
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkoxide-catalyzed addition of alkyl carbonates across alkynes – stereoselective synthesis of (E)-β-alkoxyacrylates†
Timo
Wendling
 ,
Eugen
Risto
,
Benjamin
Erb
and
Lukas J.
Gooßen
*
,
Eugen
Risto
,
Benjamin
Erb
and
Lukas J.
Gooßen
*
Fakultät für Chemie und Biochemie, Ruhr Universität Bochum, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: lukas.goossen@rub.de
First published on 14th December 2016
Abstract
Dialkyl carbonates were found to regio- and stereoselectively add to terminal alkynes in the presence of catalytic amounts of potassium methoxide. Various synthetically meaningful aryl- and heteroaryl-substituted (E)-β-alkoxyacrylates were thus obtained in high yields and with near-ideal atom economy.
Introduction
The (E)-β-methoxyacrylate subunit is found in various biologically active natural products such as dihydrokawain,1 tetronic acids,2 five- and six-membered lactones,3–10 and β-methoxyacrylate antibiotics,11–14 including melithiazol12 and haliangicin14 (Fig. 1). An E-configuration of the methoxyacrylate (MOA) moiety is decisive for biological activity.15,16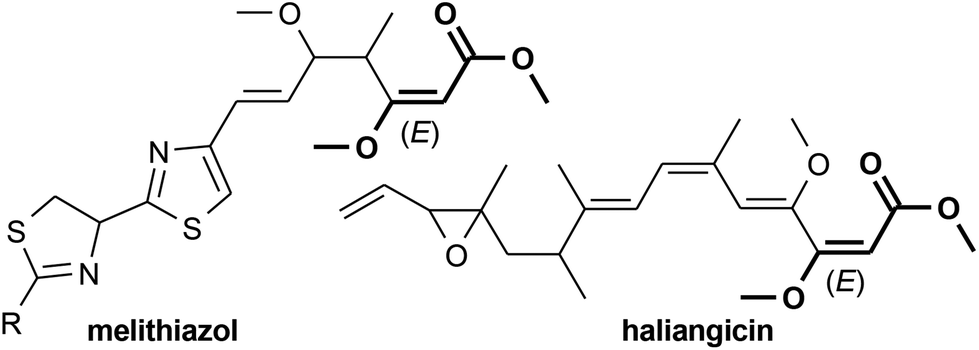 | ||
| Fig. 1 Bioactive natural compounds bearing a (E)-β-methoxyacrylate moiety. | ||
In view of their tremendous synthetic importance, sustainable, stereoselective synthetic entries to (E)-β-MOA are constantly sought. Existing synthetic strategies all have their drawbacks (Scheme 1). The addition of methanol to propargylic esters (A) suffers from the poor availability of the starting materials and calls for toxic or expensive catalysts, e.g. mercury,17 lead,18 or gold complexes, or has poor chemoselectivity.19 The photochemical coupling of sulfur ylides with chromium Fischer carbene complexes (B) gives poor stereoselectivity and produces stoichiometric amounts of sulfoxide and toxic chromium waste.20–22 Stereoselective syntheses, e.g. via Claisen condensation of a lactone with methyl formate, methylation with dimethyl carbonate and subsequent ring opening, require multiple steps.23 The palladium-catalysed methoxycarbonylation of terminal alkynes reported by Kato and Akita (C) is efficient but suffers from the use of toxic carbon monoxide and stoichiometric amounts of benzoquinone (BQ) as the oxidant.24,25
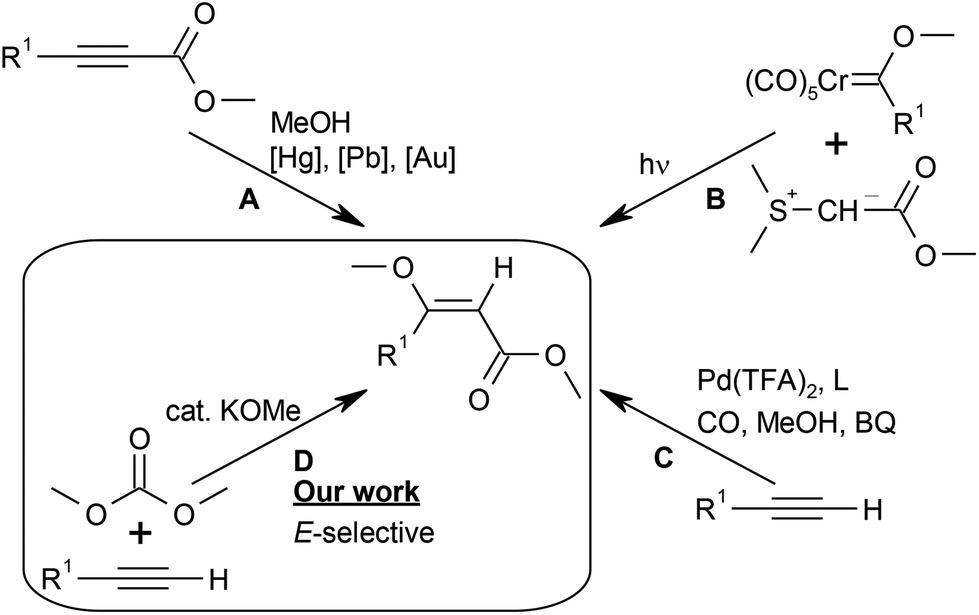 | ||
| Scheme 1 Synthetic routes to β-methoxyacrylates. | ||
From a sustainable chemistry standpoint, the addition of alkyl carbonates across terminal alkynes would represent one of the best conceivable synthetic concepts to access (E)-β-MOA.
In the proposed strategy, the alkyl carbonate would act as the source of both the alkyl carboxylate and the alkoxy group, resulting in maximal atom economy, since all atoms of the reagents are incorporated in the desired product (D). Scheme 2 shows the proposed mechanism for this transformation. In the first step, the base B− deprotonates the terminal alkyne to the acetylide (step (I)), which affords the propargylic ester after nucleophilic addition of dimethyl carbonate and elimination of the alkoxide (step (II)). The latter then adds to the C–C triple bond (step (III)), and the (E) or (Z)-products form upon protonation (IV).
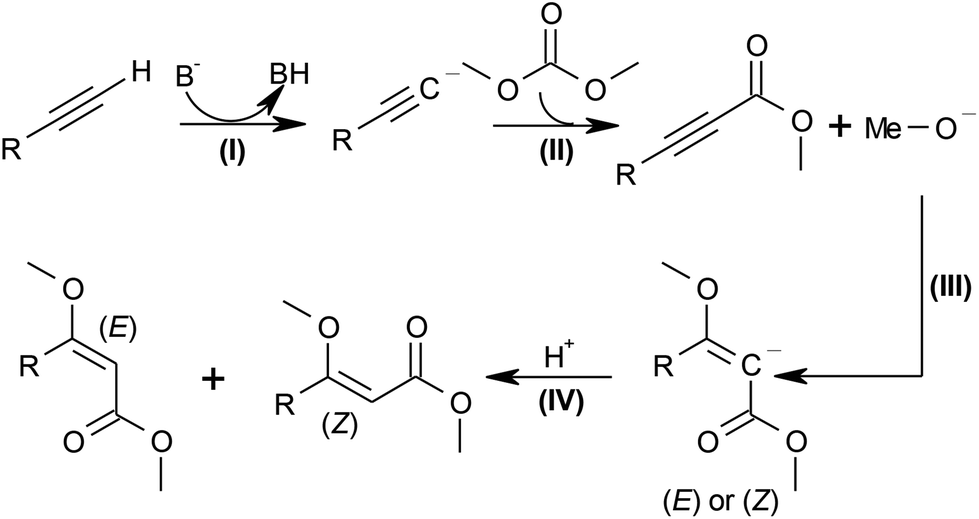 | ||
| Scheme 2 Mechanism for the β-MOA synthesis from dimethyl carbonate. | ||
Dimethyl carbonate is a “green” reagent, because it is a non-toxic, non-irritating liquid (bp. 90.3 °C) which is neither on the list of volatile organic compounds (VOCs), nor does it contribute to tropospheric ozone formation.26,27 It is used industrially as a solvent, fuel additive, and sustainable alternative to phosgene or harmful methylating agents.28 Dimethyl carbonate is accessible from methanol and carbon dioxide, potentially contributing to CO2 sequestration.28
However, the only report on its addition to alkynes dates back to 1949, and states that in the reaction of phenylacetylene with either dimethyl or diethyl carbonate, a hard-to-separate mixture of a β-alkoxyacrylate with unidentified stereochemistry and a ketal was obtained.29 In order to turn this intriguing concept into an expedient synthetic entry to (E)-β-MOA, the chemo- and stereochemistry of the reaction needed to be controlled efficiently.
Results and discussion
Using the reaction of phenylacetylene (1a) with dimethyl carbonate (2a) as a model, we systematically investigated various reaction conditions. The logical first choice for the base was potassium methoxide, since methoxide ions would be released during the process in any case (step (II)) and are known to deprotonate alkynes. In the presence of stoichiometric amounts of potassium methoxide in 2 mL dimethyl carbonate, the desired (E)-methyl 3-methoxy-3-phenylprop-2-enoate (3aa) was detected in 54% yield, along with 12% of the ketal by-product 4aa (Table 1, entry 1). By reducing the amount to 30 or 10 mol% KOMe, the yield increased to 74% or 80%, respectively, with good E/Z ratios, but 12% of ketal by-product 4aa were still formed (Table 1, entries 2 and 3).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base (mol%) | Solvent | Conv.b (%) | Yieldb (%) | ||
| 3aa | 4aa | E/Z | ||||
| a Reaction conditions: 0.50 mmol of 1a, 0.60 mmol of 2a, 10–100 mol% base, 2 mL solvent, 12 h, rt. b Conversions, yields and E/Z ratios were determined by GC using n-dodecane as internal standard. c Dropwise addition of 1.00 mmol of 1a in 0.5 mL DMSO to 1.20 mmol of 2a and 0.30 mmol of KOMe in 1 mL DMSO over 45 min, 12 h, rt. d Isolated yield. PC = propylene carbonate. | ||||||
| 1 | KOMe (100) | DMC | 100 | 54 | 12 | 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | KOMe (30) | DMC | 100 | 74 | 12 | 12:1 |
| 3 | KOMe (10) | DMC | 100 | 80 | 12 | 10:1 |
| 4 | KOMe (30) | PC | — | — | — | — |
| 5 | KOMe (30) | DMF | 100 | 70 | 2 | 13:1 |
| 6 | KOMe (30) | NMP | 100 | 76 | 6 | 11:1 |
| 7 | KOMe (30) | DMSO | 100 | 80 | 4 | 15:1 |
| 8 | KOMe (10) | DMSO | 8 | 5 | — | — |
| 9 | NaOMe (30) | DMSO | — | — | — | — |
| 10 | KOtBu (30) | DMSO | 100 | 77 | 3 | 13:1 |
| 11 | LDA (30) | DMSO | 53 | 9 | — | 7:1 |
| 12 | Cs2CO3 (30) | DMSO | — | — | — | — |
| 13 | KOMe (30) | DMSO | 100 | 89 (75) | 6 |
15:1 |
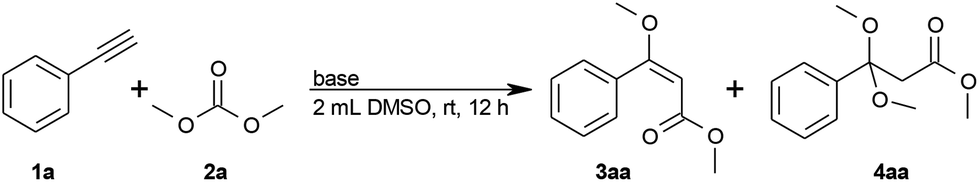
In order to minimize the amount of 4aa, which is hard to separate from the desired product 3aa, we reduced the amount of dimethyl carbonate to 1.2 equivalents and added an external solvent (Table 1, entries 4–7; see also the ESI S1, entries 3–5†). Good yields with low amounts of ketal 4aa were obtained in polar aprotic solvents (Table 1, entries 4 and 5). DMSO gave the best yield and selectivity (Table 1, entry 7). The yield of product 3aa was not improved further by increasing neither the temperature nor the amount of dimethyl carbonate (S1, entries 1 and 2†).
With less base, the conversion dropped to only 8% (Table 1, entry 8). No conversion at all was observed when sodium methoxide was used instead of the potassium salt (Table 1, entry 9). Potassium tert-butoxide afforded 77% yield of 3aa (Table 1, entry 10), whereas all other bases were ineffective (Table 1, entries 11 and 12). The amount of solvent could be reduced to 1 mL without negatively affecting the yield (S1, entry 6†). The best procedure consisted of adding the alkyne dropwise to a stirred solution of dimethyl carbonate and 30 mol% potassium methoxide in DMSO (Table 1, entry 13).
In the presence of 50 μl of water, no conversion was observed, presumably due to deactivation of the catalytic methoxide base (S1, entry 7†). The yield is also slightly decreased when the reaction is performed under air instead of a nitrogen atmosphere (S1, entry 8†).
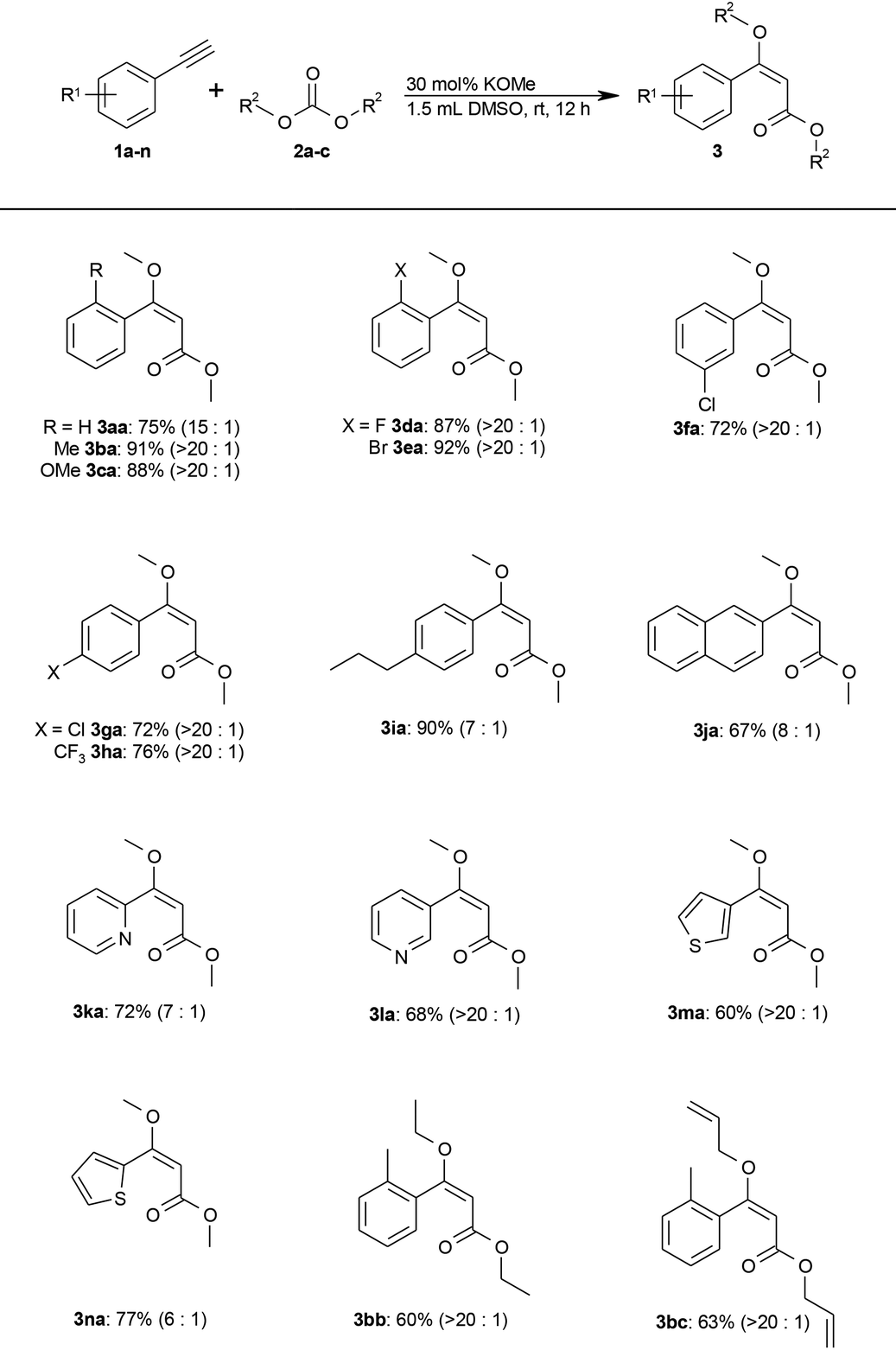
With an efficient reaction protocol in hand, we next investigated its scope using various alkynes (1) and carbonates (2). As shown in Table 2, both electron-rich and electron-deficient aromatic alkynes were selectively converted to the (E)-β-MOA (3) in good to excellent yields and high chemo- and regioselectivity.
| a Reaction conditions: Dropwise addition of 1.00 mmol of 1 in 0.5 mL DMSO to 1.20 mmol of 2 and 0.30 mmol of KOMe in 1 mL DMSO over 45 min, 12 h, rt. Isolated yield. E/Z values are given in parentheses. |
|---|
|
Common functionalities including methoxy, fluoro, bromo, chloro, and trifluoromethyl groups are tolerated. Heteroaromatic alkynes, such as pyridine or thiophene derivatives, also gave the desired products in good yields (Table 2, 3ka–3na).
The protocol also extends to other carbonates, as was shown by the addition of diethyl and diallyl carbonate (2b and 2c) to 2-ethynyltoluene (1b). Only trace amounts of methoxy-substituted by-products were observed resulting from transesterification with the methoxide base. Diphenyl carbonate gave no conversion. Instead, the methoxide undergoes quantitative transesterification with diphenyl carbonate and the resulting phenolate is not sufficiently basic to deprotonate a terminal alkyne (see step (I) in the proposed mechanism; Scheme 2).
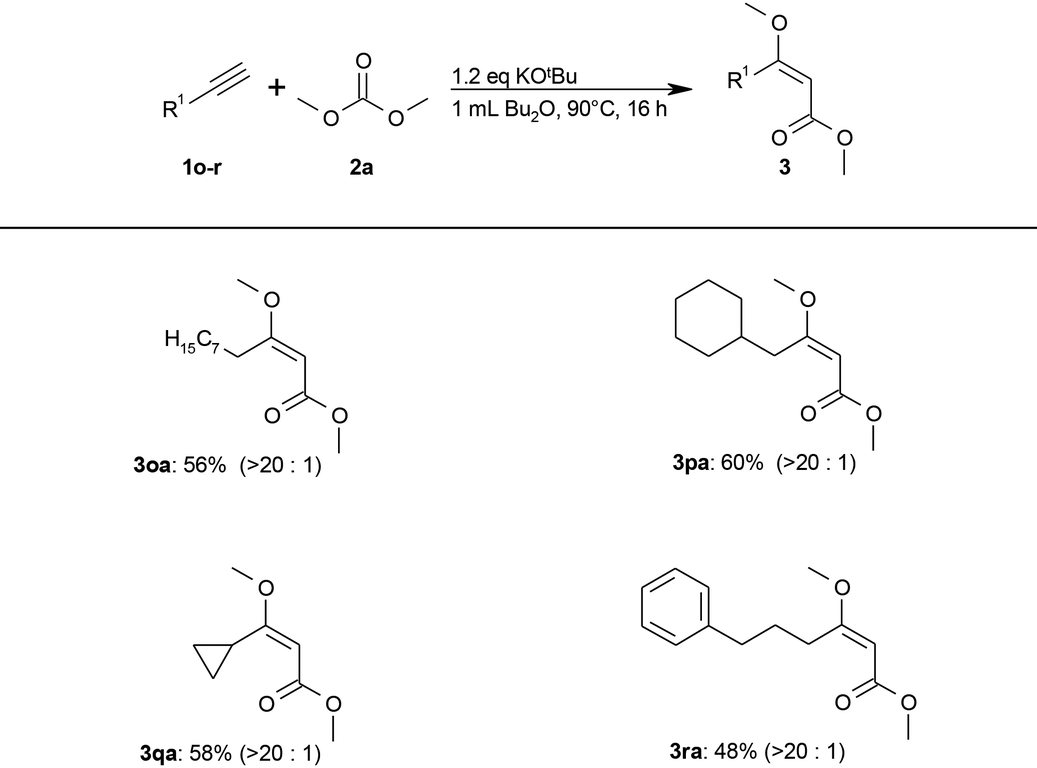
The less acidic aliphatic alkynes could not be converted under these exceptionally mild conditions. Additional screening (see ESI S2†) showed that aliphatic alkynes were converted to the desired aliphatic (E)-β-methoxyacrylates (3) in moderate yields using stoichiometric amounts of tert-butoxide in dibutyl ether at 90 °C (Table 3).
| a Reaction conditions: 1.00 mmol of 1, 3.00 mmol of 2a, 1.20 mmol of KOtBu, 1 mL Bu2O, 16 h, 90 °C. Isolated yield. E/Z values are given in parentheses. |
|---|
|
The scalability of the reaction was demonstrated on 15 mmol scale by the synthesis of ortho-bromophenyl (E)-β-MOA 3ea, which is a useful synthetic intermediate towards a variety of compounds with fungicidal activity.30 Simple distillation of the crude reaction mixture yielded 3ea in pure form and 80% yield. The E-factor (total kg waste per kg product) for the large scale synthesis of 3ea of 5.2 demonstrates the sustainability of this process (see S9† for the calculations). This is an excellent value in comparison to E-factors in chemical industry for the production of fine chemicals, which are typically in the range of 5–50.31 By recycling the solvent, the E-factor may further be reduced, ideally down to 1.5.
To elucidate the origin of the high stereoselectivity, we subjected an E/Z-mixture of 3aa to the reaction conditions. Within 12 h, the E/Z ratio changed to 15:1 in favor of the E-isomer. This points to interconversion between isomers and indicates that the E-isomer is thermodynamically more stable. The high E/Z ratio is a consequence of the unprecedentedly low reaction temperature.
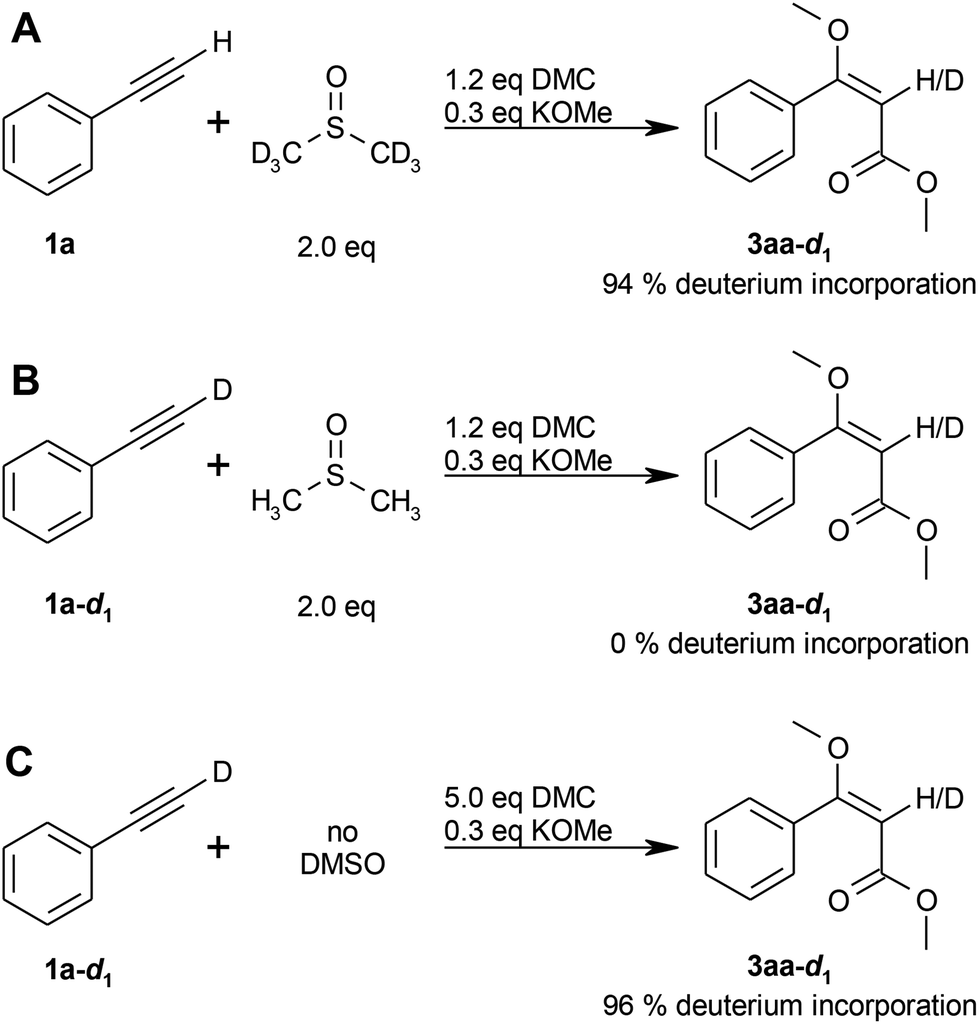
Deuterium labelling experiments were performed for further insight into the reaction mechanism, as well as to clarify the origin of the olefinic proton (see step (IV) in the proposed mechanism; Scheme 2). The reaction of phenylacetylene (1a) with dimethyl carbonate (2a) in DMSO-d6 resulted in a deuterium incorporation of 94% (Scheme 3A), whereas the reaction of d1-phenylacetylene (1a-d1) in non-deuterated DMSO showed no deuterium incorporation (Scheme 3B), thus indicating that the olefinic proton originates from the solvent.
 | ||
| Scheme 3 Deuterium labeling experiments. | ||
In contrast, the deuterium incorporation was 96% for the reaction of d1-phenylacetylene (1a-d1) in the absence of DMSO (Scheme 3C), indicating that the alkyne proton is transferred to the product in case no other proton source is available.
Conclusions
The alkoxide-catalyzed addition of alkyl carbonates to terminal alkynes represents an efficient approach for the stereoselective synthesis of synthetically valuable (E)-β-methoxyacrylates. The protocol offers a broad substrate scope covering a broad range of functionalized arenes and heteroarenes. Due to its remarkably high atom economy and low E-factor, this synthetic route is highly sustainable and interesting also for industrial applications.Acknowledgements
We thank A. Konkel for technical assistance and the Fonds der Chemischen Industrie (fellowship to T. W.) and the Deutsche Bundesstiftung Umwelt (fellowship to E. R.) for financial support.Notes and references
- A. G. van Veen, Recl. Trav. Chim. Pays-Bas, 1939, 58, 521–527 CrossRef CAS.
- A. Zografos and D. Georgiadis, Synthesis, 2006, 3157–3188 CAS.
- C. Li, M. V. Nitka, J. B. Gloer, J. Campbell and C. A. Shearer, J. Nat. Prod., 2003, 66, 1302–1306 CrossRef CAS PubMed.
- N. G. Clemo and G. Pattenden, Tetrahedron Lett., 1982, 23, 585–588 CrossRef CAS.
- K. Umezawa, M. Tachibana, C. Matsui, Y. Takeuchi and E. Suzuki, Heterocycles, 2008, 76, 1561 CrossRef.
- K. Kobayashi and T. Ui, J. Chem. Soc., Chem. Commun., 1977, 774a–774a RSC.
- M. W. Klohs, F. Keller, R. E. Williams, M. I. Toekes and G. E. Cronheim, J. Med. Pharm. Chem., 1959, 1, 95–103 CrossRef CAS.
- K. Gerth, P. Washausen, G. Höfle, H. Irschik and H. Reichenbach, J. Antibiot., 1996, 49, 71–75 CrossRef CAS PubMed.
- H. Achenbach and G. Wittmann, Tetrahedron Lett., 1970, 11, 3259–3262 CrossRef.
- T. Hashimoto, M. Suganuma, H. Fujiki, M. Yamada, T. Kohno and X. Asakawa, Phytomedicine, 2003, 10, 309–317 CrossRef CAS PubMed.
- Y. Suzuki, M. Ojika, Y. Sakagami, R. Fudou and S. Yamanaka, Tetrahedron, 1998, 54, 11399–11404 CrossRef CAS.
- F. Sasse, B. Böhlendorf, M. Herrmann, B. Kunze, E. Forche, H. Steinmetz, G. Höfle, H. Reichenbach and M. Hermann, J. Antibiot., 1999, 52, 721–729 CrossRef CAS PubMed.
- K. Gerth, H. Irschik, H. Reichenbach and W. Trowitzsch, J. Antibiot., 1980, 33, 1474–1479 CrossRef CAS PubMed.
- B. A. Kundim, Y. Itou, Y. Sakagami, R. Fudou, T. Iizuka, S. Yamanaka and M. Ojika, J. Antibiot., 2003, 56, 630–638 CrossRef CAS PubMed.
- H. Sauter, W. Steglich and T. Anke, Angew. Chem., Int. Ed., 1999, 38, 1328–1349 ( Angew. Chem. , 1999 , 111 , 1416–1438 ) CrossRef.
- RÖMPP Online, https://roempp.thieme.de/roempp4.0/do/data/RD-06-02102, (accessed April 2016).
- M. Bassetti and B. Floris, J. Chem. Soc., Perkin Trans. 2, 1988, 227–233 RSC.
- A. G. Davies and R. J. Puddephatt, J. Chem. Soc. C, 1968, 1479–1483 RSC.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 ( Angew. Chem. , 1998 , 110 , 1475–1478 ) CrossRef CAS.
- B. Alcaide, G. Dominguez, J. Rodriguez-Lopez and M. A. Sierra, Organometallics, 1992, 11, 1979–1981 CrossRef CAS.
- B. Alcaide, L. Casarrubios, G. Domínguez and M. A. Sierra, Organometallics, 1996, 15, 4612–4617 CrossRef CAS.
- R. Chaudhuri and U. Kazmaier, Synlett, 2014, 693–695 CAS.
- W. Krämer, U. Schirmer, P. Jeschke and M. Witschel, Modern Crop Protection Compounds, Wiley-VCH, Weinheim, 2011 Search PubMed.
- K. Kato, S. Motodate, T. Mochida, T. Kobayashi and H. Akita, Angew. Chem., Int. Ed., 2009, 48, 3326–3328 ( Angew. Chem. , 2009 , 121 , 3376–3378 ) CrossRef CAS PubMed.
- S. Motodate, T. Kobayashi, M. Fujii, T. Mochida, T. Kusakabe, S. Katoh, H. Akita and K. Kato, Chem. – Asian J., 2010, 5, 2221–2230 CrossRef CAS PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- Environmental Protection Agency, (EPA-HQ-OAR-2006-0948), Research Triangle Park, NC 27711, 2009.
- B. A. V. Santos, V. M. T. M. Silva, J. M. Loureiro and A. E. Rodrigues, ChemBioEng Rev., 2014, 1, 214–229 CrossRef CAS.
- W. J. Croxall and H. J. Schneider, J. Am. Chem. Soc., 1949, 71, 1257–1260 CrossRef CAS.
- H. Sauter, K. Oberdorf, H. Wingert, W. von Deyn, W. Grammenos, H. Koenig, H. Rang, F. Roehl, G. Lorenz and E. Ammermann, EP, 0525516A2, 1993 Search PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc03168d |
| This journal is © The Royal Society of Chemistry 2017 |