
Glucose promoted facile reduction of azides to amines under aqueous alkaline conditions†
Nisha
Chandna
,
Fatehjeet
Kaur
,
Shobhna
Kumar
and
Nidhi
Jain
 *
*
Department of Chemistry, Indian Institute of Technology, New Delhi-110016, India. E-mail: njain@chemistry.iitd.ac.in; Fax: +91 11 26581102; Tel: +91 11 26591562
First published on 27th July 2017
Abstract
A quick and efficient method for the reduction of azides to amines in water using D-glucose and KOH as green reagents is reported. The protocol is simple, inexpensive, scalable, and can be applied to different aromatic, heteroaromatic and sulphonyl azides. A high level of chemoselectivity is observed for azide reduction in the presence of other reducible functionalities like cyano, nitro, ether, ketone, amide and acid. The reaction gets completed in a short time (5–20 minutes), and furnishes the amines in high yield (85–99%). Unlike conventional hydrogenations, this reduction protocol does not require any metal catalyst, elaborate experimental setup or use of high-pressure equipment.
Introduction
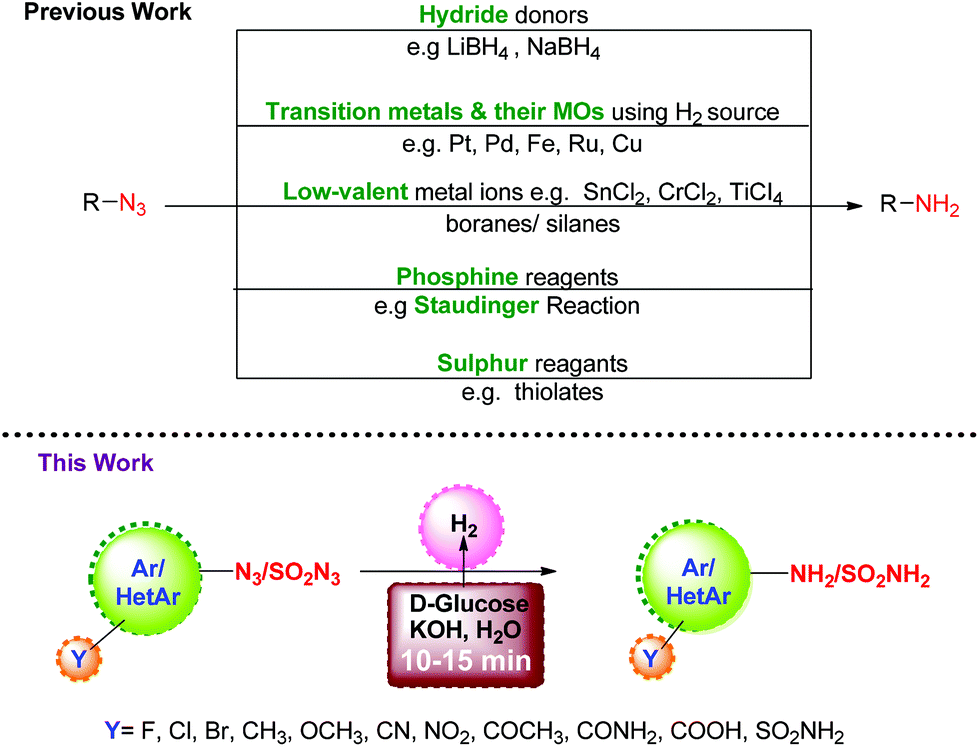
Organic azides are versatile and useful synthons which find wide applications in the synthesis of heterocycles.1,2 They can be prepared with good regio-, stereo-, and enantioselectivity and their transformation to amines provides access to a variety of organic intermediates, particularly useful in the synthesis of pharmaceuticals, carbohydrates and nucleosides.3,4 Although there are several methods available in the literature for the preparation of amines, their synthesis through the reduction of azides is one of the important protocols since azides can be easily prepared5–8 from halides and sulfonates. A number of reagents have been developed over the years to bring about this reductive transformation, and they can be typically classified as: (i) hydride donors9 such as LiAIH4 or NaBH4; (ii) H2 gas or its source in the presence of transition metals and their oxides (MOs) as catalysts;10–12 (iii) low-valent metal ions13 such as SnCl2, CrCl2, TiCl3/boranes14,15/silanes;16 (iv) phosphine reagents17 as in the Staudinger reaction; and (v) sulphur reagents18e.g. thiolates, etc. Though useful, these methods suffer from limitations with regard to chemo-selectivity in the presence of other reducible functionalities, the use of transition metals, harsh reagents, and cost efficiency. Thus, it is highly desirable to develop reduction protocols for azides which are transition metal free, chemoselective, and do not involve a toxic hydrogen source.Carbohydrates are chiral organic molecules readily available from natural and renewable resources. They are inexpensive, eco-friendly, compatible with biological systems, and soluble in water. All these properties encouraged us to explore their capability as a hydrogen source for the reduction of azides. Continuing our efforts towards greener and economical chemical protocols,19–22 we herein report the reduction of aromatic, heteroaromatic and sulfonyl azides to the corresponding amines using D-glucose and KOH under aqueous conditions.
Results and discussion
The aim to develop a transition metal-free reduction of azides motivated us initially to investigate the reaction of 4-azidobenzonitrile (1a) with 2 equiv. of D-glucose and KOH in DMF at 110 °C. As desired, 4-aminobenzonitrile (2a) was formed (Scheme 1), albeit in moderate yield (72%) (Table 1, entry 1). In a quest to improve the yield of the product, the reaction conditions were optimized. Screening of solvents showed that switching from DMF to DMSO (Table 1, entry 2) reduced the yield of 2a to 30%, along with the formation of other undesired products. Further, either no conversion or trace conversions took place in 1,4-dioxane, toluene, [Bmim]PF6, and N-methyl-2-pyrrolidone as solvents. In DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1), the yield of 2a increased to 80% (Table 1, entry 3). To eliminate the use of organic solvents completely, we treated 1a with 2 equiv. of D-glucose and KOH in a minimum amount of water at 110 °C in a pressure tube. We found that the reaction was completed in ten minutes, and this was indicated by a change in the color of the reaction mixture from yellow to brown (Table 1, entry 4). Next, we optimized the conditions with respect to base, catalyst, and temperature in water. The use of other inorganic bases like NaOH and K2CO3 gave lower yields of 2a (Table 1, entries 5 and 6). Similar low yields were seen with Na2CO3, NaOtBu, KOtBu, and CH3COONa. A control reaction in the absence of base did not yield any product even after prolonged heating for 24 h (Table 1, entry 7). Reducing the amount of KOH to 1 equiv., or raising it to 5 equiv., resulted in incomplete conversion. However, with 3 equiv. of KOH, 2a was formed in almost 99% yield (Table 1, entry 8). Optimizing the temperature revealed that the reaction performed equally well at 85 °C (Table 1, entry 9), and conversion was complete in ten minutes. The amount of D-glucose was also varied (1–3 equiv.), and it was found that 2 equivalents were necessary and sufficient for the reaction (Table 1, entry 10). To establish the role of sugars in promoting the reaction, other sugars such as maltose, fructose, D-mannose, sucrose and cellulose were tested (Table 1, entries 11–15); of them only D-mannose was found to be effective. Further, no conversion took place in the absence of added sugar (Table 1, entry 16).
:H2O (1:1), the yield of 2a increased to 80% (Table 1, entry 3). To eliminate the use of organic solvents completely, we treated 1a with 2 equiv. of D-glucose and KOH in a minimum amount of water at 110 °C in a pressure tube. We found that the reaction was completed in ten minutes, and this was indicated by a change in the color of the reaction mixture from yellow to brown (Table 1, entry 4). Next, we optimized the conditions with respect to base, catalyst, and temperature in water. The use of other inorganic bases like NaOH and K2CO3 gave lower yields of 2a (Table 1, entries 5 and 6). Similar low yields were seen with Na2CO3, NaOtBu, KOtBu, and CH3COONa. A control reaction in the absence of base did not yield any product even after prolonged heating for 24 h (Table 1, entry 7). Reducing the amount of KOH to 1 equiv., or raising it to 5 equiv., resulted in incomplete conversion. However, with 3 equiv. of KOH, 2a was formed in almost 99% yield (Table 1, entry 8). Optimizing the temperature revealed that the reaction performed equally well at 85 °C (Table 1, entry 9), and conversion was complete in ten minutes. The amount of D-glucose was also varied (1–3 equiv.), and it was found that 2 equivalents were necessary and sufficient for the reaction (Table 1, entry 10). To establish the role of sugars in promoting the reaction, other sugars such as maltose, fructose, D-mannose, sucrose and cellulose were tested (Table 1, entries 11–15); of them only D-mannose was found to be effective. Further, no conversion took place in the absence of added sugar (Table 1, entry 16).
 | ||
| Scheme 1 Various methods for the reduction of azides to amines. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Carbohydrate (equiv.) | Base (equiv.) | Temp (°C) | Solvent | Time | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol, 1 equiv.), D-glucose (2 mmol, 2 equiv.), KOH (3 mmol, 3 equiv.) were taken in water (100 μL), and stirred for 10 min at 85 °C. b Yield of 2a as determined by HPLC conversion. c 1 equiv. KOH. d 3 equiv. KOH. e 5 equiv. KOH, NR = no reaction. | ||||||
| 1. | D-Glucose (2) | KOH (2) | 110 | DMF | 10 min | 72% |
| 2. | D-Glucose (2) | KOH (2) | 110 | DMSO | 10 min | 30% |
| 3. | D-Glucose (2) | KOH (2) | 110 | DMF:water (1:1) |
10 min | 80% |
| 4. | D-Glucose (2) | KOH (2) | 110 | Water | 10 min | 90% |
| 5. | D-Glucose (2) | NaOH (2) | 110 | Water | 10 min | 62% |
| 6. | D-Glucose (2) | K2CO3 (2) | 110 | Water | 10 min | 54% |
| 7. | D-Glucose (2) | — | 110 | Water | 24 h | NR |
| 8. | D-Glucose (2) | KOH (1–5) | 110 | Water | 10 min | 70c, 99d, 65e |
| 9. | D-Glucose (2) | KOH (3) | 85 | Water | 10 min | 99% |
| 10. | D-Glucose (1 & 3) | KOH (3) | 85 | Water | 10 min | 50% & 98% |
| 11. | D-Maltose (2) | KOH (3) | 85 | Water | 10 min | 60% |
| 12. | D-Fructose (2) | KOH (3) | 85 | Water | 10 min | 58% |
| 13. | D-Mannose (2) | KOH (3) | 85 | Water | 10 min | 95% |
| 14. | Sucrose (2) | KOH (3) | 85 | Water | 10 min | NR |
| 15. | Cellulose (2) | KOH (3) | 85 | Water | 10 min | NR |
| 16. | — | KOH (3) | 85 | Water | 10 min | NR |

Next, we investigated the generality and scope of this reaction by reacting a range of azides under the optimized conditions in water. Though the azides were partly soluble in water at the beginning of the reaction, as the reaction proceeded at 85 °C, the solution turned homogeneous. Notably, a rapid completion of the reaction was observed in most cases (Table 2). Further, the work-up was simple and clean. The amines, partly soluble in water, could be easily isolated from the reaction mixture by extracting it with ethyl acetate. In most cases, the product was free from any associated impurities, and no column was required for further purification. The reduction of halogenated aryl azides yielded the corresponding amines 2c–2g in high yields without suffering any dehalogenation (Table 2). In a few cases (Table 2, compounds 2d, 2j and 2k), the reaction was found to perform better in DMF:H2O (0.5:1), though these reactions took a longer time (18–120 min) to complete. High chemoselectivity with excellent yields was observed with 4-nitro and 3-nitrophenylazides as the azide group was reduced exclusively in the presence of the nitro group yielding the corresponding nitroanilines (2h, 2i). Notably, even on using an excess (6 equiv.) of D-glucose and KOH, the azido group was reduced selectively in the presence of the nitro group. Aryl azides substituted with methoxy and methyl groups (1j and 1k) were successfully reduced though much longer reaction times were required. 4-Azidoacetophenone, 4-azidobenzoic acid and 4-azidobenzamide gave the corresponding anilines (2l–2n) without reducing the carbonyl functionality. The methodology also worked very well for the reduction of azides in the presence of a sulphonamide group (1o) as well as with 1-azidonaphthalene (1p). Further, the protocol was equally facile for heterocyclic azides, and clean reactions were observed in all the cases without affecting the heterocyclic ring (2q–2s). Unfortunately, the reaction did not work with alkyl azides, and no amine formation was seen with n-butyl azide as the substrate. In addition to aryl and heteroaryl azides, sulphonyl azides (1t–1v) could also be reduced efficiently to the corresponding amines (2t–2v) in near quantitative yields within minutes. The reduction of biologically relevant azides 5-phenylthiazol-2-azide (1w) and 5-(4-chlorobenzyl)thiazol-2-azide (1x) yielded the corresponding aminothiazoles (2w, 2x) in 95 and 96% yields, respectively. These compounds are known to display potent antifungal, antibacterial, antitubercular, and anticancer activities.23 It is noteworthy to reiterate that the reaction time required for the conversion of azides to amines is much lower compared to any of the previous methods discussed above in Scheme 1.
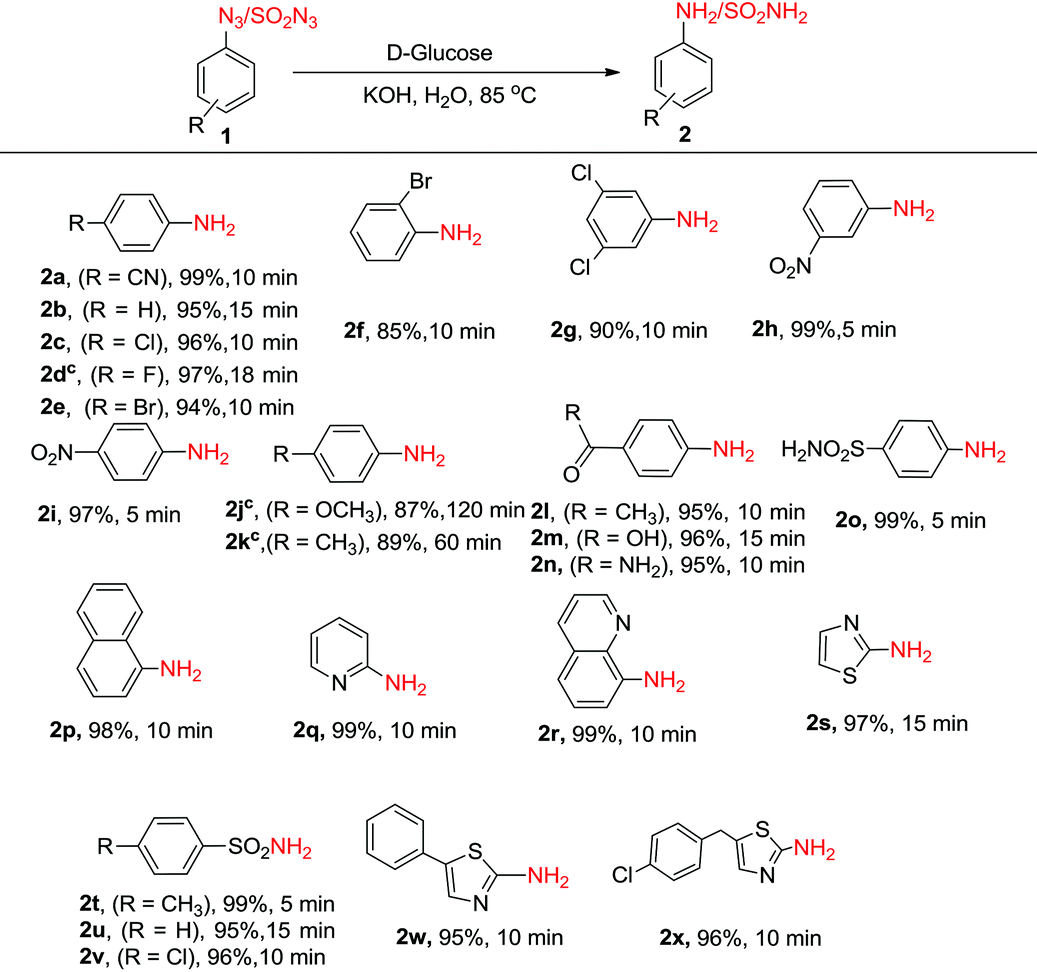
Next, the synthetic utility of the developed protocol was ascertained by carrying out the reaction on a gram scale. The reduction of 4-azidobenzonitrile (1a), 2-azidoquinoline (1r) and p-toluenesulphonyl azide (1o) starting from 10 mmol of these substrates under the optimized reaction conditions yielded the corresponding amines 2a, 2r and 2o in 95%, 93% and 97% yields, respectively.
Further, we also applied this protocol successfully to 5-(azidomethyl)-3-(3-fluoro-4-morpholinophenyl) oxazolidin-2-one (1y), which is an intermediate in the preparation of the antibiotic linezolid. The azide could be reduced using the general reaction conditions and the desired amine (2y) was isolated in 50% yield. However, K2CO3 was used instead of KOH to avoid the ring opening of oxazolidinone. Increasing the amount of glucose to 3 mmol and K2CO3 to 5 mmol enhanced the yield of 2y up to 60% (Scheme 2).
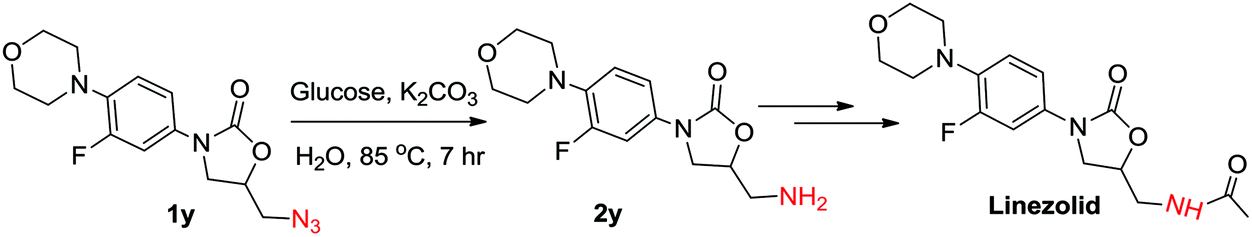 | ||
| Scheme 2 Azide reduction step in the synthesis of linezolid. | ||
To identify the source of hydrogen and understand the mechanism, the reaction of 1a was carried out in D2O instead of water (Scheme 3a). A single non-deuterated product 2a was obtained which indicated that the hydrogens of the amine were being provided by glucose which acted as a reducing agent in the reaction. Based on literature reports24–26 and the D2O experiment, we believe that the reduction of azide to amine is mediated through a hot alkaline degradation of glucose which generates hydrogen in situ along with the formation of lactate, acetate, formate and glycolate ions (Scheme 3b). To confirm this, a model reaction of 1a was carried out in D2O, and NMR analysis of the reaction mixture was performed (ESI†). The NMR data revealed that along with the formation of reduced product 2a, lactate, acetate and formate ions were also being produced in the reaction. These findings support our proposed pathway for hydrogen generation in this reaction (Scheme 3b).
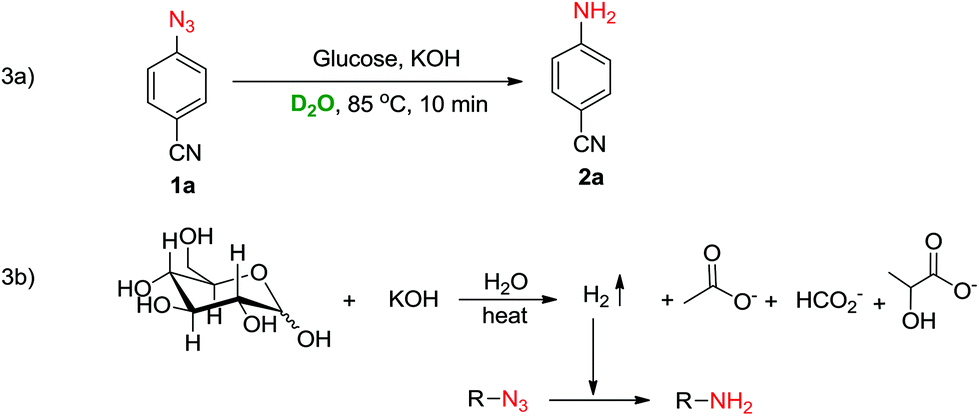 | ||
| Scheme 3 (a) Reaction in D2O; (b) proposed mechanism for reduction. | ||
Conclusions
In conclusion, a highly efficient method for the reduction of aryl(heteroaryl)azides and aryl suphonyl azides to the corresponding amines and sulphonamides employing D-glucose as the hydrogen source along with KOH in water is reported for the first time. The reagents are inexpensive, with no unpleasant odour and non-toxic. The protocol is highly chemoselective and challenging functional groups such as CN, NO2, COR, and SO2NH2 are well tolerated. The reaction is a rapid and practical way of accessing amines from aryl, heteroaryl, and suphonyl azides under metal-free conditions. The present method compares favorably to most of the earlier literature reported procedures.Acknowledgements
Dr N. C. thanks the DST for the SERB-National Post-Doctoral Fellowship. The authors thank DST-FIST for funding the ESI-HRMS facility at IIT Delhi.References
- L. Benati, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo, S. Strazzari and G. Zanardi, Org. Lett., 2002, 4, 3079–3081 CrossRef CAS PubMed.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- S. Fine, T. Nidam and V. Braude, US Pat, 7291614B2, 2007 Search PubMed.
- B. Pandey, M. G. Dave, H. M. Kothari, B. S. Shukla, S. K. Mendiratta, R. Joshi and U. Trivedi, WO2013065066A1, 2013 Search PubMed.
- P. Zanirato and S. Cerini, Org. Biomol. Chem., 2005, 3, 1508–1513 CAS.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem. – Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- O. Berger, A. Kaniti, C. T. van Ba, H. Vial, S. A. Ward, G. A. Biagini, P. G. Bray and P. M. O'Neill, ChemMedChem, 2011, 6, 2094–2108 CrossRef CAS PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- E. F. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS.
- H. Kunz and C. Unverzagt, Angew. Chem., Int. Ed. Engl., 1988, 27, 1697–1699 CrossRef.
- S. Ahammed, A. Saha and B. C. Ranu, J. Org. Chem., 2011, 76, 7235–7239 CrossRef CAS PubMed.
- S. Pagoti, S. Surana, A. Chauhan, B. Parasar and J. Dash, Catal. Sci. Technol., 2013, 3, 584–588 CAS.
- M. Bartra, P. Romea, F. Urpí and J. Vilarrasa, Tetrahedron, 1990, 46, 587–594 CrossRef CAS.
- B. C. Ranu, A. Sarkar and R. Chakraborty, J. Org. Chem., 1994, 59, 4114–4116 CrossRef CAS.
- A. M. Salunkhe, P. V. Ramachandran and H. C. Brown, Tetrahedron, 2002, 58, 10059–10064 CrossRef CAS.
- L. Benati, G. Bencivenni, R. Leardini, M. Minozzi, D. Nanni, R. Scialpi, P. Spagnolo and G. Zanardi, J. Org. Chem., 2006, 71, 5822–5825 CrossRef CAS PubMed.
- P. Molina, I. Díaz and A. Tárraga, Tetrahedron, 1995, 51, 5617–5630 CrossRef CAS.
- Y. Pei and B. O. Wickham, Tetrahedron Lett., 1993, 34, 7509–7512 CrossRef CAS.
- C. Premi and N. Jain, Eur. J. Org. Chem., 2013, 5493–5499 CrossRef CAS.
- A. Srivastava and N. Jain, Tetrahedron, 2013, 69, 5092–5097 CrossRef CAS.
- N. Chandna, N. Chandak, P. Kumar, J. K. Kapoor and P. K. Sharma, Green Chem., 2013, 15, 2294–2301 RSC.
- S. Rohilla, P. Pant and N. Jain, RSC Adv., 2015, 5, 31311–31317 RSC.
- A. Khalil, J. A. Edwards, C. A. Rappleye and W. Tjarks, Bioorg. Med. Chem., 2015, 23, 532–547 CrossRef CAS PubMed.
- A. V. Ellis and M. A. Wilson, J. Org. Chem., 2002, 67, 8469–8474 CrossRef CAS PubMed.
- L. S. Pang, A. M. Vassallo and M. A. Wilson, Fuel, 1989, 68, 253–254 CrossRef CAS.
- G. Epane, J. C. Laguerre, A. Wadouachi and D. Marek, Green Chem., 2010, 12, 502–506 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc01593c |
| This journal is © The Royal Society of Chemistry 2017 |