DOI:
10.1039/C6GC02748B
(Paper)
Green Chem., 2017,
19, 311-318
The highly selective aerobic oxidation of cyclohexane to cyclohexanone and cyclohexanol over V2O5@TiO2 under simulated solar light irradiation†
Received
1st October 2016
, Accepted 29th November 2016
First published on 29th November 2016
Abstract
The highly selective aerobic oxidation of cyclohexane to cyclohexanone and cyclohexanol (KA-oil) under benign and green conditions is still a challenging topic. In this work, V2O5@TiO2 catalysts were prepared by V species deposited on TiO2 (P25) and characterized by transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), powder X-ray diffraction (XRD), and UV-vis diffuse reflectance techniques. The selective aerobic oxidation of cyclohexane was carried out over V2O5@TiO2 catalysts with oxygen as an oxidant under simulated light irradiation. The influences of solvents, metals and mass of V species deposited on TiO2, pressure of oxygen, and reaction time on the reaction were investigated. It was found that the V species deposited on the TiO2 surface was very efficient for the photocatalytic oxidation of cyclohexane under simulated solar light irradiation. More interestingly, the selectivity of the reaction in an acetonitrile/water mixed solvent was much higher than that in other solvents. Under the optimized conditions, the selectivity to KA-oil products could be nearly 100% at a cyclohexane conversion of 18.9%. The possible pathway for the catalytic reaction was proposed.
Introduction
The highly selective catalytic oxidation of cyclohexane to high value-added chemical products is one of the most challenging subjects in chemistry1 due to the fact that the activation of the inert C–H bond and the formation of the C–O bond are very difficult.2–5 Among the oxidation products, cyclohexanone and cyclohexanol (the mixture is called ‘KA-oil’, K = ketone, A = alcohol) are key feedstocks in the manufacture of nylon-6 and nylon-6,6 polymers.4 In particular, cyclohexanone, one of the main products of this reaction, is a more important intermediate for the production of nylon-6,6 than cyclohexanol, which illustrates that a high ketone/alcohol molar ratio is required in industry.6 However, the industrial cyclohexane oxidation suffers from a complex reaction process, high temperature and pressure, low yield, and serious environmental pollution.7 In order to overcome these limitations, tremendous efforts have been made recently to improve the catalytic performance for the oxidation of cyclohexane. Photocatalytic oxidation, which is an alternative reaction technology, has received much attention due to its potential application in environmental treatment and the synthesis of fine chemicals.8 Recently, the photocatalytic oxidation of cyclohexane under visible light irradiation has been investigated using heterogeneous catalysts. For example, Tsunoji et al.9 prepared titanium(IV) acetylacetonate grafted onto the interlayer surface of the layered silicate HUS-2 and a high photocatalytic activity was obtained in the partial photocatalytic oxidation of cyclohexane. Liu et al.10 designed composites made from carbon nitride (C3N4) and Au nanoparticles for the photocatalytic oxidation of cyclohexane and a higher conversion of 10.54% was gained. More recently, new photocatalytic Cu/CQD hybrids, which were developed and applied in cyclohexane oxidation under 60 °C with TBHP as an oxidant, exhibited a good catalytic activity.11 Although these heterogeneous catalysts showed relatively high catalytic activity, there existed some problems, such as a complicated synthesis process of photocatalysts, precious metals applied in photocatalysts or the low selectivity to KA-oil products. Therefore, it is highly desirable to develop new approaches for the fabrication of photocatalysts with low cost and high selectivity to KA-oil products under mild conditions.
Among the various semi-conductor materials, TiO2 is the most efficient and common photocatalyst in the photochemical reaction under UV-light irradiation owing to its availability, low toxicity, and chemical and thermal stability.12–15 Nowadays, it is found that TiO2 as an efficient photocatalyst has been used in the photocatalytic oxidation of cyclohexane. The crystallinity, availability of holes and electrons for surface reactions, amount of surface OH-groups,16 and quantum size effect and surface state effect of TiO2 nanoparticles17 have significant influence on the selective oxidation of cyclohexane. However, the activity of TiO2 is only efficient under ultraviolet light irradiation due to its wide band gap energy (3.0 eV for rutile and 3.2 eV for anatase).18 In order to extend the working spectrum, TiO2-based materials coated with transition metal ions with a low concentration can greatly enhance the visible light absorption efficiency and reduce the recombination of the photogenerated charges by forming donor states below the conduction band (CB).6,12,15,19–21 It has been reported that noble metal Au nanoparticles immobilized on TiO2/MCM-41 could improve the photocatalytic performance for the oxidation of cyclohexane, achieving a turnover frequency (TOF) as high as 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 145 h−1 with 9.87% conversion of cyclohexane.7 Recently, the highly efficient and selective sunlight-induced photocatalytic oxidation of cyclohexane to KA-oil products was obtained on FeO@P2519 or iron oxide-modified layered titanates15 under a CO2 atmosphere. Mao and co-workers reported the selective photocatalytic oxidation of cyclohexane by molecular oxygen in the presence of HCl as an additive using visible-light-responsive hollow sulfated V-doped TS-1 photocatalysts. It was found that 5% VOSO4-modified TS-1 exhibited the highest photocatalytic performance (>14% conversion) with selectivity (>94%) to KA-oil (cyclohexanone/cyclohexanol molar ratio = 3.46).20 More recently, vanadium-substituted molybdophosphoric acids were used to catalyze the liquid phase oxidation of cyclohexane to KA-oil products by nitrous oxide with the assistance of aqueous HCl solution in acetonitrile.22 Although the activity of the photocatalytic oxidation of cyclohexane has been improved by designing different catalysts, some additives (such as a CO2 atmosphere15 or aqueous HCl solution22) were required in the reaction. So the designing of efficient catalysts with high selectivity and activity for the photocatalytic oxidation of cyclohexane without other additives is still highly desirable.
145 h−1 with 9.87% conversion of cyclohexane.7 Recently, the highly efficient and selective sunlight-induced photocatalytic oxidation of cyclohexane to KA-oil products was obtained on FeO@P2519 or iron oxide-modified layered titanates15 under a CO2 atmosphere. Mao and co-workers reported the selective photocatalytic oxidation of cyclohexane by molecular oxygen in the presence of HCl as an additive using visible-light-responsive hollow sulfated V-doped TS-1 photocatalysts. It was found that 5% VOSO4-modified TS-1 exhibited the highest photocatalytic performance (>14% conversion) with selectivity (>94%) to KA-oil (cyclohexanone/cyclohexanol molar ratio = 3.46).20 More recently, vanadium-substituted molybdophosphoric acids were used to catalyze the liquid phase oxidation of cyclohexane to KA-oil products by nitrous oxide with the assistance of aqueous HCl solution in acetonitrile.22 Although the activity of the photocatalytic oxidation of cyclohexane has been improved by designing different catalysts, some additives (such as a CO2 atmosphere15 or aqueous HCl solution22) were required in the reaction. So the designing of efficient catalysts with high selectivity and activity for the photocatalytic oxidation of cyclohexane without other additives is still highly desirable.
The solvent plays a crucial role in the selective photocatalytic oxidation of cyclohexane over heterogeneous catalytic systems. For example, a conversion of 9% and a selectivity of 63% to cyclohexanone were obtained over Ti0.99Ag0.01O2−δ catalysts for the photocatalytic oxidation of cyclohexane with oxygen as an oxidant in chloroform medium.23 While using acetonitrile as a solvent, a selectivity of 91% to cyclohexanol and cyclohexanone was obtained over CrTiSi catalysts.24 In particular, water can often be used as a green solvent in the oxidation reaction.25,26 It is reported that the addition of an appropriate amount of water into the reaction system is very beneficial for the photocatalytic oxidation,27–29 mainly for reducing the consumption of surface hydroxyl groups,27 enhancing product desorption from catalysts,28 and increasing the yield of products.29
Obviously, developing cheap, green, earth-abundant and high-performance catalytic systems to achieve high selectivity to KA-oil products is very desirable. It is well-known that transition metals play an irreplaceable role in chemical reactions.26 V and Ti elements are abundant and cheap transition metals. Here, we designed V2O5@TiO2 catalysts for the selective photocatalytic oxidation of cyclohexane to KA-oil products using oxygen as an oxidant and acetonitrile/water as a solvent under simulated solar light irradiation. The results showed that the V2O5@TiO2 catalysts in acetonitrile/water were highly effective for the photocatalytic oxidation of cyclohexane to KA-oil products with a high ketone/alcohol molar ratio.
Results and discussion
Characterization of the catalysts
In this work, a series of x-V2O5@TiO2 catalysts with different V contents were prepared using an impregnation–calcination method, in which x stands for the mass (g) of the V precursor (NH4VO3) used in the preparation of the catalysts with 1 g of TiO2. For example, 0.01-V2O5@TiO2 catalysts were fabricated using 0.01 g of NH4VO3 and 1 g of TiO2. The detailed procedures for the preparation of the catalysts are given in the Experimental section. Fig. 1 shows the characterization results of 0.01-V2O5@TiO2. It can be seen from transmission electron microscopy (TEM) images (Fig. 1a–c) that the surface of the V2O5@TiO2 catalyst became relatively rough compared to the pure TiO2, probably due to the presence of V species on the surface of TiO2. The element mapping images are shown in Fig. 1d. It is clearly shown that V elements were dispersed uniformly on the catalyst.
 |
| | Fig. 1 TEM (a) and HRTEM (b) images of 0.01-V2O5@TiO2; TEM image of TiO2 (c); EDX images of 0.01-V2O5@TiO2 (d). | |
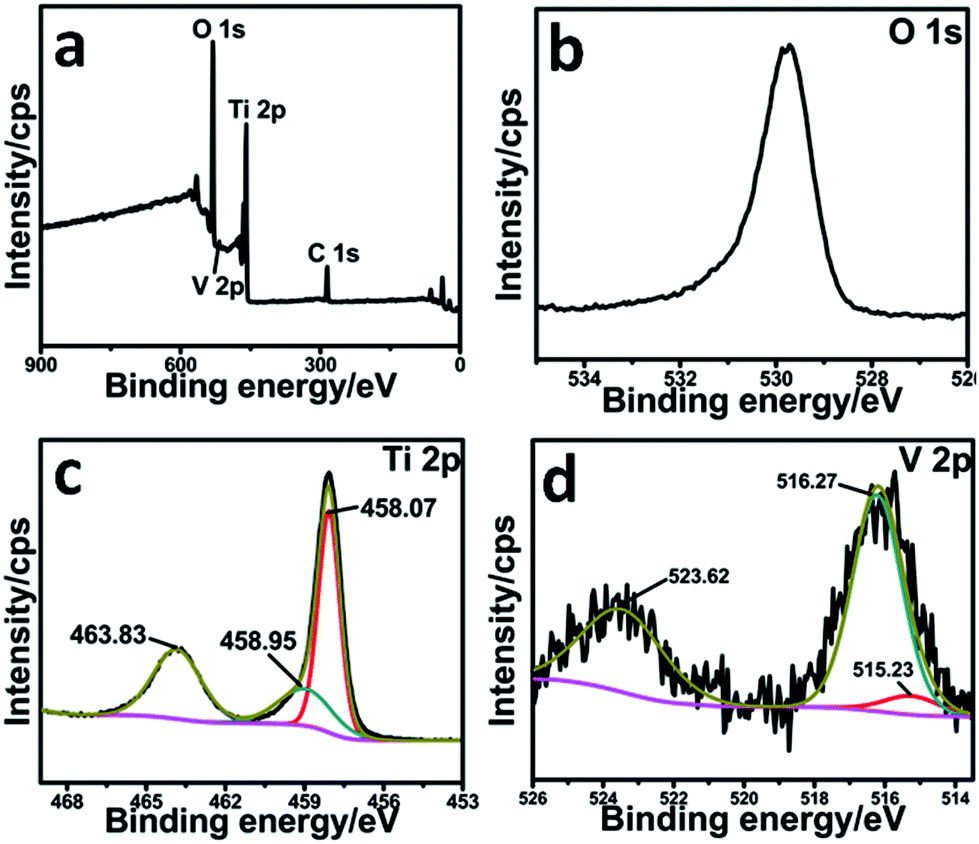
To obtain detailed information on the valence state of elements on the catalyst surfaces, X-ray photoelectron spectroscopy (XPS) characterization was performed and the spectra of the V2O5@TiO2 catalyst are shown in Fig. 2. It can be seen that the main elements in the catalyst were O(1s), Ti(2p), and V(2p). The high resolution spectrum of Ti (Fig. 2c) in the catalyst can be resolved into three peaks with binding energies of 463.83 eV, 458.07 eV and 458.95 eV. The former two were assigned to the Ti4+ species, and the last one was attributed to V-modified TiO2.20,30 From Fig. 2d, we can see the peaks of V 2p over V2O5@TiO2 located at 516.27 eV and 523.62 eV, respectively, indicating the existence of the V5+ valence state in the catalyst. The binding energy of the main peak for V 2p3/2 was lower than that of V2O5 (516.4–517.4 eV31). Besides, a small amount of the V4+ state existed in the catalysts, which can be seen from the peak of V 2p3/2 at 515.23 eV.31 It can be deduced by comparing with pure TiO2 that the binding energies of the main peaks for Ti and O were relatively high (Fig. S1 and S2†), likely due to the existence of the electron transfer or delocalization from Ti and O atoms toward V atoms in the catalysts. These results show that V species in the catalysts mainly existed in the form of V2O5.
 |
| | Fig. 2 XPS spectra of the 0.01-V2O5@TiO2 catalyst (a); high resolution spectra of O 1s (b), Ti 2p (c), and V 2p (d). | |
To further understand the crystal structure of the catalysts, XRD measurements were carried out and the XRD patterns are shown in Fig. 3. Similar to the pristine P25, the diffraction peaks of all the samples showed the crystal form of anatase TiO2 (JCPDS no. 21-1272)32 and rutile TiO2 (JCPDS no. 21-1276).33 No obvious diffraction peaks for vanadium oxide were detected at a low content of vanadium, perhaps due to good dispersity or poor crystallinity.34 However, light diffraction peaks appeared as the mass of the vanadium precursor was more than 0.03 g, which corresponds to the crystal planes of V2O5. Combining the TEM and XRD results, we can reasonably speculate that the single-component metal oxide deposition on TiO2 could adjust the surface morphology of the material and cause slight agglomeration, but would not affect its crystal form.
 |
| | Fig. 3 XRD patterns of the catalysts: P25 (1), 0.003-V2O5@TiO2 (2), 0.006-V2O5@TiO2 (3), 0.01-V2O5@TiO2 (4), 0.03-V2O5@TiO2 (5), 0.05-V2O5@TiO2 (6), 0.07-V2O5@TiO2 (7). | |
The UV-vis diffuse reflectance spectra of TiO2 and the 0.01-V2O5@TiO2 catalyst after Kubelka–Munk treatment are shown in Fig. 4. In comparison with TiO2, the absorption edge red-shift for 0.01-V2O5@TiO2 catalyst was observed, indicating that the band gap of vanadium deposited on the surface of TiO2 was narrower than that of pure P25. This might be due to the location of the V 3d orbit at the bottom of the conduction band of TiO2,35 and the d–d transition of vanadium,36 which might lead to the electron transition from the valence band (O 2p) of TiO2 to the t2g level of the V 3d atomic orbital. It is worth noting that the narrow band gap of the 0.01-V2O5@TiO2 catalyst can increase the electron excitation and the effect of photocatalytic oxidation, suggesting that V2O5 was the stronger electron withdrawing group on the catalysts.37
 |
| | Fig. 4 UV-vis diffuse reflectance spectra of TiO2 (a) and the 0.01-V2O5@TiO2 catalyst (b). | |
Effect of solvents on the photocatalytic reaction
The photocatalytic oxidation of cyclohexane was investigated in different solvents under simulated solar light irradiation using 0.01-V2O5@TiO2 as the catalyst and oxygen as the oxidant, and the results are given in Table 1. It can be seen that both the conversion and selectivity of the photocatalytic oxidation reaction were very poor when methylene dichloride/water and n-hexane/water were used as the solvents (Table 1, entries 1 and 2). This may be associated with the poor miscibility of water and the organic solvents. It is found that under similar reaction conditions the photocatalytic activity and selectivity of the reaction in acetone/water and acetonitrile/water were higher than that in methylene dichloride/water and n-hexane/water (Table 1, entries 3 and 4). In particular, the most satisfactory result was obtained with acetonitrile/water as the reaction solvent (Table 1, entry 4). It was reported that acetonitrile can suppress the formation of byproducts, i.e. CO2.28 Therefore, the cooperative effect of 0.01-V2O5@TiO2 and acetonitrile/water can promote the photocatalytic oxidation of cyclohexane to KA-oil products. Interestingly, the oxidation of cyclohexane to KA-oil products was almost completely inhibited in the absence of water (Table 1, entry 5). On increasing the volume ratio of H2O, the conversion of cyclohexane increased gradually (Table 1, entries 4 and 6–8). When the volume ratio of acetonitrile and water was less than 10/1, the selectivity to KA-oil products started to decline. So the most appropriate volume ratio of acetonitrile to water is 10/1. As will be discussed in the Reaction mechanism section, water was necessary in the reaction. Meanwhile, cyclohexanone and cyclohexanol are soluble in acetonitrile, but are insoluble in water. Thus, the addition of acetonitrile to water can enhance desorption of the products from the surface of the catalyst because they are soluble in the mixed solvent, which is favorable for enhancing the selectivity. It is found that the conversion was low when the concentration of water was too low, and the selectivity was low at a lower content of acetonitrile in the mixed solvent (Table 1, entries 5–8). Therefore, the mixed solvents with an appropriate composition showed high conversion and selectivity.
Table 1 Effect of the different solvents on the photocatalytic oxidation of cyclohexanea
| Entry |
Solvents |
V(S)/V(H2O)c |
Conversion (%) |
Selectivity (%) |
One/Olb |
| Oneb |
Olb |
Othersb |
|
Reaction conditions: cyclohexane, 0.1 mL; oxygen pressure, 2.0 MPa; 0.01-V2O5@TiO2, 0.010 g; reaction time, 4 h; reaction temperature, 30 °C; under simulated solar light irradiation.
One stands for cyclohexanone; Ol stands for cyclohexanol; others stand for small amounts of unidentified products and CO2; one/Ol stands for the ketone/alcohol molar ratio.
V(S)/V(H2O) stands for the volume ratio of the organic solvent and water. The volume of organic solvents was 1 mL.
|
| 1 |
Methylene dichloride/water |
10/1 |
2.4 |
54.8 |
23.1 |
22.1 |
2.4 |
| 2 |
n-Hexane/water |
10/1 |
1.8 |
45.2 |
20.4 |
34.4 |
2.2 |
| 3 |
Acetone/water |
10/1 |
8.5 |
70.2 |
24.5 |
5.3 |
2.9 |
| 4 |
Acetonitrile/water |
10/1 |
12.4 |
84.5 |
15.5 |
0 |
5.5 |
| 5 |
Acetonitrile |
— |
0.5 |
80.7 |
19.3 |
0 |
4.2 |
| 6 |
Acetonitrile/water |
20/1 |
4.5 |
81.2 |
18.8 |
0 |
4.3 |
| 7 |
Acetonitrile/water |
5/1 |
15.7 |
69.5 |
25.5 |
5.0 |
2.7 |
| 8 |
Acetonitrile/water |
10/3 |
20.2 |
59.4 |
24.5 |
16.1 |
2.4 |
Effect of metals on TiO2 on the photocatalytic reaction
The catalytic activity of various metals deposited on TiO2 was examined for the photocatalytic oxidation of cyclohexane and the results are shown in Table 2. The reaction did not occur without a catalyst and the catalytic activity of pure TiO2 was very low (Table 2, entries 1 and 2). Because noble metal nanoparticles, such as Pd,38,39 Au40 and Ag,23 can enhance absorption of the catalysts in the visible-light regions, Pd, Au and Ag deposited on the surface of TiO2 (Pd@TiO2, Au@TiO2 and Ag@TiO2) were investigated for the photocatalytic oxidation of cyclohexane (Table 2, entries 3–5). The results demonstrated that all of them could slightly improve the catalytic activity and ketone/alcohol molar ratio under simulated solar light irradiation, but the results were not satisfactory. It was desired that the photocatalytic oxidation of cyclohexane could be conducted over non-noble metal catalysts under solar light irradiation. So we attempted to deposit Fe or V elements on TiO2 for the photocatalytic reaction. Although Fe2O3@TiO2 as a catalyst could enhance catalytic activity under the same reaction conditions, the ketone/alcohol molar ratio was obviously lower (Table 2, entry 6). However, when V species were deposited on the surface of TiO2, the catalytic activity and ketone/alcohol molar ratio were improved significantly under the same conditions (Table 2, entry 7). One of the reasons may be that V species could promote the separation of electron–hole pairs.20 As a result, V2O5@TiO2 was a very effective catalyst for the photocatalytic oxidation of cyclohexane under simulated solar light irradiation. Meanwhile, we also conducted the experiment using light with a wavelength of <420 nm generated by the same xenon lamp with a light filter by cutting off the light with a wavelength of >420 nm. The result in Table 2 (entry 8) shows that the conversion of cyclohexane using the light from 290 to 800 nm was obviously higher than that under the light of <420 nm, indicating that the visible light also contributed to the reaction. Furthermore, we also conducted the reaction without light irradiation using 0.01-V2O5@TiO2 as catalysts and acetonitrile/water as solvents at 30 °C. It was seen that the reaction did not occur without light irradiation (entry 9 of Table 2), suggesting that light irradiation is necessary for the oxidation of cyclohexane.
Table 2 Catalytic activity of different metals deposited on the surface of TiO2 for the photocatalytic oxidation of cyclohexanea
| Entry |
Catalysts |
Conversion (%) |
Selectivity (%) |
One/Olb |
| Oneb |
Olb |
Othersb |
|
Reaction conditions: cyclohexane, 0.1 mL; oxygen pressure, 2.0 MPa; deionized water, 0.1 mL; acetonitrile, 1 mL; catalyst, 0.010 g; reaction time, 4 h; reaction temperature, 30 °C, under simulated solar light irradiation.
One stands for cyclohexanone; Ol stands for cyclohexanol; others stand for small amounts of unidentified products and CO2; one/Ol stands for the ketone/alcohol molar ratio.
Irradiated by light of <420 nm.
Without light irradiation.
|
| 1 |
— |
— |
0 |
0 |
0 |
— |
| 2 |
TiO2 |
0.9 |
72.8 |
27.2 |
0 |
2.7 |
| 3 |
0.01-Pd@TiO2 |
2.5 |
79.3 |
20.7 |
0 |
3.8 |
| 4 |
0.01-Au@TiO2 |
1.6 |
77.1 |
22.9 |
0 |
3.4 |
| 5 |
0.01-Ag@TiO2 |
2.9 |
80.2 |
19.8 |
0 |
4.1 |
| 6 |
0.01-Fe2O3@TiO2 |
8.2 |
65.3 |
34.7 |
0 |
1.8 |
| 7 |
0.01-V2O5@TiO2 |
12.4 |
84.5 |
15.5 |
0 |
5.5 |
| 8c |
0.01-V2O5@TiO2 |
7.1 |
79.5 |
20.5 |
0 |
3.0 |
| 9d |
0.01-V2O5@TiO2 |
0 |
— |
— |
— |
— |
Effect of the V content on the performance of V2O5@TiO2
We also examined the effect of the V content in the catalysts on the catalytic performance for the photocatalytic oxidation of cyclohexane under simulated solar light irradiation with oxygen as the oxidant and acetonitrile/water as the solvent, and the results are shown in Table 3. The V content on TiO2 had a considerable influence on the reaction activity and ketone/alcohol molar ratio. With increasing V content in the catalysts, the catalytic activity and ketone/alcohol molar ratio increased obviously (Table 3, entries 1–3). The 0.01-V2O5@TiO2 catalyst exhibited the highest conversion and ketone/alcohol molar ratio (Table 3, entry 3), which is attributed to the increase of the active sites on the surface of the catalysts. Further increasing the V content in the catalysts would lead to a negative effect on the activity and ketone/alcohol molar ratio (Table 3, entries 4–6). The main reason was that superabundant V in the catalysts caused agglomeration of the V species on the surface of TiO2 as known from TEM and the element mapping characterization (Fig. S3 and S4†). Therefore, in order to obtain satisfactory performance of the catalysts, a suitable content of V in the catalysts was needed for the photocatalytic oxidation of cyclohexane under simulated solar light irradiation.
Table 3 Catalytic activity of different masses of V species deposited on the surface of TiO2 for the photocatalytic oxidation of cyclohexanea
| Entry |
Catalysts |
Conversion (%) |
Selectivity (%) |
One/Olb |
| Oneb |
Olb |
Othersb |
|
Reaction conditions: cyclohexane, 0.1 mL; oxygen pressure, 2.0 MPa; deionized water, 0.1 mL; acetonitrile, 1 mL; catalyst, 0.010 g; reaction time, 4 h; reaction temperature, 30 °C; under simulated solar light irradiation.
One stands for cyclohexanone; Ol stands for cyclohexanol; others stand for amounts of unidentified products and CO2; one/Ol stands for the ketone/alcohol molar ratio.
|
| 1 |
0.003-V2O5@TiO2 |
5.0 |
79.2 |
20.8 |
0 |
3.8 |
| 2 |
0.006-V2O5@TiO2 |
8.9 |
81.4 |
18.6 |
0 |
4.4 |
| 3 |
0.01-V2O5@TiO2 |
12.4 |
84.5 |
15.5 |
0 |
5.5 |
| 4 |
0.03-V2O5@TiO2 |
10.6 |
83.0 |
17.0 |
0 |
4.9 |
| 5 |
0.05-V2O5@TiO2 |
8.4 |
81.9 |
18.1 |
0 |
4.5 |
| 6 |
0.07-V2O5@TiO2 |
7.1 |
80.1 |
19.9 |
0 |
4.0 |
Effect of oxygen pressure
The influence of oxygen pressure on the photocatalytic oxidation of cyclohexane was studied over the 0.01-V2O5@TiO2 catalyst in acetonitrile/water solvents under simulated solar light irradiation, and the results are shown in Fig. 5. It can be clearly seen that the conversion of cyclohexane continuously increased with the increase of oxygen pressure. When the pressure was less than 2.0 MPa, the selectivity to cyclohexanone slowly increased and the selectivity to cyclohexanol decreased. However, the selectivity to KA-oil products is almost 100%. These results indicated that an increasing oxygen pressure resulted in the conversion of a part of cyclohexanol to cyclohexanone. However, as the pressure exceeded 2.0 MPa, by-products began to generate, and the selectivity to KA-oil products decreased. This is understandable because excessive oxidation of KA-oil products would have occurred as the pressure was too high.
 |
| | Fig. 5 Effect of oxygen pressure on the photocatalytic oxidation of cyclohexane. Reaction conditions: cyclohexane, 0.1 mL; deionized water, 0.1 mL; acetonitrile, 1 mL; 0.01-V2O5@TiO2, 0.010 g; reaction time, 4 h; reaction temperature, 30 °C; under simulated solar light irradiation. | |
Effect of reaction time
The effect of reaction time on the photocatalytic oxidation of cyclohexane at 2.0 MPa is shown in Fig. 6. At the initial stage the conversion of cyclohexane increased quickly with an approximately constant rate, and the selectivity to the KA-oil product was almost 100% as the time was less than 8 h. The selectivity to cyclohexanone slowly increased and the selectivity to cyclohexanol slightly decreased, further suggesting the conversion of partial cyclohexanol to cyclohexanone. At a reaction time of 8 h, the conversion of cyclohexane was 18.9% and the selectivity to KA-oil products was nearly 100% and the ketone/alcohol molar ratio was 7.8. However, when the reaction time exceeded 8 h, the conversion increased with the reaction time, but the selectivity to KA-oil products decreased slowly and byproducts were formed, indicating that cyclohexanol and cyclohexanone would be transformed into by-products.
 |
| | Fig. 6 Effect of the reaction time on the photocatalytic oxidation of cyclohexane. Reaction conditions: cyclohexane, 0.1 mL; oxygen pressure, 2.0 MPa; deionized water, 0.1 mL; acetonitrile, 1 mL; 0.01-V2O5@TiO2, 0.010 g; reaction temperature, 30 °C; under simulated solar light irradiation. | |
Reaction mechanism
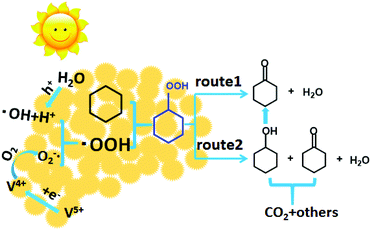
According to the literature1,20,41 and the above results, the possible mechanism for the photocatalytic oxidation of cyclohexane over the V2O5@TiO2 catalyst using oxygen as the oxidant is proposed and shown schematically in Scheme 1. The reactants were firstly absorbed on the surface of the catalysts. Under simulated solar light irradiation, the catalysts produced the electron (e−) and positive hole (h+) pairs after absorbing photons. Water could be oxidized by holes to generate H+ and ˙OH. Meanwhile, V5+ ions in the form of V2O5 capturing an electron created V4+, which could easily release and transfer the electron to O2 absorbed on the surfaces of catalysts to achieve O2˙−.20 Subsequently, H+ reacted with O2˙− to form hydroperoxyl radicals (˙OOH).20,42,43 The hydroperoxyl radicals can react with cyclohexane to yield cyclohexyl hydroperoxide, which subsequently decomposed into cyclohexanone and water (route 1) and cyclohexanone, cyclohexanol and water (route 2). On the basis of the above results, it can be deduced that the reaction rate of route 1 is faster than that of route 2 and partial cyclohexanol may be oxidized to cyclohexanone with the increase of the oxygen pressure or time. This may be attributed to the fact that the hydroxyl group of cyclohexyl hydroperoxide and cyclohexanol might form hydrogen bonding with the abundant oxygen groups on the surface of the catalyst. The interaction was beneficial for the conversion of cyclohexyl hydroperoxide and cyclohexanol to cyclohexanone, resulting in a higher ketone/alcohol molar ratio in this reaction system. However, when the reaction time exceeded 8 h or the oxygen pressure exceeded 2.0 MPa, some cyclohexanol and cyclohexanone would be transformed into by-products, leading to the decrease of KA-oil selectivity. Meanwhile, to get the evidence for the generation of free radicals in the process of the reaction, p-benzoquinone (BQ, an O2˙− radical scavenger) or triethanolamine (TEOA, a h+ radical scavenger)41 was added into the reaction system, respectively, and the photocatalytic oxidation of cyclohexane was completely inhibited, indicating that O2˙− or h+ radicals were responsible for the formation of KA-oil products. It has been reported that ˙OH radicals can react with cyclohexane to generate cyclohexyl radicals, which can be converted to cyclohexanol and further oxidize to cyclohexanone.20 In order to study the role of ˙OH radicals, we conducted the reaction by adding t-butanol (an ˙OH radical scavenger)10 in the reaction. It was demonstrated that its effect on the conversion and selectivity to KA-oil products was not obvious, suggesting that this is not the main reaction pathway in our reaction system.
 |
| | Scheme 1 Possible mechanism proposed for the photocatalytic oxidation of cyclohexane. | |
Conclusion
V2O5@TiO2 catalysts with different V contents have been prepared, in which V species are uniformly distributed on the TiO2 surface in the form of V2O5. The catalysts are efficient for the selective oxidation of cyclohexane to KA-oil products with oxygen as the oxidant under simulated solar light irradiation at room temperature. Both the content of V in the catalysts and solvent affect the reaction significantly. In particular, the selectivity to the KA-oil product was nearly 100% at the cyclohexane conversion of 18.9% when acetonitrile/water was used as the solvent. The high conversion of cyclohexane at 100% selectivity to the product, use of molecular oxygen as the oxidant and simple catalyst, and mild reaction conditions give this route potential for practical applications.
Experimental
Materials
Cyclohexane, cyclohexanone, cyclohexanol, n-hexanol, acetonitrile, acetone, n-hexane, dichloromethane, dimethyl formamide, AgNO3, NH4VO3, urea, and NaBH4 were all of analytical grade and purchased from Sinopharm Chemical Reagent Co. Ltd. HAuCl4·4H2O, Pd(NO3)2·2H2O, TiO2 (P25), anhydrous ferric chloride, p-benzoquinone, triethanolamine and dicyandiamide were obtained from Alfa Aesar and used as received. Oxygen (99.99%) was provided by the Beijing Analytical Instrument Company.
Catalyst preparation
Synthesis of x-V2O5@TiO2.
A series of V2O5@TiO2 catalysts were prepared according to the procedures reported.30 In a typical preparation, 1 g of P25 and a known amount of NH4VO3 with 30 mL of deionized water were ultrasonicated for 30 minutes. Then the mixture was stirred at 95 °C in an oil bath to remove water. The resulting solid was ground and transferred into a crucible, and then the sample was calcined from room temperature to 500 °C at a rate of 5 °C min−1 and maintained at this temperature for 4 h. Finally, a light yellow powder was obtained after cooling to room temperature and the different samples were denoted as x-V2O5@TiO2, and x represents the mass of the V precursor added.
Synthesis of other catalysts.
Similar procedures were applied in the preparation of Fe2O3@TiO2 catalysts except that FeCl3 was added as a precursor. In addition, Au@TiO2, Pd@TiO2, and Ag@TiO2 catalysts were prepared by the chemical reduction method. We take the preparation of the Au@TiO2 catalyst as an example to describe the procedures. Briefly, 1 g of TiO2 and 0.01 g HAuCl4·4H2O were added into 30 mL of deionized water, and the mixture was ultrasonicated for 30 minutes at ambient temperature. 10 mL of aqueous NaBH4 solution (20 mM) was added into the mixture under stirring. The metal ions were reduced and the metal nanoparticles were immobilized on TiO2. The mixture was washed using water and ethanol several times, and then was dried at 60 °C overnight under vacuum. The Pd@TiO2 or Ag@TiO2 catalysts were prepared using similar procedures, and the main difference was that 0.01 g of Pd(NO3)2·2H2O or AgNO3 was used.
Catalyst characterization
The morphology of the catalysts was characterized by TEM (JEOL JEM-2100F) equipped with EDX. An X-ray photoelectron spectroscopy (XPS) study was carried out using the Thermo Scientific ESCALab 250 Xi using a 300 W Al-Kα radiation. The base pressure was about 3 × 10−10 mbar in the analysis chamber. Typically, the hydrocarbon C 1s line at 284.8 eV from adventitious carbon was used for energy referencing. XRD patterns were obtained on a Rigaku D/max-2500 X-ray diffractometer using Cu-Kα radiation. UV-vis absorption spectra of the catalysts were recorded in a Perkin Elmer Lambda 35 UV/vis spectrophotometer, with BaSO4 as the reference material.
Reaction
As an example, we describe the photocatalytic reaction in the oxidation of cyclohexane over the catalyst using acetonitrile/water as the solvent. In a typical reaction, 0.1 mL of cyclohexane, appropriate amounts of acetonitrile and deionized water, and 0.01 g of the catalyst were introduced into a 8 mL cylindrical stainless-steel reactor equipped with a magnetic stirrer. There was a quartz window at the top of the reactor for light irradiation. The temperature of the reaction was kept at 30 °C with a thermostatic water bath (the quartz window was not immersed in water). After being filled with the desired amount of oxygen, the stirrer was started and the reactor was irradiated by a 500 W xenon lamp (290 nm–800 nm) for the desired reaction time. After the reaction, the gaseous products were analyzed by an Agilent 4890 gas chromatograph equipped with a TCD detector and argon as the carrier gas, with helium as the internal standard. The liquid products were detected by an Agilent 6820 gas chromatograph equipped with a flame ionization detector and a PEG-20M capillary column (30 m × 0.25 mm × 0.25 mm) using n-hexanol as the internal standard. In order to verify the free radicals generated in the reaction process, a similar reaction was conducted under optimal conditions, besides p-benzoquinone, triethanolamine or t-butanol was added into the reaction system.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21373230) and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).
Notes and references
- P. Du, J. A. Moulijn and G. Mul, J. Catal., 2006, 238(2), 342–352 CrossRef CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417(6888), 507–514 CrossRef CAS PubMed.
- J. M. Thomas, Nature, 1985, 314(6013), 669–670 CrossRef.
- N. Pal, M. Pramanik, A. Bhaumik and M. Ali, J. Mol. Catal. A: Chem., 2014, 392, 299–307 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97(8), 2879–2932 CrossRef CAS PubMed.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270(1), 16–25 CrossRef CAS.
- J. Zhou, X. Yang, Y. Wang and W. Chen, Catal. Commun., 2014, 46, 228–233 CrossRef CAS.
- W. Wu, X. He, Z. Fu, Y. Liu, Y. Wang, X. Gong, X. Deng, H. Wu, Y. Zou and N. Yu, J. Catal., 2012, 286, 6–12 CrossRef CAS.
- N. Tsunoji, Y. Ide, Y. Yagenji, M. Sadakane and T. Sano, ACS Appl. Mater. Interfaces, 2014, 6(7), 4616–4621 CAS.
- J. Liu, Y. Yang, N. Liu, Y. Liu, H. Huang and Z. Kang, Green Chem., 2014, 16, 4559–4565 RSC.
- S. Qiao, B. Fan, Y. Yang, N. Liu, H. Huang and Y. Liu, RSC Adv., 2015, 5(54), 43058–43064 RSC.
- J.-M. Herrmann, New J. Chem., 2012, 36(4), 883–890 RSC.
- J.-M. Herrmann, Catal. Today, 1999, 53(1), 115–129 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32(1), 33–177 CrossRef CAS.
- H. Hattori, Y. Ide, S. Ogo, K. Inumaru, M. Sadakane and T. Sano, ACS Catal., 2012, 2(9), 1910–1915 CrossRef CAS.
- J. T. Carneiro, A. R. Almeida, J. A. Moulijn and G. Mul, Phys. Chem. Chem. Phys., 2010, 12(11), 2744–2750 RSC.
- X. Li, G. Chen, Y. Po-Lock and C. Kutal, J. Chem. Technol. Biotechnol., 2003, 78(12), 1246–1251 CrossRef CAS.
- A. Murphy, P. Barnes, L. Randeniya, I. Plumb, I. Grey, M. Horne and J. Glasscock, Int. J. Hydrogen Energy, 2006, 31(14), 1999–2017 CrossRef CAS.
- Y. Ide, H. Hattori, S. Ogo, M. Sadakane and T. Sano, Green Chem., 2012, 14(5), 1264–1267 RSC.
- W. Zhong, T. Qiao, J. Dai, L. Mao, Q. Xu, G. Zou, X. Liu, D. Yin and F. Zhao, J. Catal., 2015, 330, 208–221 CrossRef CAS.
- J. Zhu, Z. Deng, F. Chen, J. Zhang, H. Chen, M. Anpo, J. Huang and L. Zhang, Appl. Catal., B, 2006, 62(3), 329–335 CrossRef CAS.
- J. She, Z. Fu, J. Li, B. Zeng, S. Tang, W. Wu, H. Zhao, D. Yin and S. R. Kirk, Appl. Catal., B, 2016, 182, 392–404 CrossRef CAS.
- R. Vinu and G. Madras, Appl. Catal., A, 2009, 366(1), 130–140 CrossRef CAS.
- D. Tsukamoto, A. Shiro, Y. Shiraishi and T. Hirai, J. Phys. Chem. C, 2011, 115(40), 19782–19788 CAS.
- J.-L. Cao, S.-L. Shen, P. Yang and J. Qu, Org. Lett., 2013, 15(15), 3856–3859 CrossRef CAS PubMed.
- D. Yang, T. Jiang, T. Wu, P. Zhang, H. Han and B. Han, Catal. Sci. Technol., 2016, 6(1), 193–200 Search PubMed.
- G. Martra, S. Coluccia, L. Marchese, V. Augugliaro, V. Loddo and L. Palmisano, Catal. Today, 1999, 53(4), 695–702 CrossRef CAS.
- J. T. Carneiro, C.-C. Yang, J. A. Moulijn and G. Mul, J. Catal., 2011, 277(2), 129–133 CrossRef CAS.
- V. H. Van Dijk, G. Simmelink and G. Mul, Appl. Catal., A, 2014, 470, 63–71 CrossRef CAS.
- D. Ye, R. Qu, H. Song, X. Gao, Z. Luo, M. Ni and K. Cen, Chem. Eng. J., 2016, 283, 846–854 CrossRef CAS.
- B. M. Reddy, P. M. Sreekanth and E. P. Reddy, J. Phys. Chem. B, 2002, 106, 5695–5700 CrossRef CAS.
- Y. Zhang, W. Guo, L. Wang, M. Song, L. Yang, K. Shen, H. Xu and C. Zhou, Chin. J. Catal., 2015, 36(10), 1701–1710 CrossRef CAS.
- Q. Sun, Y. Lu, H. Zhang, H. Zhao, H. Yu, J. Xu, Y. Fu, D. Yang and Y. Liu, Mater. Chem. Phys., 2012, 133(1), 253–258 CrossRef CAS.
- M. Epifani, R. Díaz, C. Force, E. Comini, T. Andreu, R. R. Zamani, J. Arbiol, P. Siciliano, G. Faglia and J. R. Morante, J. Phys. Chem. C, 2013, 117, 20697–20705 CAS.
- R. K. Dwari, D. S. Rao, A. K. Swar, P. S. R. Reddy and B. K. Mishra, Int. J. Miner., Metall. Mater., 2012, 19(11), 992–1003 CrossRef CAS.
- W. Zhao, Q. Zhong, Y. Pan and R. Zhang, Colloids Surf., A, 2013, 436, 1013–1020 CrossRef CAS.
- X. Huang, S. Zhang, H. Chen and Q. Zhong, J. Mol. Struct., 2015, 1098, 289–297 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46(8), 1900–1909 CrossRef CAS PubMed.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134(38), 15720–15723 CrossRef CAS PubMed.
- J. T. Carneiro, T. J. Savenije, J. A. Moulijn and G. Mul, J. Photochem. Photobiol., A, 2011, 217(2), 326–332 CrossRef CAS.
- M. M. Mohamed, RSC Adv., 2015, 5, 46405–46414 RSC.
- K. Ohkubo, A. Fujimotoa and S. Fukuzumi, Chem. Commun., 2011, 47, 8515–8517 RSC.
- A. Maldotti, A. Molinari, R. Amadelli, E. Carbonell and H. Garcia, Photochem. Photobiol. Sci., 2008, 7, 819–825 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS, TEM and EDX spectra of catalysts. See DOI: 10.1039/c6gc02748b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.