
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissolution of pyrite and other Fe–S–As minerals using deep eutectic solvents
Andrew P.
Abbott
 *a,
Ahmed Z. M.
Al-Bassam
a,
Alex
Goddard
a,
Robert C.
Harris
a,
Gawen R. T.
Jenkin
b,
Frazer J.
Nisbet
a and
Matthias
Wieland
a
*a,
Ahmed Z. M.
Al-Bassam
a,
Alex
Goddard
a,
Robert C.
Harris
a,
Gawen R. T.
Jenkin
b,
Frazer J.
Nisbet
a and
Matthias
Wieland
a
aDepartment of Chemistry, University of Leicester, Leicester LE1 7RH, UK. E-mail: apa1@le.ac.uk
bDepartment of Geology, University of Leicester, Leicester LE1 7RH, UK
First published on 31st March 2017
Abstract
Processing sulfur containing minerals is one of the biggest sources of acute anthropogenic pollution particularly in the form of acid mine drainage. This study attempts to show an innovative method for processing sulfide-based minerals. It is shown that pyrite can be solubilised by both electrochemical oxidation and reduction in a deep eutectic solvent (DES) Ethaline, a mixture of choline chloride and ethylene glycol. A novel method is demonstrated to investigate the redox properties of minerals using a paste made from the mineral powder in a DES. The first bulk electrochemical dissolution of pyrite is shown without the formation of H2S or SO2. It is also shown that the soluble species, including elements such as arsenic, can be recovered electrochemically which could potentially decrease acid mine drainage. The electrochemical properties of other iron–sulfur and iron–arsenic minerals are also presented and compared to those of pyrite.
Introduction
Pyrite (FeS2) is the Earth's most common sulfide1 and the dominant iron sulfide in hydrothermal and sedimentary mineral deposits, whereas pyrrhotite (a group of minerals with compositions between FeS and Fe7S8) is the dominant iron sulfide in magmatic mineral deposits. Marcasite is a metastable polymorph of pyrite that forms in some low-temperature hydrothermal systems. Apart from minor sulfuric acid production, these iron sulphide minerals are not of economic interest, yet are often present in larger proportions than the ore minerals that a deposit is being mined for. This results in large volumes of iron sulfides being discarded to the waste (tailings) during mining operations. These tailings require careful management to avoid oxidation of the iron sulfides and the production of sulfuric acid that results in acid mine drainage if not treated.2Arsenopyrite (FeAsS) can form a significant constituent in some hydrothermal ore deposits, in particular those that contain gold, whereas loellingite (FeAs2) is a relatively rare mineral formed in high temperature hydrothermal or metamorphic ore deposits. Whilst these minerals can be a source of arsenic, more generally they present challenges in ore metallurgy and require additional treatment to contain the arsenic and discard it in an environmentally acceptable form.
Whilst none of these common Fe–S–As minerals are of significant economic value they can often host economic quantities of precious metal mineral inclusions – typically gold and silver minerals in pyrite, arsenopyrite and loellingite, whereas pyrrhotite is often a host to PGE minerals.3,4 Sometimes gold and PGE may be present as nanoparticles or dissolved within the crystal lattice.4–7 Recovery of the precious metals from these sulfide concentrates will typically involve pyrometallurgy and/or hydrometallurgy. Roasting these ores releases SO2 and/or arsenic which require capture and treatment, whereas hydrometallurgy involves utilizing large volumes of aqueous solutions with reactive chemical reagents to solubilize the minerals followed by extraction and recovery. Hydrometallurgy results in large volumes of low concentration aqueous wastes that require expensive treatment and if not properly managed risks impacts on the environment through discharge of metals and toxic reagents into water courses. Therefore alternative techniques for the solubilisation of Fe–S–As minerals to liberate precious metals are desirable, in particular if they avoid the environmental drawbacks of current processes.
It has been proposed that ionic liquids could be used in several aspects of metal processing. This so-called ionometallurgical approach has recently been reviewed.8 The cost and toxicity of many ionic liquids precludes them from large scale applications such as mineral processing although they have been shown to be useful for biphasic extraction from aqueous solutions.8 Aqueous solutions of ionic liquids have also been demonstrated for the extraction of copper from chalcopyrite.9 Deep eutectic solvents (DESs) are mixtures of simple quaternary ammonium salts and hydrogen bond donors such as urea or glycerol.10 They may provide an alternative to conventional metallurgy as they use environmentally benign components and avoid the production of large volume/low concentration aqueous wastes. DESs have been shown to demonstrate relatively high solubility for transition metal oxides.11,12 It has also been shown that they can be used for direct electroreduction of copper oxide to copper13 and for the electrocatalytic dissolution and recovery of metals from alloys.14 Recently we have shown that iodine in DESs can be used to selectively solubilise minerals from ore samples.15 It has been shown that DESs can be used on a large scale to recover Zn and Pb from arc furnace dust waste.16
Here we provide the first investigation of the electrochemical behaviour of the common Fe–S–As minerals in DES. There have been numerous studies which have described the electrochemical behaviour of metal sulfides in aqueous media, in addition to the effect of pH and temperature.17–22 These studies demonstrate that the major products of oxidation of pyrite in aqueous solution are sulfate and ferrous ions, whilst other products are elemental sulfur, polysulfide, hydrogen sulfide, ferric hydroxide, iron oxide and iron(III).23,24 In another study, the reduction of pyrite in aqueous solution produces FeS, S2− and hydrogen.25
In this study it is shown that DESs can be used to solubilise iron sulfide based minerals through novel redox chemistry. These can be processed using pastes to minimise the amount of lixiviant (digesting solvent) used. We demonstrate here how the electrochemistry of the Fe–S–As minerals in DES compares and contrasts with the aqueous system and how elements from ores can be efficiently solubilised and recovered.
Experimental
The DES used in this study was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of choline chloride (HOC2H4N(CH3)3Cl) and ethylene glycol (HOCH2CH2OH) (both from Aldrich, 99%) which were stirred at 70 °C until a homogeneous solution was observed. It is referred to in the text by the trade name Ethaline. The water content was less than 1 wt% (thermogravimetry) but the addition of up to 5 wt% water had a negligible effect on the voltammetric response.
:2 molar ratio of choline chloride (HOC2H4N(CH3)3Cl) and ethylene glycol (HOCH2CH2OH) (both from Aldrich, 99%) which were stirred at 70 °C until a homogeneous solution was observed. It is referred to in the text by the trade name Ethaline. The water content was less than 1 wt% (thermogravimetry) but the addition of up to 5 wt% water had a negligible effect on the voltammetric response.
All minerals used were natural samples. SEM EDX analyses showed pyrite, marcasite and arsenopyrite to be stoichiometric. The calculated error in the stoichiometry on deposits was found to be ±0.05 for Fe, ±0.01 for S, ±0.01 for As, ±0.14 for C and ±0.10 for O. The pyrrhotite appears to have a composition at the maximum vacancy possible (Fe0.86S). The loellingite sample contains ∼30 mol% substitution by safflorite (CoAs2) with minor S (∼3 mol% S/(S + As)). The loellingite sample also contains some inclusions of a mineral with the chemistry of moderite [(Co,Fe)As] but no nickel was found in this particular sample. The arsenopyrite, loellingite and pyrrhotite samples contain minor amounts of silicates that are inert in Ethaline. The α-sulfur sample was from Sigma Aldrich (99.9%).
The mineral electrodes were made by two different methods. In the first method a single crystal of the mineral was connected to a copper wire using silver conductive paint (RS). This was made into an electrode by casting the sulfide particle in a non-conductive resin. A smooth electrode surface was obtained by using an ATA SAPHIR 520 polisher with multiple grades of sand paper and then polished with various abrasives down to 0.05 μm alumina slurry. Residual alumina was removed by sonication in distilled water for 10 minutes. The second method involved grinding the mineral into a fine powder (which was sieved to ensure particle sizes of 1–5 μm) and then mixing it with a few drops of Ethaline to make a paste that was then painted onto a Pt flag electrode.19
All electrochemical experiments were carried out using an Autolab-potentiostat which is controlled by the GPES programme. A three-electrode system was used for all experiments. The working electrode was either a mineral cast in epoxy resin (approximate 1 mm2 area) or mineral paste on a 1 cm2 Pt flag electrode. The reference electrode was a silver wire immersed in 0.1 M AgCl in Ethaline contained in a glass tube which was separated from the electrochemical cell by a Vycor glass frit. A 1 cm2 Pt flag electrode was used as the counter electrode. Electrochemical quartz crystal microbalance studies (EQCM) were carried out using a GAMRY EQCM using Gamry 5.65v controlled software with a 10 MHz gold coated quartz crystal resonator.
UV-Vis spectroscopy was carried out using a Shimadzu 1601 UV-Vis spectrometer. The optical quartz cells used in this study were 10 mm wide with 2 ml capacity. A Zeta-20 3D Optical Profiler was used to measure the etch depth of a pyrite surface during the chronocoulometry experiments.
Results and discussion
Cyclic voltammetry
The electrochemical behaviour of pyrite was measured using cyclic voltammetry for an individual pyrite crystal. Fig. 1a shows the voltammogram for a single grain of pyrite as a working electrode in Ethaline as the electrolyte at a sweep rate of 50 mV s−1. Ethaline was chosen as it has a lower viscosity and a higher conductivity than most other DESs. It can be seen that a broad oxidation wave occurs with two maxima at ca. +0.20 V and +0.31 V. A broad reduction peak is also obtained with a maximum at ca. −0.53 V. The signals will clearly be made up of contributions from the FeII/III couple and the complex redox chemistry of the S22− anion.1,9,16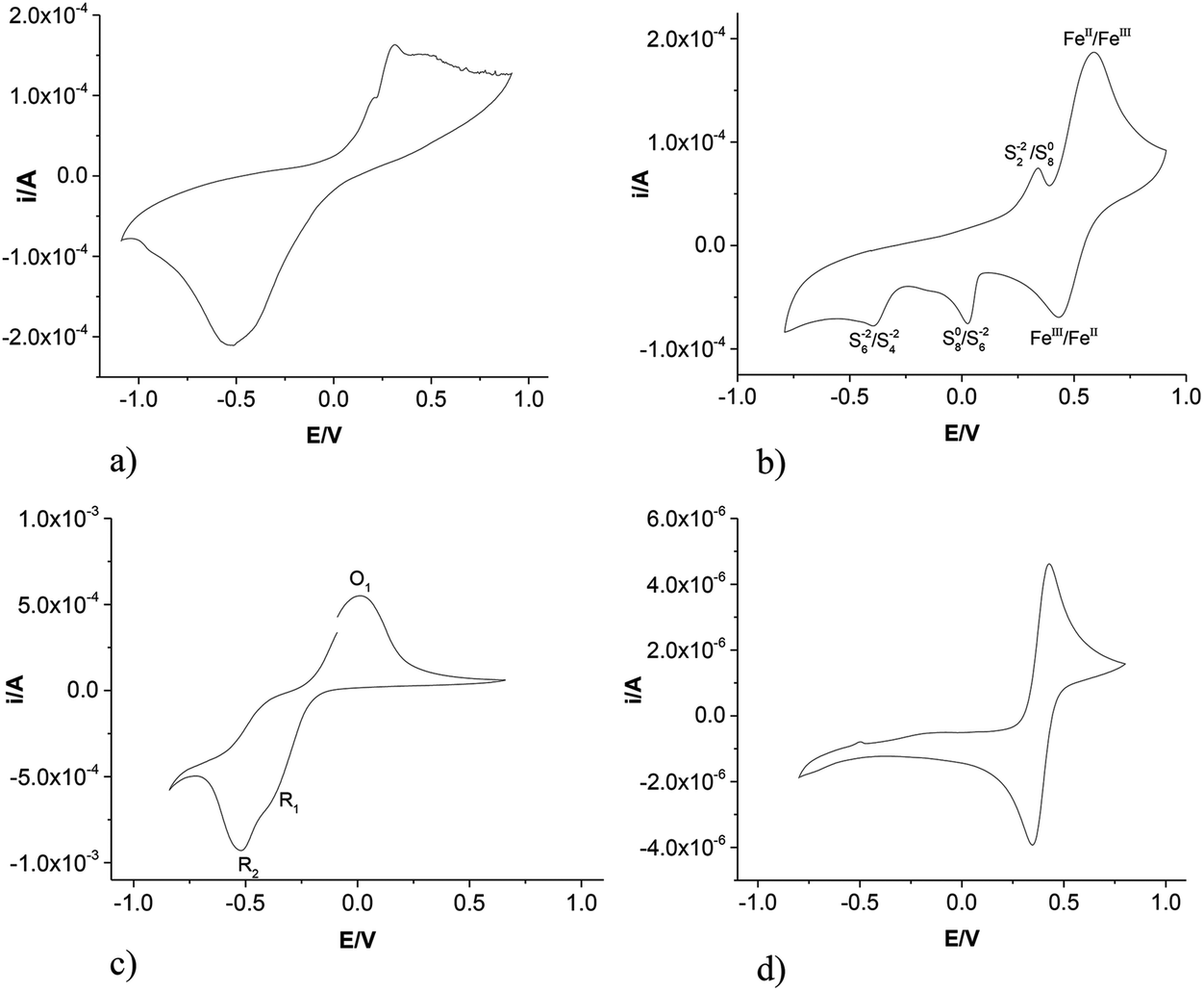 | ||
| Fig. 1 Cyclic voltammogram of (a) single pyrite crystal, (b) pyrite paste on Pt, (c) sulfur paste on Pt and (d) FeCl2 (0.1 M) on a Pt electrode, all in Ethaline at 20 °C at a scan rate of 50 mV s−1 all using a Ag/AgCl reference electrode. | ||
Mineral samples can have a variety of impurities and different crystal faces can exhibit anisotropic behaviour which can complicate the appearance of the cyclic voltammogram. The minerals themselves show relatively poor conductivity so a large crystal can induce a resistive artefact in the voltammogram.19
To circumvent the resistive issues a novel method was devised to determine the electrochemical properties of pyrite. A sample of pyrite was ground in a pestle and mortar and the fine powder was mixed with Ethaline to make a thick paste. This paste was then painted onto a Pt flag electrode. This technique has recently been demonstrated to be effective for the dissolution and electrochemical recovery of galena (PbS) and the signal was shown to be relatively independent of the mineral loading.26 The use of a paste to dissolve and recover elements from ores ensures the minimum volume of solvent (lixiviant). The solute is processed from saturated solutions ensuring small distances over which mass transport occurs.
Fig. 1b shows the cyclic voltammogram of the FeS2 coated Pt flag electrode in Ethaline. It can clearly be seen that the cyclic sweep voltammetry of ground pyrite has five separate peaks. The cyclic sweep was started from 0 V and swept in an anodic direction to 1.0 V. The potential was then swept to −1.0 V then back to 0 V at a scan rate of 50 mV s−1. Two peaks can be observed on the anodic scan. These are inferred to be due to the S22−/S80 at ca. 0.45 V and the FeII/FeIII at ca. 0.7 V (vide infra). On the cathodic scan three peaks can be observed. The first reduction peak at ca. 0.51 V is most probably the reduction of FeIII to FeII whilst the second and third reduction peaks may be related to the reduction of sulfur species.
To prove the origin of each of the peaks observed with pyrite the electrochemistry of sulfur paste, painted onto a Pt electrode, was studied in Ethaline and the results are shown in Fig. 1c. The cyclic voltammogram has one anodic peak O1 (ca. 0.0 V) and two cathodic peaks marked R1 and R2 (at about −0.39 V and −0.52 V) respectively. It should, however, be noted that there is a pronounced shoulder in the anodic sweep between −0.5 and −0.3 V which is probably the reverse process for R2. The cyclic voltammogram of sulfur has also been reported by Manan et al.27 who used a Pt electrode to study sulfur electrochemistry but using an ionic liquid, 1-butyl-3-methyl-imidazolium dicyanamide as the electrolyte. They assigned the two cathodic processes to
| R1: 3S8 + 8e− → 4S62−, | (1) |
| R2: 2S62− + 2e− → 3S42−. | (2) |
A direct comparison of the redox potentials is difficult due to the differences in the liquid junction potential and the reference electrode, however the shapes and relative differences between O1, R1 and R2 are very similar to those reported by Manan confirming the validity of the assumption.
Fig. 1d shows the cyclic voltammogram for FeCl2 dissolved in Ethaline. The classical one electron oxidation and reduction is what would be expected for a reversible redox couple. Comparing Fig. 1d with 1b it is logical to infer the peak assignments for iron based species shown in Fig. 1b. The redox processes for sulfur shown in Fig. 1c are not as well defined as those shown in Fig. 1b but this is to be expected given the poor conductivity of elemental sulfur and the difference can clearly be ascribed to a resistive artefact.
The mechanism of pyrite reduction and oxidation in 1 M HCl has been studied by several groups.14,28,29 Pyrite oxidation consists of three steps: a cathodic reaction, an electron transfer and an anodic reaction. If oxygen is the oxidant then the cathodic reaction is the reduction of oxygen, which is followed by electron transfer to the iron centre followed by a reaction of the sulfur with water and oxygen to make the sulfate anion, the overall reaction being:
| FeS2 + 3.5O2 + H2O → Fe2+ + 2H+ + 2SO42−. | (3) |
This is confirmed from the iron–sulfur Pourbaix diagram which shows sulfate as the most stable species at more positive over-potentials.30
Electrochemical quartz crystal microbalance (EQCM)
Although the above assignments seem logical it does not provide any information about the phases of oxidation and reduction products. EQCM can be used to measure small changes in mass with the charge that flows through the cell. This is monitored using changes in the oscillating frequency of the quartz crystal. Pyrite paste was painted onto the gold electrode on the quartz crystal resonator and the electrode was cycled from −0.8 V to +0.8 V. A plot of charge passed vs. mass change can be seen in Fig. 2. It can be seen that on the anodic scan there is a mass loss associated with the peak marked FeII/III in Fig. 1b. By measuring the change in mass with charge the species dissolving can be estimated. If the process is 100% current efficient then the slope should equal the molar mass divided by the number of electrons times the Faraday constant (Faraday's Law). If the soluble species is FeIII originating from a 1 electron oxidation then the slope should be 5.8 × 10−4 g C−1. The data from Fig. 2 show the slope is 6.3 × 10−4 g C−1, which is close to the theoretical value. It should however be noted that there are other alternative loss processes such as the loss of FeS2+ which would correspond to a slope of 4.6 × 10−4 g C−1 or FeS22+ which would be 6.3 × 10−4 g C−1; exactly the same as that observed. It is important therefore to get an idea of speciation from UV-Vis spectroscopy.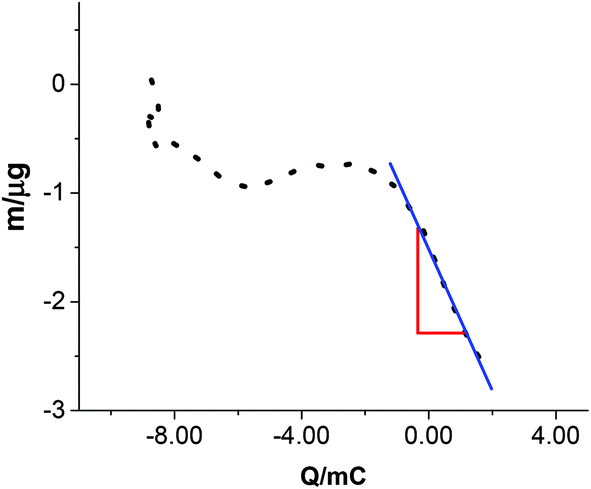 | ||
| Fig. 2 Mass versus charge plot of pyrite powder pasted on a Au coated quartz crystal as the working electrode in Ethaline at 20 °C. | ||
Bulk electrolysis
Bulk electrolysis was carried out on the pyrite paste electrode to investigate the solubility of the oxidised and reduced products in Ethaline. Fig. 1b shows that pyrite has both oxidative and reductive signals. Bulk electrolysis was carried out in a two-electrode set up, applying 2 V between the electrodes for 24 hours; the first experiment held the mineral paste at a cathodic potential and the second held it at an anodic potential. At the end of this period the Ethaline solution had turned yellow/brown where the electrode had been held at a positive potential. This suggests that the iron dissolved as Fe3+ and this ties in with the EQCM result, which shows the same. When the solution was left to stand exposed to air a brown precipitate formed after about 3 days which is likely to be an oxide or hydrated oxide of iron formed from the reaction with absorbed atmospheric moisture (Fig. 3). The solution obtained when the sample was held at a negative potential turned a grey/blue colour as shown in Fig. 3. It was thought that FeS could be formed upon reduction and while FeS is soluble in Ethaline, giving a solution of the same colour, the speciation in solution is unknown. | ||
| Fig. 3 Photograph showing a sample of Ethaline before electrolysis (centre) after both reduction of pyrite (left) and oxidation of pyrite (right) at a constant potential of 2 V. Both samples had been allowed to stand for a total of 3 days after electrolysis. | ||
UV-Vis spectroscopy
UV-Vis spectroscopy was used to investigate the speciation of the soluble species formed upon oxidative and reductive dissolutions in Ethaline. The spectrum obtained from the oxidative dissolution of pyrite in Ethaline was compared with the spectra of FeCl2 and FeCl3 standard solutions in Ethaline, and can be seen in Fig. 4a. It has previously been shown by using EXAFS that most transition metal halide salts dissolved in Ethaline exist as a tetrachlorometallate anion.31 It was found that FeCl2 dissolves to form FeCl42− however the EXAFS spectrum of FeCl3 in Ethaline shows evidence of some oxygen ligands probably originating from ethylene glycol. Fig. 4a however suggests that the solution obtained from the oxidation of pyrite does not correlate well with that for the dissolution of either FeCl3 or FeCl2 suggesting that the soluble species is not a simple tetrachloroferrate. It is likely therefore that a different ligand is attached to the iron, most probably sulfur or oxygen. The identity of the soluble species will be studied in the near future using EXAFS. | ||
| Fig. 4 Comparison of UV-Vis spectra of the FeCl3 in Ethaline and with the solution obtained from (a) the anodic dissolution and (b) the cathodic dissolution of pyrite ore in Ethaline at 20 °C. | ||
The same process was carried out for the reductive dissolution of pyrite and the results are shown in Fig. 4b. While FeS2 is insoluble in Ethaline, FeS shows limited solubility. The peak maximum of FeS occurs at about 275 nm. Solutions obtained from both the anodic and cathodic dissolution of pyrite have absorbance maxima at about this wavelength. Again EXAFS will be used to investigate the species formed from cathodic dissolution.
In situ chronocoulometry and 3D microscopy
An in situ method has been utilized to monitor the electrochemical dissolution of pyrite. A sample was made with several randomly oriented crystals of pyrite connected to a copper sheet electrode and the whole material was encased in a non-conducting resin before being polished back to expose the randomly oriented pyrite crystals. The electrically connected pyrite crystals were held at a constant potential of +1.2 V vs. Ag/Ag+ for 4 hours and the sample was then analysed using optical profilometry to measure the different effects and etch rates on different crystal orientations of pyrite during anodic dissolution in Ethaline. Fig. 5 shows an image of a sample of these crystals before and after they were electrochemically polarised. The dark area between the crystals is an electrically insulating resin. It can be seen that there is a certain degree of anisotropy with regard to the dissolution rates. Crystal B is largely undissolved whereas A, C and D are dissolved to different extents. Crystal A has dissolved to a depth of approximately 50 μm, whereas C and D have dissolved by approximately 10–20 μm. One explanation for this could be that the orientation of the exposed crystal faces caused dissolution at different rates i.e. there is a degree of anisotropy to the dissolution rate. | ||
| Fig. 5 3D colour optical microscopy images of pyrite grains (a) before and (b) after anodic polarisation at +1.2 V for 4 hours, (c) cross-sectional profile across the line. | ||
To test whether anisotropic etching occurs with pyrite an array of randomly oriented pyrite crystals (>20) were attached to a nickel plate using a conducting adhesive and then set in an insulating resin (Buehler epoxy cure 2 resin with its epoxy hardener in a 4:1 volume ratio). The sample was then polished using a diamond abrasive to ensure a flat surface. This was then analysed using XRD and the results are shown in Fig. 6. As would be expected from a randomly oriented natural mineral a variety of crystal faces are observed. The sample was put into Ethaline and held at +1.2 V vs. Ag/Ag+ for 4 hours. Following that period the sample was removed, washed with water and dried. The sample was re-analysed using XRD and the results are also shown in Fig. 6. The data were normalised to the [200] signal and it can be seen that following dissolution the relative ratios of the peaks have changed. The signals for the diagonal crystal planes e.g. [311], [321], [111] and [222] are significantly decreased. This would be expected since these faces would have the largest proportion of energetically unfavourable atoms. It is proposed that these diagonal crystal faces are preferentially etched below the analysis surface as shown in Fig. 5a such that the XRD signal is much lower for them, leaving predominantly the [200] face. It can clearly be seen from Fig. 6 that anisotropic electrochemical etching of pyrite does occur in Ethaline. This observation helps to explain the differences between the electrochemical response for pyrite shown in Fig. 1a and b as the former has only one crystal plane exposed to the electrolyte whereas the latter has a variety.
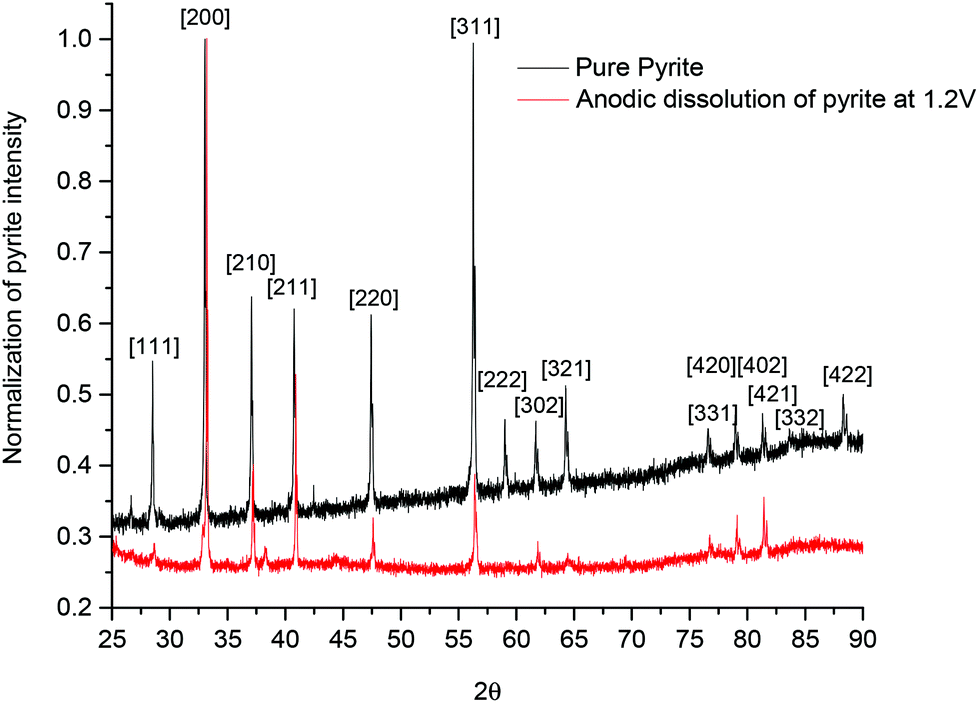 | ||
| Fig. 6 Normalised XRD spectra of a pyrite sample before and after etching in Ethaline at +1.0 V for 4 hours. | ||
Other iron–sulfur and iron–arsenic minerals
Fig. 7 shows a comparison of the voltammetric responses of five iron–sulfur and iron–arsenic minerals. In all cases the FeII/III redox couple is clearly discernible at about +0.5 V as a reversible, diffusion controlled process. The redox potentials are relatively unaffected by the nature of the mineral. Relatively little is known about the electrochemical properties of the minerals studied here. Almeida and Giannetti studied arsenopyrite by mixing it with carbon powder and making it into an electrode and electrochemically oxidising it in an aqueous acetate buffer solution.32 A relatively poorly resolved voltammogram was obtained which contained most of the redox processes shown in Fig. 7d. | ||
| Fig. 7 Cyclic voltammogram of (a) pyrite FeS2, (b) marcasite FeS2, (c) pyrrhotite (Fe1−xS), (d) arsenopyrite (FeAsS) and (e) loellingite (FeAs2) paste on Pt both in Ethaline at 20 °C at a scan rate of 10 mV s−1. | ||
The electrochemical response for the sulfur or arsenic component is clearly different for each mineral. Comparing pyrite and marcasite which are chemically the same (both FeS2) it can be seen that the relative peak heights for the iron based redox couple compared to the sulfur peaks are significantly different. For pyrite the sulfur signal is significantly smaller than that for iron whereas the reverse is the case for marcasite. Pyrite has a cubic crystal structure rather than an orthorhombic crystal structure found in marcasite.33 In contrast, pyrrhotite has a relatively weak redox signal for sulfur. This could be due to it being predominantly S2− rather than S22− which could be why the cathodic signal at −0.1 V is so weak. In arsenopyrite the anionic component is nominally (SAs)2− with the iron being principally in the FeII form.34 In addition these materials have significantly different band gaps in the pure state; pyrite is 0.95 eV whereas marcasite is 0.34 eV and arsenopyrite is 1.19 eV. It would therefore be expected that marcasite would be easier to oxidise than either of the other two minerals.
Bulk anodic electrolysis of all of the minerals shown in Fig. 7 resulted in coloured solutions as shown in Fig. 8. Although the dissolution for all of the minerals should result in the formation of FeIII it is evident that the solutions all have a different colour which would not be the case if iron was formed [FeCl4]−. In the extreme, loellingite forms a green solution on dissolution, which is characteristic of Fe–As complexes such as those formed in the so-called green rust.35 It seems logical therefore that upon dissolution the iron and chalcogenide remain bonded to each other in the soluble complex. The colour could also arise from other metal impurities e.g. Co. There is some evidence of cobalt being incorporated in the deposit obtained on the cathode (<3 wt%) but this is too small to colour the solution. We characterised Co in Ethaline solutions but the characteristic peak was absent from the UV-Vis analysis of the loellingite solution.
 | ||
| Fig. 8 Solutions obtained from the bulk anodic electrolysis of five minerals in Ethaline following electrolysis at a constant current density of 5 mA cm−2 (except for marcasite which was 2.5 mA cm−2) for 16 h. | ||
Bulk electrolysis was carried out in the cell as reported previously.19 The anodic material was iridium coated titanium which has been found to be anodically stable in the DES and the cathodic material was a nickel sheet. After electrolysis of the mineral paste on the anodically polarised electrode for 16 h it was found that for all minerals a black deposit was obtained on the nickel cathode and the morphology (SEM) and composition (EDX) of the deposits are shown in Fig. 9.
 | ||
| Fig. 9 SEM images and elemental compositions (EDX) of cathode deposits obtained after the electrolysis of (a) pyrite (FeS2), (c) pyrrhotite (Fe1−xS), (d) arsenopyrite (FeAsS) and (e) loellingite (FeAs2) following electrolysis at a current density of 5 mA cm−2. Whilst the deposition of (b) marcasite (FeS2) was obtained at a current density of 2.5 mA cm−2 in Ethaline at 50 °C for 20 h. | ||
All of the deposits contained iron but with different amounts of sulfur, arsenic, oxygen and carbon. None of the samples are magnetic showing that it is not elemental iron which is deposited. The electropolishing of stainless steel has previously been studied in the same DES and it was found that when the iron was oxidised through anodic polarisation a brown solid was formed in solution. This was found to be an iron glycolate species and it is proposed that the deposits shown in Fig. 9 are likely to contain glycolates as ligands.36 If the carbon and oxygen only came from the glycolate ligand, it would be expected that the C:O ratio would be 1. In all cases the C:O ratio is <1 meaning that some iron oxide must also be present.
The deposit obtained from loellingite contains a high As:Fe ratio whereas the S:Fe ratio for the deposit obtained from pyrite is much lower. This shows that when the mineral dissolves under anodic polarisation the metal enters the solution with the sulfur or arsenic still attached but some exchange clearly occurs in solution. This confirms the idea that the EQCM result shown in Fig. 2 corresponds to the pyrite possibly forming [FeS2]2+ when it dissolves in the DES. The electrolysis of arsenopyrite results in only As and no S in the deposit showing that arsenic is a stronger ligand than sulfur and becomes incorporated into the deposit more easily.
For this methodology to be used on a practical scale the DES would have to be recovered and recycled. This can be done by adding an equal volume of water to DES. This reduces the viscosity and allows the paste to be filtered. The addition of water precipitates any remaining iron in solution leaving a colourless aqueous solution of DES. The water can be removed by evaporation. This has been demonstrated for numerous electrochemical processes on a pilot plant scale (>500 kg) and is described more fully in the literature.37
Conclusions
In the present study, the influence of Ethaline on the electrochemical behaviour of pyrite (FeS2) was examined at ambient temperature with the aim of developing a non-acidic dissolution protocol. It is shown that mineral electrochemistry can be studied by painting a paste of the mineral in a DES onto an electrode. The cyclic voltammogram of natural pyrite in Ethaline was observed to have two anodic peaks and three cathodic peaks. The two anodic peaks could be probably attributed to the formation of elemental sulfur and Fe(III), whereas the three cathodic peaks are related to the reduction of Fe(III) to Fe(II) with the remaining peaks attributed to the reduction of sulfur. A method for achieving bulk mineral dissolution and elemental recovery using electrolysis has been demonstrated. Anodic polarisation of pyrite led to bulk dissolution although this was highly anisotropic, with some sections showing dissolution rates of 12 μm h−1.The electrochemical properties of 5 iron–sulfur and iron–arsenic minerals were studied and shown to exhibit similar responses for the iron component but the sulfur and arsenic components differed with mineral stoichiometry. Anodic bulk dissolution of all of these minerals led to their solubilisation. Most understandably yielded brown solutions but loellingite produced a green solution showing that the arsenic remained as a ligand on the iron after dissolution. Bulk electrolysis also showed that iron based deposits could be obtained on the cathode surface. The use of pastes for mineral processing decreases the lixiviant volume and ensures minimal volume of DES usage.
Acknowledgements
This work was supported by the NERC Minerals Security of Supply (SoS) grant NE/M010848/1 Tellurium and Selenium Cycling and Supply (TeaSe). AZMA-B would like to thank the Ministry of Higher Education in Iraq for funding a studentship.References
- D. J. Vaughan and J. R. Craig, Sulfide ore mineral stabilities, morphologies and intergrowth textures, in Geochemistry of hydrothermal ore deposits, ed. H. L. Barnes, John Wiley & Sons, New York, 3rd edn, 1997, ch. 8, pp. 367–434 Search PubMed.
- R. Murphy and D. R. Strongin, Surf. Sci. Rep., 2009, 64, 1–45 CrossRef CAS.
- S. W. Knipe, R. P. Foster and C. J. Stanley, Trans. Inst. Min. Metall., Sect. B, 1992, 101, B83–B88 CAS.
- D. A. Holwell and I. McDonald, Platinum Met. Rev., 2010, 54, 26–36 CrossRef CAS.
- N. J. Cook and S. L. Chryssoulis, Can. Mineral., 1990, 28, 1–16 CAS.
- C. S. Palenik, S. Utsunomiya, M. Reich, S. E. Kesler, L. Wang and R. C. Ewing, Am. Mineral., 2004, 89, 1359–1366 CrossRef CAS.
- G. Pal'yanova, Y. Mikhlin, K. Kokh, N. Karmanov and Y. Seryotkin, J. Alloys Compd., 2015, 649, 67–75 CrossRef.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- D. Godfrey, J. H. Bannock, O. Kuzmina, T. Welton and T. Albrecht, Green Chem., 2016, 18, 1930–1937 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. Rasheed and P. Shikotra, Inorg. Chem., 2005, 44, 6497 CrossRef CAS PubMed.
- Q. B. Zhang and Y. X. Hua, Phys. Chem. Chem. Phys., 2014, 16, 27088–27095 RSC.
- A. P. Abbott, R. C. Harris, F. Holyoak, G. Frisch, J. Hartley and G. R. T. Jenkin, Green Chem., 2015, 17, 2172–2179 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- A. P. Abbott, J. Collins, I. Dalrymple, R. C. Harris, R. Mistry, F. Qiu, J. Scheirer and W. R. Wise, Aust. J. Chem., 2009, 62, 341–347 CrossRef CAS.
- C. Almeida and B. Giannetti, Phys. Chem. Chem. Phys., 2003, 5, 604–610 RSC.
- A. Elsherief, Miner. Eng., 2002, 15, 215–223 CrossRef CAS.
- M. Nicol, R. Paul and J. Diggle, Electrochim. Acta, 1978, 23, 635–639 CrossRef CAS.
- T. Biegler, D. Rand and R. Woods, in Trends in Electrochemistry, Springer, 1977, pp. 291–302 Search PubMed.
- C. L. M. Almeida and B. F. Giannetti, J. Electroanal. Chem., 2003, 553, 27–34 CrossRef CAS.
- I. Cisneros-González, M. T. Oropeza-Guzmán and I. González, Electrochim. Acta, 2000, 45, 2729–2741 CrossRef.
- P. R. Holmes and F. K. Crundwell, Geochim. Cosmochim. Acta, 2000, 64, 263–274 CrossRef CAS.
- A. Chandra and A. R. Gerson, Surf. Sci. Rep., 2010, 65, 293–315 CrossRef CAS.
- W. Zhao, H. Zhu, Z.-M. Zong, J.-H. Xia and X.-Y. Wei, Fuel, 2005, 84, 235–238 CrossRef CAS.
- A. P. Abbott, F. Bevan, M. Baeuerle, R. C. Harris and G. R. T. Jenkin, Electrochem. Commun., 2017, 76, 20–23 CrossRef CAS.
- N. S. Manan, L. Aldous, Y. Alias, P. Murray, L. J. Yellowlees, M. C. Lagunas and C. Hardacre, J. Phys. Chem. B, 2011, 115, 13873–13879 CrossRef CAS PubMed.
- J. D. Rimstidt and D. J. Vaughan, Geochim. Cosmochim. Acta, 2003, 67, 873–880 CrossRef CAS.
- M. Eghbalnia and D. G. Dixon, J. Solid State Electrochem., 2013, 17, 235–267 CrossRef CAS.
- M. Bouroushian, Electrochemistry of Metal Chalcogenides, Monographs in Electrochemistry, Springer-Verlag, Berlin, Heidelberg, 2010 Search PubMed.
- J. M. Hartley, C.-M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- C. M. V. B. Almeida and B. F. Giannetti, Phys. Chem. Chem. Phys., 2003, 5, 604–610 RSC.
- I. Dodony, M. Posfai and P. R. Buseck, Am. Mineral., 1996, 81, 119–125 CAS.
- R. A. Jones and H. Wayne Nesbitt, Am. Mineral., 2002, 87, 1692–1698 CrossRef CAS.
- S. Thoral, J. Rose, J. M. Garnier, A. van Geen, P. Refait, A. Traverse, E. Fonda, D. Nahon and J. Y. Bottero, Environ. Sci. Technol., 2005, 39, 9478–9485 CrossRef CAS PubMed.
- A. P. Abbott, K. J. McKenzie and K. S. Ryder, Metal dissolution processes in ionic Liquids, in Electrodeposition of Metals from Ionic Liquids, ed. F. Endres, A. P. Abbott and D. MacFarlane, Wiley VCH, 2008 Search PubMed.
- A. P. Abbott, Process Scale-up, in Electrodeposition of Metals from Ionic Liquids, ed. F. Endres, A. P. Abbott and D. MacFarlane, Wiley VCH, 2nd edn, 2017 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |