
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A laboratory-scale annular continuous flow reactor for UV photochemistry using excimer lamps for discrete wavelength excitation and its use in a wavelength study of a photodecarboxlyative cyclisation†
Erica N.
DeLaney
a,
Darren S.
Lee
a,
Luke D.
Elliott
b,
Jing
Jin
a,
Kevin I.
Booker-Milburn
b,
Martyn
Poliakoff
*a and
Michael W.
George
*ac
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: mike.george@nottingham.ac.uk; martyn.poliakoff@nottingham.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
cDepartment of Chemical and Environmental Engineering, University of Nottingham Ningbo China, 199 Taikang East Road, Ningbo 315100, China
First published on 23rd November 2016
Abstract
This paper describes a new annular reactor for continuous UV photochemistry, which uses easily interchangeable excimer lamps of different wavelengths. The reactor has narrow clearance to form thin films of material for efficient irradiation of molecules. Its use is demonstrated by investigating the effect of discrete wavelength lamps (222, 282 and 308 nm) on the reaction of potassium N-phthalimidobutanoate 1. The ability of the reactor to be integrated into multistep processes is illustrated by combining it with an Amberlyst scavenger and a solid acid catalyst, NbOPO4, to access a second product 3 that is obtained in a single telescoped process. The tricyclic scaffold in 3 is a motif found in several biologically active compounds and has possibilities as a synthon for new pharmaceutical products.
Introduction
Photochemistry offers highly selective, atom economical reactions and can be combined with catalytic conditions with the generation of little or no waste. Indeed, the green credentials of photochemistry were realised over 100 years ago when Giacomo Ciamician published The Photochemistry for the future and predicted the value of manufacturing chemicals using light.1 Thus, there is an increased activity in photochemistry particularly to maximise the usage of light as a green and sustainable ‘traceless’ reagent.2–10 This involves both the development of new reactors and technologies together with new synthetic approaches.11–20 At present there are many groups working on different aspects of photochemistry, and several are focused on developing greener photochemical methodologies12,13,15,17 and using sustainable less toxic solvents and reagents.21–25 The need for more efficient technology to carry out photochemistry is also an area that is receiving more attention.Flow chemistry is often seen as a technique that encompasses many aspects of green chemistry. Furthermore continuous reactors offer opportunities to address real problems which occur when scaling up batch photochemistry. As batch reactors become larger, the Beer–Lambert Law dictates that light penetration will be significantly reduced towards the centre of the vessel, meaning that over-irradiation of molecules and formation of by-products can occur. Combining flow chemistry with photochemistry opens up opportunities to access photochemical reactors that can be optimised to avoid over irradiation, overcome the light penetration problems and have high efficiencies. Numerous effective continuous reactor designs have been reported in the literature; slug flow,26,27 spinning disc,28–30 falling film,31,32 vortex,33 bubble column,34 high pressure,21 FEP tubular,35 parallel tubular36 and our recently described design based around a rotary evaporator.37
In addition, flow chemistry can reduce safety risks by employing smaller reactors that generate only small quantities of reactive intermediates at any one time, which are deemed unsafe for larger batch operations, e.g. peroxo compounds. Many chemical reactions have been realised in flow, and more complex systems have been developed that incorporate downstream processing to completely automate the reactions, work-up and isolation of the product. This has been demonstrated on several complex natural product targets.38–42 In this paper, we describe a new type of photochemical flow reactor based around three interchangeable annular excimer lamps that each have a different fixed excitation wavelength (308 nm – XeCl, 282 nm – XeBr or 222 nm – KrCl) and illustrate this approach in the streamlined synthesis of a tricyclic scaffold.
There are a wide variety of light sources for photochemistry, particularly for visible light.18 The choice is rather more limited for UV photochemical reactions, especially if a particular wavelength is needed. Reactions are often carried out using medium or low pressure Hg arc lamps, which offer a relatively inexpensive and effective option and are often considered the standard light source for UV photochemical reactions.18 High pressure Hg lamps emit a broad range of visible and UV wavelengths as well as heat, which must be removed by powerful cooling. Frequently, the most intense emission wavelengths of Hg lamps do not match well with the absorption of the targeted molecular chromophore. This can lead to competing formation of by-products and/or product degradation.19 This problem can be addressed in part by using wavelength specific filters, e.g. by using pyrex glassware18 but then more energy is wasted. Phosphor coatings which absorb the UV radiation and reemit less energetic photons at specific wavelengths can harness the unwanted UV but multiple lamps and long irradiation times are often required for scale-up.43 Tunable lasers have been used in the past to explore the wavelength selective irradiation of molecules; however these reactions are often carried out on analytical scales as the scaling up of a laser based system is often not practicable.44
Excimer lamps, such as those used in our reactor, offer a valuable alternative to the use of traditional Hg based lamps because they are intense narrow band UV emitters45 and have none of the problems inherent to the use of Hg. They also have good electrical efficiency further adding to their green credentials. Access to single wavelengths permits more selective irradiation of a molecule as the absorbance of interest can be irradiated solely. Excimer lamps have been used in the past in a few chemical reactions,46,47 but they are most often used in the purification of water48,49 or photocuring.50 In this work we employ our novel continuous UV reactor to establish the most effective irradiation wavelength for a photodecarboxylative C–C coupling reaction for constructing tricyclic scaffolds of potential interest to the pharmaceutical industry.
Results and discussion
Reactor design
The lamps used in this study are cylindrical with a bore of 16 mm diameter down the centre. The reactor sits within this bore. Apart from the entrance and exit holes the entire lamp is enclosed, thereby largely eliminating the hazards associated with scattered UV radiation, which are associated with most Hg lamps. Each lamp works at a single wavelength, (308, 282, or 222 nm). When running, the inside surface of the bore reaches ca. 250 °C.The principal design challenge is how to cool the reactor to prevent heating of the solution while avoiding UV absorption by the coolant. An additional challenge is that, ideally, the reactor and all the feeds (coolant, reaction mixture, and thermocouple) should enter from the same end so that the reactor can be easily withdrawn from the lamp and switched between the three lamps, thereby changing the excitation wavelength without dismantling and reassembling the reactor.
The final version of our reactor is shown in Fig. 1. Our design places the cooling medium inside the reactor rather than placing the cooling between the light source and substrate. Since the light does not have to travel through the cooling medium, there was no requirement for the medium to be UV-transparent, allowing a greater temperature range (i.e. <0 °C) to be used. The cold finger is constructed of stainless steel and is a close fit inside a quartz jacket (essentially a long horizontal test tube) with narrow clearance to provide a thin annular volume for irradiation of the reaction mixture (Fig. 1a and b). The substrate feed pipe (1/16 in. o.d.) runs the entire length of the cold finger to deliver the reaction mixture right at the far end of the quartz jacket. The substrate is then irradiated as it flows back through the annular space between the quartz jacket and the surface of the cold finger to an exit tube.
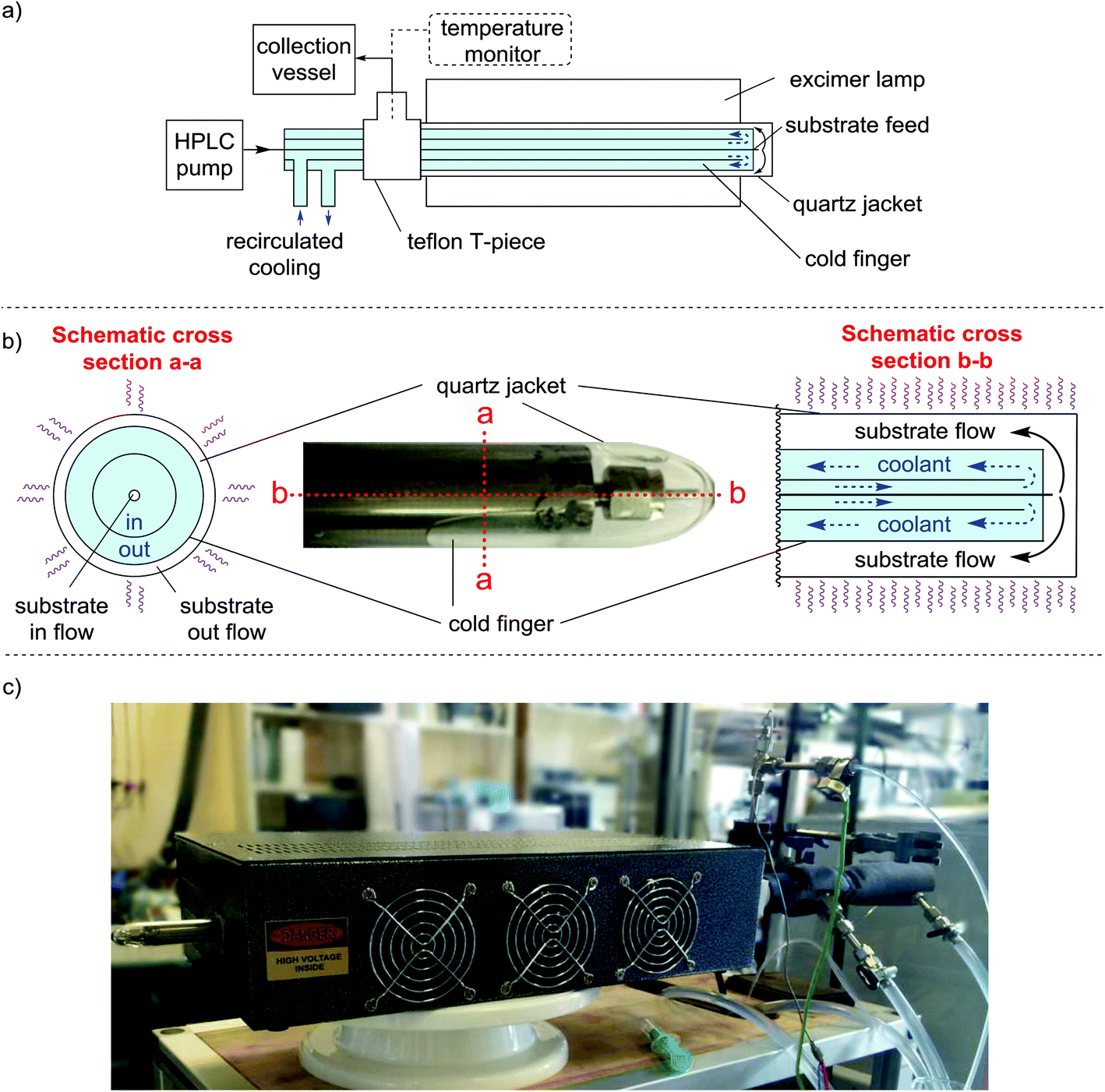 | ||
| Fig. 1 (a) Simplified schematic of our reactor highlighting the ‘tubes within a tube’ design (not to scale). The clearance between the cold finger and the quartz jacket is approx. 120 μm, creating a continuously flowing thin film. The overall irradiation volume of the reactor is 1.37 mL, whilst the system volume is approx. 10 mL. The temperature is monitored directly after the flow exits the quartz jacket. A Teflon T-piece is used to provide a leak free seal to the quartz jacket, the collection pipe is also attached at this point. (b) Photograph showing the tip of the cold finger inside the quartz jacket. (The protruding nut enables the cold finger to be dismantled and rebuilt by the user). The Schematic shows a cross section view of the cold finger with the direction of flow within the tubes (not to scale). The coolant flow is in the same direction as the substrate because, given the dimensions of the pipes this causes less back-pressure than flowing in the opposite direction. (c) Photograph of the whole reactor setup inside one of the excimer lamps. Each lamp consists of the excimer gas discharge tube, power supply and cooling all of which are housed inside a metal box which helps to contain stray UV light (see ESI† for further details). | ||
UV study
Here, we demonstrate the use of our reactor with the photodecarboxylative cyclisation of potassium N-phthalimidobutanoate 1 and show how different excitation wavelengths affect the formation of 2 (Scheme 1). In a later section, we describe how this reaction can be integrated with the acid-catalysed dehydration of 2 to 3. The photodecarboxylation was first reported by Griesbeck and has subsequently been developed by Oelgemöller.2,43,51–55 It is an example of a C–C bond forming reaction, an important class of reaction in synthetic chemistry. Typically C–C bond formation employs transition metal catalysis and/or the use of stoichiometric organometallic reagents, i.e. Grignard reagents. More recently visible photocatalytic routes to forming C–C bonds have emerged,7,13 but the use of UV light circumvents the need for a photocatalyst as direct excitation of the substrate can be achieved. This work builds upon the pioneering work of Griesbeck who used a falling film reactor surrounding a 3 kW excimer lamp.56 More recently the reaction was shown to be more productive when exposed to broadband UV (i.e. when irradiating through quartz instead of pyrex).36 We have chosen this reaction to illustrate the versatility of our new design that allows us to rapidly switch excitation wavelength and assess the productivity of the reaction at the different wavelengths.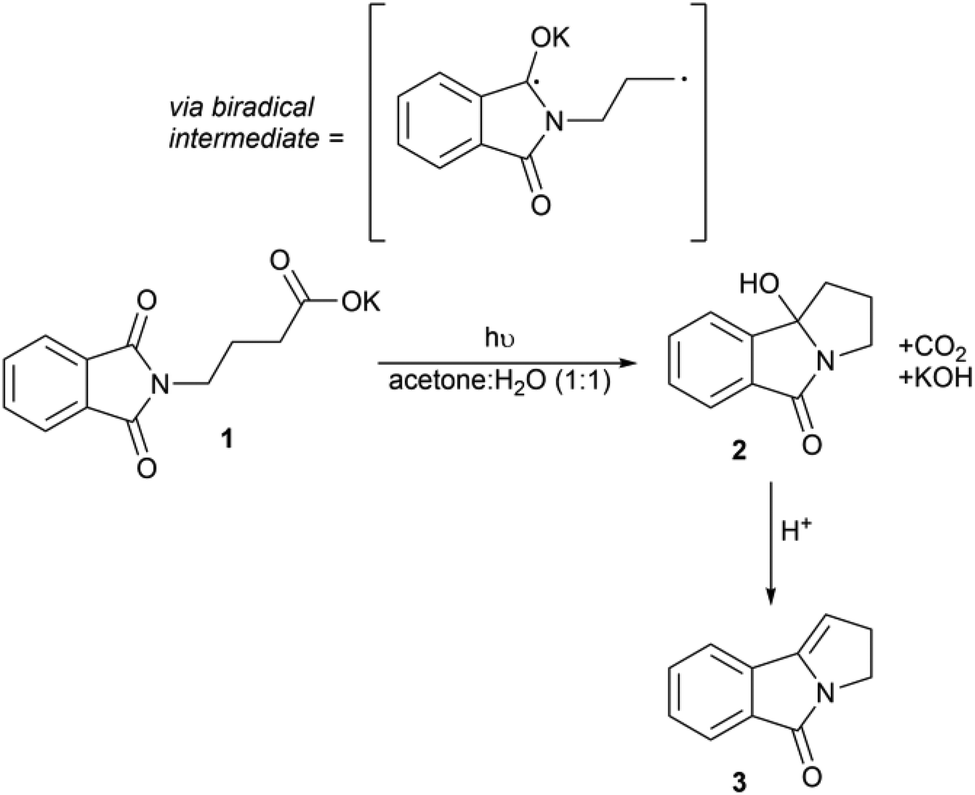 | ||
| Scheme 1 The photodecarboxylation of 1 and subsequent cyclisation to 2via intermediate radical C–C coupling, followed by acid-catalysed dehydration to yield pyrrolizidine 3. | ||
We used aqueous acetone, the solvent previously used for this reaction,57 which provides a good balance between environmental acceptability and solvent power.58–60 Although acetone has an absorption at ca. 280 nm, that absorption is weak compared to the bands of 1. Therefore, in this case, acetone maybe a triplet sensitiser; as it has been used in this role previously but establishing the extent requires further investigation.57,61 Furthermore the miscibility of acetone with water enabled us to use aqueous mixtures to ensure 1 was completely dissolved (i.e. up to 0.2 M in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone:H2O). Above 0.3 M, the mixture became biphasic and some precipitation of 1 and 2 occurred.
:1 acetone:H2O). Above 0.3 M, the mixture became biphasic and some precipitation of 1 and 2 occurred.
Fig. 2a shows the UV-Visible spectrum of 1 with the previously reported62 absorption bands at 220 nm and 295 nm. Using the three excimer lamps, one can target either of these absorptions individually to establish which excitation wavelength is the most productive for obtaining 2. Fig. 2b shows the absorption of the photo-product 2, which absorbs strongly at wavelengths <250 nm and more weakly between 250 nm and 300 nm. Thus 2 will strongly absorb light from the KrCl lamp, more weakly absorb light from the XeBr lamp and barely absorb light from the XeCl lamp at all.
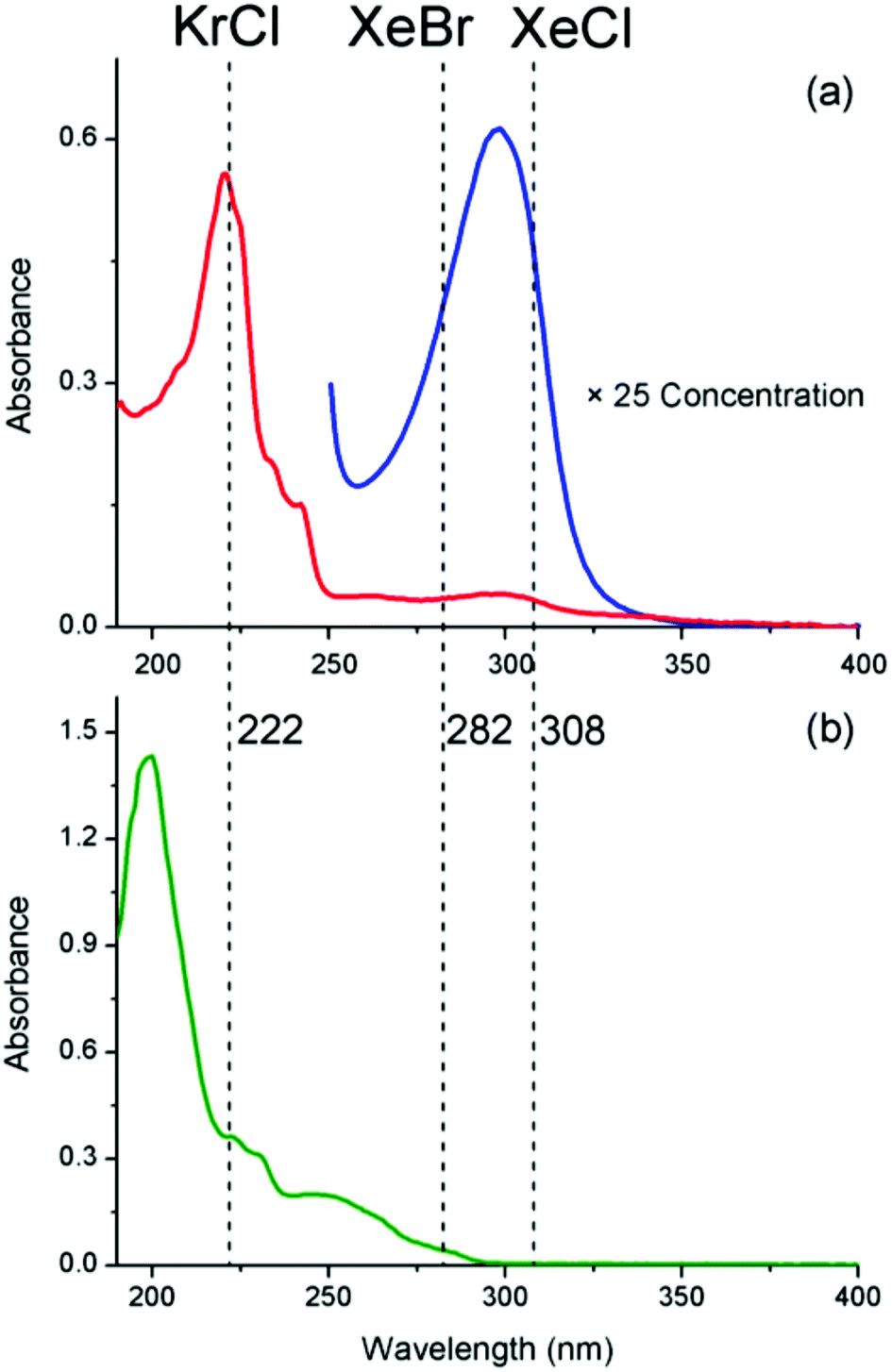 | ||
| Fig. 2 (a) Electronic absorption spectra of 1 at 2.5 × 10−4 M (blue) and 1.0 × 10−5 M (red) in 1:1 MeCN:H2O (pathlength = 1 cm). (b) Electronic absorption spectrum of photo product 2 (5.0 × 10−5 M) in 1:1 MeCN:H2O (pathlength = 1 cm). Note that the solutions used for the photochemistry are far more concentrated. Both spectra are overlaid with the emission wavelengths of the excimer lamps used in this study. | ||
Table 1 summarises the more important experimental results. The following points are clear from the table.
:H2O
| Entry | Conc. (M) | Flow rate (mL min−1) | Yield 2a (%) |
STYb (mmol h−1 mL−1) |
|---|---|---|---|---|
| a Yield determined by 1H NMR integrations using 1,3,5-trimethoxybenzene as an internal standard. b Space time yield is determined by [(substrate concentration × flow rate × yield × 60)/irradiation volume (1.37 mL)]. c Despite all three lamps drawing the same power, each lamp emits a different number of photons per unit area according to the manufacturers figures (KrCl – 222 nm: 21 mW cm−2 or 2.34 × 1016 photons per s per cm2, XeBr – 282 nm: 36 mW cm−2 or 5.11 × 1016 photons per s per cm2, XeCl – 308 nm: 49 mW cm−2 or 7.57 × 1016 photons per s per cm2). | ||||
| 222 nm irradiationc | ||||
| 1 | 0.1 | 0.2 | 37 | 0.32 |
| 2 | 0.1 | 0.5 | 28 | 0.54 |
| 3 | 0.1 | 1.0 | 16 | 0.72 |
| 4 | 0.1 | 1.5 | 8 | 0.55 |
| 5 | 0.2 | 0.2 | 26 | 0.45 |
| 6 | 0.2 | 0.5 | 16 | 0.69 |
| 7 | 0.2 | 1.2 | 9 | 0.82 |
| 8 | 0.2 | 1.5 | 9 | 1.18 |
| 282 nm irradiationc | ||||
| 9 | 0.1 | 0.2 | 97 | 0.85 |
| 10 | 0.1 | 0.5 | 58 | 1.26 |
| 11 | 0.1 | 1.0 | 42 | 1.85 |
| 12 | 0.1 | 1.5 | 38 | 2.47 |
| 13 | 0.2 | 0.2 | 74 | 1.29 |
| 14 | 0.2 | 0.5 | 55 | 2.39 |
| 15 | 0.2 | 1.0 | 35 | 3.09 |
| 16 | 0.2 | 1.5 | 28 | 3.72 |
| 308 nm irradiationc | ||||
| 17 | 0.1 | 0.2 | >99 | 0.88 |
| 18 | 0.1 | 0.5 | 87 | 1.91 |
| 19 | 0.1 | 1.0 | 68 | 2.99 |
| 20 | 0.1 | 1.5 | 43 | 2.80 |
| 21 | 0.2 | 0.2 | 94 | 1.64 |
| 22 | 0.2 | 0.5 | 54 | 2.35 |
| 23 | 0.2 | 1.0 | 32 | 2.83 |
| 24 | 0.2 | 1.5 | 20 | 2.58 |
(a). At all wavelengths, the highest yield of 2 is obtained at the lowest flow rates both at 0.1 and 0.2 M concentrations of 1.
(b). Even at the slowest flow rates, the yields with the KrCl (222 nm) are significantly lower than with the other two lamps. This is probably due to a combination of two factors: absorption by photoproduct 2 and the lower spectral irradiance of this lamp. Bearing in mind that spectral irradiance of KrCl is between 1/2 and 1/3 of the other two lamps, one can see that the STYs per photon are surprisingly similar for all three lamps. Compare entries 1–3 with 9–11 and 17–14. This suggests that the absorption by 2 is relatively unimportant in affecting the yield.
(c). Lower yields but higher STYs can be obtained at higher flow rates. Indeed, the highest STY is obtained with 282 nm and 1.5 mL min−1 corresponding to a yield of only 28% at a residence time of <1 min, entry 16.
(d). Although the 308 nm lamp has the highest spectral irradiance (i.e. produces most photons), the maximum STY appears to be somewhat lower. Compare entries 22–24 with entries 14–16.
(e). In general doubling the concentration of substrate increases the STY in all cases but the increase is nearly always rather less than ×2.
Thus the reaction works well in this reactor. Even at the highest flow rates, however, the flow is unlikely to be turbulent and therefore inner-filter effects may come into play at higher concentrations. Mixing within the film could probably be increased by patterning the surface of the cold finger but this was not attempted. Slight fouling of the reactor occurred at 222 nm and the slowest flow rate, which was observed as a pale yellow film that could be cleaned using DMSO. Nevertheless, in most cases no fouling was observed over periods of 8 hours of running.
To investigate the possible effects of acetone as a sensitiser for the reaction, we repeated the experiments using MeCN:H2O, which has recently been employed for this reaction.36 As this solvent mixture is transparent to UV wavelengths >200 nm, only 1 and 2 will be directly subjected to irradiation. Table 2 outlines the results of the reaction at the three excitation wavelengths using MeCN:H2O (1:1) as the reaction solvent. Comparing Tables 1 and 2, it can be seen that at 0.2 M (Table 2, entries 5–8, 13–16 and 21–24) the yields are close to those obtained using acetone:H2O; this is observed across all wavelengths for this concentration. However at the lower concentration (0.1 M) with MeCN at 308 nm (Table 2, entries 17–20) the yields are generally lower than with acetone; this may well be due to sensitisation. At 282 nm (Table 2, entries 10–12) the yield of 2 was higher compared to the experiments in acetone:H2O, except at the slowest flow rate (Table 2, entry 9) where the yield was 15% lower than with acetone:H2O possibly because at this low concentration of 1 the effect of sensitisation in acetone:H2O will be more pronounced. At 222 nm (Table 2, entries 1–4), the yields appear to be higher in MeCN than in acetone. However in some experiments following prolonged irradiation (i.e. at the slowest flow rate) there was significant mass loss and reactor fouling which resulted in a drop in the observed yield (Table 2, entry 1).
:H2O
| Entry | Conc. (M) | Flow rate (mL min−1) | Yield 2a (%) |
STYb (mmol h−1 mL−1) |
|---|---|---|---|---|
| a Yield determined by 1H NMR integrations using 1,3,5-trimethoxybenzene as an internal standard. b Space time yield is determined by [(substrate concentration × flow rate × yield × 60)/irradiation volume (1.37 mL)]. c Despite all three lamps drawing the same power, each lamp emits a different number of photons per unit area according to the manufacturers figures (KrCl – 222 nm: 21 mW cm−2 or 2.34 × 1016 photons per s per cm2, XeBr – 282 nm: 36 mW cm−2 or 5.11 × 1016 photons per s per cm2, XeCl – 308 nm: 49 mW cm−2 or 7.57 × 1016 photons per s per cm2). | ||||
| 222 nm irradiationc | ||||
| 1 | 0.1 | 0.2 | 83 | 0.73 |
| 2 | 0.1 | 0.5 | 54 | 1.18 |
| 3 | 0.1 | 1.0 | 34 | 1.49 |
| 4 | 0.1 | 1.5 | 22 | 1.45 |
| 5 | 0.2 | 0.2 | 22 | 0.39 |
| 6 | 0.2 | 0.5 | 19 | 0.83 |
| 7 | 0.2 | 1.2 | 17 | 1.49 |
| 8 | 0.2 | 1.5 | 11 | 1.45 |
| 282 nm irradiationc | ||||
| 9 | 0.1 | 0.2 | 82 | 0.72 |
| 10 | 0.1 | 0.5 | 74 | 1.62 |
| 11 | 0.1 | 1.0 | 61 | 2.67 |
| 12 | 0.1 | 1.5 | 35 | 2.30 |
| 13 | 0.2 | 0.2 | 77 | 1.35 |
| 14 | 0.2 | 0.5 | 63 | 2.76 |
| 15 | 0.2 | 1.0 | 39 | 3.42 |
| 16 | 0.2 | 1.5 | 26 | 3.42 |
| 308 nm irradiationc | ||||
| 17 | 0.1 | 0.2 | 89 | 0.78 |
| 18 | 0.1 | 0.5 | 78 | 1.71 |
| 19 | 0.1 | 1.0 | 50 | 2.19 |
| 20 | 0.1 | 1.5 | 26 | 1.71 |
| 21 | 0.2 | 0.2 | 96 | 1.68 |
| 22 | 0.2 | 0.5 | 53 | 2.32 |
| 23 | 0.2 | 1.0 | 32 | 2.80 |
| 24 | 0.2 | 1.5 | 22 | 2.89 |
The co-products of the reaction are CO2 and KOH, which presumably react to form potassium bicarbonate, KHCO3. Since carbonic acid is a weak acid, this will be at least partially hydrolysed under our reaction conditions. Although neither the K+ nor CO2 was quantified, the apparent pH was measured by indicator paper as being approximately 10.
In summary, the conversion of 1 to 2 is very clean with no by-products apart from CO2 and KOH. The transformation is ‘reagent-less’ in the sense that nothing is added aside from photons. The solvent system of acetone/water, as developed in previous studies47,57,63,64 is environmentally relatively benign.58–60 However MeCN:H2O is also viable and has already been shown to be effective on large scales.36 We now demonstrate how this photochemical step can be integrated into a single flow process to make 3.
Integrating photochemistry into a multi-step process
Traditional processes are often carried out in a step-wise manner, with a work-up and isolation step after each reaction, a practice that often leads to large quantities of waste being generated. Continuous flow chemistry potentially offers savings in both solvent and waste by simply integrating reactors, so that multiple steps can be carried out with a single substrate feed. Photochemical reactions are particularly suitable for integrating into sequential reactions because they usually do not require additional reagents, which might complicate subsequent steps.The formation of 3 from 1 is a good example of a reaction sequence to be integrated because the intermediate product 2 can be dehydrated to product 3 by treatment with acid (Scheme 1). This dehydration has been briefly reported previously63 and we confirmed that isolated samples of 2 could be converted to 3 in a batch process with HCl in the same mixture of acetone and water as used for the photochemical reaction. Flow processes are much simpler using solid acids. An unsuccessful attempt was made to replace the HCl in the batch reaction with Amberlyst 15, presumably because the Amberlyst was an insufficiently strong acid. By contrast NbOPO4 was much more successful. This phosphate is a relatively under-utilised solid acid that exhibits high levels of Brønsted acidity (similar to ca. 90% H2SO4)65,66 coupled with Lewis acidic sites that enable a range of reactions to be catalysed on its surface.67–69
The next stage was an attempt to integrate the acid-catalysed conversion of 2 → 3 with the photochemical conversion of 1 → 2 under the conditions that gave the highest conversion (Table 1, entry 17; 308 nm, 0.1 M, 0.2 mL min−1). Initially, the overall process was unsuccessful with samples collected in the first 10 minutes of the reaction showing only 3% conversion to 3. Unsurprisingly, the low conversion was due the rapid deactivation of the solid acid caused by the presence of the KOH by-product which turned out to be neutralising the acidic sites. Therefore we decided to exploit the lower acidity of Amberlyst as a means of removing the KOH before the reaction stream encountered the stronger acid, NbOPO4.
Scheme 2 summarises the overall reactor chain. The product emerging from the photochemical reactor passes through a bed of Amberlyst 15 and then through a third reactor containing a small quantity of NbOPO4. This third reactor was oriented so that there was upward flow of the solution to prevent the finely powdered NbOPO4 from compacting and causing a back-pressure build up in the photochemical reactor. Using this setup, 3 was obtained in 79% conversion from the three coupled steps.
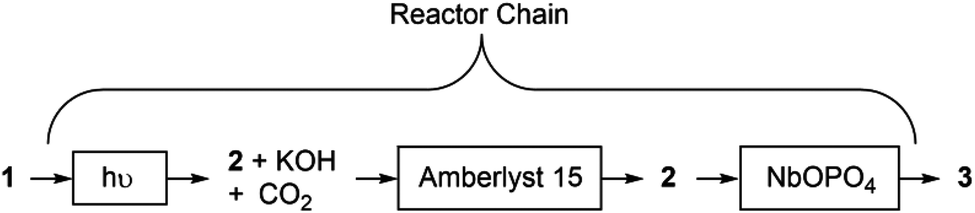 | ||
| Scheme 2 Daisy-chained process from 1 to 3 with three coupled reactors: Reactor 1. UV reactor for the generation of 2 + KOH; Reactor 2. In-line quench to remove KOH generated; Reactor 3. Catalytic NbOPO4 for dehydration of 2 to 3. | ||
Obviously one cannot use Amberlyst indefinitely as a sacrificial scavenger of KOH because the acid sites will eventually become saturated. However, it would be relatively simple to have two Amberlyst reactors in parallel so that one reactor could be used to scavenge KOH while the other was being refilled or preferably regenerated. Nevertheless, a single reactor was sufficient to demonstrate the principle of the integrated process for a period of 10 hours. Eventually the conversion of 3 began to fall from 79% to 15% but this appeared to be the results of channelling in the NbOPO4 rather than saturation of the Amberlyst.
Conclusion
A new UV photochemical reactor that employs rapidly interchangeable narrowband excimer lamps has been described. The reactor has been used to establish the most efficient wavelength and conditions for a photoinduced decarboxylative C–C coupling reaction. At 308 nm, when the reaction is at full conversion, the STY of the 0.2 M reaction is approximately twice that of the 0.1 M reaction for both acetone/water and MeCN/water. This implies there is a significant transmission of UV due to the weak absorption of 1, however at the lower concentration in MeCN the yields were generally lower than with acetone perhaps suggesting a mild sensitisation effect for acetone. At 282 nm, when comparing the reactions at similar conversions for both acetone and MeCN, the 0.2 M reaction also shows consistently higher STY than the 0.1 M reaction. There is little difference in STY between acetone and MeCN at similar conversions. When the STY's at similar conversions are normalised according to the spectral irradiances of the different lamps, that value differs surprisingly little for the three wavelengths studied which is in part due to the photochemical robustness of the aromatic amide 2. We provided a simple demonstration of a new annular reactor for continuous UV photochemistry showing that the interchangeable excimer lamps of different wavelengths can be used to optimise photochemical processes and that daisy chaining this UV reactor with further reactors allows multiple chemical transformations to be carried out in a single streamlined process. Furthermore, the value of NbOPO4 has been demonstrated as a solid acid catalyst to replace strong liquid acids such as trifluoroacetic acid. From the green point of view excimer lamps are energy efficient and have fewer safety issues then Hg lamps and, in the way used here, generate almost no stray UV light. Furthermore, the snug fit of the reactor within the lamp appears to virtually eliminate the generation of ozone by eliminating airflow within the bore of the lamp. We are continuing to explore the efficiency of new photochemical reactions using the continuous excimer UV reactor and study their reactivity with different wavelengths of UV irradiation as this allow us to easily pinpoint the optimal conditions for a photochemical reaction whilst suppressing detrimental side-reactions.Experimental
Warning: UV radiation can be harmful. Full experimental and reactor details are provided in the ESI.† Example procedure for the irradiation of 1: the reactor was flushed with 50 mL of a mixture of degassed acetone–water (1:1 by volume). The circulating bath was set to 30 °C and allowed to stabilise at that temperature. 4-Phthalimidobutyric acid (7.0 g, 30.0 mmol) was suspended in deionised water (75 mL). K2CO3 (2.08 g, 15.0 mmol) was added and the resulting mixture was stirred until completely dissolved. Acetone (75 mL) was added and stirred for 10 minutes, giving a 0.2 M solution with respect to 1. The solution was degassed with argon for 10 minutes and pumped through the reactor at 0.2 mL min−1 under irradiation by the corresponding lamp (222, 282 or 308 nm). Samples were taken at one flow rate before changing to the next flow rate, allowing for equilibration of the system before taking the next samples. Two system volumes (i.e. 2 × 10 mL) were allowed to flow through the rig at each flow rate before collection.
Acknowledgements
We thank the EPSRC for support (EP/L021889/1) and to CBMM for a studentship to J. Jin and for supplying Nb catalyst used in this study. We also thank Dr S. M. Avdeev of IHCE SB RAS, Tomsk, for advice on the excimer lamps, and Dr Z. Amara for helpful discussions. Additionally we thank C. Dixon, M. Dellar, R. Wilson, P. Fields, D. Lichfield and M. Guyler for technical support at the University of Nottingham.Notes and references
- G. Ciamician, Science, 1912, 36, 385–394 CAS.
- M. Oelgemöller, J. Chin. Chem. Soc., 2014, 61, 743–748 CrossRef.
- D. Ravelli, S. Protti, P. Neri, M. Fagnoni and A. Albini, Green Chem., 2011, 13, 1876–1884 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- E. E. Coyle, K. Joyce, K. Nolan and M. Oelgemöller, Green Chem., 2010, 12, 1544–1547 RSC.
- S. Protti and M. Fagnoni, Photochem. Photobiol. Sci., 2009, 8, 1499–1516 CAS.
- S. Protti, D. Dondi, M. Fagnoni and A. Albini, Green Chem., 2009, 11, 239–249 RSC.
- M. Oelgemöller, C. Jung and J. Mattay, Pure Appl. Chem., 2007, 79, 1939–1947 CrossRef.
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC.
- A. Albini and M. Fagnoni, in New Methodologies and Techniques for a Sustainable Organic Chemistry, ed. A. Mordini and F. Faigl, Springer Netherlands, Dordrecht, 2008, pp. 279–293 Search PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- M. Oelgemöller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed.
- M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. – Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322–5325 CrossRef CAS PubMed.
- I. G. T. M. Penders, Z. Amara, R. Horvath, K. Rossen, M. Poliakoff and M. W. George, RSC Adv., 2014, 5, 6501–6504 RSC.
- D. Noutsias, I. Alexopoulou, T. Montagnon and G. Vassilikogiannakis, Green Chem., 2012, 14, 601–604 RSC.
- X. Han, R. A. Bourne, M. Poliakoff and M. W. George, Chem. Sci., 2011, 2, 1059–1067 RSC.
- Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489–495 CrossRef CAS PubMed.
- F. Levesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef CAS PubMed.
- T. Horie, M. Sumino, T. Tanaka, Y. Matsushita, T. Ichimura and J. Yoshida, Org. Process Res. Dev., 2010, 14, 405–410 CrossRef CAS.
- T. Van Gerven, G. Mul, J. Moulijn and A. Stankiewicz, Chem. Eng. Process., 2007, 46, 781–789 CrossRef CAS.
- D. D. Dionysiou, G. Balasubramanian, M. T. Suidan, A. P. Khodadoust, I. Baudin and J. M. Laîné, Water Res., 2000, 34, 2927–2940 CrossRef CAS.
- K. Barberis and C. R. Howarth, Ozone: Sci. Eng., 1991, 13, 501–519 CrossRef CAS.
- O. Shvydkiv, C. Limburg, K. Nolan and M. Oelgemöller, J. Flow Chem., 2012, 2, 52–55 CrossRef CAS.
- K. Jähnisch and U. Dingerdissen, Chem. Eng. Technol., 2005, 28, 426–427 CrossRef.
- M. N. Gandy, C. L. Raston and K. A. Stubbs, Chem. Commun., 2015, 51, 11041–11044 RSC.
- A. Yavorskyy, O. Shvydkiv, C. Limburg, K. Nolan, Y. M. C. Delaure and M. Oelgemöller, Green Chem., 2012, 14, 888–892 RSC.
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, Org. Process Res. Dev., 2016, 20, 1806–1811 CrossRef CAS.
- C. A. Clark, D. S. Lee, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2016, 20, 1792–1798 CrossRef CAS.
- J. T. Hodgkinson, W. R. J. D. Galloway, S. Saraf, I. R. Baxendale, S. V. Ley, M. Ladlow, M. Welch and D. R. Spring, Org. Biomol. Chem., 2011, 9, 57–61 CAS.
- C. F. Carter, H. Lange, D. Sakai, I. R. Baxendale and S. V. Ley, Chem. – Eur. J., 2011, 17, 3398–3405 CrossRef CAS PubMed.
- M. Baumann, I. R. Baxendale, M. Brasholz, J. J. Hayward, S. V. Ley and N. Nikbin, Synlett, 2011, 1375–1380 CAS.
- I. R. Baxendale, C. M. Griffiths-Jones, S. V. Ley and G. K. Tranmer, Synlett, 2006, 427–430 CAS.
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566–2568 RSC.
- O. Shvydkiv, S. Gallagher, K. Nolan and M. Oelgemöller, Org. Lett., 2010, 12, 5170–5173 CrossRef CAS PubMed.
- D. M. E. Davies, C. Murray, M. Berry, A. J. Orr-Ewing and K. I. Booker-Milburn, J. Org. Chem., 2007, 72, 1449–1457 CrossRef CAS PubMed.
- U. Kogelschatz, Atomic and Molecular Pulsed Lasers V, 2004, vol. 5483, pp. 272–286 Search PubMed.
- M. Oelgemöller, P. Cygon, J. Lex and A. G. Griesbeck, Heterocycles, 2003, 59, 669–684 CrossRef.
- A. G. Griesbeck, W. Kramer and M. Oelgemöller, Green Chem., 1999, 1, 205–208 RSC.
- G. Matafonova and V. Batoev, Chemosphere, 2012, 89, 637–647 CrossRef CAS PubMed.
- E. A. Sosnin, T. Oppenländer and V. F. Tarasenko, J. Photochem. Photobiol., C, 2006, 7, 145–163 CrossRef CAS.
- S. Beke, F. Anjum, L. Ceseracciu, I. Romano, A. Athanassiou, A. Diaspro and F. Brandi, Laser Phys., 2013, 23, 035602 CrossRef.
- S. Josland, S. Mumtaz and M. Oelgemöller, Chem. Eng. Technol., 2016, 39, 81–87 CrossRef.
- H. M. Pordanjani, C. Faderl, J. Wang, C. A. Motti, P. C. Junk and M. Oelgemöller, Aust. J. Chem., 2015, 68, 1662–1667 CrossRef CAS.
- M. Oelgemöller, S. Gallagher and K. McCarthy, Processes, 2014, 2, 158–166 CrossRef.
- O. Shvydkiv, K. Nolan and M. Oelgemöller, Beilstein J. Org. Chem., 2011, 7, 1055–1063 CrossRef CAS PubMed.
- Y. Lee, D. Ahn, K. Lee, A. R. Kim, D. J. Yoo and M. Oelgemöller, Tetrahedron Lett., 2011, 52, 5029–5031 CrossRef CAS.
- A. G. Griesbeck, N. Maptue, S. Bondock and M. Oelgemöller, Photochem. Photobiol. Sci., 2003, 2, 450–451 CAS.
- A. G. Griesbeck, A. Henz, W. Kramer, J. Lex, F. Nerowski, M. Oelgemöller, K. Peters and E. Peters, Helv. Chim. Acta, 1997, 80, 912–933 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- A. D. Curzons, D. C. Constable and V. L. Cunningham, Clean Products and Processes, 1999, vol. 1, pp. 82–90 Search PubMed.
- A. G. Griesbeck, Chimia, 1998, 52, 272–283 CAS.
- A. G. Griesbeck and H. Gorner, J. Photochem. Photobiol., A, 1999, 129, 111–119 CrossRef CAS.
- A. G. Griesbeck, A. Henz, K. Peters, E.-M. Peters and H. G. von Schnering, Angew. Chem., Int. Ed., 1995, 34, 474–476 CrossRef CAS.
- A. G. Griesbeck, A. Henz, J. Hirt, V. Ptatschek, T. Engel, D. Löffler and F. W. Schneider, Tetrahedron, 1994, 50, 701–714 CrossRef CAS.
- J. C. G. Da Silva, M. D. Vieira, W. D. Andrade and A. C. B. Dos Santos, J. Mater. Sci., 2005, 40, 4455–4460 CrossRef CAS.
- P. Carniti, A. Gervasini, S. Biella and A. Auroux, Chem. Mater., 2005, 17, 6128–6136 CrossRef CAS.
- W. Weng, M. Davies, G. Whiting, B. Solsona, C. J. Kiely, A. F. Carley and S. H. Taylor, Phys. Chem. Chem. Phys., 2011, 13, 17395–17404 RSC.
- P. Carniti, A. Gervasini, S. Biella and A. Auroux, Catal. Today, 2006, 118, 373–378 CrossRef CAS.
- S. Okazaki and N. Wada, Catal. Today, 1993, 16, 349–359 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02888h |
| This journal is © The Royal Society of Chemistry 2017 |