
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The greening of peptide synthesis†
Stefan B.
Lawrenson
 ,
Roy
Arav
and
Michael
North
*
,
Roy
Arav
and
Michael
North
*
Green Chemistry Centre of Excellence, Department of Chemistry, University of York, York, Y10 5DD, UK. E-mail: Michael.north@york.ac.uk
First published on 2nd March 2017
Abstract
The synthesis of peptides by amide bond formation between suitably protected amino acids is a fundamental part of the drug discovery process. However, the required coupling and deprotection reactions are routinely carried out in dichloromethane and DMF, both of which have serious toxicity concerns and generate waste solvent which constitutes the vast majority of the waste generated during peptide synthesis. In this work, propylene carbonate has been shown to be a green polar aprotic solvent which can be used to replace dichloromethane and DMF in both solution- and solid-phase peptide synthesis. Solution-phase chemistry was carried out with Boc/benzyl protecting groups to the tetrapeptide stage, no epimerisation occurred during these syntheses and chemical yields for both coupling and deprotection reactions in propylene carbonate were at least comparable to those obtained in conventional solvents. Solid-phase peptide synthesis was carried out using Fmoc protected amino acids on a ChemMatrix resin and was used to prepare the biologically relevant nonapeptide bradykinin with comparable purity to a sample prepared in DMF.
Introduction
Peptides are central compounds in the pharmaceutical industry, providing both final medications and lead compounds for the preparation of peptidomimetic or non-peptidic pharmaceuticals.1 Therapeutic peptides are recognised as being extremely specific in their binding to in vivo targets, resulting in them often being highly potent and very selective, whilst having very few negative side effects.2,3 They are therefore expected to play a major role in the future of drug discovery.4 Currently there are more than 60 peptide based medicines approved for use by the US Food & Drug Administration, around 140 peptide drugs currently in clinical trials and over 500 in preclinical trials.2,5 Consequently, the peptide medicines market is currently worth an estimated US$ 14.1 billion, and is predicted to grow to around US$ 25.4 billion by 2018.2 As such, there is a resurgence of interest in the synthesis of synthetic peptides.The synthesis of peptides by amide bond formation between partially protected amino acid derivatives is however, one of the most wasteful and least green chemical processes.6 Numerous auxiliary reagents in the form of protecting groups and coupling agents are required,7 giving the process low atom efficiency. Additionally, peptide synthesis is almost universally carried out in toxic solvents such as DMF or DMF/dichloromethane mixtures.8 These issues are amplified in solid-phase peptide synthesis7,9 where large excesses of reagents are used and resins are washed multiple times with toxic solvents. The use of polar aprotic solvents such as DMF or NMP is a particular concern as these solvents are known to be reprotoxic,10 cause environmental problems when present in waste11 and are two of the six chemicals of highest concern under EU REACh (Registration, Evaluation and Authorisation of Chemicals) regulations.12 As a result, green solvent selection guides have repeatedly highlighted the need to replace DMF with a greener alternative in all chemical processes.13,14
As for most chemical transformations,10 by far the largest source of waste in either solution- or solid-phase peptide synthesis is the solvent, but only a few attempts have previously been made to avoid the use of DMF. Acetonitrile15,16 and THF15 have been used as replacements for DMF in peptide bond formations and acetonitrile has also been used as a solvent for Fmoc deprotections in solid-phase peptide synthesis,14 but neither of these are considered to be green solvents.12,13 Recently, the use of 2-MeTHF as a solvent for coupling reactions during solid-phase peptide synthesis was reported though the Fmoc deprotection steps were still performed in DMF.17 2-MeTHF is a sustainable but not very green solvent due to concerns about its effect on health, safety and the environment.12,13 Whilst this paper was being written, the use of a mixture of 2-MeTHF and ethyl acetate for both coupling and Fmoc deprotection steps was reported.18 However, in some cases elevated temperatures were required and the production of mixed solvent waste is undesirable as it complicates solvent recycling especially when the solvents have virtually identical boiling points (77 and 78 °C for ethyl acetate and 2-MeTHF respectively). There are also a few reports of use of water as a solvent for solid-phase peptide bond formations using coupling agents19 or enzymes20 and of solvent free non-resin supported peptide synthesis by ball-milling.21 However, the development of a green solvent for both solution- and solid-phase peptide synthesis remains an unsolved challenge.
Cyclic carbonates, especially propylene carbonate (PC) 1 and ethylene carbonate (EC) 2 are remarkably polar aprotic compounds with dipole moments and dielectric constants higher than those of conventional polar aprotic solvents22 (Table 1). Unlike conventional polar aprotic compounds, they contain only carbon, hydrogen and oxygen and so generate no NOx or SOx on incineration and PC is known to be non-toxic and is used in topically applied medical products23 and cosmetics.24 PC is also less expensive than DMF25 and unlike DMF is not significantly hygroscopic.26 Cyclic carbonates, including PC and EC, can be prepared by the 100% atom economical reaction between epoxides and carbon dioxide (Scheme 1),27 a reaction which is currently attracting considerable attention due to its ability to utilize waste carbon dioxide in chemical synthesis.28 PC, EC and other cyclic carbonates are already produced commercially29 and have a number of applications, including as electrolytes in lithium ion batteries.30 PC was ranked as the greenest solvent in the latest green solvents guide13 and has previously been used as a solvent for cycloaddition reactions31 and for metal catalysed20,32 and organocatalysed33 transformations. Therefore we decided to investigate the use of PC and EC as solvents for both coupling and deprotection steps in both solution- and solid-phase peptide synthesis using a range of common protecting groups and coupling agents.
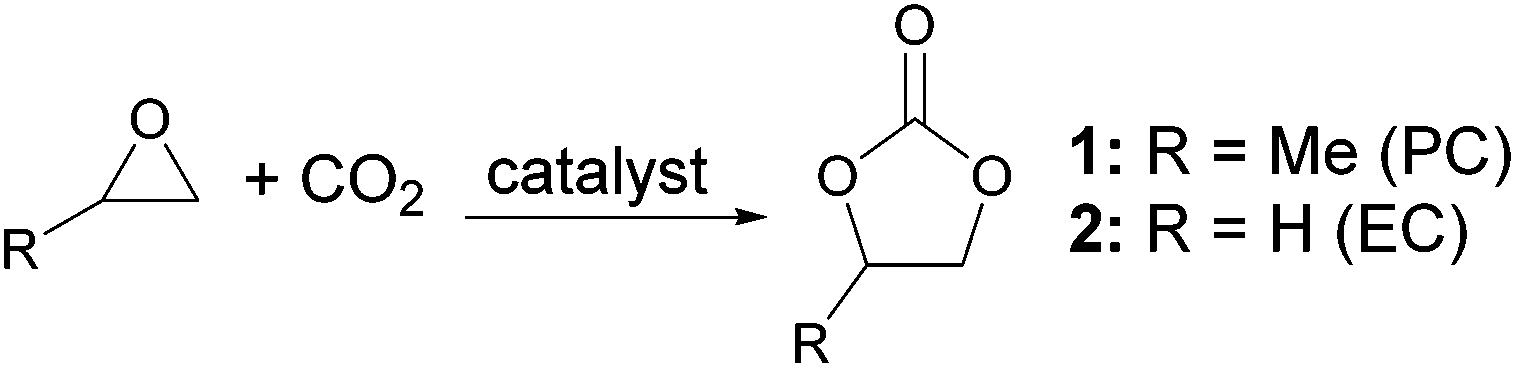 | ||
| Scheme 1 Synthesis of cyclic carbonates. | ||
| Solvent | Bp (Mp) (°C) | Dipole moment (D) | Dielectric constant |
|---|---|---|---|
| DMF | 153 | 3.8 | 38 |
| NMP | 204 | 4.1 | 32 |
| MeCN | 82 | 3.4 | 38 |
| DMSO | 189 | 3.9 | 47 |
| PC 1 | 242 | 4.9 | 65 |
| EC 2 | 248 (36) | 4.9 | 90 |
Results and discussion
To investigate the feasibility of peptide synthesis in cyclic carbonate solvents, the known34 coupling reaction between Boc-alanine 3a and phenylalanine benzyl ester 4a was studied (Scheme 2) and the results are presented in Table 2. Entry 1 gives the literature conditions29 using a conventional dichloromethane and DMF solvent system. Entry 2 shows that changing the coupling agent, additive and base to give a less costly and homogeneous reaction whilst still using the conventional solvent system results in a 10% reduction in the yield of dipeptide 5a. However, as shown in entry 3, simply changing the solvent to PC virtually restored the yield of dipeptide 5a to the literature value and use of EC as solvent (entry 4) gave a significantly higher yield of 5a than that obtained in the conventional solvent system. However, whilst other reactions have been successfully carried out in EC at ambient temperature,28 this peptide coupling had to be carried out at 40 °C to keep the reaction mixture liquid. Therefore, the coupling reaction in PC was further optimised (entry 5) and this allowed the literature yield of dipeptide 5a to be exceeded. To allow a direct comparison between PC and conventional solvent systems to be made, the use of EDC, HOBt and diisopropylethylamine in a dichloromethane and DMF mixed solvent and in just DMF was also carried out (entries 6 and 7). In both cases the yield of peptide 5a was inferior to that obtained in PC.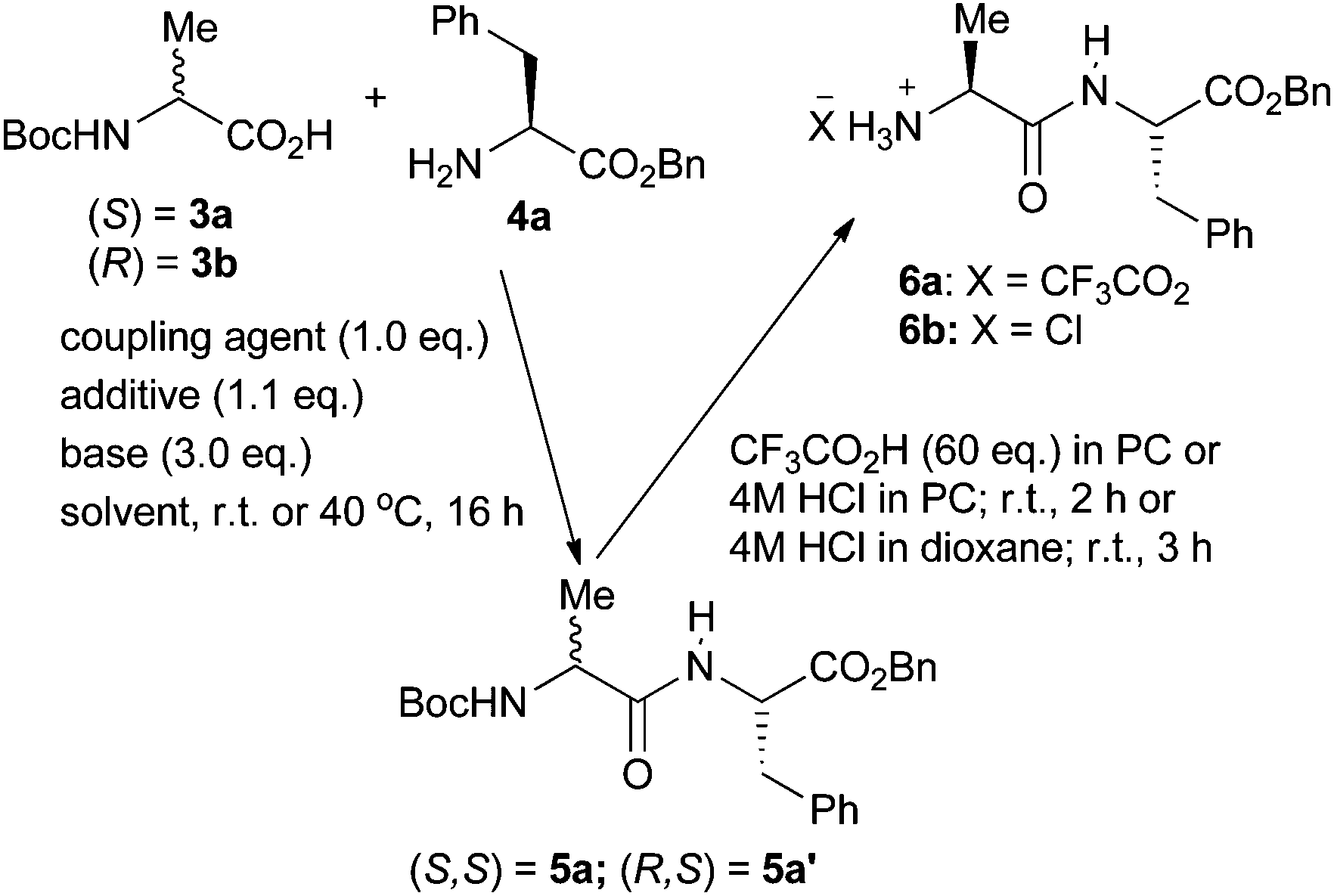 | ||
| Scheme 2 Synthesis of dipeptides 5-6a,b in PC. | ||
| Entry | Coupling agent/additive | T (°C) | Base | Solvent | Yield (%) |
|---|---|---|---|---|---|
| a EDC = N-ethyl-N-(3-dimethylaminopropyl)carbodiimide; DCC = N,N-dicyclohexylcarbodiimide; HOAt = 1-hydroxy-7-azabenzotriazole; HOBt = hydroxybenzotriazole; PFP = pentafluorophenol. | |||||
| 1 | EDC/HOAt | 20 | NaHCO3 | CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF :DMF |
86 (ref. 29) |
| 2 | DCC/PFP | 20 | Et3N | CH2Cl2:DMF |
76 |
| 3 | DCC/PFP | 20 | Et3N | PC | 83 |
| 4 | DCC/PFP | 40 | Et3N | EC | 96 |
| 5 | EDC/HOBt | 20 | iPr2EtN | PC | 93 |
| 6 | EDC/HOBt | 20 | iPr2EtN | CH2Cl2:DMF |
81 |
| 7 | EDC/HOBt | 20 | iPr2EtN | DMF | 83 |
| 8 | EDC/HOBt | 70 | iPr2EtN | DMF | 78 |
| 9 | EDC/HOBt | 70 | iPr2EtN | PC | 82 |
It is becoming increasingly common to carry out peptide synthesis at elevated temperatures, often with microwave heating.35 Therefore, the synthesis of peptide 5a in both DMF and PC was also carried out at 70 °C in a microwave reactor (entries 8 and 9). In both cases high yields of peptide 5a were again obtained. No evidence for reaction of amine 4a with the propylene carbonate was seen in these reactions even though there is literature precedent for the reaction of amines with cyclic carbonates.36 A control experiment was therefore carried out in which amine 4a was dissolved in PC and heated to 70 °C in a microwave reactor in the absence of any other reagents. Even under these conditions, only 1% reaction between the amine and solvent was detected. Thus, reaction of amino acid derivatives with PC does not occur to any significant extent under the reaction conditions used for peptide synthesis, even at 70 °C and cannot compete with peptide synthesis.
Activated amino acid derivatives generated during peptide synthesis are prone to racemisation.7 To demonstrate that no such racemisation occurs when the coupling reactions are carried out in PC, diastereomeric dipeptide 5a′ was prepared from Boc-(R)-alanine 3b and ester 4a in 87% yield using the conditions of Table 1, entry 5. HPLC analysis of dipeptides 5a and 5a′ as well as a 1:1 mixture of the two dipeptides confirmed that they had stereochemical purities of greater than 99:1 even when the peptide synthesis was carried out at 70 °C.37
The other key step for solution-phase peptide synthesis is selective removal of the N-terminal Boc protecting group to allow subsequent chain elongation. This is usually achieved by use of a strong acid such as hydrogen chloride or trifluoroacetic acid in solvents such as 1,4-dioxane, dichloromethane, THF, acetonitrile or DMF. Therefore, the use of PC as solvent for Boc deprotection reactions was also investigated. Initially, hydrogen chloride was generated in PC and used to convert fully protected dipeptide 5a into the hydrochloride salt of Ala-Phe-OBn in 86% yield. However, as a solution of hydrogen chloride in PC is not commercially available, it was more convenient to use trifluoroacetic acid in PC and this converted fully protected dipeptide 5a into trifluoroacetate salt 6a in 99% yield. In contrast, use of 4 M hydrogen chloride in dioxane gave the corresponding chloride salt 6b in just 93% yield. Thus, both coupling and deprotection reactions could be carried out in PC in higher yield than the yields obtained using DMF, dichloromethane and dioxane.
To illustrate the generality of this process, five fully protected tetrapeptides 9a–e were prepared as shown in Scheme 3. Peptide sequences 5-9a,b contain only unhindered and unfunctionalised amino acids. However, sequences 5-9c–e were chosen to include the sterically hindered amino acids valine and aminoisobutyric acid (Aib), functionalised amino acids with their functional groups unprotected (methionine and tryptophan) and functionalised amino acids with their functional groups protected using standard protecting groups (serine, lysine, aspartic acid and glutamine). All five tetrapeptides were successfully prepared using PC as the only solvent. To further prove that no racemisation of the activated amino acids occurs during peptide synthesis in PC, diastereomeric dipeptide 5b′ and tripeptide 7a′ were prepared using Boc-(R)-leucine and HPLC analysis of the diastereomers 5b/5b′ and 7a/7a′ again showed stereochemical purities of greater than 99:1.37 Finally, to show that the methodology was compatible with the palladium on carbon used to remove benzyl based protecting groups, tripeptide 7a was subjected to hydrogenolysis to give acid 10a in 96% yield and then treated with trifluoroacetic acid in PC to give fully deprotected tripeptide 11a in quantitative yield.
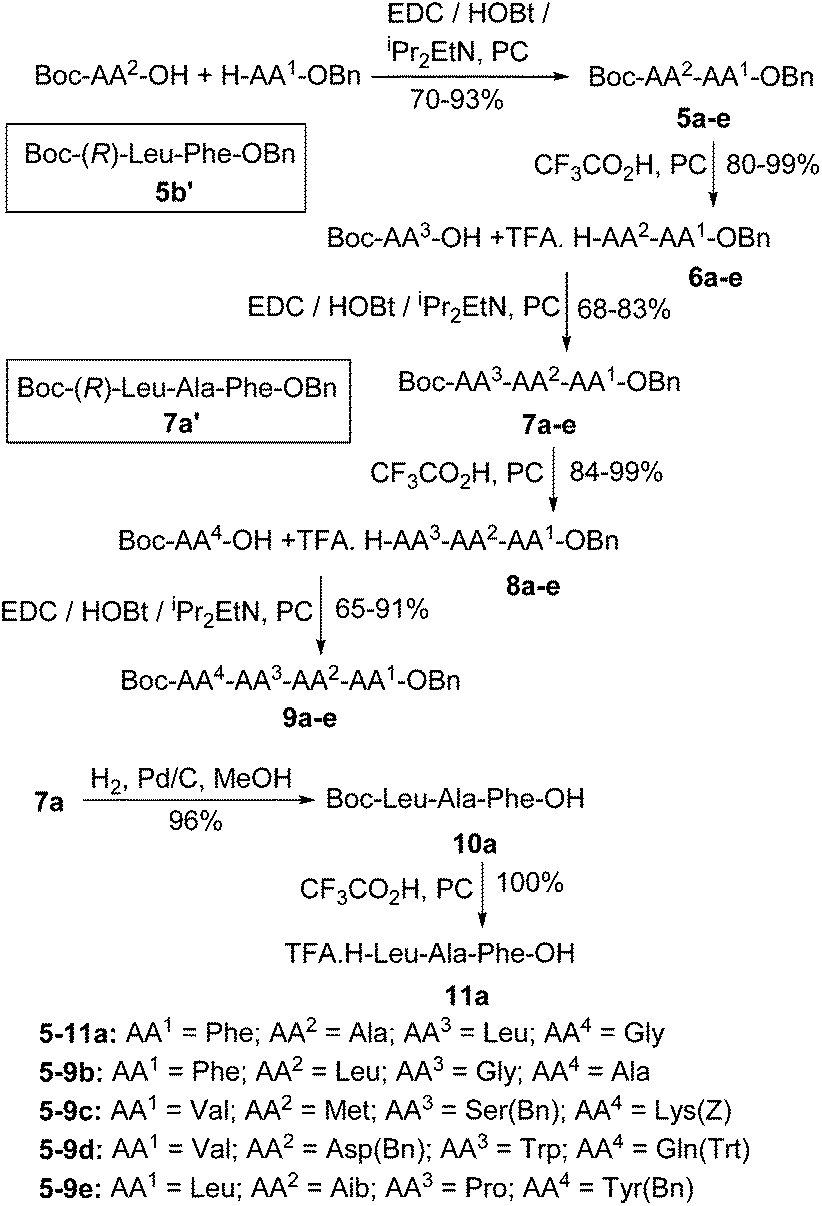 | ||
| Scheme 3 Synthesis of tetrapeptides in PC. | ||
Having demonstrated that PC could be used as a solvent for solution-phase peptide synthesis, its application to solid-phase peptide synthesis was investigated. This presented a new challenge: the solvent must swell the resin upon which the peptide is being constructed. We have recently reported a study of the swelling of various resins used for solid-phase synthesis in over 20 green solvents including PC and EC.38 This showed that polystyrene based resins such as 2% cross linked polystyrene (Merrifield resin39) did not swell adequately in PC. However, a cross-linked polyethylene glycol (ChemMatrix resin40) did swell (to 6.0 mL g−1) in PC which is well above the 4.0 mL g−1 required for a resin to be considered as well swollen.41 If a resin swells too much in a particular solvent, this can also be a problem for solid-phase peptide synthesis as it can cause blockages and result in over-filling of resin holding vessels. It is thus advantageous that whilst PC swelled ChemMatrix resin to 6.0 mL g−1 this is less than the 7.6–7.8 mL g−1 swelling observed in DMF and NMP.38 Hence, all solid-phase peptide synthesis was carried out on ChemMatrix resin.
The synthesis of the tripeptide TFA·H-Leu-Ala-Phe-OH 11a was chosen as a test sequence and its solid-phase preparation was undertaken in both DMF and PC (Scheme 4). The synthesis started with commercially available ChemMatrix resin preloaded with an acid-labile HMPB (4-(4-hydroxymethyl-3- methoxyphenoxy)butyric acid) linker42 and a phenylalanine residue with a free amino group. Each coupling reaction was carried out using three equivalents each of an Fmoc-protected amino acid, HBTU43 (3-[bis(dimethylamino)methyliumyl]-3H-benzotriazol-1-oxide hexafluorophosphate) and HOBt and six equivalents of diisopropylethylamine. At the end of each coupling and deprotection reaction, the resin was only washed with the solvent being used for the synthesis rather than with a range of solvents (usually including DMF, dichloromethane and methanol) to ensure that the use of non-green solvents was completely avoided. The tripeptide was cleaved from the resin by treatment with trifluoroacetic acid in the presence of TIPS (triisopropylsilane) and water as cation scavengers,44 then precipitated from and triturated with diethyl ether. Analysis of both samples by 1H NMR spectroscopy showed that TFA·H-Leu-Ala-Phe-OH 11a had been successfully prepared in both solvents.
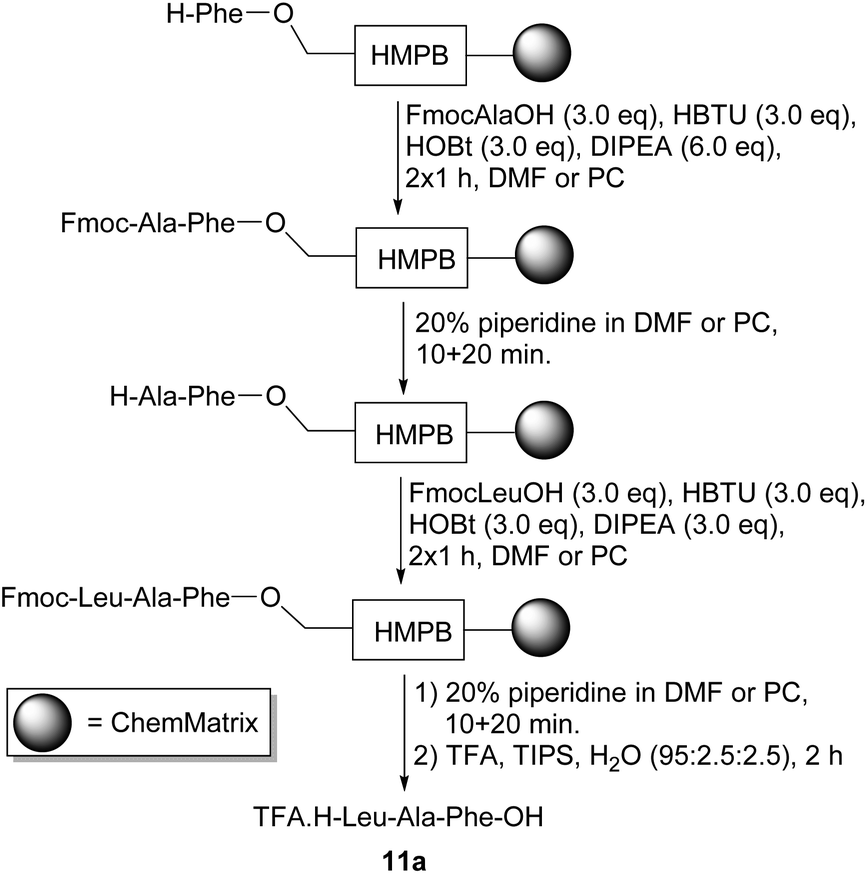 | ||
| Scheme 4 Solid-phase synthesis of tetrapeptide 11a. | ||
Having demonstrated that PC could be used as a solvent for solid-phase peptide synthesis, the synthesis of a biologically relevant peptide was undertaken. The naturally occurring nonapeptide bradykinin (H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH) which is a vasodilator the concentration of which is increased by ACE-inhibitors45 was selected as a suitable test as it is of moderate length and contains a mixture of functionalised and unfunctionalised amino acids. The synthesis of bradykinin starting from H-Arg-HMPB-ChemMatrix was carried out using the coupling and deprotection reactions given in Scheme 5.37
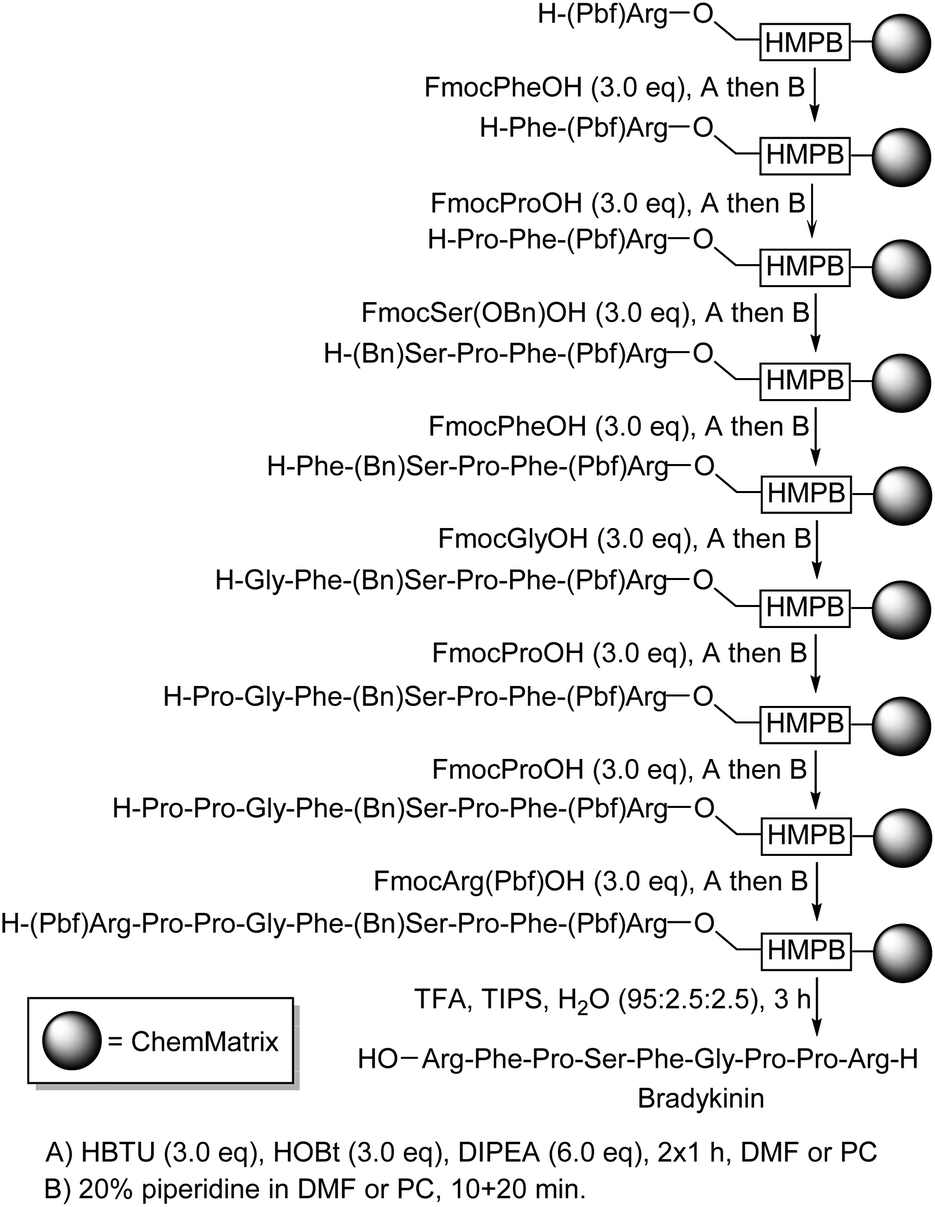 | ||
| Scheme 5 Solid-phase synthesis of bradykinin in PC. | ||
The cleaved peptide was precipitated into cold diethyl ether, triturated and lyophilised. To allow a comparison with conventional methodology to be undertaken, the synthesis was also carried out in DMF and both synthetic samples were compared with commercially available bradykinin by HPLC-UV-HRMS (Fig. 1).37 The bradykinin samples prepared in PC and DMF were found to have purities of 77 and 79% respectively, showing that use of PC as solvent had no significant detrimental effect on the synthesis. The sample prepared in DMF (Fig. 1B) contained a single impurity peak which overlapped with the bradykinin peak and so would be difficult to separate by preparative HPLC. Analysis of the mass spectrum of this impurity peak showed that it was actually a mixture of the premature chain termination sequences H-Ser-Pro-Phe-Arg-OH and H-Pro-Phe-Arg-OH. In contrast, this impurity was not present in the sample prepared in PC (Fig. 1C) and the impurity peaks that were present were well separated from the bradykinin peak, thus facilitating purification by preparative HPLC if desired.
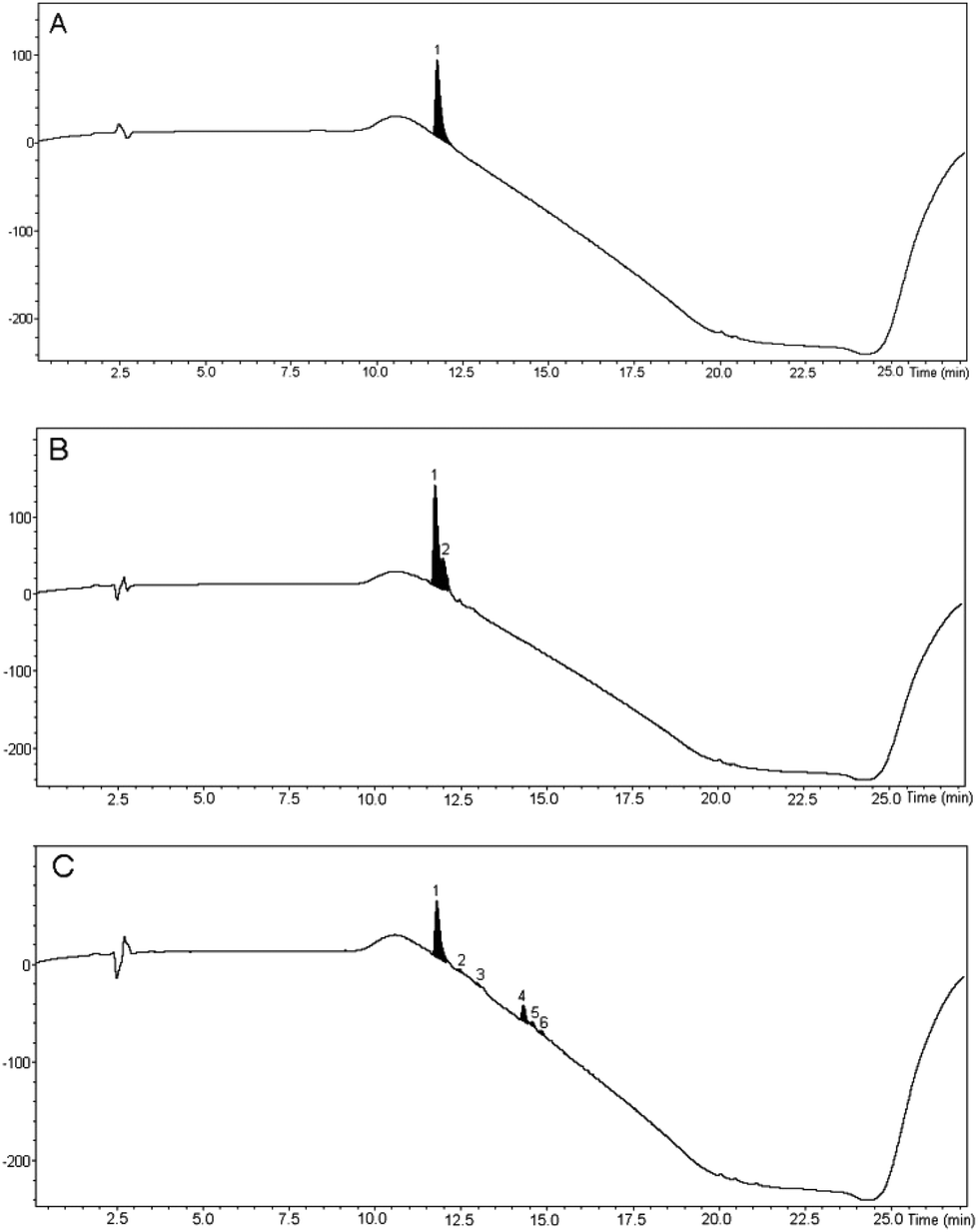 | ||
| Fig. 1 HPLC-UV traces of bradykinin. A: Commercial sample, B: sample prepared in DMF, C: sample prepared in PC. Chromatograms were eluted with a gradient from 1:9 MeCN:H2O to 1:1 MeCN:H2O.37 | ||
Experimental
Details of instrumentation are given in the ESI.†General procedure for peptide coupling reactions in PC
To a suspension of an N-Boc-amino acid (1.0 eq.) and an amino acid or peptide benzyl ester (1.0 eq.) in PC (5 mL mmol−1), at 0 °C, was added a solution of HOBt (1.1 eq.) and iPr2EtN (3.0 eq.) in a minimal quantity of PC. EDC (1.1 eq.) was added dropwise and the reaction mixture was allowed to stir at room temperature for 16 h. The reaction mixture was then diluted using EtOAc (50 mL) and washed with 1 M HClaq (3 × 25 mL), saturated Na2CO3 (3 × 25 mL) and H2O (3 × 25 mL). The organic layer was dried (MgSO4), filtered and concentrated in vacuo. Any residual PC was removed via short path distillation. Purification details for each peptide and characterising data are given in the ESI.†General procedure for Boc deprotections in PC
An N-Boc-peptide benzyl ester (1.0 eq.) was dissolved in a minimum amount of PC and trifluoroacetic acid (60 eq.) was added. The reaction mixture was allowed to stir for 3 h. at room temperature before being concentrated in vacuo. Any residual PC was removed via short path distillation. Characterising data for each deprotected peptide are given in the ESI.†Synthesis of Boc-Ala-Phe-OBn 5a in PC in a microwave reactor
N-Boc-alanine (189 mg, 1.0 mmol, 1.0 eq.), phenylalanine benzyl ester (292 mg, 1.0 mmol, 1.0 eq.) and HOBt (149 mg, 1.0 mmol, 1.1 eq.) were combined in a G10 microwave vial. PC (2 mL) and iPr2EtN (523 μL, 3.0 mmol, 6.0 eq.) were then added. The vial was cooled to 0 °C and EDC (195 μL, 1.1 mmol, 1.1 eq.) was added dropwise. The reaction mixture was then heated at 70 °C for 3 hours using an Anton Parr Monowave 300 microwave reactor. The reaction was then diluted with EtOAc (50 mL) and washed with 1 M HCl (50 mL), sat. Na2CO3 (50 mL) and brine (50 mL). The organic fraction was dried (MgSO4), filtered and concentrated in vauco. Residual PC was removed via short path distillation. The residue was purified using flash column chromatography (40:60, EtOAc:petroleum ether) to give N-Boc-Ala-Phe-OBn as a white solid (352 mg, 82%).
Procedure for Boc deprotection of dipeptide 5a using HCl in PC
Boc-Ala-Phe-OBn 5a (50 mg, 0.117 mmol) was dissolved in PC (2.34 mL). MeOH (0.40 mL, 9.8 mmol) was added and the solution cooled to 0 °C. Acetyl chloride (0.67 mL, 9.36 mmol) was added dropwise and the solution allowed to stir at room temperature for 2 h. Then, PC was removed by short path distillation. The residue was suspended in cold Et2O and triturated to give HCl·Ala-Phe-OBn as a white solid (46 mg, 86%).General procedure for solid-phase peptide synthesis in PC
The synthesis of peptides on a solid-phase was carried out in a 5 mL syringe fitted with a polypropylene frit. Preloaded ChemMatrix-HMPB resin (200 mg, 0.6 mmol g−1) was first washed with PC (3 × 5 mL) and then swollen in PC (3.5 mL) for 1 h. In a separate vial, an Fmoc-amino acid (0.36 mmol, 3.0 eq.), HBTU (137 mg, 0.36 mmol, 3.0 eq.), HOBt (49 mg, 0.36 mmol, 3.0 eq.) and DIPEA (125 μL, 0.72 mmol, 6.0 eq.) were combined in PC (3.6 mL). After 3 min, this preactivated amino acid solution was added to the resin. Coupling reactions were performed for 1 h. at ambient temperature and carried out in duplicate. Following each coupling reaction the resin was washed using PC (3 × 5 mL). Fmoc-deprotections were performed in duplicate (10 min followed by 20 min) using a freshly prepared solution of 20% (v/v) piperidine in PC (10–12 mL g−1). Following deprotection the resin was washed using PC (3 × 5 mL). Once the full sequence was constructed and the final Fmoc-deprotection had been performed, the resin was dried under reduced pressure. The peptide was then cleaved from the resin using TFA:TIPS:H2O (95:2.5:2.5) (8–10 mL g−1) for 2 h. at ambient temperature. The resin was removed by filtration and the peptide precipitated into cold Et2O, triturated and lyophilised. Characterising data for the peptides prepared in this way are given in the ESI.†
Conclusions
Propylene carbonate 1 has been shown to be a green replacement for reprotoxic amide based solvents which are widely used in peptide synthesis. Both solution- and solid-phase peptide synthesis can be carried out in propylene carbonate using acid and base labile amine protecting groups respectively. No significant racemisation of the activated amino acids occurs in propylene carbonate and the viability of solid-phase peptide synthesis in propylene carbonate was demonstrated by the synthesis of the nonapeptide bradykinin.Acknowledgements
The authors thank EPSRC (grant number EP/M506680/1) for a studentship to SL.Notes and references
- (a) Y. Liu and T. Liu, Curr. Med. Chem., 2012, 19, 3982–3999 CrossRef CAS PubMed; (b) S. Lohan and G. S. Bisht, Mini-Rev. Med. Chem., 2013, 13, 1073–1088 CrossRef CAS PubMed; (c) J. Vabeno, B. E. Haug and M. M. Rosenkilde, Future Med. Chem., 2015, 7, 1261–1283 CrossRef CAS PubMed; (d) R. Domalaon, G. G. Zhanel and F. Schweizer, Curr. Top. Med. Chem., 2016, 16, 141–155 CrossRef.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- (a) A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS PubMed; (b) J. P. Tam, Proc. Natl. Acad. Sci. USA, 1988, 85, 5409–5413 CrossRef CAS PubMed.
- (a) D. Goodwin, P. Simerska and I. Toth, Curr. Med. Chem., 2012, 19, 4451–4461 CrossRef CAS PubMed; (b) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- F. Albericio and H. G. Kruger, Future Med. Chem., 2012, 4, 1527–1531 CrossRef CAS PubMed.
- For examples of previous work on improving the efficiency of peptide synthesis using flow reactors see: (a) O. Flögel, J. D. C. Codée, D. Seebach and P. H. Seeberger, Angew. Chem., Int. Ed., 2006, 45, 7000–7003 CrossRef PubMed; (b) M. D. Simon, P. L. Heider, A. Adamo, A. A. Vinogradov, S. K. Mong, X. Li, T. Berger, R. L. Policarpo, C. Zhang, Y. Zou, X. Liao, A. M. Spokoyny, K. F. Jensen and B. L. Pentelute, ChemBioChem, 2014, 15, 713–720 CrossRef CAS PubMed; (c) I. M. Mándity, B. Olasz, S. B. Ötvös and F. Fülöp, ChemSusChem, 2014, 7, 3172–3176 CrossRef PubMed.
- (a) F. Alberico, Biopolymers, 2000, 55, 123–239 CrossRef; (b) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS; (c) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- Methods of Organic Synthesis: Synthesis of Peptides and Peptidomimetics, ed. M. Goodman, A. Felix, L. Moroder and C. Toniolo, Thieme, Stuttgart, 2004, vol. E22a Search PubMed.
- (a) E. Atherton and R. C. Sheppard, in Solid phase peptide synthesis a practical approach, OUP, Oxford, 1989 Search PubMed; (b) I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- G. L. Kennedy, Crit. Rev. Toxicol., 2012, 42, 793–826 CrossRef CAS PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- Regulation (EC) 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH); candidate list of substances of very high concern for authorisation, http://echa.europa.eu/candidate-list-table (accessed July 20, 2016).
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- G. A. Acosta, M. del Fresno, M. Paradis-Bas, M. Rigau-DeLlobet, S. Côté, M. Royo and F. Albericio, J. Pept. Sci., 2009, 15, 629–633 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393–2398 CAS.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419–426 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814 CrossRef CAS.
- (a) A. S. Galanis, F. Albericio and M. Grøtli, Org. Lett., 2009, 11, 4488–4491 CrossRef CAS PubMed; (b) K. Hojo, A. Hara, H. Kitai, M. Onishi, H. Ichikawa, Y. Fukumori and K. Kawasaki, Chem. Cent. J., 2011, 5, 49 CrossRef CAS PubMed; (c) R. De Marco, A. Tolomelli, A. Greco and L. Gentilucci, ACS Sustainable Chem. Eng., 2013, 1, 566–569 CrossRef CAS; (d) K. Hojo, N. Shinozaki, K. Hidaka, Y. Tsuda, Y. Fukumori, H. Ichikawa and J. D. Wade, Amino Acids, 2014, 46, 2347–2354 CrossRef CAS PubMed.
- (a) R. V. Ulijn, B. Baragaña, P. J. Halling and S. L. Flitsch, J. Am. Chem. Soc., 2002, 124, 10988–10989 CrossRef CAS PubMed; (b) R. Haddoub, M. Dauner, F. A. Stefanowicz, V. Barattini, N. Laurent and S. L. Flitsch, Org. Biomol. Chem., 2009, 7, 665–670 RSC ; Water enzyme.
- (a) V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318–9321 CrossRef CAS PubMed; (b) J. Bonnamour, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2013, 15, 1116–1120 RSC; (c) J. G. Hernández and E. Juaristi, J. Org. Chem., 2010, 75, 7107–7111 CrossRef PubMed; (d) J. M. Landeros and E. Juaristi, Eur. J. Org. Chem., 2017, 687–694 CrossRef CAS.
- (a) R. Payne and I. E. Theodorou, J. Phys. Chem., 1972, 76, 2892–2900 CrossRef CAS; (b) L. B. Silva and L. C. G. Freitas, J. Mol. Struct. (THEOCHEM), 2007, 806, 23–34 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- All work in this paper used 99% grade solvents and 2016 prices from Thermo Fisher Scientific were £9.5 and £17.4 for 250 g of PC and DMF respectively.
- T. Fujinaga and K. Izutsu, Pure Appl. Chem., 1971, 27, 273–280 CrossRef CAS.
- (a) M. North, ARKOVIC, 2012,(part (i)), 610–628 Search PubMed; (b) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef; (c) A. C. Kathalikkattil, R. Babu, J. Tharun, R. Roshan and D.-W. Park, Catal. Surv. Asia, 2015, 19, 223–235 CrossRef CAS; (d) B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS PubMed; (e) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed; (f) B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang and S.-J. Zhang, Green Chem., 2015, 17, 108–122 RSC; (g) V. D'Elia, J. D. A. Pelletier and J.-M. Basset, ChemCatChem, 2015, 7, 1906–1917 CrossRef; (h) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- (a) I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212–215 RSC; (b) M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163–4170 RSC; (c) A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- (a) W. J. Peppel, Ind. Eng. Chem., 1958, 50, 767–770 CrossRef CAS; (b) M. Yoshida and M. Ihara, Chem. – Eur. J., 2004, 10, 2886–2893 CrossRef CAS PubMed.
- (a) K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed; (b) S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS; (c) V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–326 RSC; (d) B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- (a) C. E. P. Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 7372–7386 RSC; (b) J. S. B. Forero, E. M. D. Carvalho, J. J. Junior and F. M. Silva, Curr. Org. Synth., 2015, 102–107 CrossRef CAS.
- (a) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed; (b) M. North and M. Omedes-Pujol, Tetrahedron Lett., 2009, 50, 4452–4454 CrossRef CAS; (c) P. Lenden, P. M. Ylioja, C. González-Rodríguez, D. A. Entwistle and M. C. Willis, Green Chem., 2011, 13, 1980–1982 RSC; (d) B. Yu, Z.-F. Diao, C.-X. Guo, C.-L. Zhong, L.-N. He, Y.-N. Zhao, Q.-W. Song, A.-H. Liu and J.-Q. Wang, Green Chem., 2013, 15, 2401–2407 RSC; (e) B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320–324 RSC; (f) H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS; (g) P. Neubert, S. Fuchs and A. Behr, Green Chem., 2015, 17, 4045–4052 RSC.
- (a) M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862–865 CrossRef CAS PubMed; (b) W. Clegg, R. W. Harrington, M. North, F. Pizzato and P. Villuendas, Tetrahedron: Asymmetry, 2010, 21, 1262–1271 CrossRef CAS; (c) M. North and P. Villuendas, Org. Lett., 2010, 12, 2378–2381 CrossRef CAS PubMed.
- J. Nam, D. Shin, Y. Rew and D. L. Boger, J. Am. Chem. Soc., 2007, 129, 8747–8755 CrossRef CAS PubMed.
- (a) B. Bacsa, K. Horváti, S. Bösze, F. Andreae and C. O. Kappe, J. Org. Chem., 2008, 73, 7532–7542 CrossRef CAS PubMed; (b) B. Bacsa, S. Bösze and C. O. Kappe, J. Org. Chem., 2010, 75, 2103–2106 CrossRef CAS PubMed.
- S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef.
- Data given in ESI.†.
- S. Lawrenson, M. North, F. Peigneguy and A. Routledge, Green Chem., 2017, 19, 952–962 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef PubMed.
- R. Santini, M. C. Griffith and M. Qi, Tetrahedron Lett., 1998, 39, 8951–8954 CrossRef CAS.
- B. Riniker, A. Flörsheimer, H. Fretz, P. Sieber and B. Kamber, Tetrahedron, 1993, 49, 9307–9320 CrossRef CAS.
- R. Knorr, A. Trzeciak, W. Bannwarth and D. Gillessen, Tetrahedron Lett., 1989, 30, 1927–1930 CrossRef CAS.
- D. A. Pearson, M. Blanchette, M. L. Baker and C. A. Guindon, Tetrahedron Lett., 1989, 30, 2739–2742 CrossRef CAS.
- M. Perez, A. Adam and G. Molinaro, J. Clin. Basic Cardiol, 2001, 4, 39–46 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterising data including copies of NMR spectra and HPLC traces. See DOI: 10.1039/c7gc00247e |
| This journal is © The Royal Society of Chemistry 2017 |