
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of thiols into sulfonyl halogenides under aerobic and metal-free conditions†‡
Marjan
Jereb
 * and
Luka
Hribernik
* and
Luka
Hribernik
Faculty of Chemistry and Chemical Technology, Večna pot 113, 1001 Ljubljana, Slovenia. E-mail: marjan.jereb@fkkt.uni-lj.si; Fax: +38 61 241 9144; Tel: +386 1 479 8577
First published on 19th April 2017
Abstract
An environmentally benign, metal-free synthesis of sulfonyl chlorides and bromides from thiols in the presence of ammonium nitrate, an aqueous solution of HCl and HBr and oxygen as a terminal oxidant was developed. The reactivity of various substituted thiophenols, benzylic-, aliphatic- and heteroaromatic thiols was examined. Ammonium nitrate served as a source of nitrogen oxides (NO/NO2), which are the crucial players in a redox-catalytic cycle. Sulfonyl chlorides and bromides were isolated without extraction and “filtered” over a short pad of silica gel; the use of solvents was greatly reduced in comparison with traditional isolation and purification. A “one-pot” protocol for the conversion of thiol into sulfonamide is also demonstrated. Scale-up experiments on the preparation of sulfonyl chloride and bromide are shown. A possible reaction pathway is discussed.
Introduction
Green chemistry has become an increasingly important subject in organic chemistry.1 In an era of serious climate changes, humanity will be forced to strengthen endeavors in the direction of sustainable development. Reducing the use of hazardous reagents and solvents, minimizing the production of harmful waste, and enhancing safety, cost- and atom efficiency, with renewable resources in operationally simple processes are the prime targets in this regard.2Oxidation reactions are common chemical transformations employing a vast number of oxidizing agents. Several metal- and halogen-based oxidants are toxic and hazardous; moreover, they produce considerable amounts of hazardous waste. Although often efficient, such reagents need safer and non-toxic replacements. Environmentally benign oxidants, such as an aqueous solution of hydrogen peroxide3 and air or oxygen, are “green” alternatives that produce no dangerous waste, and are welcome from an economic point of view.4 Hence, aerobic transformation is a current hot topic in chemistry.5
Organic sulfonyl halogenides are versatile intermediates in chemical, agrochemical, and medicinal chemistry. They can be converted into numerous different sulfonyl derivatives i.e. amides, hydrazides, azides, cyanides, sulfonates, sulfinates, sulfones, and others. One unique and interesting feature of SO2 moiety-containing molecules is the extrusion of sulfur dioxide, generating reactive and hard-to-prepare intermediates.6 The extrusion of sulfur dioxide from cyclic sulfones leads to ring-contracted products that may not be readily obtained by other methods.7 Sulfonyl chlorides may undergo diverse desulfitative cross-couplings;8,9 they could serve as arylating agents.10–12 Sulfones are a noteworthy type of compound in organocatalysis due to their stereoelectronic properties and the possibility of subsequent transformations;13 in addition, they are essential in the Julia-type olefination reactions.14 3-[18F]fluoropropanesulfonyl chloride was utilized for the preparation of radiolabeled sulfonamides that can be used as imaging agents for positron emission tomography.15 [35S]aryl sulfonyl chlorides could be transformed into the corresponding sulfonamides, important radioligands to study biological functions, and have certain advantages over tritiated and iodinated markers.16,17 A sulfonamide moiety is extremely important because it is present in sulfa drugs,18 a broad family of antimicrobial and antibacterial agents. Sulfonyl azides are particularly important in cycloaddition reactions,19 while sulfonyl hydrazides,20 sulfonyl chlorides,21 and sulfinates22 could serve as odorless surrogates for thiophenols as well as for sulfonylations.23
Sulfonyl chlorides could be prepared in several different ways. Oxidative chlorination of thiols was frequently applied to synthetic pathways using several combinations of oxidants and chloride sources, i.e. NCS/Bu4NCl,24 TCCA/BnMe3NCl,25–27 chloramine-T/Bu4NCl,28 DCH/BnMe3NCl,29 H2O2/ZrCl4,30 H2O2/TiCl4,31 POCl3/H2O2 in SDS micelles,32 H2O2/SOCl2,33 NaOCl/HCl,34 PCBS or TCBDA/BnMe3NCl,35 nitrate salt with TMSCl36 or sulfuryl chloride,37 and oxone/KX.38 In addition, the sole oxidants may play a dual role also as a source of chloride i.e. NaOCl·5H2O,39 NCS,40 DCDMH,41 and ClO2.42 There are several methods for the synthesis of sulfonyl chlorides from miscellaneous reactants. These include chlorination of sodium sulfonates with Ph3P·Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 or cyanuric chloride,44 chlorination of sulfonic acids with cyanuric chloride,39 1,3,5-triazo-2,4,6-triphosphorine 2,2,4,4,6,6-hexachloride (TAPC),45 and Cl3CCN/PPh3.46 It can be achieved by reaction of disulfides with NCS,47 oxidation of the biologically relevant sulfides48 with chlorine on an industrial scale,49 and transformation of S-alkylisothiourea salts with NaClO2,50 NaClO51 and NCS.52 In addition, sulfonyl chlorides can be prepared from other different sulfur functionalities,53–55 by oxidation of triisopropylsilanylsulfanyls with KNO3/SO2Cl2,56 oxidation/chlorination of thioacetates,57 reaction of the Grignard reagents with SO258 and DABSO,59 and transformation of the lithiated aromatics with SO2 and NCS,60 and others. Meerwein's batch approach61 was successfully modified by chlorosulfonation with SO2/CuCl2 of an in situ generated diazonium salt by using a continuous flow reactor.62
43 or cyanuric chloride,44 chlorination of sulfonic acids with cyanuric chloride,39 1,3,5-triazo-2,4,6-triphosphorine 2,2,4,4,6,6-hexachloride (TAPC),45 and Cl3CCN/PPh3.46 It can be achieved by reaction of disulfides with NCS,47 oxidation of the biologically relevant sulfides48 with chlorine on an industrial scale,49 and transformation of S-alkylisothiourea salts with NaClO2,50 NaClO51 and NCS.52 In addition, sulfonyl chlorides can be prepared from other different sulfur functionalities,53–55 by oxidation of triisopropylsilanylsulfanyls with KNO3/SO2Cl2,56 oxidation/chlorination of thioacetates,57 reaction of the Grignard reagents with SO258 and DABSO,59 and transformation of the lithiated aromatics with SO2 and NCS,60 and others. Meerwein's batch approach61 was successfully modified by chlorosulfonation with SO2/CuCl2 of an in situ generated diazonium salt by using a continuous flow reactor.62
Chlorosulfonation of arenes can also be performed directly by reacting with ClSO3H, SO2Cl2, or a mixture of SO2 and Cl2; however, these reagents are extremely hazardous, irritable, and highly reactive. Reagents like ClSO3H, PCl5 or SOCl2 can convert sulfonic acid into sulfonyl chlorides; however, these agents are noxious and their use may be dangerous due to the possible internal pressure.63 Phenyl chlorosulfate was found to be an excellent source of [SO2Cl]+ species in a Pd-catalyzed Suzuki–Miyaura cross-coupling reaction.64 Several other halogenating agents and oxidants, i.e. NCS, 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), cyanuric chloride, trichloroisocyanuric acid (TCCA), 1,3,5-triazo-2,4,6-triphosphorine 2,2,4,4,6,6-hexachloride (TAPC), oxone and Ph3P·Cl2 are efficient reagents; however, they are associated with a generation of a considerable amount of waste. A sustainable synthetic method for the preparation of sulfonyl halogenides embracing an environmentally benign oxidant and the natural form of the halogen is therefore highly desired. Here, we report on a metal-free synthesis of sulfonyl halogenides from thiols in the presence of ammonium nitrate, an aqueous solution of HCl or HBr and oxygen as a terminal oxidizer.
Results and discussion
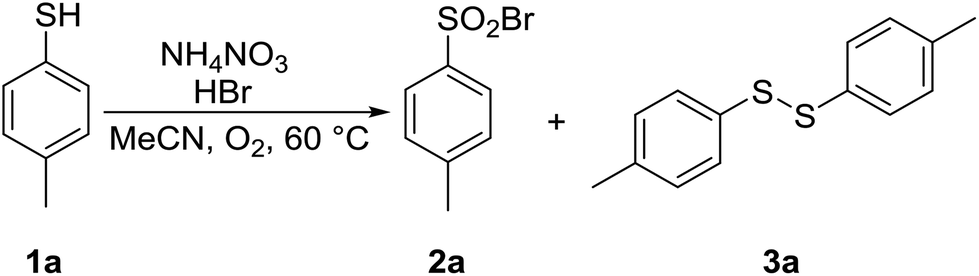
4-Methylthiophenol 1a was selected as a model substrate to optimize the reaction conditions. It was allowed to react in a selected solvent in the presence of HBr (48% aqueous solution, 1.1 equiv.) and NH4NO3 (0.2 equiv.) at 60 °C for 1.25 h. The results of the solvent screening experiments are summarized in Table 1 and briefly discussed as follows. Whereas compound 1a remained completely unreacted in water (entry 1), a 7% conversion into disulfide 3a was observed in methanol (entry 2). Aprotic and rather non-polar DCM was also found to be ineffective (entry 3). The full conversion of 1a and encouraging selectivity was noted in acetic acid, affording 33% of disulfide 3a and 67% of sulfonyl bromide 2a (entry 4). Finally, the reaction in acetonitrile took place with a quantitative conversion of 1a, yielding exclusively and in excellent isolated yield the desired sulfonyl bromide 2a (entry 5).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | 2a | 3a | Conv.b (%) |
| a Reaction conditions: 1a (124 mg, 1 mmol), solvent (5 mL), NH4NO3 (16 mg, 0.2 mmol), HBr (48% aqueous solution, 186 mg, 1.1 mmol), 1.25 h, balloon of O2. b Relative distribution of products determined by 1H NMR. c Isolated yield. | |||||
| 1 | H2O | 60 | 0 | 0 | 0 |
| 2 | MeOH | 60 | 0 | 7 | 7 |
| 3 | DCM | 40 | 0 | 6 | 6 |
| 4 | AcOH | 60 | 67 | 33 | 100 |
| 5 | MeCN | 60 | 100 | 0 | 100 (93)c |
With acetonitrile as the solvent of choice, we examined the effect of the reaction temperature. The experiments were conducted at three different temperatures under otherwise identical reaction conditions, and the results are summarized in Table 2. At room temperature and 40 °C, a modest 6% and 24% conversion of thiophenol 1a into disulfide 3a was noted (entries 1 and 2). Interestingly, raising the temperature to 60 °C resulted in a full conversion into the target sulfonyl bromide 2a, which could be isolated in 93% yield, setting 60 °C as the temperature of choice (entry 3) for further experiments.
Next, we examined the effect of concentration of thiophenol 1a in the reaction mixture, and the results are collected in Table 3. The transformation of one mmol of 1a in various amounts of MeCN was examined. Whereas the reaction in 1 mL of the solvent proved to be inefficient (entry 1), the use of a considerably larger amount of MeCN (20 mL) resulted in 100% conversion into 2a (entry 2). Some additional screening indicated that the amount of MeCN could be reduced to as low as 5 mL with no loss in the outcome (entry 3). This concentration (0.2 M) of 1a in MeCN was thus selected for further experiments.
In the next step, the effect of various atmospheres on the transformation of 1a was examined (Table 4). Reaction under the atmosphere of pure oxygen proceeded with complete selectivity towards the desired sulfonyl bromide 2a (entry 1). Air atmosphere contains approximately 20% oxygen; however, it was considerably less efficient, affording disulfide 3a as the only product (entry 2). The transformation of 1a under a nitrogen atmosphere took place with 23% conversion into disulfide 3a as the sole product (entry 3).
The role of ammonium nitrate (AN) was also examined, and the results are summarized in Table 5. To demonstrate its role, the functionalization of 1a was performed under the optimized reaction conditions as indicated above in the presence of different amounts of AN. The increasing amount of AN, ranging between 0 and 0.1 equiv. relative to 1a, increased the conversion into disulfide 3a (entries 1–3). Surprisingly, a complete turn in selectivity was observed when using 0.2 equiv. of AN, affording full conversion of thiophenol 1a into sulfonyl bromide 2a (entry 4).
In principle, one equiv. of HBr should be a sufficient amount for the complete conversion of 1a into 2a. The experimental work, however, demonstrated that a small excess of acid is beneficial and that 1.1 equiv. of 48% aqueous solution of HBr provided an optimal performance. The above screening through the reaction conditions revealed the optimal concentration and solvent (0.2 M of 1a in MeCN), temperature (60 °C), atmosphere (O2 balloon) and additives (0.2 equiv. of AN and 1.1 equiv. of 48% HBr), which were used in the subsequent experiments.
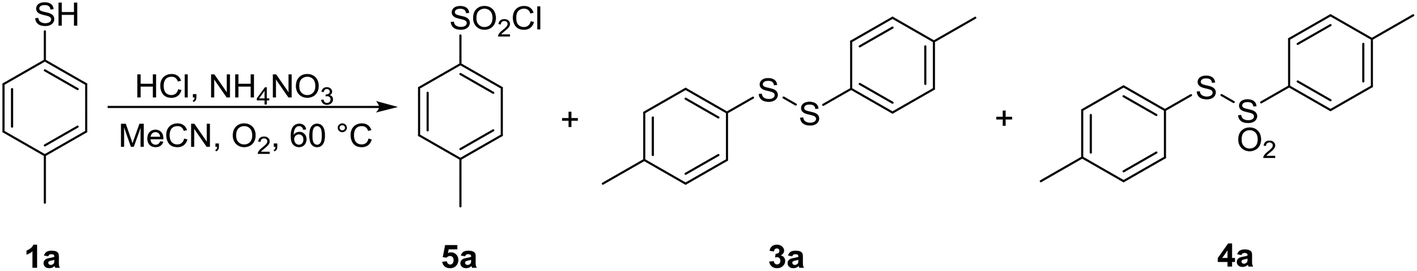
Having optimized parameters for aerobic bromination of thiophenol 1a, the experiments were directed towards aerobic chlorination with HCl. For this reaction, the amounts of AN and HCl were reoptimized, and the results are summarized in Table 6. An optimal (100%) conversion into the desired sulfonyl chloride 5a was achieved by using 1.0 equiv. of AN and 1.8 equiv. of 37% aqueous solution of HCl. Here again, in the absence of AN only trace amounts of 1a reacted to form disulfide 3a, confirming the importance of AN (entry 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | AN (equiv.) | HCl (equiv.) | 5a | 3a | 4a | Conv.b (%) |
| a Reaction conditions: 1a (124 mg, 1 mmol), MeCN (5 mL), NH4NO3 (up to 80 mg, 1.0 mmol), HCl (37% aqueous solution, up to 197 mg, 2.0 mmol), 1 h, balloon of O2. b Relative distribution of products determined by 1H NMR. c Isolated yield. | ||||||
| 1 | 0 | 1.8 | 0 | 4 | 0 | 4 |
| 2 | 0.5 | 2.0 | 64 | 10 | 26 | 100 |
| 3 | 1.0 | 1.5 | 89 | 0 | 11 | 100 |
| 4 | 1.0 | 1.8 | 100 | 0 | 0 | 100 (84)c |
Due to the higher oxidation potential, the chloride ion is expected to be less reactive compared to the bromide ion; explaining higher amounts of HCl and AN required for the transformation (compare Tables 5 and 6). However, although by employing 0.5 equiv. of AN and 2.0 equiv. of HCl full conversion of 1a could be achieved, the selectivity was disappointing with sulfonyl chloride 5a, disulfide 3a and thiosulfonate 4a in the ratio 64:10:26 (entry 2). It appeared that a larger excess of HCl could not compensate for the lower amount of AN. An additional optimization finally returned the optimal reaction conditions for the selective formation of sulfonyl chloride 5a (entries 3 and 4).
In the next step, we explored the substrate scope. Several aromatic and aliphatic thiols were subjected to the reaction conditions, optimized for the preparation of sulfonyl bromides, and the results are summarized in Table 7. Thiophenols 1a–1d bearing electron-donating groups yielded the corresponding sulfonyl bromides 2a–2d in moderate to high yield (entries 1–4). Transformation proceeded well with electron-deficient thiophenols also (entries 5–13) furnishing the related sulfonyl bromides in good to high yields. No significant substituent effect could be observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | 1 | t (h) | 2 | Yieldb (%) |
| a Reaction conditions: 1 (1 mmol), MeCN (5 mL), NH4NO3 (16 mg, 0.2 mmol), HBr (48% aqueous solution, 186 mg, 1.1 mmol), balloon of O2. b Isolated yield. | |||||
| 1 | 4-Me-C6H4- | 1a | 1.25 | 2a | 93 |
| 2 | 4-i-Pr-C6H4- | 1b | 1.5 | 2b | 89 |
| 3 | 2,4-Di-Me-C6H3- | 1c | 1.5 | 2c | 49 |
| 4 | 4-MeO-C6H4- | 1d | 1.5 | 2d | 75 |
| 5 | 3-MeO-C6H4- | 1e | 1.5 | 2e | 78 |
| 6 | C6H5- | 1f | 1.0 | 2f | 74 |
| 7 | 4-Cl-C6H4- | 1g | 1.5 | 2g | 73 |
| 8 | 2,5-Di-Cl-C6H3- | 1h | 1.25 | 2h | 97 |
| 9 | 3,4-Di-Cl-C6H3- | 1i | 2.5 | 2i | 73 |
| 10 | 2-F-C6H4- | 1j | 2.0 | 2j | 67 |
| 11 | 4-F-C6H4- | 1k | 3.0 | 2k | 73 |
| 12 | 2,4-Di-F-C6H3- | 1l | 1.5 | 2l | 64 |
| 13 | 3-CF3-C6H4- | 1m | 1.5 | 2m | 81 |
| 14 | C6F5- | 1n | 3.5 | 2n | 67 |
| 15 | 2-Naphthyl- | 1o | 1.5 | 2o | 70 |
| 16 | 1-Octyl | 1p | 4.0 | 2p | 71 |
| 17 | Cyclohexyl- | 1q | 4.0 | 2q | 84 |
Difluoro- and dichlorothiophenols (entries 8, 9 and 12) exhibited good reactivity, and we decided to test pentafluorothiophenol 1n (entry 14). The corresponding sulfonyl bromide 2n was isolated in a good yield under considerably milder conditions than known in the literature.65,66 2-Naphthalenethiol 1o yielded the related sulfonyl bromide 2o in 70% yield (entry 15). In addition, the reactivity of aliphatic thiols 1p and 1q was examined. Both thiols produced the desired sulfonyl bromides 2p and 2q in good yields (entries 16 and 17). The reaction was exemplified on a heterocyclic coumarine derivative 1r. The related sulfonyl bromide 2r was isolated in 49% yield, Scheme 1. The acidic-labile lactone functionality did not interfere in spite of the acidic reaction conditions and the elevated temperature.
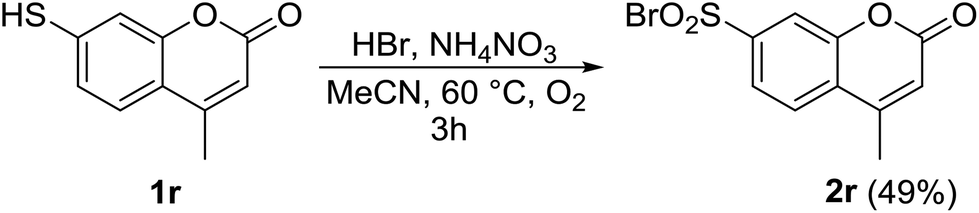 | ||
| Scheme 1 Aerobic functionalization of coumarine derivative 1r. | ||
Reaction times for aliphatic thiols are longer compared to the aromatic thiols, indicating that nucleophilicity is not the most important parameter determining the reactivity in this transformation. By taking into account the relative insensitivity of the reaction rate on the substituents in the case of aryl thiols, acidity can also be ruled out. This suggests that radical species may play a role in the reaction mechanism. It is based on the fact that the thiophenoxy radicals are more stabilized than the thioalkoxy counterparts, tentatively explaining an increased reactivity of aryl- over alkyl thiols.
The substrate scope was explored for the transformation of thiols 1 into the corresponding sulfonyl chlorides 5; the results are presented in Table 8.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | 1 | t (h) | 5 | Yieldb (%) |
| a Reaction conditions: 1 (1 mmol), MeCN (5 mL), NH4NO3 (80 mg, 1.0 mmol), HCl (37% aqueous solution, 178 mg, 1.8 mmol), balloon of O2. b Isolated yield. | |||||
| 1 | 4-Me-C6H4- | 1a | 1.0 | 5a | 84 |
| 2 | 4-i-Pr-C6H4- | 1b | 1.5 | 5b | 63 |
| 3 | 2,4-Di-Me-C6H3- | 1c | 3.0 | 5c | 89 |
| 4 | 3,5-Di-Me-C6H3- | 1cc | 2.0 | 5cc | 75 |
| 5 | 2-MeO-C6H4- | 1dd | 3.25 | 5dd | 79 |
| 6 | 3-MeO-C6H4- | 1e | 2.5 | 5e | 84 |
| 7 | C6H5- | 1f | 3.0 | 5f | 87 |
| 8 | 4-Cl-C6H4- | 1g | 2.25 | 5g | 74 |
| 9 | 2,5-Di-Cl-C6H3- | 1h | 5.0 | 5h | 55 |
| 10 | 3,4-Di-Cl-C6H3- | 1i | 3.5 | 5i | 55 |
| 11 | 2-F-C6H4- | 1j | 2.0 | 5j | 75 |
| 12 | 4-F-C6H4- | 1k | 3.25 | 5k | 77 |
| 13 | 2,4-Di-F-C6H3- | 1l | 3.5 | 5l | 73 |
| 14 | 3-CF3-C6H4- | 1m | 2.5 | 5m | 66 |
| 15 | 2-Naphthyl- | 1o | 2.0 | 5o | 73 |
| 16 | 1-Octyl | 1p | 4.5 | 5p | 67 |
| 17 | Cyclohexyl- | 1q | 3.5 | 5q | 83 |
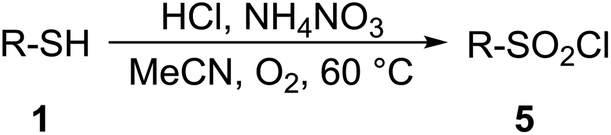
As evident from Table 8, chlorination of thiols 1 is more sluggish than bromination and there is no apparent substituent effect. This is consistent with the above-mentioned chlorination and bromination results. Both electron deficient and electron rich as well as aromatic and aliphatic substrates afforded the desired sulfonyl chlorides 5 in good yields. The results suggest that chlorination and bromination reaction pathways are similar.
Unlike aromatic and aliphatic thiols, benzylic thiols turned out to be somehow more specific substrates in the transformation with HBr/AN, exhibiting considerably higher reactivity.
For this reason, the reactions were performed under slightly modified conditions, at 40 °C with 0.4 equiv. of AN and 2.2 equiv. of HBr. Higher temperatures and lower amounts of reagents gave rise to complex reaction mixtures. Nevertheless, 4-methoxybenzyl thiol 6a was transformed into two unexpected products, 7a and 8a. Apparently, oxidation into aldehyde 8a and ipso-substitution into 7a is accompanied by an electrophilic aromatic substitution ortho to the activating methoxy group. In contrast, 4-chlorobenzyl thiol 6b yielded the corresponding disulfide 7b, and only 3-(trifluoromethyl)benzyl thiol 6c furnished the expected sulfonyl bromide 7c in a moderate 22% yield.
As discussed above, under certain conditions, oxidative halogenation of thiol 1a gave rise to disulfide 3a and thiosulfonate 4a (Tables 1–6). To shed light on the reaction mechanism, the above-mentioned presence of disulfide 3a and thiosulfonate 4a prompted us to prepare and react three disulfides 3a–g and thiosulfonate 4g. As shown in Schemes 3 and 4, upon reaction with HCl or HBr in the presence of AN both types of the substrates afforded the corresponding sulfonyl halogenides, with no other products being detected. This suggested that disulfides 3 and/or thiosulfonates 4 are potential intermediates in the reaction pathway from 1 to 2 or 5.
 | ||
| Scheme 2 Aerobic functionalization of benzylic thiols 6a–c with HBr/AN. | ||
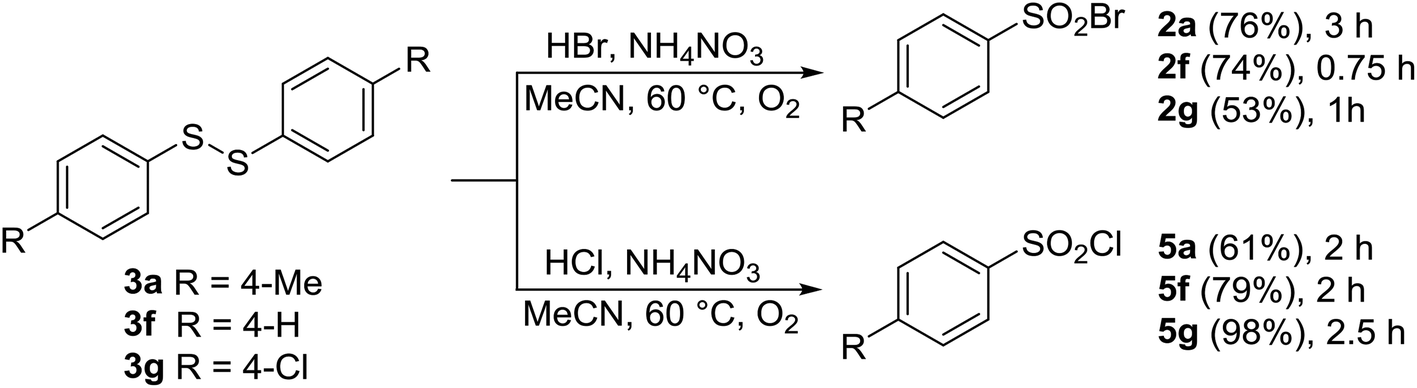 | ||
| Scheme 3 Aerobic oxidative transformation of disulfides 3a, f, g with HX/AN. | ||
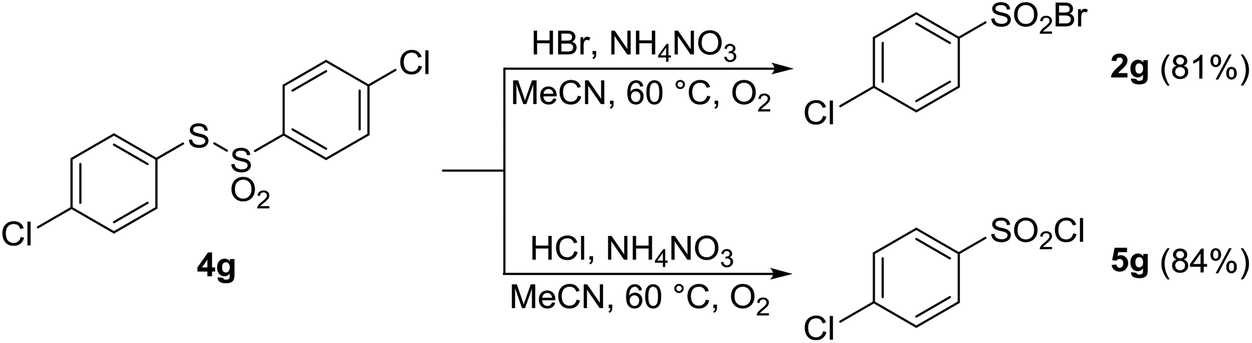 | ||
| Scheme 4 Aerobic transformation of thiosulfonate 4g with HX/AN. | ||
The transformation of 1a under the optimized reaction conditions was then carried out with HBr/AN in the presence of various inhibitors and traps, including 2,2,6,6-tetramethylpiperidine oxil (TEMPO), 2,6-di-tert-butyl-4-methylphenol (BHT) and o-dinitrobenzene (o-DNB) in equimolar amounts. The results are presented in Table 9.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Inhibitor | 2a | 3a | 4a | Conv.b (%) |
| a Reaction conditions: 1a (37 mg, 0.3 mmol), MeCN (2.5 mL), inhibitor (0.3 mmol), NH4NO3 (5 mg, 0.06 mmol), HBr (48% aqueous solution, 56 mg, 0.33 mmol), 1.25 h, balloon of O2. b Relative distribution of products determined by 1H NMR. c 64% of an unknown product was observed, and it was transformed into 4a during column chromatography on silica gel. | |||||
| 1 | — | 100 | 0 | 0 | 100 |
| 2 | TEMPO | 36 | 0 | 0 | 100c |
| 3 | BHT | 71 | 17 | 12 | 100 |
| 4 | o-DNB | 94 | 0 | 6 | 100 |
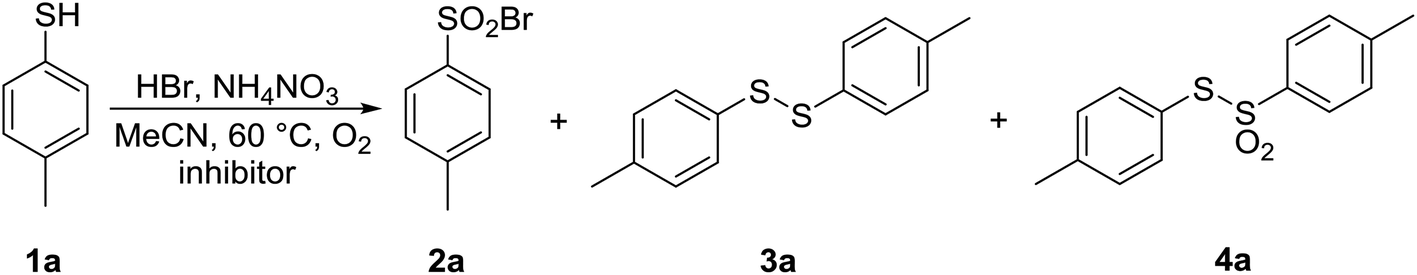
The reaction course in the presence of TEMPO (entry 2) was noticeably different from a reaction without TEMPO (entry 1). Full conversion of 1a was noted, and only 36% of the targeted product 2a was formed, signifying that radical intermediates play one of the key roles in this transformation. In addition, 64% of an unknown product was observed, which was transformed into thiosulfonate 4a during the attempt of isolation. A potential explanation is presented in Scheme 5. Although the effect of BHT was less pronounced, it significantly modified the distribution of the products (Table 9, entry 3). o-DNB as an electron scavenger made little impact, but its influence on the course of the reaction was in favor of the involvement of single-electron transfer and radical intermediates (Table 9, entry 4).
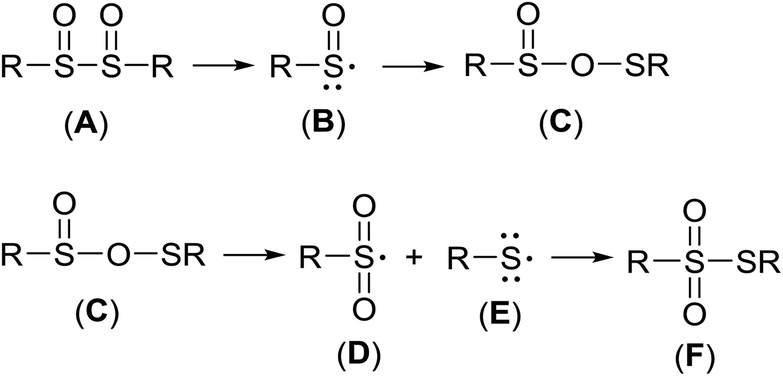 | ||
| Scheme 5 Transformation of vic-disulfoxide (A) into thiosulfonate (F). | ||
As established earlier,67 it is reasonable to expect that vic-disulfoxide (A) decomposes to a sulfinyl radical (B), and the latter recombines into OS-sulfenyl sulfinate (C). The unstable species (C) fragments into sulfenyl- (D) and sulfonyl radical (E) that recombine into thiosulfonate (F), Scheme 5.
One of the potential pathways for the formation of disulfide 3a from 1a could be via sulfenyl halogenide (RSX), a source of RS+. As compounds RSX are highly reactive and difficult to isolate, experiments were performed to test their presence in the reaction mixture. Thus, thiol 1a was treated with HBr/AN under the optimized reaction conditions in the presence of 1 equiv. of 1,1-diphenylethene (DPE), a potential scavenger of electrophilic species.
When TLC noted the complete consumption of 1a, the reaction was quenched and the crude reaction mixture was analyzed by 1H NMR spectroscopy. It revealed the full conversion of DPE, and the presence of three compounds. One of those, exhibiting a singlet resonance in the aliphatic region at approximately δ 4.5 ppm (minor component in the spectrum), was tentatively assigned to the addition product (G). The other two compounds, having singlet resonances in the olefinic region at δ 6.8 ppm were assigned to the addition–elimination products (H) (major component) and (I) (Scheme 6). Sulfonyl bromide 2a could only be detected in trace amounts. To reduce the complexity of the above crude reaction mixture, it was allowed to react with potassium carbonate in refluxing DCM for 6 h. Subsequent 1H NMR analysis showed the disappearance of the resonance belonging to (G), whereas those to (H) and (I) remained present. GC-MS analysis revealed the presence of the main product (H) with m/z 302, and two minor products, 1-bromo-2,2-diphenylethene (I) (m/z 258) and disulfide 3a (m/z 246). Detection of product (H) gives a strong indication of the presence of electrophilic sulfenyl bromide. Product (I) confirmed the formation of electrophilic bromine species. Another positive indication of the electrophilic bromonium species is the above-mentioned aromatic ring bromination of 6a into 7a and 8a (Scheme 2).
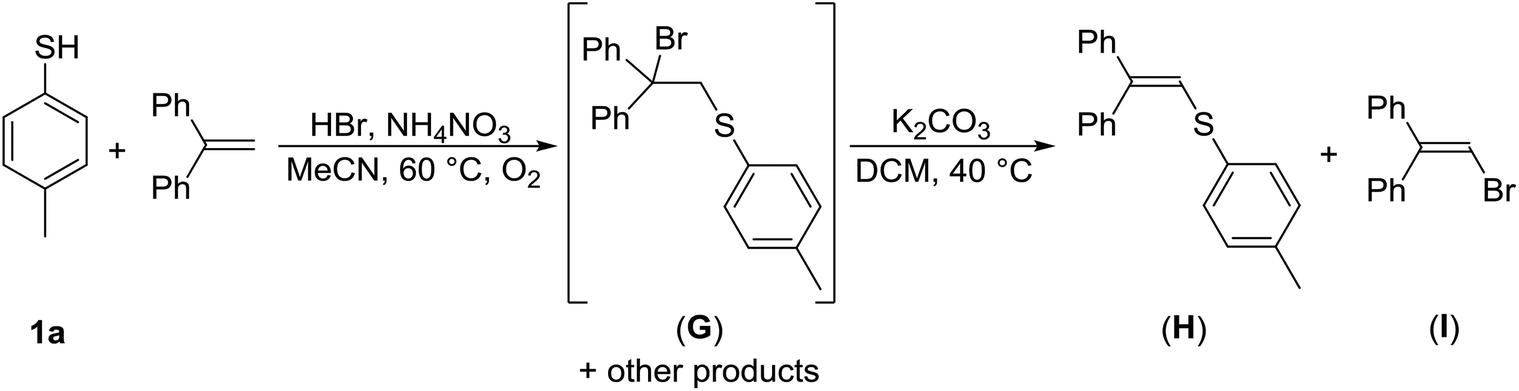 | ||
| Scheme 6 Trapping of electrophilic intermediates with DPE. | ||
In bromination of thiol 1a into sulfenyl bromide 2a, p-tolylsulfenyl bromide is likely an intermediate. To confirm this hypothesis, in independent experiments, 1a was upon treatment with NBS and NCS transformed into p-tolylsulfenyl bromide and chloride, respectively, which were then further treated with oxygen in the presence of ammonium nitrate at 60 °C. p-Tolylsulfenyl bromide was completely converted into sulfonyl bromide 2a, whereas the oxidation of p-tolylsulfenyl chloride into sulfonyl chloride 5a proceeded with lower conversion and selectivity. It could be concluded that RSX may play a role as an intermediate in this transformation.
A potential reaction cycle, proposed in Scheme 7 is similar to the one proposed by Madabhushi.38 Ammonium nitrate decomposition in the acidic medium yielded NO/NO2, which are the key players in the catalytic cycle. Indeed, during the experiments it was possible to observe several consecutive appearances and disappearances of brown gas.
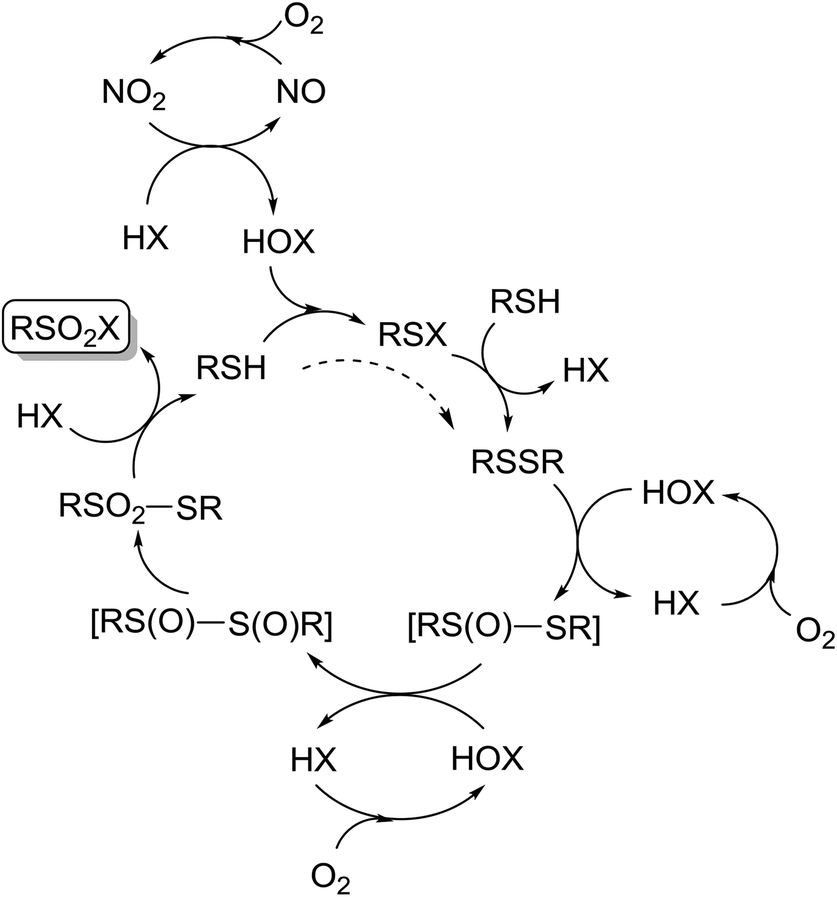 | ||
| Scheme 7 Proposed reaction pathways for the transformation of thiols into sulfonyl halogenides. | ||
NO2 served as an oxidizer for halogenide ions into the electrophilic halogen species, and the latter might be involved in oxidation processes to give the final sulfonyl halogenides. Oxidation of NO into NO2 took place with oxygen as the terminal oxidizer. Thiol probably reacted first with the electrophilic halogenic species that is formed from HX, thus giving RSX. Thiol could react with RSX, yielding disulfide and HX. A direct formation of disulfide from thiols and without RSX could not be completely ruled out;68 however, the reaction could in part take place directly and partly over RSX formation. Disulfide is further likely oxidized into thiosulfonate with the involvement of the potential intermediates as shown in Scheme 5. An experiment in favor of these intermediates may possibly be seen in Table 9, entry 2, where an unknown product was transformed into thiosulfonate 4a during column chromatography. Thiosulfonate is finally transformed into the target sulfonyl halogenide by attack of halogenide ions, while the concomitantly formed RSH enters a new cycle.
Sulfonyl halogenides are versatile synthons for the preparation of sulfonyl amides, which are very useful in pharmacy and human and veterinary medicine. One of our goals was a “one-pot” transformation of thiols into sulfonamides, Scheme 8.
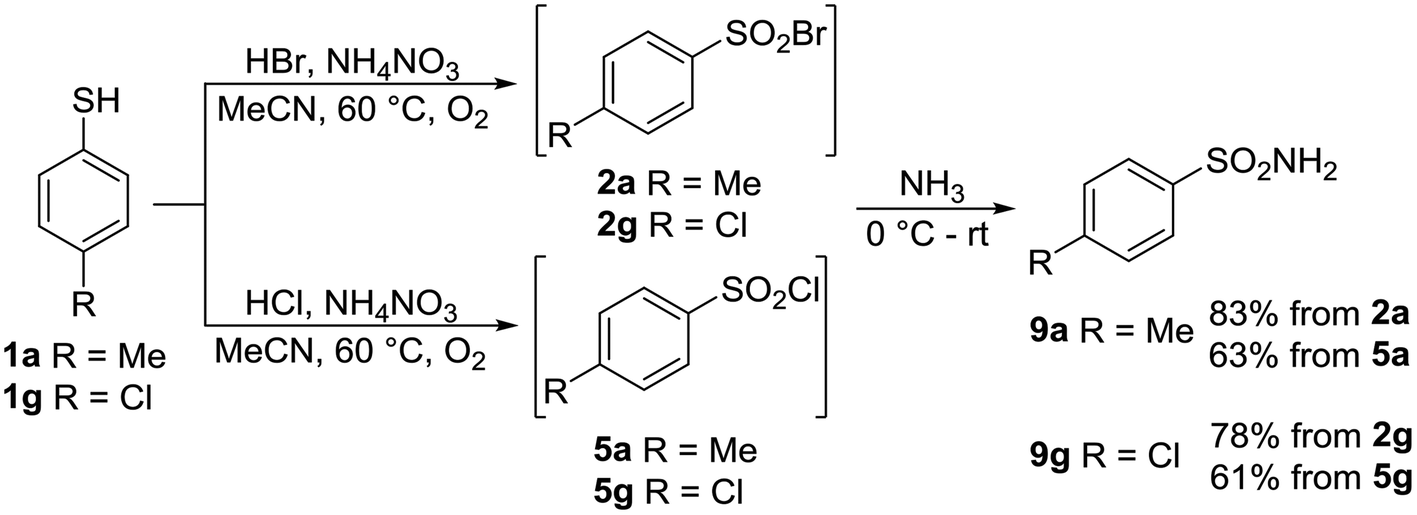 | ||
| Scheme 8 “One-pot” conversion of thiols to sulfonamides. | ||
4-Methylthiophenol 1a and 4-chlorothiophenol 1g were transformed into the corresponding sulfonyl bromides 2a and 2g and sulfonyl chlorides 5a and 5g, and were treated further without isolation with the aqueous solution of ammonia. Sulfonamides 9a and 9g were successfully isolated; however, the yields of sulfonamides from sulfonyl bromides were appreciably higher (Scheme 8).
Finally, we explored the reaction scale-up with HBr and HCl systems. Thiol 1a (20 mmol) was solubilized in 20 mL of MeCN and stirred at 60 °C in the presence of 0.2 equiv. of AN (4 mmol) and 1.1 equiv. of 48% aqueous solution of HBr. The reaction was complete in 3 h as judged by TLC. Reactions on a one mmol scale were conducted with higher amounts of MeCN (5 mL) in order to assure relatively short reaction times. In the case of scale-up, we decided to reduce the amount of solvent at the cost of a reasonably longer reaction time. We were pleased to see that the reaction was complete in 3 h in comparison with 1.25 h on a one mmol scale. Several cycles of NOx were observed with appearances and disappearances of brown gas. The balloon was replaced with a new balloon of oxygen when the cycling ceased. Two replacements of balloons with oxygen were made. The solvent was removed under reduced pressure, and 40 mL of water was added and solid filtered. The air-dried solid was “filtered” over a short pad of silica gel, and 56% of 2a was isolated.
Scale-up in the case with HCl/AN was also carried out on 20 mmol of 1a in 20 mL of MeCN with 1.8 equiv. of 36% aqueous solution of HCl and 1.5 equiv. of AN. Similarly as in the case of HBr, the lower amount of solvent was used, and the reaction was complete in 4.5 h. On a one mmol scale, the reaction was complete in 1 h. The reaction course was similar to the previous example. Two replacements of balloons with oxygen were made. The solvent was evaporated, and 20 mL of methyl-t-butyl ether (MTBE) and a few drops of water were added. The resulting mixture was cooled in an ice-bath, carefully neutralized with solid NaHCO3, dried with anhydrous sodium sulfate and filtered. The filtrate was concentrated, and “filtered” over a short pad of silica gel. Sulfonyl chloride 5a was isolated in 52% yield.
Conclusion
Environmentally benign, metal-free aerobic oxidation of thiols into sulfonyl bromides and chlorides with the aqueous solutions of HBr and HCl in the presence of AN was developed. Inorganic reactants were used in their natural form – bromide, chloride, nitrate and oxygen. Halogenated solvents were not used. The reacting system consisted of NOx gases, a crucial actor, while oxygen was the terminal oxidizer. The HBr/AN system exhibited higher reactivity than the HCl/AN system due to the more favorable oxidation potential of the bromide ion. Consequently, transformations in the case of HBr/AN required lower amounts of reactants: 0.2 equivalents of AN and 1.1 equivalents of HBr, thus contributing noticeably to the atom economy. The transformations produced low amounts of non-hazardous inorganic waste, while there was practically no organic waste in contrast to the numerous known methods for the preparation of sulfonyl halogenides. Acetonitrile was removed by distillation and could be reused. Aryl- and alkyl-substituted thiols furnished the corresponding sulfonyl halogenides as sole products, whereas benzyl thiols exhibited higher reactivity and products with different selectivity. An important contribution to green chemistry principles was made by isolation and purification. Sulfonyl halogenides were isolated without extraction and column chromatography. Owing to the excellent reaction selectivity and the purity of the crude products, “filtration” over a short pad of silica gel was done. Avoidance of both classic operations saved considerable amounts of solvents. The reaction pathways were examined and a catalytic cycle was proposed. Disulfides and thiosulfonates as potential intermediates could yield the sulfonyl halogenides under the studied reaction conditions. Electrophilic sulfur species RS+ and HOX were also the likely intermediates according to the isolated products. Radicals are likely to be important intermediates in this transformation, and the substituents have a rather low influence on the reactivity of the aryl-substituted thiols. Comparison of the reactivity of alkyl- and aryl thiols may be an indication that nucleophilicity was not the main factor governing the reactivity. Little differences in the reactivity of aryl thiols indicated that acidity was not the decisive factor determining the reactivity. It appears that stabilization of the thiophenoxy radical is a more important parameter. “One-pot” transformation of thiol into sulfonamide via sulfonyl bromide and chloride was demonstrated. Scale-up preparation of sulfonyl bromide and chloride was also carried out.Acknowledgements
We would like to express our gratitude to Prof. A. Togni, Dr J. Charpentier, E. Pietrasiak and Mass Spectrometry Laboratory at ETH Zurich, Switzerland for their kind, swift and generous response with HRMS analyses. Prof. J. Cerkovnik for GC-MS, Mrs T. Stipanovič and Prof. J. Svete for the elemental combustion analyses and the Slovenian Research Agency (P1-0134 and P1-0230) for financial support are gratefully acknowledged.Notes and references
- (a) M. Lancaster, Green Chemistry: An Introductory Text, RSC, Cambridge, 2002 Search PubMed; (b) Handbook of Green Chemistry and Technology, ed. J. Clark and D. Macquarrie, Blackwell Science, Oxford, 2002 Search PubMed; (c) Green Chemical Reactions, ed. P. Tundo and V. Esposito, Springer, Dordrecht, 2008 Search PubMed.
- (a) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed; (b) R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC; (c) Green Chemistry: Designing Chemistry for the Environment, ed. P. T. Anastas and T. C. Williamson, ACS Symposium Series 626, Washington, DC, 1996 Search PubMed; (d) Green Chemical Syntheses and Processes, ed. P. T. Anastas, L. G. Heine and T. C. Williamson, ACS Symposium Series 767, Washington, DC, 2000 Search PubMed.
- See, for example: (a) N. D. Litvinas, B. H. Brodsky and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 4513–4516 CrossRef CAS PubMed; (b) G. De Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC; (c) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef PubMed; (d) A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef PubMed; (e) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS PubMed; (f) M. Jereb, M. Zupan and S. Stavber, Chem. Commun., 2004, 2614–2615 RSC; (g) M. Jereb, Green Chem., 2012, 14, 3047–3052 RSC.
- See, for example: (a) R. A. Sheldon, I. W. C. E. Arends, G.-J. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed; (b) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC; (c) H. Miyaura and S. Kobayashi, Acc. Chem. Res., 2014, 47, 1054–1066 CrossRef PubMed; (d) A. N. Vedernikov, Acc. Chem. Res., 2012, 45, 803–813 CrossRef CAS PubMed; (e) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS PubMed; (f) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed; (g) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed; (h) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed; (i) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- See, for example: (a) Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 548–552 CrossRef CAS PubMed; (b) S. Handa, J. C. Fennewald and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 3432–3435 CrossRef CAS PubMed; (c) H. Huang, J. Huang, Y.-M. Liu, H.-Y. He, Y. Cao and K.-N. Fan, Green Chem., 2012, 14, 930–934 RSC; (d) R. Prebil, G. Stavber and S. Stavber, Eur. J. Org. Chem., 2014, 395–402 CrossRef CAS; (e) X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059–6066 CrossRef CAS PubMed.
- (a) H.-T. Yang, G.-W. Wang, Y. Xu and J.-C. Huang, Tetrahedron Lett., 2006, 47, 4129–4131 CrossRef CAS; (b) K. Wojciechowski and K. Dolatowska, Tetrahedron, 2005, 61, 8419–8422 CrossRef CAS.
- See, for example: (a) C. Wei, K.-F. Mo and T.-L. Chan, J. Org. Chem., 2003, 68, 2948–2951 CrossRef CAS PubMed; (b) M. G. H. Vicente, M. T. Cancilla, C. B. Lebrilla and K. M. Smith, Chem. Commun., 1998, 2355–2356 RSC.
- C. M. R. Volla and P. Vogel, Angew. Chem., Int. Ed., 2008, 47, 1305–1307 CrossRef PubMed.
- S. Zhang, X. Zeng, Z. Wei, D. Zhao, T. Kang, W. Zhang, M. Yan and M. Luo, Synlett, 2006, 1891–1894 CAS.
- L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC.
- Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188–9190 RSC.
- M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686–11688 RSC.
- (a) M. Nielsen, C. B. Jacobsen, N. Holub, M. W. Paixão and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 2668–2679 CrossRef CAS PubMed; (b) A.-N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018–2033 RSC.
- (a) E. Pfund, T. Lequeux and D. Gueyrard, Synthesis, 2015, 1534–1546 CAS; (b) B. Chatterjee, S. Bera and D. Mondal, Tetrahedron: Asymmetry, 2014, 25, 1–55 CrossRef CAS.
- R. Löser, S. Fischer, A. Hiller, M. Köckerling, U. Funke, A. Maisonial, P. Brust and J. Steinbach, Beilstein J. Org. Chem., 2013, 9, 1002–1011 CrossRef PubMed.
- M. A. Wallace, C. E. Raab, D. C. Dean and D. G. Melillo, J. Labelled Compd. Radiopharm., 2005, 48, 275–283 CrossRef CAS.
- C. E. Raab, J. W. Butcher, T. M. Connolly, J. Karczewski, N. X. Yu, S. J. Staskiewicz, N. Liverton, D. C. Dean and D. G. Melillo, Bioorg. Med. Chem. Lett., 2006, 16, 1692–1695 CrossRef CAS PubMed.
- See, for example: (a) M. R. Caira, Mol. Pharmaceutics, 2007, 4, 310–316 CrossRef CAS PubMed; (b) D. Mandloi, S. Joshi, P. V. Khadikar and N. Khosla, Bioorg. Med. Chem. Lett., 2005, 15, 405–411 CrossRef CAS PubMed; (c) K. Namba, X. Zheng, K. Motoshima, H. Kobayashi, A. Tai, E. Takahashi, K. Sasaki, K. Okamoto and H. Kakuta, Bioorg. Med. Chem., 2008, 16, 6131–6144 CrossRef CAS PubMed; (d) G. L. Backes, D. M. Neumann and B. S. Jursic, Bioorg. Med. Chem., 2014, 22, 4629–4636 CrossRef CAS PubMed.
- See, for example: (a) B. T. Worrell, S. P. Ellery and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 13037–13041 CrossRef CAS PubMed; (b) B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- See, for example: (a) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929–4932 CrossRef CAS PubMed; (b) T.-T. Wang, F.-L. Yang and S.-K. Tian, Adv. Synth. Catal., 2015, 357, 928–932 CrossRef CAS; (c) J. Chen, J. Mao, Y. He, D. Shi, B. Zou and G. Zhang, Tetrahedron, 2015, 71, 9496–9500 CrossRef CAS.
- See, for example: (a) Y. Chen, F. Xiao, H. Chen, S. Liu and G.-J. Deng, RSC Adv., 2014, 4, 44621–44628 RSC; (b) S. Guo, W. He, J. Xiang and Y. Yuan, Tetrahedron Lett., 2015, 56, 2159–2162 CrossRef CAS.
- See, for example: (a) F. Xiao, H. Xie, S. Liu and G.-J. Deng, Adv. Synth. Catal., 2014, 356, 364–368 CrossRef CAS; (b) Y.-M. Lin, G.-P. Lu, C. Cai and W.-B. Yi, Org. Lett., 2015, 17, 3310–3313 CrossRef CAS PubMed; (c) H. Rao, P. Wang, J. Wang, Z. Li, X. Sun and S. Cao, RSC Adv., 2014, 4, 49165–49169 RSC.
- See, for example: (a) J.-K. Qiu, W.-J. Hao, D.-C. Wang, P. Wei, J. Sun, B. Jiang and S.-J. Tu, Chem. Commun., 2014, 50, 14782–14785 RSC; (b) S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496–4499 RSC; (c) T.-T. Wang, F.-X. Wang, F.-L. Yang and S.-K. Tian, Chem. Commun., 2014, 50, 3802–3805 RSC; (d) W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Org. Lett., 2015, 17, 3552–3555 CrossRef CAS PubMed; (e) Y. Tang, Y. Zhang, K. Wang, X. Li, X. Xu and X. Du, Org. Biomol. Chem., 2015, 13, 7084–7090 RSC.
- H. Veisi, R. Ghorbani-Vaghei, S. Hemmati and J. Mahmoodi, Synlett, 2011, 2315–2320 CrossRef CAS.
- J. D. Bonk, D. T. Amos and S. J. Olson, Synth. Commun., 2007, 37, 2039–2050 CrossRef CAS.
- H. Veisi, A. Sedrpoushan, S. Hemmati and D. Kordestani, Phosphorus, Sulfur Silicon Relat. Elem., 2012, 187, 769–775 CrossRef CAS.
- S. Hemmati, M. M. Mojtahedi, M. S. Abaee, Z. Vafajoo, S. G. Saremi, M. Noroozi, A. Sedrpoushan and M. Ataee, J. Sulfur Chem., 2013, 4, 347–357 CrossRef.
- B. Maleki, S. Hemmati, R. Tayebee, S. Salemi, Y. Farokhzad, M. Baghayeri, F. M. Zonoz, E. Akbarzadeh, R. Moradi, A. Entezari, M. R. Abdi, S. S. Ashrafi, F. Taimazi and M. Hashemi, Helv. Chim. Acta, 2013, 96, 383–386 CrossRef.
- H. Veisi, Bull. Korean Chem. Soc., 2012, 33, 383–386 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and M. Soheilizad, Synlett, 2009, 2773–2776 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and D. Khaledian, Tetrahedron Lett., 2012, 53, 354–358 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and J. Abbasi, Synthesis, 2012, 316–322 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and M. Soheilizad, J. Org. Chem., 2009, 74, 9287–9291 CrossRef CAS PubMed.
- S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed.
- H. Veisi, R. Ghorbani-Vaghei and J. Mahmoodi, Bull. Korean Chem. Soc., 2011, 32, 3692–3695 CrossRef CAS.
- G. K. S. Prakash, T. Mathew, C. Panja and G. A. Olah, J. Org. Chem., 2007, 72, 5847–5850 CrossRef CAS PubMed.
- Y. J. Park, H. H. Shin and Y. H. Kim, Chem. Lett., 1992, 21, 1483–1486 CrossRef.
- S. Madabhushi, R. Jillella, V. Sriramoju and R. Singh, Green Chem., 2014, 16, 3125–3131 RSC.
- T. Okada, H. Matsumuro, T. Iwai, S. Kitagawa, K. Yamazaki, T. Akiyama, T. Asawa, Y. Sugiyama, Y. Kimura and M. Kirihara, Chem. Lett., 2015, 44, 185–187 CrossRef.
- A. R. Massah, S. Sayadi and S. Ebrahimi, RSC Adv., 2012, 2, 6606–6616 RSC.
- Y.-M. Pu, A. Christensen and Y.-Y. Ku, Tetrahedron Lett., 2010, 51, 418–421 CrossRef CAS.
- O. M. Lezina, S. A. Rubtsova and A. V. Kuchin, Russ. J. Org. Chem., 2011, 47, 1249–1251 CrossRef CAS.
- T. Kataoka, T. Iwama, T. Setta and A. Takagi, Synthesis, 1998, 423–426 CrossRef CAS.
- G. Blotny, Tetrahedron Lett., 2003, 44, 1499–1501 CrossRef CAS.
- K. Bahrami, Synlett, 2011, 2671–2674 CrossRef CAS.
- O. Chantarasriwong, D. O. Jang and W. Chavasiri, Tetrahedron Lett., 2006, 47, 7489–7492 CrossRef CAS.
- M. Kirihara, S. Naito, Y. Nishimura, Y. Ishizuka, T. Iwai, H. Takeuchi, T. Ogata, H. Hanai, Y. Kinoshita, M. Kishida, K. Yamazaki, T. Noguchi and S. Yamashoji, Tetrahedron, 2014, 70, 2464–2471 CrossRef CAS.
- C. Wang, C. Hamilton, P. Meister and C. Menning, Org. Process Res. Dev., 2007, 11, 52–55 CrossRef CAS.
- N. Barnwell, P. Cornwall, D. Horner, J. Knott and J. Liddon, Org. Process Res. Dev., 2010, 14, 278–288 CrossRef CAS.
- Z. Yang, Y. Zheng and J. Xu, Synlett, 2013, 2165–2169 CAS.
- Z. Yang, B. Zhou and J. Xu, Synthesis, 2014, 225–229 Search PubMed.
- Z. Yang and J. Xu, Synthesis, 2013, 1675–1682 CAS.
- See, for example: (a) A. Nishiguchi, K. Maeda and S. Miki, Synthesis, 2006, 4131–4134 CrossRef CAS; (b) L. Kværnø, M. Werder, H. Hauser and E. M. Carreira, Org. Lett., 2005, 7, 1145–1148 CrossRef PubMed; (c) V. Percec, T. K. Bera, B. B. De, Y. Sanai, J. Smith, M. N. Holerca, B. Barboiu, R. B. Grubbs and J. M. J. Fréchet, J. Org. Chem., 2001, 66, 2104–2117 CrossRef CAS PubMed.
- H. Sohmiya, T. Kimura, M. Fujita and T. Ando, Tetrahedron, 1998, 54, 13737–13750 CrossRef CAS.
- Y. Joyard, C. Papamicaël, P. Bohn and L. Bischoff, Org. Lett., 2013, 15, 2294–2297 CrossRef CAS PubMed.
- Y. Gareau, J. Pellicelli, S. Laliberté and D. Gauvreau, Tetrahedron Lett., 2003, 44, 7821–7824 CrossRef CAS.
- M. C. F. Monnee, M. F. Marijne, A. J. Brouwer and R. M. J. Liskamp, Tetrahedron Lett., 2000, 41, 7991–7995 CrossRef CAS.
- R. Pandya, T. Murashima, L. Tedeschi and A. G. M. Barrett, J. Org. Chem., 2003, 68, 8274–8276 CrossRef CAS PubMed.
- H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876–4878 CrossRef CAS PubMed.
- P. Vedsø, P. H. Olesen and T. Hoeg-Jensen, Synlett, 2004, 892–894 CrossRef.
- H. Meerwein, G. Dittmar, R. Göllner, K. Hafner, F. Mensch and O. Steinfort, Chem. Ber., 1957, 90, 841–852 CrossRef CAS.
- L. Malet-Sanz, J. Madrzak, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2010, 8, 5324–5332 CAS.
- C. Ferri, Reaktionen der organischen Synthese, Thieme, Stuttgart, 1978 Search PubMed.
- J. R. DeBergh, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10638–10641 CrossRef CAS PubMed.
- V. E. Platonov, R. A. Bredikhin, A. M. Maksimov and V. V. Kireenkov, J. Fluorine Chem., 2010, 131, 13–16 CrossRef CAS.
- R. A. Bredikhin, D. O. Usatenko, A. M. Maksimov and V. E. Platonov, Procedia Chem., 2015, 15, 265–271 CrossRef CAS.
- F. Freeman, Chem. Rev., 1984, 84, 117–135 CrossRef CAS.
- W. A. Pryor, D. F. Church, C. K. Govindan and G. Crank, J. Org. Chem., 1982, 47, 159–161 CrossRef.
Footnotes |
| † Dedicated to Professor Emeritus Marko Zupan on the occasion of his 70th anniversary. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc00556c |
| This journal is © The Royal Society of Chemistry 2017 |