and they are potentially recovered and reused. For these reasons, organic carbonates are increasingly exploited in industrial processes. As stated by the Nobel Laureate Noyori,267 pursuing practical elegance in chemical synthesis is pivotal and “without attention to what is now called green chemistry, chemical manufacturing will be unsustainable in this century”. With this perspective, the great interest of the scientific community in dialkyl carbonates hints that we might be going in the right direction.
Abbreviations
| AAA | Asymmetric alkyl allylation |
| A-ADOTA+ | tris(Dialkylamino)-azadioxatriangulenium carbenium ion |
| Ar | Aryl |
| BAc2 | Base-catalysed acyl cleavage bimolecular mechanism |
| BAl2 | Base-catalysed alkyl cleavage bimolecular mechanism |
| [BMIm] | 1-n-Butyl-3-methylimidazolium |
| BPs | By-products |
| BTPP | tert-Butylimino-tris(pyrrolidino)-phosphorane |
| Bu | Butyl |
| b.p. | Boiling point |
| CMCH | Methoxycarbonylcyclohexanone |
| CMCP | Carboxymethylcyclopentanone |
| Cod | 1,5-Cyclooctadiene |
| Cp | Cyclopentadienyl |
| CSZ | Conventional sulfated zirconia |
| CTAB | Cetyl trimethyl ammonium bromide |
| c.-f. | Continuous flow |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DAC(s) | Dialkyl carbonate(s) |
| Dba | Dibenzylideneacetone |
| DBnC | Dibenzyl carbonate |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DC | Dicarboxymethyl isosorbide |
| DEC | Diethyl carbonate |
| DEF | Diethyl formamide |
| Dipp | 2,6-Diisopropylphenyl |
| DMA | Adipic dimethylester |
| DMAc | Dimethylacetamide |
| Dmdba | Bis(3,5-dimethoxybenzylidene)acetone |
| DMAP | 4-(N,N-Dimethlyamino)pyridine |
| DMDMS | Dimethoxydimethylsilane |
| DMC | Dimethyl carbonate |
| DME | Dimethyl ether |
| DMF | Dimethyl formamide |
| DMI | Dimethyl isosorbide |
| DMIi | Dimethyl isomannide |
| DMIm | Dimethyl isoiodide |
| DMP | Pimelic dimethyl ester |
| DMPU | 1,3-Dimethyl-2-oxohexahydropyrimidine |
| DMSO | Dimethyl sulfoxide |
| DPC | Diphenyl carbonate |
| EC | Ethylene carbonate |
| [EMIm]Cl | 1-Ethyl-3-methylimidazolium |
| EPR | Electron paramagnetic resonance |
| Et | Ethyl |
| FT-IR | Fourier transform-infrared spectroscopy |
| GC | Glycerol carbonate |
| GC-MS | Gas Chromatography Mass Spectrometry |
| GL-PTC | Gas–Liquid Phase-Transfer Catalysis |
| HMDS | Hexamethyldisilylazane |
| HSBA | Hard–Soft Acid–Base theory |
| ILs | Ionic liquids |
| L | Ligand |
| LL-PTC | Liquid–Liquid Phase-Transfer Catalysis |
| LDA | Lithium diisopropylamide |
| LDBB | Lithium 4,4′-di-tert-butylbiphenylide |
| MBC | Methyl-butyl carbonate |
| Me | Methyl |
| MMC | Magnesium methyl carbonate |
| MO | Methyl 2-methyl-3-oxopentanoate |
| MOM | Methoxymethyl |
| MP | Methyl propionate |
| MPD | m-Phenylenediamine |
| MPTMS | 3-Mercaptopropyltrimethoxysilane |
| MSN | Bifunctional mesoporous silica nanoparticle |
| MSZ | Mesoporous sulfated zirconia |
| OPD | o-Phenylenediamine |
| Ph | Phenyl |
| PMC | Potassium methyl carbonate |
| PMBO | p-Methoxybenzyl |
| Pr | Propyl |
| PT | Phase transfer |
| PTSA | p-Toluenesulfonic acid |
| MEC | Methyloctyl ether |
| MOC | Methyloctyl carbonate |
| MS | Mass |
| MTMS | Methoxytrimethylsilane |
| Ms | Mesyl |
| NMR | Nuclear magnetic resonance |
| PEG | Polyethylene glycol |
| PBR | Peripheral benzodiazepine receptor |
| PivOH | Pivalic acid |
| Py | Pyridine |
| RON | Research octane number |
| TADMAP | 3-(2,2,2-Triphenyl-1-acetoxyethyl)-DMAP |
| TBAC | Tetrabutylammonium chloride |
| TBAF | Tetrabutylammoniumfloride |
| TBAT | Tetrabutylammonium difluorotriphenylsilicate |
| TBU | 1,5,7-Triazabicyclo-[4.4.0]dec-5-ene |
| TEA | Triethylamine |
| TES | Triethylsilyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
| TMS | Trimethylsilyl |
| Ts | Tosyl |
| UV | Ultraviolet |
| wt% | Weight percentage |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to acknowledge the Ministry of Education, Universities and Research (MIUR) for financial support: Innovazione di prodotto e di processo per una manutenzione, conservazione e restauro sostenibile e programmato del patrimonio culturale—(SCN_00520) project Smart Cities and Communities and Social Innovation on Cultural Heritage.References
- (a) P. Tundo, L.-N. He, E. Lokteva and C. Mota, Chemistry Beyond Chlorine, Springer, 2016 Search PubMed; (b) I. Garcia-Herrero, R. M. Cuéllar-Franca, V. M. Enríquez-Gutiérrez, M. Alvarez-Guerra, A. Irabien and A. Azapagic, ACS Sustainable Chem. Eng., 2016, 4, 2088 CrossRef CAS.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed.
- (a) S. Grego, F. Aricò and P. Tundo, Pure Appl. Chem., 2014, 469, 108 Search PubMed; (b) S. Grego, F. Aricò and P. Tundo, Org. Process Res. Dev., 2013, 17, 679 CrossRef CAS.
- (a) W. Tai-Teh, J. Huang, N. Arrington and G. J. Dill, J. Agric. Food Chem., 1987, 35, 823 CrossRef; (b) T. Kato, K. Suzuki, J. Takahashi and K. Kamoshita, J. Pestic. Sci., 1984, 9, 489 CrossRef CAS; (c) F. Rivetti, U. Romano and M. Sasselli, U.S. Patent, 4514339, 1985 Search PubMed; (d) M. Aresta and E. Quaranta, Chem. Technol., 1997, 27, 32 CAS; (e) F. A. Dawodu, O. O. Ayodele, J. Xin and S. Zhang, Renewable Energy, 2014, 68, 581 CrossRef CAS.
- (a) P. Tundo, F. Trotta and G. Moraglio, Italian Pat, 20159A/90C, 1990 Search PubMed; (b) P. Loosen, P. Tundo and M. Selva, U.S, 5278533, 1994 Search PubMed; (c) P. Fuertes, M. Ibert, E. Josien, P. Tundo and F. Aricò, US, 8399601B2, 2013 Search PubMed P. Fuertes, M. Ibert, E. Josien, P. Tundo and F. Aricò, EU, WO2011/039483A1, 2013 Search PubMed (d) M. Selva and P. Tundo, patent, 03029005.0, 2007 Search PubMed; (e) H. Bevinakatti, C. Newman, S. Ellwood, P. Tundo, F. Aricò and M. Schroeder, US, 8536349B2, 2013 Search PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS PubMed.
- U. Romano, F. Rivetti and N. Di Muzio, U.S. Patent, 4318862, 1979 Search PubMed.
- K. Nisihra, K. Mizutare and S. Tanaka, (UBE Industries, Japan). EP Patent Appl, 425197, 1991 Search PubMed.
- M. Tojo and H. Miyaji, EP, 1760059B1, 2007 Search PubMed.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS; (b) R. G. Pearson and J. Songstad, J. Am. Chem. Soc., 1967, 89, 1827 CrossRef CAS; (c) P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219 CrossRef CAS PubMed; (d) H. Tse-Lok, Chem. Rev., 1975, 75, 1 CrossRef; (e) J. L. Gàzquez and F. J. Mèndez, J. Phys. Chem., 1994, 98, 4591 CrossRef; (f) G. Klopman, Chemical reactivity and reaction Paths, Wiley, New York, 1974 Search PubMed; (g) G. Klopman, J. Am. Chem. Soc., 1968, 90, 223 CrossRef CAS; (h) R. G. Pearson, Inorg. Chem., 1988, 27, 734 CrossRef CAS; (i) R. G. Pearson, J. Am. Chem. Soc., 1988, 110, 7684 CrossRef CAS; (j) K. W. Eggers and A. T. Cocks, Helv. Chim. Acta, 1973, 56, 1516 CrossRef; (k) D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493 CrossRef CAS; (l) T. L. Gresham, J. E. Jansen, F. W. Shaver, J. T. Gregory and W. L. Beears, J. Am. Chem. Soc., 1948, 70, 1004 CrossRef CAS; (m) T. L. Gresham, J. E. Jansen, F. W. Shaver, M. R. Frederik, F. T. Fiedorek, R. A. Bankert, J. T. Gregory and W. L. Beears, J. Am. Chem. Soc., 1952, 74, 1323 CrossRef CAS; (n) T. L. Gresham, J. E. Jansen, F. W. Shaver and J. T. Gregory, J. Am. Chem. Soc., 1948, 70, 999 CrossRef CAS; (o) T. L. Gresham, J. E. Jansen, F. W. Shaver and R. A. Bankert, J. Am. Chem. Soc., 1949, 71, 661 CrossRef CAS; (p) R. S. Davidson, W. H. H. Gunther, S. M. Waddington-Feather and B. Lythgoe, J. Chem. Soc., 1964, 4907 RSC; (q) D. E. L. Carrington, K. Clarke and R. M. Scrowston, J. Chem. Soc. C, 1971, 74, 3262 RSC; (r) A. C. Pierce and M. M. Joulliè, J. Org. Chem., 1962, 27, 3968 CrossRef CAS; (s) F. G. Bordwell, Organic Chemistry, Macmillian, New York, 1963, p. 218 Search PubMed; (t) A. J. Parke, Adv. Phys. Org. Chem., 1967, 5, 173 CrossRef; (u) R. F. Hudson and G. Klopman, J. Chem. Soc., 1964, 5 RSC; (v) P. Tundo, F. Aricò, A. E. Rosamilia, M. Rigo, A. Maranzana and G. Tonachini, Pure Appl. Chem., 2009, 81, 1971 CrossRef CAS.
- A. Dibenedetto, M. Aresta, P. Giannoccaro, C. Pastore, I. Pápai and G. Schubert, Eur. J. Inorg. Chem., 2006, 908 CrossRef CAS.
- (a) F. Aricò, P. Tundo, A. Maranzana and G. Tonachini, ChemSusChem, 2012, 5, 1578 CrossRef PubMed; (b) F. Aricò, M. Chiurato, J. Peltier and P. Tundo, Eur. J. Org. Chem., 2012, 3223 CrossRef; (c) P. Tundo, F. Aricò, G. Gauthier, L. Rossi, A. E. Rosamilia, H. S. Bevinakatti, R. L. Sievert and C. P. Newman, ChemSusChem, 2010, 3, 566 CrossRef CAS PubMed.
- E. Gershonov, I. Columbus and Y. Zafrani, J. Org. Chem., 2009, 74, 329 CrossRef CAS PubMed.
- (a) D. Chaturvedi and S. Ray, Monatsh. Chem., 2006, 137, 127 CrossRef CAS; (b) M. Carafa and E. Quaranta, Mini-Rev. Org. Chem., 2009, 6, 168 CrossRef CAS; (c) D. Chaturvedi, Curr. Org. Chem., 2011, 15, 1593 CrossRef CAS.
- (a) M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110 RSC; (b) O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431 RSC; (c) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459 RSC.
- F. W. Swamer and C. R. Hauser, J. Am. Chem. Soc., 1950, 72, 1352 CrossRef CAS.
- C. Prandi and P. Venturello, J. Org. Chem., 1994, 59, 5458 CrossRef CAS.
- V. H. Wallingford, A. H. Homeyer and D. M. Jones, J. Am. Chem. Soc., 1941, 63, 2252 CrossRef CAS.
- R. Levine and C. R. Hauser, J. Am. Chem. Soc., 1944, 66, 1768 CrossRef CAS.
- M. Selva, C. A. Marques and P. Tundo, Gazz. Chim. Ital., 1993, 123, 515 CAS.
- D. Wu, X. Fu, F. Xiao, J. Li, N. Zhao, W. Wei and Y. Sun, Catal. Commun., 2008, 9, 680 CrossRef CAS.
- P. Tundo, Pure Appl. Chem., 2001, 73, 1117 CrossRef CAS.
- Z. Chen and D. Wu, Ind. Eng. Chem. Res., 2011, 50, 12343 CrossRef CAS.
- S. Memoli, P. Tundo and M. Selva, WO, 0214257(A2), 2002 Search PubMed.
- (a) P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565 CrossRef CAS; (b) P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1989, 28, 881 CrossRef CAS; (c) P. Tundo, F. Trotta and G. Moraglio, J. Chem. Soc., Perkin Trans. 1, 1989, 1070 RSC.
- (a) D. Wu and Z. Chen, Ind. Eng. Chem. Res., 2009, 48, 6287 CrossRef CAS; (b) W. Dudu, C. Zhi, J. Zhenbin and S. Li, Sci. China: Chem., 2012, 55, 380 CrossRef.
- G.-S. Zhang, M.-M. Zhu, Q. Zhang, Y.-M. Liu, H.-Y. He and Y. Cao, Green Chem., 2016, 18, 2155 RSC.
- E. Deseke, W. Hoyer and E. Winterfeldt, Tetrahedron, 1996, 52, 14577 CrossRef CAS.
- F. Radner, A. Rassat and C.-J. Hersvall, Acta Chem. Scand., 1996, 50, 146 CrossRef CAS.
- R. W. Carling, J. S. Clark and A. B. Holmes, J. Chem. Soc., Perkin Trans. 1, 1992, 83 RSC.
- S. Gao, Y. Q. Tu, X. Hu, S. Wang, R. Hua, Y. Jiang, Y. Zhao, X. Fan and S. Zhang, Org. Lett., 2006, 8, 2373 CrossRef CAS PubMed.
- D. Damour, M. Barreau, V. Fardin, F. Dhaleine, M. Vuilhorgne and S. Mignani, Synlett, 1998, 153 CrossRef CAS.
- P. A. Aristoff, P. D. Johnson and A. W. Harrison, J. Am. Chem. Soc., 1985, 107, 7967 CrossRef CAS.
- D. F. Taber, R. B. Sheth and W. Tian, J. Org. Chem., 2009, 74, 2433 CrossRef CAS PubMed.
- H. E. Zimmerman and G. A. Sereda, J. Org. Chem., 2003, 68, 283 CrossRef CAS PubMed.
- M. Watanabe, T. Abe and N. Harada, J. Org. Chem., 1997, 62, 2992 CrossRef CAS PubMed.
- J. Vebrel and R. Carrie, Bull. Soc. Chim. Fr., 1982, 2, 161 Search PubMed.
- A. P. Kozikowski, Q. Ding, A. Saxena and B. P. Doctor, Bioorg. Med. Chem. Lett., 1996, 6, 259 CrossRef CAS.
- S. Sekhar Mandal, J. Chakraborty and A. De, J. Chem. Soc., Perkin Trans. 1, 1999, 2639 RSC.
- M. Cushman, D. Nagarathnam, D. L. Burg and R. L. Geahlen, J. Med. Chem., 1991, 34, 798 CrossRef CAS PubMed.
- J.-C. Jung, Y.-J. Jung and O.-S. Park, Synth. Commun., 2001, 31, 1195 CrossRef CAS.
- (a) G. Koller, Chem. Ber., 1927, 60, 1108 CrossRef; (b) M. Topolski, J. Org. Chem., 1995, 60, 5588 CrossRef CAS.
- T. X. Nguyen, M. Dakanali, L. Trzoss and E. A. Theodorakis, Org. Lett., 2011, 13, 3308 CrossRef CAS PubMed.
- D. Sengupta and R. V. Venkateswaran, J. Chem. Res., 1984, 372 CAS.
- R. R. Briese and J. M. McElvain, J. Chem. Res., 1933, 66, 1697 Search PubMed.
- P.-Y. Michellys, P. Maurin, L. Toupet, H. Pellissier and M. Santelli, J. Org. Chem., 2001, 66, 115 CrossRef CAS PubMed.
- M. Anzini, A. Cappelli, S. Vomero, G. Giorgi and L. Thierry, J. Med. Chem., 1996, 39, 4275 CrossRef CAS PubMed.
- Y. Takeuchi, H. Iwashita, K. Yamada, M. Gotaishi, N. Kurose, T. Koizumi, K. Kabuto and T. Kometani, Chem. Pharm. Bull., 1995, 43, 1668 CrossRef CAS.
- K. Matsumoto, M. Suzuki and M. Miyoshi, J. Org. Chem., 1973, 38, 2094 CrossRef CAS PubMed.
- A. E. Chichibabin, Ber., 1905, 38, 561 CrossRef.
- J. W. Terpstra and A. M. Van Leusen, J. Org. Chem., 1986, 51, 230 CrossRef CAS.
- D. L. Boger and C. E. Brotherton, J. Org. Chem., 1984, 49, 4050 CrossRef CAS.
- P. W. Shum and A. P. Kozikowski, Tetrahedron Lett., 1990, 31, 6785 CrossRef CAS.
- P. Kurach, S. Luliński and J. Serwatowski, Eur. J. Org. Chem., 2008, 3171 CrossRef CAS.
- Y. Uozumi, H. Kyota, K. Kato, M. Ogasawara and T. Hayashi, J. Org. Chem., 1999, 64, 1620 CrossRef CAS PubMed.
- Y. S. Park and P. Beak, Tetrahedron, 1996, 52, 12333 CrossRef CAS.
- K. Durka, P. Kurach, S. Luliński and J. Serwatowski, Eur. J. Org. Chem., 2009, 4325 CrossRef CAS.
- R. J. Anderson and J. C. Morris, Tetrahedron Lett., 2001, 42, 8697 CrossRef CAS.
- X. Cheng, S.-F. Zhu, X.-C. Qiao, P.-C. Yan and Q.-L. Zhou, Tetrahedron, 2006, 62, 8077 CrossRef CAS.
- W. W. Moyer and C. S. Marvel, Org. Synth., 1931, 11, 98 CrossRef.
- T. J. Reddy, T. Iwama, H. J. Halpern and V. H. Rawal, J. Org. Chem., 2002, 67, 4635 CrossRef CAS PubMed.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255 CrossRef CAS.
- B. W. Laursen and T. J. Sørensen, J. Org. Chem., 2009, 74, 3183 CrossRef CAS PubMed.
- (a) M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377 CrossRef CAS; (b) M. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 2474 CrossRef CAS PubMed.
- J. Monot, A. Solovyev, H. Bonin-Dubarle, É. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 122, 9352 CrossRef.
- M. Stiles, J. Am. Chem. Soc., 1959, 81, 2598 CrossRef CAS.
- H. Finkbeiner, J. Am. Chem. Soc., 1964, 86, 961 CrossRef CAS.
- R. C. A. Isaacs, M. J. Di Grandi and S. J. Danishefsky, J. Org. Chem., 1993, 58, 3938 CrossRef CAS.
- H. A. Duong, T. M. Nguyen, N. Z. B. Rosman and L. J. L. Tan, Synthesis, 2014, 1881 CrossRef.
- (a) J. March and M. B. Smith, March's Advanced Organic Chemistry, Wiley, New York, 5th edn, 2001 Search PubMed; (b) G. Lamoureux and C. Agüero, ARKIVOC, 2009, 251 CAS; (c) H. Feuer and J. Hooz, The Ether Linkage, Patai, 1967, p. 445 Search PubMed.
- (a) K. S. Gates, Chem. Res. Toxicol., 2009, 22, 1747 CrossRef CAS PubMed; (b) Y. Mishina, E. M. Duguid and C. He, Chem. Rev., 2006, 106, 215 CrossRef CAS PubMed.
- (a) K. S. Gates, Chem. Res. Toxicol., 2009, 22, 1747 CrossRef CAS PubMed; (b) Y. Mishina, E. M. Duguid and C. He, Chem. Rev., 2006, 106, 215 CrossRef CAS PubMed.
- (a) H. M. Bolt and B. Gansewendt, Crit. Rev. Toxicol., 1993, 23, 237 CrossRef CAS PubMed; (b) L. A. Pokier, G. D. Stoner and M. B. Shimkin, Cancer Res., 1975, 35, 1411 Search PubMed; (c) J. Mc Cann, E. Choi, E. Yamasaki and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 5135 CrossRef CAS.
- (a) X. Wu, Y. J. Park, K. S. Ryu, S. H. Chang and Y. Hong, U.S. Patent, 20040105809, 2004 Search PubMed; (b) J. T. Carey, J. Thomas and H. Oumar-Mahamat, US, 5962379, 1999 Search PubMed; (c) A.-A. Shaik and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef; (d) P. Tundo, F. Aricò, A. E. Rosamilia, S. Grego and L. Rossi, in Green Chemical Reactions, ed. P. Tundo and V. Esposito, Springer, Dordrecht, 2006, p. 213 Search PubMed.
- P. Tundo, S. Memoli, D. Hérault and K. Hill, Green Chem., 2004, 6, 609 RSC.
- (a) M. H. J. Wijnen, J. Phys. Chem., 1961, 34, 1465 CrossRef CAS; (b) A. Greco, F. Rivetti and U. Romano, Enichem Synthesis S.p.A., BE Pat, 1006039, 1994 Search PubMed; (c) P. Tundo, S. Memoli, D. Herault and T. Loehl, EP, 1050524(A2), 2000 Search PubMed; (d) Y. Fu, H. Zhu and J. Shen, Thermochim. Acta, 2005, 434, 88 CrossRef CAS; (e) P. Tundo, F. Arico, A. E. Rosamilia and S. Memoli, Green Chem., 2008, 10, 1182 RSC; (f) T. Wajima, Energy Environ. Res., 2014, 4, 3 Search PubMed; (g) P. Tundo, S. Memoli, D. Herault and K. Hill, Green Chem., 2004, 6, 609 RSC; (h) D. B. Pattison, J. Am. Chem. Soc., 1957, 79, 3455 CrossRef CAS.
- M. Selva, V. Benedet and M. Fabris, Green Chem., 2012, 14, 188 RSC.
- (a) G. Flèche and M. Huchette, Starch/Staerke, 1986, 38, 26 CrossRef; (b) M. A. Andrews, K. K. Bhatia and P. Fagan, U.S., 6689892, Feb. 10, 2004.
- (a) F. J. Hopton and G. H. S. Thomas, Can. J. Chem., 1969, 47, 2395 CrossRef CAS; (b) Y. Zhu, M. Durand, V. Molinier and J. M. Aubry, Green Chem., 2008, 10, 532 RSC.
- A. J. Sanborn and S. J. Howard, US, 2009/253920A1, 2009 Search PubMed.
- F. Aricò, A. S. Aldoshin and P. Tundo, ChemSusChem, 2017, 10, 53 CrossRef PubMed.
- C. Hou, Y. Chen and W. Li, Carbohydr. Res., 2012, 355, 87 CrossRef CAS PubMed.
- M. Selva, E. Militello and M. Fabris, Green Chem., 2008, 10, 73 RSC.
- S. X. Chong, H. A. Wahab and H. H. Abdallah, Comput. Mater. Sci., 2012, 55, 217 CrossRef CAS.
- (a) V. A. Nasadyuk, E. V. Fedevich, Y. A. Pazderskii and I. I. Moiseev, Izv. Akad. Nauk SSSR, Ser. Khim., 1989, 787 ( Chem. Abstr. , 1989 , 111 , 194208 ) CAS; (b) H. King and E. V. Wright, J. Chem. Soc., 1939, 1168 RSC.
- (a) Y. Lee and I. Shimizu, Synlett, 1998, 1063 CrossRef CAS; (b) Z. L. Shen, X. Z. Jiang, W. M. Mo, X. B. Hu and N. Sun, Green Chem., 2005, 7, 97 RSC; (c) F. Rajabi and M. R. Saidi, Synth. Commun., 2004, 34, 4179 CrossRef CAS.
- (a) Y. Ono, Pure Appl. Chem., 1996, 68, 367 CrossRef CAS; (b) Y. Ono, Appl. Catal., A, 1997, 155, 133 CrossRef CAS.
- (a) M. B. Talawar, T. M. Jyothi, T. Raja, B. S. Rao and P. D. Sawant, Green Chem., 2000, 2, 266 RSC; (b) T. M. Jyothi, T. Raja, M. B. Talawar and B. S. Rao, Appl. Catal., A, 2001, 211, 41 CrossRef CAS.
- (a) T. Weidlich, M. Pokorný, Z. Padělková and A. Růžička, Green Chem. Lett. Rev., 2007, 1, 53 CrossRef CAS; (b) T. Weidlich, L. Dusek, B. Vystrcilova, A. Eisner, P. Svec and A. Růžička, Appl. Organomet. Chem., 2012, 26, 293 CrossRef CAS.
- (a) N. Komblum, R. Seltzer and P. Haberfield, J. Am. Chem. Soc., 1963, 85, 1148 CrossRef; (b) O. A. Reutov and A. C. Kurts, Russ. Chem. Rev., 1977, 46, 1040 CrossRef; (c) M. Fernando, S. Quici and P. Tundo, J. Org. Chem., 1983, 48, 199 CrossRef.
- M. Lissel, S. Schmidt and B. Neumann, Synthesis, 1986, 382 CrossRef CAS.
- S. Ouk, S. Thiebaud, E. Borredon, P. Legars and L. Lecomte, Tetrahedron Lett., 2002, 43, 2661 CrossRef CAS.
- N. Notari, F. Mizia and F. Rivetti, US, 5849955, 1998, CAS No 129: 54182m Search PubMed.
- (a) J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156 RSC; (b) P. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS PubMed; (c) P. A. Z. Suarez, J. E. L. Dullius, S. Einlft, R. F. De Souza and J. Dupont, Polyhedron, 1996, 15, 1217 CrossRef CAS.
- Z. L. Shen, X. Z. Jiang, W. M. Mo, B. X. Hu and N. Sun, Green Chem., 2005, 7, 97 RSC.
- S. Ouk, S. Thiebaud, E. Borredon and P. L. Gars, Appl. Catal., A, 2003, 241, 227 CrossRef CAS.
- S. K. Kabra, M. Huuhtanen, R. L. Keiski and G. D. Yadav, React. Chem. Eng., 2016, 1, 330 CAS.
- G. Barcelo, D. Grenouillat, J.-P. Senet and G. Sennyey, Tetrahedron, 1990, 46, 1839 CrossRef CAS.
- (a) R. Bernini, F. Crisante and M. C. Ginnasi, Molecules, 2011, 16, 1418 CrossRef CAS PubMed; (b) M. Hosny, K. Dhar and J. P. N. Rosazza, J. Nat. Prod., 2001, 64, 462 CrossRef CAS PubMed.
- J.-Q. Nie, H.-W. Chen, Q.-H. Song, B. Liao and Q.-X. Guo, Energy Fuels, 2010, 24, 5722 CrossRef CAS.
- Q. H. Song, J. Q. Nie, M. G. Ren and Q. X. Guo, Energy Fuels, 2009, 23, 3307 CrossRef CAS.
- A. K. Kinage, S. P. Gupte, R. K. Chaturvedi and R. V. Chaudhari, Catal. Commun., 2008, 9, 1649 CrossRef CAS.
- A. M. Truscello, C. Gambarotti, M. Lauria, S. Auricchio, G. Leonardi, S. U. Shisodia and A. Citterio, Green Chem., 2013, 15, 625 RSC.
- P. Tundo, Continuous Flow Methods in Organic Synthesis, Ellis Horwood Limited, 1991 Search PubMed.
- (a) P. Tundo, A. E. Rosamilia and F. Aricò, J. Chem. Educ., 2010, 87, 1233 CrossRef CAS; (b) G. Newman and K. Jensen, Green Chem., 2013, 15, 1456 RSC.
- Z.-H. Fu and Y. Ono, Catal. Lett., 1993, 21, 43 CrossRef CAS.
- T. Beutel, J. Chem. Soc., Faraday Trans., 1998, 94, 985 RSC.
- B. Xue, K. Jia, J. Xu, N. Liu, P. Liu, C. Xu and Y. Li, React. Kinet., Mech. Catal., 2012, 107, 435 CrossRef CAS.
- U. Tilstam, Org. Process Res. Dev., 2012, 16, 1150 CrossRef CAS.
- W.-C. Shieh, S. Dell and O. Repiĉ, Org. Lett., 2001, 3, 4279 CrossRef CAS PubMed.
- (a) M. L. Laurila, N. A. Magnus and M. A. Staszak, Org. Process Res. Dev., 2009, 13, 1199 CrossRef CAS; (b) T. Ulf, Org. Process Res. Dev., 2012, 16, 1974 CrossRef.
- Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1998, 166, 419 CrossRef CAS.
- Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1998, 166, 425 CrossRef CAS.
- Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1999, 176, 201 CrossRef CAS.
- (a) T. M. Jyothi, T. Raja, M. B. Talawar, K. Sreekumar, S. Sugunan and B. S. Rao, Synth. Commun., 2000, 30, 3929 CrossRef CAS; (b) T. M. Jyothi, T. Raja, M. B. Talawar and B. S. Rao, Appl. Catal., A, 2011, 211, 41 CrossRef.
- P. Tundo, E. Trotta, G. Moraglio and E. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565 CrossRef CAS.
- M. S. Newman and J. A. Celia, J. Org. Chem., 1974, 39, 214 CrossRef.
- G. Iori and U. Romano, UK Patent Appl. GB, 2026484, 1980 Search PubMed.
- M. Vijayaraj and C. S. Gopinath, J. Catal., 2006, 243, 376 CrossRef CAS.
- Y. Tamura, T. Saito, H. Ishibashi and M. Ikeda, Synthesis, 1975, 641 CrossRef CAS.
- M. Lissel, S. Schmidt and B. Neumann, Synthesis, 1986, 382 CrossRef CAS.
- P. Tundo, E. Trotta, G. Moraglio and E. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565 CrossRef CAS.
- X. Jian-Gang, W. Chengyao, Q. Jing, Z. Li-Min and C. Branford, White Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 31 Search PubMed.
- X. Jian-Gang, Q. Jing, L. Shu-Bai, Z. Yan and Z. Li-Min, Synth. Commun., 2011, 41, 871 CrossRef.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464 CrossRef CAS PubMed.
- (a) P. Tundo, Pure Appl. Chem., 2000, 72, 1793 CAS; (b) P. Tundo, M. Selva, A. Perosa and S. Memoli, J. Org. Chem., 2002, 67, 1071 CrossRef CAS PubMed; (c) P. Tundo, M. Selva and A. Bomben, Checked by: P. Belica, R. Koehler and S. Wolff, Org. Synth., 2004, 10, 640 Search PubMed; (d) P. Belica, R. Koehler and S. Wolff, Org. Synth., 1999, 76, 169 CrossRef.
- (a) P. Tundo, E. Trotta and G. Moraglio, J. Chem. Soc., Perkin Trans. 1, 1989, 1070 RSC; (b) P. Tundo, M. Selva and C. A. Marques, in ACS Symposium Ser. No.626, Green Chemistry, Designing Chemistry for the Environment, ed. P. T. Anastas and T. C. Williamson, American Chemical Society, Washington, 1996, p. 81 Search PubMed.
- C. Venkatesan, M. Chidambaram and A. P. Singh, Appl. Catal. A, 2005, 292, 344 CrossRef CAS.
- D. A. White, Synth. Commun., 1977, 8, 559 CrossRef.
- A. Caretto, M. Noè, M. Selva and A. Perosa, ACS Sustainable Chem. Eng., 2014, 2, 2131 CrossRef CAS.
- (a) J. E. Bercaw, N. Hazari, J. A. Labinger, V. J. Scott and G. J. Sunley, J. Am. Chem. Soc., 2008, 130, 1198 Search PubMed; (b) J. E. Bercaw, P. L. Diaconescu, R. H. Grubbs, N. Hazari, R. D. Kay, J. A. Labinger, P. Mehrkhodavandi, G. E. Morris, G. J. Sunley and P. Vagner, Inorg. Chem., 2007, 46, 11371 CrossRef CAS PubMed.
- (a) J. H. Ahn, B. Temel and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 3814 CrossRef CAS PubMed; (b) D. A. Simonetti, J. H. Ahn and E. Iglesia, J. Catal., 2011, 277, 173 CrossRef CAS; (c) B. J. Daniel, B. P. Gracey, D. J. Law, B. M. Maunders, J. G. Sunley and J. C. Van Der Waal, WO, 2009063177, 2009 Search PubMed.
- G. G. Armitage, M. L. M. Bonati, N. Guo, S. Gaemers and J. W. Shabaker, Catal. Lett., 2013, 143, 370 CrossRef CAS.
- (a) T. Baba, M. Fujiwara, A. Oosaku, A. Kobayashi, R. G. Deleon and Y. Ono, Appl. Catal., A, 2002, 227, 1 CrossRef CAS; (b) R. G. Deleon, A. Kobayashi, T. Yamauchi, J. Ooishi, T. Baba, M. Sasaki and F. Hiarata, Appl. Catal., A, 2002, 225, 43 CrossRef; (c) T. Baba, A. Kobayashi, Y. Kawanami, K. Inazu, A. Ishikawa, T. Echizenn, K. Murai, S. Aso and M. Inomata, Green Chem., 2005, 7, 159 RSC; (d) M. Curini, F. Epifano, F. Maltese and O. Rosati, Tetrahedron Lett., 2002, 43, 4895 CrossRef CAS; (e) M. Distaso and E. Quaranta, J. Catal., 2008, 253, 278 CrossRef CAS.
- P. Tundo, S. Bressanello, A. Loris and G. Sathicq, Pure Appl. Chem., 2005, 77, 1719 CrossRef CAS.
- J. F. Bunnett and G. T. Davis, J. Am. Chem. Soc., 1960, 82, 665 CrossRef CAS.
- W. P. Jencks and J. Carriuolo, J. Am. Chem. Soc., 1960, 82, 675 CrossRef CAS.
- A. Loris, A. Perosa, M. Selva and P. Tundo, J. Org. Chem., 2004, 69, 3953 CrossRef CAS PubMed.
- Z. Zheng, T. Wu and X. Zhou, Chem. Commun., 2006, 1864 RSC.
- (a) R. S. Varma and V. V. Namboodiri, Chem. Commun., 2001, 643 RSC; (b) P. Bonhote, A. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS PubMed.
- Z. Q. Zheng, J. Wang, T. H. Wu and X. P. Zhou, Adv. Synth. Catal., 2007, 349, 1095 CrossRef CAS.
- (a) M. Ue, M. Takeda, T. Takahashi and M. Takehara, Electrochem. Solid-State Lett., 2002, 6, A119 CrossRef; (b) G. W. Earl, D. E. Weisshaar, D. Paulson, M. Hanson, J. Uilk, D. Wineinger and S. Moeckly, J. Surfactants Deterg., 2005, 8, 325 CrossRef CAS.
- B. Oelkers and J. Sundermeyer, Green Chem., 2011, 13, 608 RSC.
- M. Selva and P. Tundo, Tetrahedron Lett., 2003, 44, 8139 CrossRef CAS.
- (a) M. Aresta and E. Quaranta, Tetrahedron, 1991, 47, 9489 CrossRef CAS; (b) M. Aresta, A. Dibenedetto, E. Quaranta, M. Boscolo and R. Larsson, J. Mol. Catal. A: Chem., 2001, 174, 7 CrossRef CAS; (c) A. Dibenedetto, M. Aresta, C. Fragale and M. Narracci, Green Chem., 2002, 4, 439 RSC.
- M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2003, 68, 7374 CrossRef CAS PubMed.
- F. Bonino, A. Damin, S. Bordiga, M. Selva, P. Tundo and A. Zecchina, Angew. Chem., Int. Ed., 2005, 44, 4774 CrossRef CAS PubMed.
- W. J. Ebenezer, M. G. Hutchings, K. Jones, D. A. Lambert and I. Watt, Tetrahedron Lett., 2007, 48, 1641 CrossRef CAS.
- (a) P. F. Gordon and P. Gregory, Organic Chemistry in Colour, Springer, Berlin, 1987 Search PubMed; (b) H. Zollinger, Color Chemistry, VHCA, Zurich and Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- G. Semple, H. Ryder, D. P. Rooker, A. R. Batt, D. A. Kendrick, M. Szelke, M. Ohta, M. Satoh, A. Nishida, S. Akuzawa and K. Miyata, J. Med. Chem., 1997, 40, 331 CrossRef CAS PubMed.
- (a) R. Juarez, A. Padilla, A. Corma and H. Garcia, Catal. Commun., 2009, 10, 472 CrossRef CAS.
- K. Sreekumar, T. M. Jyothi, T. Mathew, M. B. Talawar, S. Sugunan and B. S. Rao, J. Mol. Catal. A: Chem., 2000, 159, 327 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275 RSC.
- M. Selva, A. Perosa, P. Tundo and D. Brunelli, J. Org. Chem., 2006, 71, 5770 CrossRef CAS PubMed.
- (a) M. Lissel, Liebigs Ann. Chem., 1987, 77 CrossRef CAS; (b) H. Jansen in der War and M. Lissel, Z. Naturforsch., 1989, 44b, 863 Search PubMed; (c) H. Jansen in der War and M. Lissel, Z. Chem., 1989, 29, 253 Search PubMed; (d) M. Lissel and A. R. Rohani-Dezfuli, Chem.-Ztg., 1987, 111, 83 CAS.
- S. Ouk, S. Thiébaud, E. Borredon and B. Chabaud, Synth. Commun., 2005, 35, 3021 CrossRef CAS.
- W.-C. Shieh, M. Lozanov, M. Loo, O. Repič and T. J. Blacklock, Tetrahedron Lett., 2003, 44, 4563 CrossRef CAS.
- W.-C. Shieh, M. Lozanov and O. Repič, Tetrahedron Lett., 2003, 44, 6943 CrossRef CAS.
- W. Xiao, C. Q. Wenjun, T. Wu, Y. Wu, L. Dai, X. Wang and C. Song, Chem. Lett., 2010, 39, 1112 CrossRef CAS.
- X. Jiang, A. Tiwari, M. Thompson, Z. Chen, T. P. Cleary and T. B. K. Lee, Org. Process Res. Dev., 2001, 5, 604 CrossRef CAS.
- S.-Y. Zhao, H.-Q. Zhang, D.-Q. Zhang and Z.-Y. Shao, Synth. Commun., 2012, 42, 128 CrossRef CAS.
- I. Shimizu and Y. Lee, Synlett, 1998, 1063 Search PubMed.
- M. L. Laurila, N. A. Magnus and M. A. Staszak, Org. Process Res. Dev., 2009, 13, 1199 CrossRef CAS.
- E. Quaranta, M. Carafa and F. Trani, Appl. Catal., B, 2009, 91, 380 CrossRef CAS.
- (a) A. B. Taleb and C. Jenner, J. Mol. Catal., 1993, 84, L121 CrossRef; (b) A. B. Taleb and G. Jenner, J. Mol. Catal., 1993, 84, L131 CrossRef.
- V. V. Rekha, M. V. Ramani, A. Ratnamala, V. Rupakalpana, G. V. Subbaraju, C. Satyanarayana and C. S. Rao, Org. Process Res. Dev., 2009, 13, 769 CrossRef CAS.
- (a) V. G. Devulapelli and H.-S. Weng, Catal. Commun., 2009, 10, 1711 CrossRef CAS; (b) X. Song and A. Sayari, Catal. Rev., 1996, 38, 329 CrossRef CAS; (c) G. D. Yadav and J. J. Nair, Microporous Mesoporous Mater., 1999, 33, 1 CrossRef CAS.
- (a) L. Goossen and A. Dohring, Adv. Synth. Catal., 2003, 345, 943 CrossRef CAS; (b) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, E. Marcantoni, P. Melchiorre, P. Palazzi and L. Sambri, Eur. J. Org. Chem., 2006, 4429 CrossRef CAS.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464 CrossRef CAS PubMed.
- (a) R. Kirumakki, N. Nagaraju, K. V. R. Chary and S. Narayanan, Appl. Catal., A, 2003, 248, 161 CrossRef; (b) S. R. Kirumakki, N. Nagaraju, K. V. V. S. B. S. R. Murthy and S. Narayanan, Appl. Catal., A, 2002, 226, 175 CrossRef CAS.
- (a) D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448 CrossRef CAS; (b) Y. Zheng, J. Li, N. Zhao, W. Wei and Y. Sun, Microporous Mesoporous Mater., 2006, 92, 195 CrossRef CAS.
- W.-C. Shieh, S. Dell and O. Repic, J. Org. Chem., 2002, 67, 2188 CrossRef CAS PubMed.
- (a) M. Carafa, E. Mesto and E. Quaranta, Eur. J. Org. Chem., 2011, 2458 CrossRef CAS; (b) M. Carafa, F. Iannone, V. Mele and E. Quaranta, Green Chem., 2012, 14, 3377 RSC.
- W.-C. Shieh, S. Dell and O. Repic, Tetrahedron Lett., 2002, 43, 5607 CrossRef CAS.
- F. Tanasa, M. Onciu and C. Chiriac, Rev. Roum. Chim., 2003, 48, 869 CAS.
- K. N. Chau, F. Duus and T. N. Le, Green Chem. Lett. Rev., 2013, 6, 89 CrossRef CAS.
- (a) Y. Nitta and Y. Arakawa, Chem. Pharm. Bull., 1985, 33, 1380 CrossRef CAS; (b) A. Etienne, J. Vincent, G. Lonchambon and R. Garreau, Bull. Soc. Chim. Fr., 1977, 483 CAS.
- (a) E. Suzuki, M. Akiyama and Y. Ono, J. Chem. Soc., Chem. Commun., 1992, 136 RSC; (b) Y. Ono, M. Akiyama and E. Suzuki, Chem. Mater., 1993, 5, 442 CrossRef CAS; (c) Y. Ono, Appl. Catal., A, 1997, 155, 133 CrossRef CAS.
- L. N. Lewis, F. J. Schattenmann, T. M. Jordan, J. C. Carnahan, W. P. Flanagan, R. J. Wroczynski, J. P. Lemmon, J. M. Anostario and M. A. Othon, Inorg. Chem., 2002, 41, 2608 CrossRef CAS PubMed.
- M. Akiyama, E. Suzuki and Y. Ono, Inorg. Chim. Acta, 1993, 207, 259 CrossRef CAS.
- M. Okamoto, S. Suzuki and E. Suzuki, Appl. Catal., A, 2004, 261, 239 CrossRef CAS.
- Y. Ono, A. Kusano, H. Hatayama and E. Suzuki, J. Mater. Chem., 1997, 7, 2049 RSC.
- M. F. Carroll, J. Chem. Soc., 1940, 704 RSC.
- (a) I. Shimizu, T. Yamada and J. Tsuji, Tetrahedron Lett., 1980, 21, 3199 CrossRef CAS; (b) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387 CrossRef.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292 CrossRef CAS.
- I. Shimizu and J. Tsuji, J. Am. Chem. Soc., 1982, 104, 5844 CrossRef CAS.
- (a) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (b) B. M. Trost, Tetrahedron, 2015, 71, 5708 CrossRef CAS PubMed; (c) N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821 CrossRef CAS.
- (a) R. M. McFadden and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 7738 CrossRef CAS PubMed; (b) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS PubMed; (c) M. Nakamura, A. Hajra, K. Endo and E. Nakamura, Angew. Chem., 2005, 117, 7414 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7248 ) CrossRef.
- (a) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 17180 CrossRef CAS PubMed; (b) J. T. Mohr, D. C. Behenna, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2005, 44, 6924 CrossRef CAS PubMed; (c) S. Bouzbouz and B. Kirschleger, Synthesis, 1994, 714 CrossRef CAS.
- J. Tsuji, I. Shimizu, I. Minami, Y. Ohashi, T. Sugiura and K. Takahashi, J. Org. Chem., 1985, 50, 1523 CrossRef CAS.
- A. T. Dickschat, F. Behrends, S. Surmiak, M. Weiß, H. Eckert and A. Studer, Chem. Commun., 2013, 49, 2195 RSC.
- I. Shimizu, Y. Ohashi and J. Tsuji, Tetrahedron Lett., 1983, 24, 3865 CrossRef CAS.
- J. Tsuji, I. Minami and I. Shimizu, Chem. Lett., 1983, 12, 1325 CrossRef.
- (a) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092 CrossRef CAS PubMed; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545 CrossRef CAS; (c) M. Luparia, M. T. Oliverira, D. Audisio, F. Frebault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed.
- (a) B. M. Trost, T. R. Verhoeven and J. M. Fortunak, Tetrahedron Lett., 1979, 20, 2301 CrossRef; (b) J. A. Keith, D. C. Behenna, N. Sherden, J. T. Mohr, S. Ma, S. C. Marinescu, R. J. Nielsen, J. Oxgaard, B. M. Stoltz and W. A. Goddard, J. Am. Chem. Soc., 2012, 134, 19050 CrossRef CAS PubMed.
- (a) Y. Castanet and F. Petit, Tetrahedron Lett., 1979, 20, 3221 CrossRef; (b) X.-H. Li, B.-H. Zheng, C.-H. Ding and X.-L. Hou, Org. Lett., 2013, 15, 6086 CrossRef CAS PubMed.
- D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 15044 CrossRef CAS PubMed.
- (a) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 2846 CrossRef CAS PubMed; (b) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343 CrossRef CAS PubMed.
- M. Seto, J. L. Roizen, B. M. Stoltz and M. Brian, Angew. Chem., Int. Ed., 2008, 47, 6873 CrossRef CAS PubMed.
- B. M. Trost and Y. Zhang, Chem. – Eur. J., 2011, 17, 2916 CrossRef CAS PubMed.
- S.-C. Sha, J. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602 CrossRef CAS PubMed.
- A. Millán, A. G. Campaña, B. Bazdi, D. Miguel, L. Álvarez de Cienfuegos, A. M. Echavarren and J. M. Cuerva, Chem. – Eur. J., 2011, 17, 3985 CrossRef PubMed.
- (a) S.-I. Sasaoka, T. Yamamoto, H. Kinoshita, K. Inomata and H. Kotake, Chem. Lett., 1985, 14, 315 CrossRef; (b) B. M. Trost and K. M. Pietrusiewicz, Tetrahedron Lett., 1985, 26, 4039 CrossRef CAS.
- A. G. Campaça, B. Bazdi, N. Fuentes, R. Robles, J. M. Cuerva, J. E. Oltra, S. Porcel and A. M. Echavarren, Angew. Chem., 2008, 120, 7625 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7515 ) CrossRef.
- (a) B. Schmidt and S. Nave, Adv. Synth. Catal., 2006, 348, 531 CrossRef CAS; (b) B. M. Trost and D. L. Van Vraken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (c) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (d) R. Stragies and S. Blechert, J. Am. Chem. Soc., 2000, 122, 9584 CrossRef CAS.
- B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053 CrossRef CAS PubMed.
- (a) C. Welter, R. M. Moreno, S. Streiff and G. Helmchen, Org. Biomol. Chem., 2005, 3, 3266 RSC; (b) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247 CrossRef CAS.
- (a) M. Gärtner, M. Jäkel, M. Achatz, C. Sonnenschein, O. Tverskoy and G. Helmchen, Org. Lett., 2011, 13, 2810 CrossRef PubMed; (b) M. Jaekel, J. Qu, T. Schnitzer and G. Helmchen, Chem. – Eur. J., 2013, 19, 16746 CrossRef CAS PubMed.
- (a) P. A. Evans and J. E. Robinson, Org. Lett., 1999, 1, 1929 CrossRef CAS PubMed; (b) P. A. Evans, J. E. Robinson and K. K. Moffett, Org. Lett., 2001, 21, 3269 CrossRef.
- N. Gao, X.-W. Guo, S.-C. Zheng, W.-K. Yang and X.-M. Zhao, Tetrahedron, 2012, 68, 9413 CrossRef CAS.
- X. Qian, A. Auffrant, A. Felouat and C. Gosmini, Angew. Chem., Int. Ed., 2011, 50, 10402 CrossRef CAS PubMed.
- H. He, W. B. Liu, L. X. Dai and S. L. You, J. Am. Chem. Soc., 2009, 131, 8346 CrossRef CAS PubMed.
- H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed.
- B. M. Trost and A. Martos-Redruejo, Org. Lett., 2009, 11, 1071 CrossRef CAS PubMed.
- C. Zhao, Z. Tan, Z. Liang, W. Deng and H. Gong, Synthesis, 2014, 1901 Search PubMed.
- (a) Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Org. Biomol. Chem., 2012, 10, 8786 RSC; (b) K. Ghabili, P. S. Agutter, M. Ghanei, K. Ansarin, Y. Panahi and M. M. Shoja, Crit. Rev. Toxicol., 2011, 41, 384 CrossRef CAS PubMed; (c) F. R. Tang and W. K. Loke, Crit. Rev. Toxicol., 2012, 42, 688 CrossRef CAS PubMed; (d) K. Ghabili, K. Ansarin, M. M. Shoja, P. S. Agutter and M. Ghanei, J. Appl. Toxicol., 2010, 7, 627 CrossRef PubMed; (e) K. Marshall Jr., J. Am. Med. Assoc., 1919, 73, 684 CrossRef; (f) K. E. Jackson, Chem. Rev., 1934, 15, 425 CrossRef CAS; (g) R. C. Malhotra, K. Ganesan, K. Sugendran and R. V. Swamy, Def. Sci. J., 1999, 49, 97 CrossRef CAS; (h) R. J. Duchovic and J. A. Vilensky, J. Chem. Educ., 2007, 84, 944 CrossRef CAS.
- (a) O. Kamm and J. H. Waldo, J. Am. Chem. Soc., 1921, 43, 2223 CrossRef CAS; (b) Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Org. Biomol. Chem., 2012, 10, 8786 RSC.
- (a) Q.-Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Inorg. Chem., 2012, 51, 760 CrossRef CAS PubMed; (b) O. F. Erdem, A. Silakov, E. Reijerse, W. Lubitz, K.-G. S. Lennart, P. Huang, S. Ott and M. Stein, Angew. Chem., Int. Ed., 2011, 50, 1439 CrossRef CAS PubMed; (c) C. Fliedel, A. Sabbatini and P. Braunstein, Dalton Trans., 2010, 39, 8820 RSC; (d) P. A. Ulmann, A. M. Brown, M. V. Ovchinnikov, C. A. Mirkin, A. G. Di Pasquale and A. L. Rheingold, Chem. – Eur. J., 2007, 13, 4529 CrossRef CAS PubMed.
- (a) V. Mai and L. R. Comstock, J. Org. Chem., 2011, 76, 10319 CrossRef CAS PubMed; (b) I. Konstantinova, K. Bukhryakov, Y. Gezentsvey and M. Krasavin, Lett. Org. Chem., 2011, 8, 628 CrossRef; (c) R. P. Y. Choy, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2011, 76, 80 CrossRef PubMed; (d) N. E. Shevchenko, V. G. Nenajdenko and E. S. Balenkova, Synthesis, 2003, 1191 CAS; (e) J. Fang, B. H. Wallikewitz, F. Gao, G. Tu, C. Muller, G. Pace, R. H. Friend and T. S. Huck, J. Am. Chem. Soc., 2011, 133, 683 CrossRef CAS PubMed; (f) L. Wang, Y. Wen, J. Liu, J. Zhou, C. Li and C. Wie, Org. Biomol. Chem., 2011, 9, 2648 RSC.
- (a) I. C. Papaconstantinou, M. A. Fousteris, A. I. Koutsourea, G. N. Pairas, A. D. Papageorgiou and S. S. Nikolaropoulos, Anticancer Drugs, 2013, 24, 52 CrossRef CAS PubMed; (b) A. G. Morphosys, J. Amersdorfer, S. Steidl, M. Winderlich, S. Krohn and L. Rojkjaer, Patent WO, 2013/24097, 2013 Search PubMed; (c) M. C. S. Barnes, H. J. Dennison, S. S. Flack, J. A. Lumley, P. S. Pang and K. C. Spencer, Patent WO, 2011/27156, 2011 Search PubMed; (d) V. Moneo Ocana, G. Santamaria Nunez, L. F. Garcia Fernandez, C. M. Galmarini, M. J. Guillen Navarro and P. M. Aviles Marin, Patent WO, 2012/62920, 2012 Search PubMed; (e) S. A. Laufer and S. Margutti, J. Med. Chem., 2008, 51, 2580 CrossRef CAS PubMed; (f) G. Ahn, A. Couture, P. Grandclaudon, A. Ryckebusch, N. Schifano-Faux, J.-F. Goossens, B. Baldeyrou and A. Lansiaux, Med. Chem. Lett., 2011, 21, 2259 CrossRef CAS PubMed; (g) C. B. Phippen and C. S. P. McErlean, Tetrahedron Lett., 2011, 52, 1490 CrossRef CAS.
- E. Block, Reactions of organosulfur compounds, Academic Press, New York, 1978, pp. 141–145 Search PubMed.
- F. Aricò and P. Tundo, Pure Appl. Chem., 2016, 88, 3 Search PubMed.
- C. Chiappe, A. Sanzone and J. P. Dyson, Green Chem., 2011, 13, 1437 RSC.
- (a) S. Winstein, E. Clippinger, A. H. Fainberg, R. Heck and G. C. Robison, J. Am. Chem. Soc., 1956, 78, 328 CrossRef CAS; (b) H. Kessler and M. Feigel, Acc. Chem. Res., 1982, 15, 2 CrossRef CAS; (c) J. L. Fry, C. J. Lancelot, L. K. M. Lam, J. M. Harris, R. C. Bingham, D. J. Raber, R. E. Hall and P. V. R. Schleyer, J. Am. Chem. Soc., 1970, 92, 2538 CrossRef CAS.
- (a) F. Aricò, S. Evaristo and P. Tundo, RSC Adv., 2014, 4, 31071 RSC; (b) A. Perosa, M. Selva, P. Tundo and F. Zordan, Synlett, 2000, 272 CrossRef CAS.
- (a) M. H. Benn and K. A. Vinod Singh, Can. J. Chem., 1986, 64, 940 CrossRef CAS; (b) M. Gibbs, J. Am. Chem. Soc., 1935, 57, 1137 CrossRef.
- F. Aricò, I. Udrea, M. Crisma and P. Tundo, ChemPlusChem, 2015, 80, 471 CrossRef.
- F. Aricò, A. S. Aldoshin and P. Tundo, ACS Sustainable Chem. Eng., 2016, 4, 2843 CrossRef.
- Y. Imai, J. Kitazawa, T. Sato, N. Tajima, R. Kuroda, Y. Matsubara and Z. Yoshida, Tetrahedron, 2007, 63, 1995 CrossRef CAS.
- (a) J. W. Benbow and R. Katoch-Rouse, J. Org. Chem., 2001, 66, 4965 CrossRef CAS PubMed; (b) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 5767 CrossRef CAS PubMed.
- H. B. Bode and A. Zeeck, Phytochemistry, 2000, 54, 597 CrossRef CAS PubMed.
- H. Kikuzaki, S. Kayano, N. Fukutsuka, A. Aoki, K. Kasamatsu, Y. Yamasaki, T. Mitani and N. Nakatani, J. Agric. Food Chem., 2004, 52, 344 CrossRef CAS PubMed.
- (a) S. Apers, A. Vlietinck and L. Pieters, Phytochem. Rev., 2003, 2, 201 CrossRef CAS; (b) L. Pieters, S. Van Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemière, J. Med. Chem., 1999, 42, 5475 CrossRef CAS PubMed.
- J. S. Shin, Y. M. Kim, S. S. Hong, H. S. Kang, Y. J. Yang, D. K. Lee, B. Y. Hwang, J. S. Ro and M. K. Lee, Arch. Pharmacal Res., 2005, 28, 1337 CrossRef CAS.
- R. M. Pinder and J. H. Wieringa, Med. Res. Rev., 1993, 13, 259 CrossRef CAS PubMed.
- Y. Takano, M. Takano and T. L. Yaksh, Eur. J. Pharmacol., 1992, 219, 465 CrossRef CAS PubMed.
- R. Zhou, G. Luo and A. Ewing, J. Neurosci., 1994, 14, 2402 CAS.
- A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, 2010 Search PubMed.
- F. Aricò, S. Evaristo and P. Tundo, Green Chem., 2015, 17, 1176 RSC.
- H. H. Orvis and K. K. Kimura, Del. Med. J., 1968, 40, 237 CAS.
- K. Torfgård and J. Ahlner, Cardiovasc. Drugs Ther., 1994, 8, 701 CrossRef.
- W. A. Parker and G. L. MacKinnon, Drug Intell. Clin. Pharm., 1981, 15, 806 CrossRef CAS PubMed.
- S. K. Roy and J. Tang, Org. Process Res. Dev., 2000, 4, 170 CrossRef CAS.
- C. R. McElroy, F. Aricò, F. Benetollo and P. Tundo, Pure Appl. Chem., 2011, 84, 707 CrossRef.
- (a) C. R. McElroy, F. Aricò and P. Tundo, Synlett, 2012, 23, 1809 CrossRef CAS; (b) F. Aricò, S. Bravo, M. Crisma and P. Tundo, Pure Appl. Chem., 2016, 88, 227 Search PubMed.
- F. Aricò, U. Toniolo and P. Tundo, Green Chem., 2012, 14, 58 RSC.
- W. Lossen, Justus Liebigs Ann. Chem., 1872, 161, 347 CrossRef.
- O. Kreye, S. Wald and M. A. R. Meier, Adv. Synth. Catal., 2013, 355, 81 CrossRef CAS.
- W. Steglich and G. Höfle, Tetrahedron Lett., 1970, 11, 4727 CrossRef.
- (a) C. D. Campbell, C. Concellón and A. D. Smith, Tetrahedron: Asymmetry, 2011, 22, 797 CrossRef CAS; (b) D. Uraguchi, K. Koshimoto, S. Miyake and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 5567 CrossRef CAS PubMed.
- S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va and E. Vedejs, J. Am. Chem. Soc., 2006, 128, 925 CrossRef CAS PubMed.
- T. A. Duffey, S. A. Shaw and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14 CrossRef CAS PubMed.
- C. A. Marques, M. Selva, P. Tundo and F. Montanari, J. Org. Chem., 1993, 58, 5765 CrossRef CAS.
- S. Blechert, Synthesis, 1989, 71 CrossRef CAS.
- M. Barontini, I. Proietti Silvestri, V. Nardi, P. Bovicelli, L. Pari, F. Gallucci, R. Spezia and G. Righi, Tetrahedron Lett., 2013, 54, 5004 CrossRef CAS.
- D. J. Cram and R. Davis, J. Am. Chem. Soc., 1949, 71, 3871 CrossRef CAS.
- Y. Tsuji and J. P. Richard, J. Phys. Org. Chem., 2016, 29, 557 CrossRef CAS PubMed.
- (a) J. T. D. Cross, R. Hunter and V. R. Stimson, Aust. J. Chem., 1976, 29, 1477 CrossRef CAS; (b) G. G. Smith and B. L. Yates, J. Org. Chem., 1965, 30, 434 CrossRef CAS.
- A. S. Gordon and W. P. Norris, J. Phys. Chem., 1965, 69, 3013 CrossRef CAS.
- G. G. Smith, D. A. K. Jones and R. Taylor, J. Org. Chem., 1963, 28, 3547 CrossRef.
- C. D. Hurd and F. H. Blunck, J. Am. Chem. Soc., 1938, 60, 2419 CrossRef CAS.
- L. Tschugaeff, Ber., 1899, 32, 3332 CrossRef CAS.
- R. Notario, J. Quijano, C. Sánchez and E. Vélez, J. Phys. Org. Chem., 2005, 18, 134 CrossRef CAS.
- (a) D. B. Bigley and C. M. Wren, J. Chem. Soc., Perkin Trans. 2, 1972, 926 RSC; (b) G. G. Smith, K. K. Lum, J. A. Kirby and J. Posposil, J. Org. Chem., 1969, 34, 2090 CrossRef CAS.
- (a) D. B. Bigley and C. M. Wren, J. Chem. Soc., Perkin Trans. 2, 1972, 2359 RSC; (b) D. B. Bigley, C. Brown and R. H. Weatherliead, J. Chem. Soc., Perkin Trans. 2, 1976, 701 RSC.
- D. B. Pattison, J. Am. Chem. Soc., 1957, 79, 3455 CrossRef CAS.
- R. Noyori, Chem. Commun., 2005, 1807 RSC.
| This journal is © The Royal Society of Chemistry 2018 |