
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A metagenomics approach for new biocatalyst discovery: application to transaminases and the synthesis of allylic amines†
Damien
Baud‡
a,
Jack W. E.
Jeffries‡
b,
Thomas S.
Moody
c,
John M.
Ward
*b and
Helen C.
Hailes
 *a
*a
aDepartment of Chemistry, UCL, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk
bDepartment of Biochemical Engineering, UCL, Bernard Katz Building, London, WC1E 0AH, UK. E-mail: j.ward@ucl.ac.uk
cAlmac, Department of Biocatalysis & Isotope Chemistry, 20 Seagoe Industrial Estate, Craigavon, BT63 5QD, N. Ireland, UK
First published on 11th January 2017
Abstract
Transaminase enzymes have significant potential for the sustainable synthesis of amines using mild aqueous reaction conditions. Here a metagenomics mining strategy has been used for new transaminase enzyme discovery. Starting from oral cavity microbiome samples, DNA sequencing and bioinformatics analyses were performed. Subsequent in silico mining of a library of contiguous reads built from the sequencing data identified 11 putative Class III transaminases which were cloned and overexpressed. Several screening protocols were used and three enzymes selected of interest due to activities towards substrates covering a wide structural diversity. Transamination of functionalized cinnamaldehydes was then investigated for the production of valuable amine building blocks.
Introduction
The use of biocatalytic strategies holds significant potential for the sustainable synthesis of fine chemicals and pharmaceuticals. One important class of enzymes is the transaminases (TAms, EC 2.6.1) that can convert ketones and aldehydes into the corresponding amines in high yields and selectivities under mild aqueous reaction conditions.1,2 TAms catalyse the amino group transfer from an amine donor to a ketone, or aldehyde amine acceptor, using pyridoxal-5′-phosphate (PLP) as co-factor.3 The first step of the reaction is the transfer of the amino group of the amine donor resulting in the formation of pyridoxamine-5′-phosphate and release of the amine donor as a ketone or aldehyde. The second step is the regeneration of PLP with the transfer of the amino group to the amine acceptor which is released as an amine.1,4 Notably, TAms are an interesting alternative to chemical synthetic methods from an environmental and economic point of view and have been reported in the synthesis of a number of (R)- or (S)-pharmaceutical ingredients or biologically active compounds.5Currently TAms have significant potential for the synthesis of an increasing number of amine products. Although a number of TAms have been reported and are now available commercially there is a need to identify new TAms with variation in the primary sequence for future applications including enzyme engineering projects: a unique starting point has the potential to ensure more diverse enzymes are available. In addition new TAms not encumbered by the current patent space would be very valuable. Here our approach has been to use a metagenomics mining strategy for new TAm discovery.
Metagenomics is the concept of processing and analysing DNA extracted from an environment as if it were a single large genome.6 A frequently quoted statistic suggests that on average only 0.1–1% of bacteria are culturable from any given niche, depending on the complexity and the understanding of said niche.7,8 Attempts have been made to address this shortfall between cultivatable bacteria and the diversity known to be present. Extracted DNA can be fragmented and ligated into large Bacterial Artificial Chromosome (BAC) clone libraries. Once the environmental DNA is in a suitable host such as E. coli the BACs can be screened for desired activity using plate screens or amplified to a level compatible with Sanger sequencing methods of analysis. Such BAC and Fosmid libraries are currently used to interrogate metagenomic samples to discover new enzyme activities or new examples of enzymes with existing activities, although this is a strategy that requires suitable high-throughput screens.9,10
With the advent of high throughput sequencing technologies, raw DNA extracted from the environment can be used for sequence based analysis without the need for amplification in BAC libraries. Most of the research using such sequence based analysis has been aimed towards using microbial 16S based taxonomy to understand the range and diversity in a microbiome. More recently it has been used to observe how the microbiome changes with different inputs or host alterations, interactions between organisms and between organisms and the host, with protein annotations being used primarily to infer nutrient use and nutrient flow through the niche.11–13
More recently it has been used to observe how the microbiome changes with different inputs or host alterations, interactions between organisms and between organisms and the host, with protein annotations being used primarily to infer nutrient use and nutrient flow through the niche.11–13
Sequence data is used less frequently as an aid to enzyme discovery, most often by providing consensus sequences for primer design.14,15 The strategy applied here uses a recently reported sequence-directed enzyme retrieval method involving the identification of individual open reading frames (ORFs) with subsequent specific primer design to each non redundant example of a desired enzymes class.16 From amino acid sequence alignment there are six groups of TAms.1 The Class III TAms are characterized by broad substrate acceptance and high regio- and stereoselectivities and have been used in a wide range of applications for the synthesis of single isomer chiral amines.1,2 Here, the interrogation of an in silico metagenomic library identified 15 full length non redundant Class III transaminases of which 11 were successfully expressed and used in screens to identify substrate profiles.
Results and discussion
Protein selection and production
DNA, extracted from combined oral tongue scrapings from nine volunteers, was sequenced using the Roche 454 Titanium platform.16 Sequencing returned 1.182 million reads with a mean sequence length of 385 base pairs (bp). This raw data was submitted to bioinformatics analysis for quality control to remove corrupted sequences, trim corrupted ends and filter out human sequences. After this the data set contained 1048 million reads with a mean length of 410 bp. This reduced data set was assembled using MIRA for the creation of a contiguous (contig) read library, where individual reads were joined into longer stretches of DNA with Spoiler detection in MIRA to prevent chimeric contigs from joining. Sequencing of the DNA sample was of sufficient depth to create 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 971 contigs containing 76.8 Mbase pairs with the largest contig being 42 kbp and 2347 contigs were over 5 kb giving a large library of contigs containing several full length open reading frames (ORFs).11
971 contigs containing 76.8 Mbase pairs with the largest contig being 42 kbp and 2347 contigs were over 5 kb giving a large library of contigs containing several full length open reading frames (ORFs).11
The contig library was analysed with the Genemark software to identify and mark ORFs. These putative protein-coding stretches of DNA were then scanned by the Pfam standalone tool. This process marked the ORFs with a Pfam ID linked to a specific protein family or enzyme class. Because of this all ORFs of a desired enzyme class, provided they have a Pfam ID, within the contig library could be extracted.
The Pfam ID for Class III TAms was used to extract all ORFs marked from the contig library. 53 ORFs were identified of which 15 were full length and non-redundant. Enzymes identified from the metagenome were to be cloned using conventional restriction cloning techniques. To this end the sequences for retrieval were scanned for internal restriction sites. Primer pairs where designed for enzymes including an Nde1 restriction site in the forward primer and various restriction enzyme sites in the reverse primer depending on any endogenous restriction sites. 11 out of 15 sequences were successfully retrieved from the metagenome. Blunt end PCR product was ligated into the PCR capture vector PCR-Blunt and through a series of restrictions and ligation was cloned into the pET29a expression vector. The pET29a vector containing the enzyme DNA sequences was transformed into the BL21*DE3 pLysS strain of E. coli for expression. The 11 putative enzymes were named with pQR numbers (see ESI†) and analysed by SDS-PAGE. All of the enzymes had a good level of expression except pQ1108 with a lower level of total expression of soluble and insoluble protein (Fig. 1, B) and pQR1117 (Fig. 1, K) with very little total expression. pQR1112 was completely insoluble (Fig. 1, F1 (total protein), F2 (soluble fraction)) and pQR1114 was partially insoluble (Fig. 1, H1 (total protein), H2 (soluble fraction)).
 | ||
| Fig. 1 SDS gels of induced TAms. Two samples for each enzyme was loaded onto the gels, total protein fraction and soluble fraction respectively (1 = total protein fraction and 2 = soluble fraction). 5 μL of sample was loaded into each well (from a mixture of 10 μL of protein sample and 30 μL of loading dye). Protein markers 10–250 kDa (broad range ladder NEB). Enzyme molecular weights were calculated using the online ExpPASy ProtParam tool and are given in kDa after the plasmid name. (A) Empty vector (B) pQR1108 – 52; (C) pQR1109 – 46.2; (D) pQR1110 – 50.4; (E) pQR1111 – 47.5; (F) pQR1112 – 44.6; (G) pQR1113 – 52.2; (H) pQR1114 – 54; (I) pQR1115 – 47.4; (J) pQR1116 – 47.9; (K) pQR1117 – 48.6; (L) pQR1118 – 48.4. | ||
Screening of 11 potential TAms
The 11 potential TAms were then screened as crude cell lysates using three different methods (Scheme 1): an (S)-α-methylbenzylamine ((S)-MBA 1) amine donor assay against 21 aldehydes and ketones (Fig. 2A) with detection of acetophenone 2 formation by HPLC at 250 nm (Scheme 1A); a benzaldehyde 3 amine acceptor screening method based on benzylamine 4 formation detected by HPLC at 210 nm (Scheme 1B) with five classical amine donors (Fig. 2B), and a copper sulfate assay based on the detection of alanine 6 by formation of a complex with copper,17 monitored with a spectrophotometer at 595 nm using sodium pyruvate 5 as amine acceptor (Scheme 1C) and nine amine donors (Fig. 2C). | ||
| Scheme 1 Screening assays performed with the TAms. (A) (S)-MBA 1a screening reaction conditions: 5 mM aldehyde or ketone, 25 mM (S)-MBA 1a, 1 mM, KPi pH 7.5 100 mM, enzyme as crude cell lysate 0.2–0.4 mg mL−1, 18 h, 30 °C, 500 rpm. (B) Benzaldehyde screening reaction conditions: 5 mM benzaldehyde 3, 25 mM amine donor, 1 mM, KPi pH 7.5 100 mM, enzyme as crude cell lysate 0.2–0.4 mg mL−1, 18 h, 30 °C, 500 rpm. (C) Copper sulfate screening reaction conditions: 15 mM sodium pyruvate 5, 20 mM amine donor, 1 mM KPi pH 7.5 100 mM, enzyme as crude lysate 0.2–0.4 mg mL−1, 1 h, 30 °C, 500 rpm. | ||
 | ||
| Fig. 2 Substrates screened with assays (A) ((S)-MBA 1 and HPLC detection of 2), (B) (benzaldehyde 3 and HPLC detection of 4), and (C) (pyruvate 5 and spectrophotometric detection of alanine 6 after treatment with CuSO4). | ||
Three of the 11 TAm enzymes, pQR1108, pQR1113 and pQR1114 were found to accept a wide range of structurally diverse aldehydes and ketones 7–25 from the (S)-MBA screening, and selected results are shown in Fig. 3. pQR1108 showed the highest activity towards both linear and cyclic substrates. For the cyclic substrates, the best conversion yields were obtained with benzaldehyde 3 (61%) and cinnamaldehyde 15 (44%). Modification or absence of the aromatic ring led to significantly lower conversions with for example 4-hydroxybenzaldehyde 13 and cyclohexanecarboxaldehyde 10 (22% and 24% conversion yields, respectively). More hindered aromatic substrates like 1-indanone 14 resulted in lower conversion yields (8%). Cyclohexanone 7 was also well accepted by pQR1108 (27%) and modification of the six-membered ring led to a lower activity for 2-methylcyclohexanone 8 (4%) or no activity for the enone cyclohex-2-en-1-one 9. When using linear substrates, the length of the carbon chain seemed to be important: a 42% conversion yield was obtained with pQR1108 and butanal 25 but 11% for octanal 24. Functionalized aldehydes or ketones were also accepted, for example when using L-erythrulose 20 (9% yield) and glycoaldehyde 21 (9% yield) (Fig. 3). Linear ketones such as 2-heptanone 22 and 2-butanone 23 were not accepted. In order to confirm that the activity was due to the overexpressed TAm an assay for each (positive) substrate was performed in triplicate with E. coli cells containing an empty vector: no activity was observed. The benzaldehyde screening with five classical amine donors ((S)-MBA 1a, (R)-MBA 1b, (S)-alanine 6a, (R)-alanine 6b and isopropylamine 26) did not give rise to any new activities: benzylamine formation was only observed when using (S)-MBA. This highlighted that all three enzymes exhibited (S)-stereoselectivity. For the copper sulfate screening, with sodium pyruvate as amine acceptor and nine different amine donors 4, 27–34, no activity was observed, consistent with the (S)-MBA screen where pyruvate 5 was not accepted as a substrate. The remaining 8 enzymes showed negligible activity against the substrates screened. This reflected for pQR1117 poor enzyme expression, pQR1112 low levels of soluble protein formed, and for pQR1109, pQR1110, pQR1111, pQR1115, pQR1116, pQR1118 most likely that preferred donors/acceptors were not screened. Using a combination of the Bradford assay and SDS page densitometry to calculate protein loading, specific activities for pQR1108, pQR1113 and pQR1114 were investigated for two substrates 3 and 10.
 | ||
| Fig. 3 Screening results of three of the TAms against 12 selected substrates. Screening conditions: 5 mM aldehyde or ketone, 25 mM (S)-MBA 1a, 1 mM KPi (pH 7.5, 100 mM), enzyme as crude cell lysate 0.2–0.4 mg mL−1, 18 h, 30 °C, 500 rpm. Conversion yields (Scheme 1A assay via detection of 2) were obtained from three independent experiments and varied by <±4%. | ||
pQR1108 had the highest specific activities of 1.7 μmol min−1 mg−1, and 0.4 μmol min−1 mg−1 for 3 and 10 respectively. pQR1113 had an activity of 0.1 μmol min−1 mg−1 for 10. It was not possible to calculate specific activities for pQR1113 with 3 and pQR1114 with 3 and 10 due to low activity over the course of the reaction.
Sequence homology of the 11 potential TAms was compared18,19 against 4 reported TAms from Vibrio fluvialis (Vf-TAm),20Chromobacterium violaceum DSM30191 (Cv-TAm),21Pseudomonas putida KT2440 (Pp-TAm)22 and Klebsiella pneumonia JS2F (Kp-TAm).23 Interestingly, pQR1113 and pQR1114 were found to have high sequence homology (93% identity) in agreement with the screening results. pQR1108 was found to have reasonably high sequence homology with Kp-TAm (55% identity plus 23% similarity) (Fig. 4). The three enzymes had less than 30% identity to Cv-TAm and Vf-TAm. The remaining 8 enzymes which had shown negligible activity had less than 30% identity with the 4 reported TAms (see Table 1 ESI† for sequence homology comparison to Cv-TAm). pQR1109, pQR1110, pQR1111, pQR1115, pQR1116 and pQR1118 had between 70–95% homology with each other and it is likely that these TAms require a different amine donor. Similarly pQR1113, pQR1114 and pQR1117 form a cluster of TAms that are closely related to each other and are likely to have other donor specificity.
 | ||
| Fig. 4 Amino acid sequences of the metagenomics TAms and 4 TAms from identified species (Kp-TAm, Pp-TAm, Cv-TAm and Vf-TAm) were aligned and used to generate a maximum likelihood phylogenetic tree (bootstrap values calculated from 1000 replicates).19,24 | ||
The important active site residues of pQR1108, pQR1113 and pQR1114 were also compared with the same 4 reported TAms. Nine residues are considered as very important in the Cv-TAm dimer and are coloured in red for the key residues of the first monomer and in green for the key residues of the second monomer (Fig. 5).25 Most residues were conserved in the 3 new enzymes. For example, Lys288 (using numbering of amino acids based on the sequence of Cv-TAm) is involved in the formation of a covalent bond with the PLP. Asp259, Ser121 and Tyr153 which are known to be important for the coordination of the PLP in the active site are also all fully conserved in our 3 enzymes. The residue Asp259 is involved in hydrogen bonding with the pyridine ring of the PLP and Ser121 and Tyr153 have roles in the phosphate group coordination of the PLP.
 | ||
| Fig. 5 Mulitple alignment of the active metagenomics enzymes with Kp-TAm, Pp-TAm, Cv-TAm and Vf-TAm. Residues important for substrate and PLP binding in the Cv-TAm dimer are shown in red for the key residues in the first monomer and in green for the key residues in the second monomer.26 Lysine 288 crucial for Schiff base formation is represented with a red star. | ||
Reaction optimisation
The main parameters influencing the rate of a biocatalytic transamination (temperature, buffer, substrate and co-factor concentrations, pH, co-solvents,) were studied with enzyme pQR1108, pQR1113 and pQR1114 using (S)-MBA 1a with cyclohexanecarboxaldehyde 10 (5 mM) as amine acceptor in order to identify the preferred reaction conditions. The results are shown in Fig. 6. Regarding the buffer, a slightly higher conversion was obtained with pQR1108 and PIPES, while HEPES gave a much lower conversion: due to the low cost of potassium phosphate buffer this was used in subsequent experiments. For (S)-MBA 1a concentration, with pQR1108 no inhibition was observed when using up to 50 mM, and for the 3 enzymes 25 mM of 1a was preferred so was used. A slight increase in conversion yield was also observed for pQR1108 when lowering the potassium phosphate (KPi) buffer concentration to 50 mM (27% yield compared to 24% yield at 100 mM) and by operating at pH 8.0 (30% yield against 24% yield at pH 7.5). For pQR1113 and pQR1114 a pH of 7.5 and 50 mM KPi was preferred. | ||
| Fig. 6 Effect of various parameters for the transamination of cyclohexanecarboxaldehyde 10. Conversion yields (Scheme 1A assay via detection of 2), were obtained from experiments in triplicate and varied by <±4%. | ||
Using these preferred conditions it was established that the 3 enzymes were not affected by addition of a co-solvent (10% v/v). Interestingly, the conversion with pQR1108 increased with addition of co-solvents DMSO or MeOH (30% and 29% yields compared to 24% without cosolvent) perhaps reflecting increased substrate solubility. The best reaction temperature observed was 30 °C for the 3 enzymes, and a PLP concentration in the range of 1–1.5 mM was required (data not shown). With the following improved conditions, (S)-MBA 1a 25 mM, amine acceptor 10 5 mM, PLP 1 mM, KPi buffer pH 8 and 50 mM, and DMSO as co-solvent (10% v/v) and pQR1108 as cell lysate (0.3 mg mL−1) for 18 hours gave a 41% conversion yield.
Synthesis of allylic amines
The transamination of conjugated aldehydes including cinnamaldehyde and functionalised derivatives could give access to allylamine building blocks for the synthesis of analogues of drugs such as the antifungal Naftin® (naftifine).26,27 They could also be used for the synthesis of cinnamylamine derivatives: indeed several related compounds such as cinnamamides have shown promising activities against breast cancer cells.28,29Transaminases have been reported for the synthesis of a wide range of amines, however little has been described on the acceptance of conjugated aldehydes/ketones other than the use of cinnamaldehyde 15 as an amine acceptor with Cv-TAm and Vf-TAm.21,30,31 Since pQR1108 demonstrated good activity towards cinnamaldehyde 15 in initial screens it was used with a range of conjugated aromatic aldehydes 35–41, 3-phenylpropanal 42 to investigate the acceptance of a non-conjugated analogue, and several conjugated aliphatic analogues 43–47 to generate allylic amines (Table 1). The previously optimized reaction conditions were used with (S)-MBA 1a as the amine donor. Cinnamaldehyde 15 was most readily accepted in comparable conversion yields to those reported with Cv-TAm.21 Other cinnamaldehyde analogues were accepted other than (E)-3-(4-(dimethylamino)phenyl)acrylaldehyde 38 probably due the dimethylamine electron donating group reducing the electrophilicity of the aldehyde. Modification to the aromatic ring with other electron donating groups including ortho- and para-methoxy and para-methyl moieties, 35, 39 and 37 respectively, gave rise to lower yields (39%–58%) than with cinnamaldehyde again reflecting the lower electrophilicity of the aldehyde. (E)-3-(4-Bromophenyl)acrylaldehyde 36 was also readily accepted in 48% conversion yield. Increasing the steric bulk of the amine acceptor and addition of a methyl group or bromine at the alkene C-2 position led to a slight decrease in conversion yield compared to cinnamaldehyde: (2E)-2-methyl-3-phenylpropenal 40 and (2Z)-2-bromo-3-phenylpropenal 41 were accepted in 57% and 54% yield. The non-conjugated aldehyde 42 gave a conversion yield similar to the substituted cinnamaldehydes (56%). For the linear conjugated aldehydes 43–47, acrolein 43 was accepted although the use of crotonaldehyde 44 gave rise to higher yields. 3-Methylcrotonaldehyde 45, 2-pentenal 46 and 2-methyl-2-pentenal 47 were also accepted but in slightly lower conversion yields than for crotonaldehyde 44. The ease of generating such allylic amines using transaminases is extremely useful and has not been reported to date: it provides a sustainable synthetic approach avoiding the use of metal catalysts or toxic reducing agents.
|
|
||
|---|---|---|
| Aldehyde | Conversiona | |
| Reaction conditions: (S)-MBA 25 mM, amine acceptor 5 mM, PLP 1 mM, KPi buffer 50 mM at pH 8, DMSO as co-solvent (10% v/v) and pQR1108 cell lysate (1.2 mg mL−1), 500 rpm for 18 hours.a Conversions (Scheme 1A assay via detection of 2) were obtained from three independent experiments and varied by <±4%.b Quantified using the amine product and HPLC.c Isolated yield after scale-up. | ||
| 15 |

|
72% |
| 75%b | ||
| 35 |
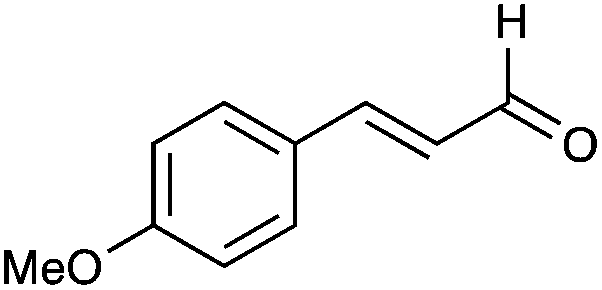
|
39% |
| 36 |
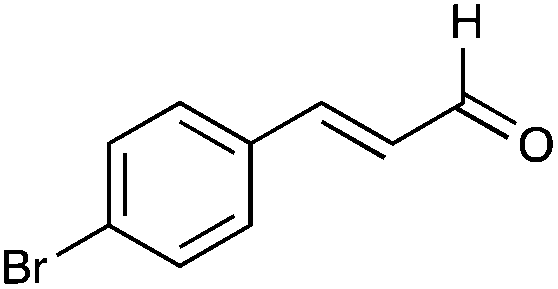
|
48% |
| 37 |

|
49% |
| 38 |

|
0% |
| 39 |

|
58% |
| 57%b | ||
| 84%c of 48 | ||
| 40 |

|
57% |
| 41 |

|
54% |
| 42 |
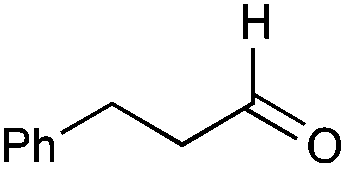
|
56% |
| 58%b | ||
| 43 |
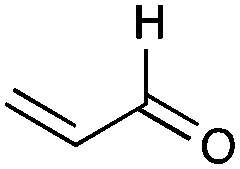
|
35% |
| 44 |

|
49% |
| 45 |
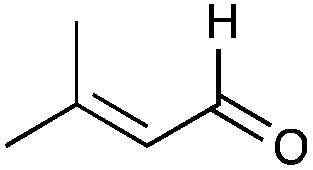
|
45% |
| 46 |

|
37% |
| 47 |

|
37% |

To demonstrate the applicability of using pQR1108 further a preparative scale reaction was carried out with the optimised reaction conditions and 2-methoxycinnamaldehyde 39. After 18 hours a quantitative conversion based on acetophenone formation was observed possibly due to an improved reaction when stirring on the larger scale. After purification, amine 48 was isolated in 84% yield.
Conclusions
The present study demonstrates the efficiency of combining metagenomics for new enzyme discovery and screening to identify new transaminases. Metagenomics allows the mining of enzymes based on their protein sequence from in silico libraries of enzymes. It can thus uncover diverse enzymes that may have not been detected in microwell or agar plate screens. Eleven putative transaminases have been screened against several substrates and three new transaminases were identified as being active towards a broad range substrates. The biocatalytic potential of the new TAms was illustrated with the sustainable synthesis of allylic amines using pQR1108: such amine building blocks can be used as precursors for the synthesis of analogues of several drugs. We are currently expanding the enzyme discovery approach using other metagenomic samples and a range of important biocatalytic enzyme families.Experimental
General information
The solvents and chemicals were purchased from Sigma Aldrich or Fluorochem and were used as supplied. NMR spectra were recorded on a Bruker Spectrometer AMX300. Chemical shifts (in ppm) are quoted relative to tetramethylsilane and referenced to residual pronated solvent. Coupling constants (J) are measured in hertz (Hz) and multiplicities for 1H NMR coupling are shown as s (singlet), d (doublet), t (triplet), and m (multiplet). HPLC analyses were performed on an Agilent 1260 Infinity provided with a 1260 VWD detector. Mass spectrometry analyses were performed at the UCL Chemistry Mass Spectrometry Facility using a Finnigan MAT 900XP mass spectrometer. Accurate mass determination was performed by the peak-matching method.Enzyme selection and production
Sequences generated by the Roche 454 titanium platform were passed to the online platform MG-Rast, redundant sequences were removed, sequences filtered against human reference sets and dereplicated. The filtered output of MG-Rast was further analysed using the Galaxy platform to assign a Phred quality score to each nucleotide base. All sequence fragments that had a minimum score of 20 and length of 50 nucleotides were carried on for further analysis. The MIRA assembler software was used to take these individual sequences reads and assemble them into larger contiguous reads. The ORFs within these contigs were marked using the Genemark software and subsequently marked with a Pfam ID using the Pfam standalone tool.Contigs that contained ORFs marked with the Pfam identifier for Class III TAms were collected and visualised using the Artemis DNA viewer. ORFS that were truncated by being at the start or end of the contig were discarded. DNA corresponding to full length ORFs was excised. Due to inaccuracies in the in silico sequences stemming from the sequencing technology, ORFs comprising the whole sequence were often fragmented across the three reading frames. Due to this DNA sequence downstream of the suspected stop codon was collected to ensure the full length of the coding sequence was taken forward for analysis. Collected DNA for the ORFs was aligned and visualised using the MEGA software. Multiple sequences were highly similar at the amino acid level, for these matching sequences one example was chosen to take forward for primer design.
PCR primers were designed for the 15 identified enzymes. Due to inconsistencies in the in silico DNA sequences, terminal primers were designed beyond the suspected 3′ end of the enzyme. Primers were designed to contain an NdeI restriction site in the forward primer and an NcoI, XhoI or BglII site in the terminal primer depending on restriction sites within the coding sequence. PCR was carried out on the metagenomics DNA sample using NEB Phusion PCR kit. All reactions were carried out following the standard protocol given in the kit with 52 ng of metagenomic DNA per reaction and a primer concentration of 5 pmol per reaction. Successful PCR was followed by gel electrophoresis with PCR products cut out of the gel and DNA recovered using Qiagen Gel extraction columns. Blunt ended PCR products were ligated into a PCR-Blunt capture vector and transformed into Top10 cloning cells. Top10 cells harbouring the plasmid were grown overnight in 5 mL of Terrific Broth with kanamycin 50 μg mL−1. Plasmids were extracted using Qiagen miniprep kits and sent for sequencing to confirm the 3′ end of the coding sequence of the enzymes with confidence. A second round of terminal primers were designed to remove the stop codon and introduce a restriction site. PCR was carried out using these primers and the PCR capture vector containing the PCR product from the first round of PCR. The same process of cloning as described above was carried out on the 2nd round of PCR products. Plasmid carrying the PCR product from this second round of cloning was purified and digested with Nde1 and the requisite restriction enzymes to generate cohesive ends. The digested plasmid was run on a gel and the cohesive ended insert cut out and extracted from the gel. The digestion fragments were ligated into pET29a, digested to generate matching cohesive ends, and transformed into BL21* DE3 pLysS cells.
Small scale enzyme expression and preparation
The TAms in E. coli BL21(DE3)pLysS were added to terrific broth medium (5 mL) containing kanamycin (50 μg mL−1) and chloramphenicol (25 μg mL−1) and incubated overnight at 37 °C: 1 mL of the overnight culture was then used to inoculate terrific broth (100 mL containing the same concentration of antibiotic) which was grown at 37 °C until an OD600 of 0.6 was reached at which point they were induced with 1 mM IPTG and grown for a further 5 h at 25 °C. The cells were harvested by centrifugation, the supernatant removed, and the cell pellet resuspended in potassium phosphate buffer (KPi; 5 mL of 0.1 M KPi pH 7.5, 1 mM PLP) and flash frozen in liquid nitrogen. The frozen pellet was placed on a freeze dryer. 25 mg of freeze dried cells were resuspended in the same 0.1 M KPi buffer pH 7.5 (1 mL) as before and sonicated. The lysed freeze dried cells were spun down and the enzyme concentration was determined using the Bradford method and then diluted if necessary to obtain an enzyme concentration of between 2.0 and 4.0 mg mL−1.Initial (S)-MBA screening method
(S)-MBA 1a (25 mM) and PLP (1 mM) in KPi buffer (pH 7.5, 100 mM), and the substrate (5 mM in water other than for cinnamaldehyde 15 and 1-indanone 14 when DMSO was used) were added in a 96 well-plate to give a total volume of 180 μL. The reaction was started by the addition of 20 μL of clarified cell extract (final concentration of 0.2–0.4 mg mL−1). After incubation for 18 h at 30 °C and 500 rpm, the reaction was quenched with 1 μL of TFA and denatured protein was removed by centrifugation. The supernatant (180 μL) was diluted with water (540 μL) and analysed by HPLC using an ACE 5-C18300 column (150 × 4.6 mm) with UV detection at 250 nm. The concentration of acetophenone produced was determined using a linear gradient 15%–72% over 10 minutes at 1 mL min−1 (A = water with 0.1% of TFA and B = acetonitrile) with subtraction of a negative control without amine acceptor for all substrates. The acetophenone produced eluted at a retention time of 8.8 minutes. Results were verified in triplicate against a negative control without amine acceptor following the same procedure.Benzaldehyde screening method
The amine donor (25 mM) and PLP (1 mM) in KPi buffer (pH 7.5, 100 mM), and benzaldehyde (5 mM in water) were added in a 96 well plate to give a total volume of 180 μL. The reaction was started by the addition of 20 μL of clarified cell extract (final concentration of 0.2–0.4 mg mL−1). After incubation for 18 h at 30 °C and 500 rpm, the reaction was quenched with 1 μL of TFA and denatured protein was removed by centrifugation. The supernatant (180 μL) was diluted with water (540 μL) and analysed by HPLC using a ACE 5-C18 300 column (150 × 4.6 mm) with UV detection at 210 nm. The concentration of benzylamine was determined using a gradient 15%–72% B over 10 minutes at 1 mL min−1 (A = water with 0.1% of TFA and B = acetonitrile). The benzylamine produced eluted at a retention time of 3.4 minutes.CuSO4/MeOH screening method
Preparation of the CuSO4/MeOH solution: 300 mg of copper(II) sulfate pentahydrate was dissolved in 500 μL of water and 30 mL of MeOH. Sodium pyruvate (25 mM) and PLP (1 mM) in KPi buffer (pH 7.5, 100 mM) and the substrate (5 mM in water) were added in a 96 well plate to give a total volume of 180 μL. The reaction was started by the addition of 20 μL of clarified cell extract (final concentration of 0.2–0.4 mg mL−1). After incubation for 1 h at 30 °C and 500 rpm, 40 μL of the prepared staining solution was added and the reaction mixture was then centrifuged (1 minute at 9000 rpm) to remove the insoluble phosphate–copper complex. The supernatant was then added to a 96 well-plate and absorbance was measured at 595 nm with a spectrometer SpectraMax Plus 384 from Molecular Devices. ΔOD was then calculated between the assay and a negative control without amine acceptor. This value was compared with ΔOD calculated for Cv-TAm and benzylamine as an amine donor and used as a reference (for 100% of relative activity). The limit of sensitivity was established to be 10% of relative activity.(S)-MBA screening with preferred conditions for conjugated aldehydes
All substrates were solubilized in DMSO to give a final concentration of 10% v/v. (S)-MBA (25 mM) and PLP (1 mM) in KPi (pH 8.0, 50 mM) and the aldehyde (5 mM in water) were added to a 96 well plate to give a total volume of 140 μL. The reaction was started by the addition of 60 μL of clarified cell extract of pQR1108 (final concentration of 1.2 mg mL−1). After an incubation for 18 h at 30 °C and 500 rpm, the reaction was quenched with TFA (1 μL, 0.5% v/v) and denatured protein was removed by centrifugation. The supernatant (180 μL) was diluted with water (540 μL) and analysed by HPLC using a ACE 5-C18 300 column (150 × 4.6 mm) with UV detection at 250 nm. Concentrations of acetophenone were determined using a linear gradient 15%–72% B over 10 minutes at 1 mL min−1 (A = water with 0.1% of TFA and B = acetonitrile) with subtraction of a negative control without amine acceptor for all the substrates. The acetophenone produced eluted at a retention time of 8.8 minutes.Preparative scale reaction
The enzymatic reaction was performed using a total volume of 100 mL, containing (S)-MBA 1 (25 mM, 2.5 mmol, 302 mg) as amine donor, (E)-3-(2-methoxyphenyl)acrylaldehyde 39 (5 mM, 0.5 mmol, 82 mg) as amine acceptor, PLP (1 mM), KPi buffer pH 8.0 (50 mM), cell lysate of pQR1108 (1.2 mg mL−1) and DMSO as co-solvent (10% v/v) at 30 °C and 500 rpm for 18 h. The reaction mixture was acidified and centrifuged. The supernatant was extracted with Et2O (3 × 100 mL) then basified with NaOH (2 M) and re-extracted with Et2O (3 × 100 mL). The combined organic extracts were concentrated in vacuo. The product was added to water with 0.1% TFA (8 mL) and purified by preparative HPLC (Varian ProStar model 210) using a Dr Maisch GmbH C8 Reprosil Gold 200 column (150 × 10 mm, 5 μm) with UV detection at 210 nm with a linear gradient 5%–50% B over 10 minutes at 6 mL min−1 (A = water with 0.1% of TFA and B = acetonitrile with 0.1% of TFA). Compound 48 eluted with a retention time of 5.4 minutes to afford compound 48 as a white solid (116 mg, 84%). M.p. 217 °C (decomposed); 1H NMR (300 MHz; D2O) δ 7.52 (1H, d, J = 7.5 Hz, 6-H), 7.32–7.43 (1H, m, 4-H), 6.98–7.09 (3H, m, 3 H, 5 H,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh), 6.30 (1H, dt, J = 15.6 and 6.9 Hz, CHCHPh), 3.86 (s, 3H, OCH3), 3.76 (2H, d, J = 6.9 Hz, CH2N); 13C (75 MHz; D2O with MeOH as a standard, TFA salt signals not described) δ 157.0, 131.5, 130.6, 127.9, 125.0, 121.8 (signals superimposed), 112.6 (CH), 56.2 (CH3), 42.2 (CH2); m/z (CI+) 164 ([M]+, 60%), 147 (100); HRMS (CI+) found [M]+ 164.1071; C16H15N3O4 requires 164.1070.
CHPh), 6.30 (1H, dt, J = 15.6 and 6.9 Hz, CHCHPh), 3.86 (s, 3H, OCH3), 3.76 (2H, d, J = 6.9 Hz, CH2N); 13C (75 MHz; D2O with MeOH as a standard, TFA salt signals not described) δ 157.0, 131.5, 130.6, 127.9, 125.0, 121.8 (signals superimposed), 112.6 (CH), 56.2 (CH3), 42.2 (CH2); m/z (CI+) 164 ([M]+, 60%), 147 (100); HRMS (CI+) found [M]+ 164.1071; C16H15N3O4 requires 164.1070.
Acknowledgements
Funding from the Biotechnology and Biosciences Research Council (BBSRC) (BB/L007444/1) for D. B. and J. W. E. J. and ALMAC is gratefully acknowledged. Furthermore, we thank the UCL Mass Spectrometry and NMR Facilities in the Department of Chemistry.References
- J. M. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914 CrossRef CAS.
- For reviews: (a) D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324 CrossRef CAS PubMed; (b) M. S. Malik, E. S. Park and J. S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163 CrossRef CAS PubMed; (c) S. Mathew and H. Yun, ACS Catal., 2012, 2, 993 CrossRef CAS; (d) M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965 CrossRef CAS PubMed.
- R. A. John, Biochim. Biophys. Acta, 1995, 1248, 81 CrossRef.
- C. Sayer, R. J. Martinez-Torres, N. Richter, M. N. Isupov, H. C. Hailes, J. A. Littlechild and J. M. Ward, FEBS J., 2014, 281, 2240 CrossRef CAS PubMed.
- For example: (a) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305 CrossRef CAS PubMed; (b) T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772 CrossRef CAS PubMed; (c) N. Richter, R. C. Simon, W. Kroutil, J. M. Ward and H. C. Hailes, Chem. Commun., 2014, 50, 6098 RSC; (d) C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215 CrossRef CAS; (e) E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernandez and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1937 CrossRef CAS; (f) B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852 RSC; (g) M. T. Gundersen, P. Tufvesson, E. J. Rackham, R. C. Lloyd and J. M. Woodley, Org. Process Res. Dev., 2016, 20, 602 CrossRef CAS.
- J. Handelsman, M. R. Rondon, S. F. Brady, J. Clardy and R. M. Goodman, Chem. Biol., 1998, 5, 245 CrossRef.
- B. Temperton and S. J. Giovannoni, Curr. Opin. Microbiol., 2012, 15, 605 CrossRef CAS PubMed.
- V. Torsvik, L. Øvreås and T. F. Thingstad, Science, 2002, 296, 1064 CrossRef CAS PubMed.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed.
- T. Narihiro, A. Suzuki, K. Yoshimune, T. Hori, T. Hoshino, I. Yumoto, A. Yokota, N. Kimura and Y. Kamagata, Microbes Environ., 2014, 29, 154 CrossRef PubMed.
- V. Lazarevic, K. Whiteson, S. Huse, D. Hernandez, L. Farinelli, M. Østerås, J. Schrenzel and P. François, J. Microbiol. Methods, 2009, 79, 266 CrossRef CAS PubMed.
- S. Jacquiod, S. Demanèche, L. Franqueville, L. Ausec, Z. Xu, T. O. Delmont, V. Dunon, C. Cagnon, I. Mandic-Mulec, T. M. Vogel and P. Simonet, J. Biotechnol., 2014, 190, 18 CrossRef CAS PubMed.
- J. S. McLean, Front. Cell. Infect. Microbiol., 2014, 4, 98 Search PubMed.
- N. Itoh, K. Isotani, Y. Makino, M. Kato, K. Kitayama and T. Ishimota, Enzyme Microb. Technol., 2014, 55, 140 CrossRef CAS PubMed.
- J. G. Owen, B. V. B. Reddy, M. Ternei, Z. Charlop-Powers, P. Y. Calle, J. H. Kim and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11797 CrossRef CAS PubMed.
- J. W. E. Jeffries, N. Dawson, C. Orengo, T. S. Moody, D. J. Quinn, H. C. Hailes and J. M. Ward, ChemistrySelect, 2016, 1, 2217 CrossRef CAS.
- B. Y. Hwang and B. G. Kim, Enzyme Microb. Technol., 2004, 34, 429 CrossRef CAS.
- C. Notredame, D. G. Higgins and J. Heringa, J. Mol. Biol., 2000, 302, 205 CrossRef CAS PubMed.
- S. Guindon, J. Dufayard, V. Lefort, M. Anisimova, W. Hordijk and O. Gascuel, Syst. Biol., 2010, 59, 307 CrossRef CAS PubMed.
- J. S. Shin, J. W. Wang and B. G. Kim, Appl. Microbiol. Biotechnol., 2003, 61, 463 CrossRef CAS PubMed.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628 CrossRef CAS.
- M. Espinosa-Urgel and J. L. Ramos, Appl. Environ. Microbiol., 2001, 11, 5219 CrossRef PubMed.
- J. S. Shin and B. G. Kim, Biosci., Biotechnol., Biochem., 2001, 65, 1782 CrossRef CAS PubMed.
- D. C. Higgins and J. Heringa, J. Mol. Biol., 2000, 302, 205 CrossRef PubMed.
- M. S. Humble, K. E. Cassimjee, M. Håkansson, Y. R. Kimbung, B. Walse, V. Abedi, H. J. Federsel, P. Berglund and D. T. Logan, FEBS J., 2012, 279, 779 CrossRef CAS PubMed.
- E. G. Evans, I. G. James, R. A. Seaman and M. D. Richardson, Br. J. Dermatol., 1993, 129, 437 CrossRef CAS PubMed.
- C. T. Lee, Y. C. Giam and T. Tan, Singapore Med. J., 1987, 28, 429 CAS.
- A. R. Germain, L. C. Carmody, P. P. Nag, B. Morgan, L. Verplank, C. Fernandez, E. Donckele, Y. Feng, J. R. Perez, S. Dandapani, M. Palmer, E. S. Lander, P. B. Gupta, S. L. Schreiber and B. Munoz, Bioorg. Med. Chem. Lett., 2013, 23, 1834 CrossRef CAS PubMed.
- Y. Xiong, T. Ye, M. Wang, Y. Xia, N. Wang, X. Song, F. Wang, L. Liu, Y. Zhu, F. Yang, Y. Wei and L. Yu, Cell. Physiol. Biochem., 2014, 34, 1863 CrossRef CAS PubMed.
- J. S. Shin and B. G. Kim, J. Org. Chem., 2002, 67, 2848 CrossRef CAS PubMed.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02769e |
| ‡ Joint first authors. |
| This journal is © The Royal Society of Chemistry 2017 |