
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquids that form adducts with alcohols†
H. Q. Nimal
Gunaratne
*,
Philip
McCarron
and
Kenneth R.
Seddon
The QUILL Research Centre, School of Chemistry and Chemical Engineering, The Queen's University of Belfast, Stranmillis Road, Belfast BT9 5AG, UK. E-mail: n.gunaratne@qub.ac.uk; quill@qub.ac.uk
First published on 24th October 2016
Abstract
A class of ionic liquids with a built-in active ketone functionality is shown to form adducts with structurally diverse alcohols. The positions of the equilibria for adduct formation are shown to be dependent on the structure of the alcohols. How these functionalised ionic liquids could be applied in altering vapour phase compositions of alcohols and in separations of perfumery alcohols are demonstrated, here.
Alcohols are, in general, less reactive nucleophiles than alkoxides, amines or thiols towards electrophiles. Most often they react with highly reactive electrophilic reagents in conjunction with added bases or other catalysts due to their low nucleophilicity.1 Organic reagents that strongly react/interact with alcohols without the aid of catalysts are few and far between.2 In contrast, amines and thiols would readily react with electrophiles such as Michael acceptors, aldehydes and some activated ketones, demonstrating their superior reactivity based on nucleophilicity of the reacting atoms (i.e. N or S).3 It is challenging to design ketone functionalised reagents that would readily react with alcohols to form a hemi- or di-acetal in the absence of an acid catalyst. If ketones could be designed to form selective adducts with a range of different alcohols, this could pave the way towards their separation and/or altering the vapour-phase composition of a mixture of alcohols. It would also be desirable to have reagents having negligible vapour pressure and appropriate phase behaviour, so that the separation process is straightforward, devoid of any contamination from the said reagent. Since ionic liquids possess such unique qualities as negligible vapour pressures, thermal stability and structural diversity,4 they were worthy of investigation as reagent candidates. Although there are myriad reports in the literature on the use of entrainment for the separation of organic mixtures,5 the role of ionic liquids as entrainment agents for the separation of organic mixtures is underdeveloped.5,6 These methods take into account various weak intermolecular forces that exist between organic molecules (or mixtures thereof) and the appropriately designed ionic liquids, to disrupt the intermolecular forces and introduce new cation and/or anion interactions with the molecules (such as dipolar, hydrogen bond and/or van der Waals forces).7 An ionic liquid could be designed to interact with one component of multicomponent mixture preferentially to other components, selectively binding the molecule of interest, and ‘freeing up’ the remaining components. However, as the interactions induced by such ionic liquids would be intrinsically weak, they would lead to less effective separations. If, however, covalent interactions could be induced, this would lead to a much higher intrinsic recognition and binding. To our knowledge, this ‘reactive’ selection of a single component from a multicomponent mixture has been given less, or no, attention. Here, we have explored the design of a new class of ionic liquids in order to interact selectively with certain class of alcohols, allowing a separation process by modulating their vapour pressures. We have employed a similar strategy of using reactive ionic liquids for applications ranging from malodorous gas scavenging, metal ions separations, dye removal, and controlled release of perfumes.8,9
In this communication, we describe the synthesis of a simple ionic liquid system (1) where the cation is embedded in a cyclohexane ring bearing a ketone functionality (Fig. 1). The carbonyl group in (1) is activated by the presence of the positive charge in the vicinity of the functional group where the activation is likely to be caused by inductive effects10 imparted by the quaternary ammonium group in the ring (cf. the original studies by Hammett11). Though these ionic liquids (1) are devoid of charge delocalisation through a π-system to transmit the effect of the positive charge from the quaternary nitrogen to the carbonyl group,9 it provides an interesting example to explore the inductive influence of the ‘localised’ positive charge.
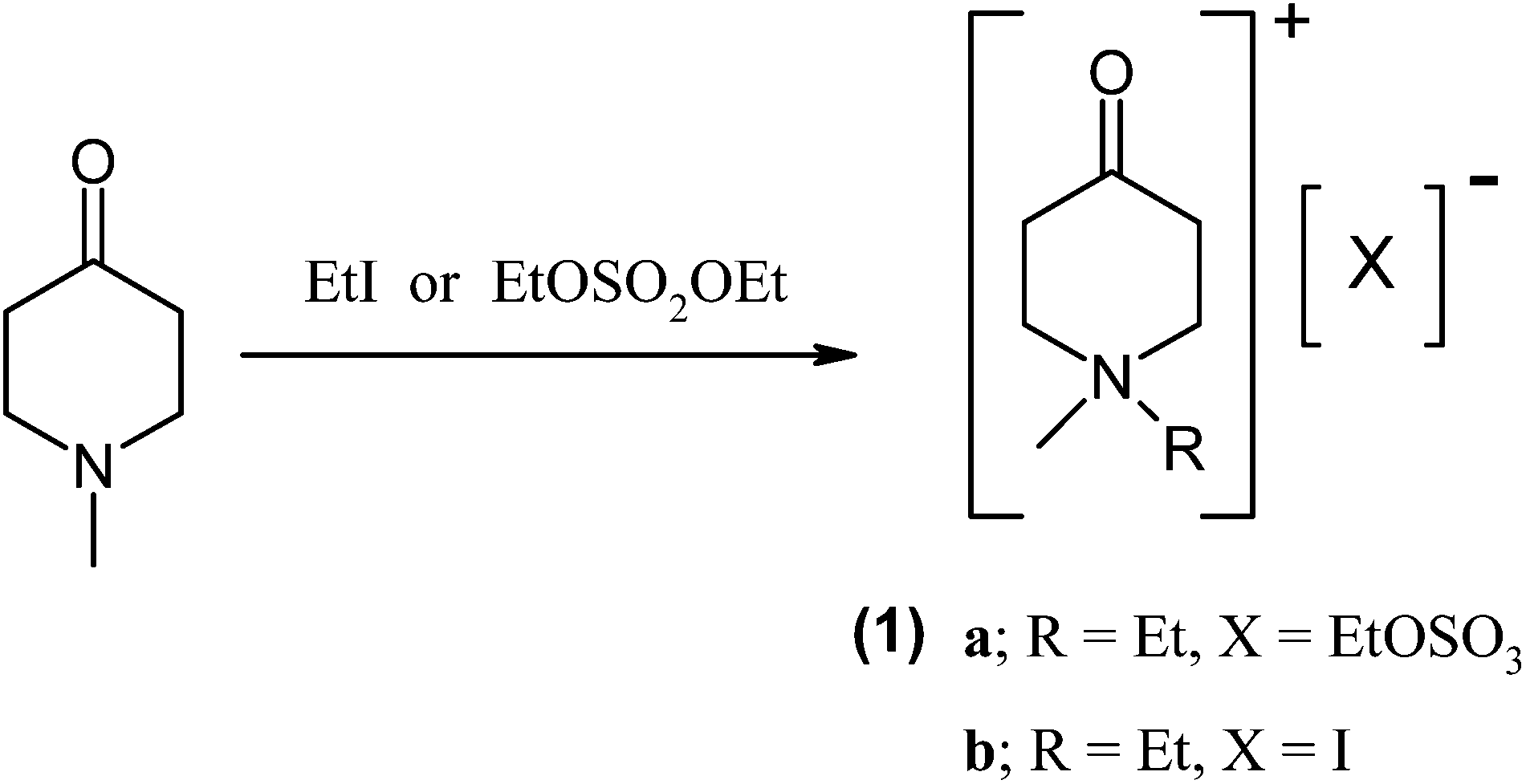 | ||
| Fig. 1 Synthetic scheme for ionic liquids used in this study. | ||
The syntheses of (1) was carried out by treating commercially available 1-methylpiperid-4-one with either iodoethane or diethyl sulfate (or dimethyl sulfate) yielding (1a) as a viscous liquid (Tg −34 °C) or (1b) as a pale yellow solid (Tm 94 °C). It is noteworthy that all synthetic procedures were simple and straightforward, producing pure materials with high yields.
The reaction of (1) with alcohols was examined, using methanol as a model case, by means of 1H and 13C NMR spectroscopy in CD3OD, and in dmso-d6 for comparison. The reactivity of (1) towards water was also explored for comparison. 13C NMR spectra of (1a) in dmso-d6, CD3OD and D2O are shown in Fig. 2, and 1H NMR spectra are shown in Fig. 3. The 13C NMR spectra clearly show that the signal for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group at δ 200.8 (in dmso-d6) disappears completely when spectra are run in either CD3OD or D2O, indicating the complete formation of the hemiacetal or the hydrate, respectively (see Fig. 4). It should be stressed here that no other catalyst was needed for these reactions to take place, demonstrating the exceptional reactivity of the ketone functionality of the cation. In stark contrast, the uncharged ketone (1-methylpiperid-4-one) neither reacts with methanol nor water within a week.
O group at δ 200.8 (in dmso-d6) disappears completely when spectra are run in either CD3OD or D2O, indicating the complete formation of the hemiacetal or the hydrate, respectively (see Fig. 4). It should be stressed here that no other catalyst was needed for these reactions to take place, demonstrating the exceptional reactivity of the ketone functionality of the cation. In stark contrast, the uncharged ketone (1-methylpiperid-4-one) neither reacts with methanol nor water within a week.
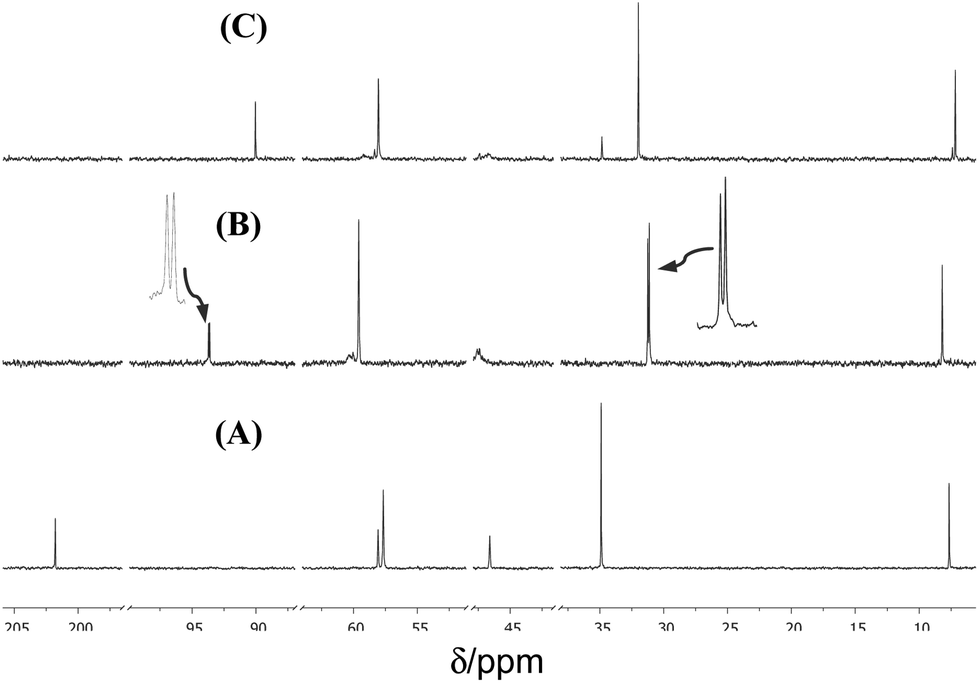 | ||
| Fig. 2 Partial 13C NMR (100.6 MHz; 25 °C) spectra of (1a; 15 mM) in: (A) dmso-d6; (B) CD3OD; expansions are shown next to two peaks; and (C) D2O. The solvent peaks were removed for clarity. | ||
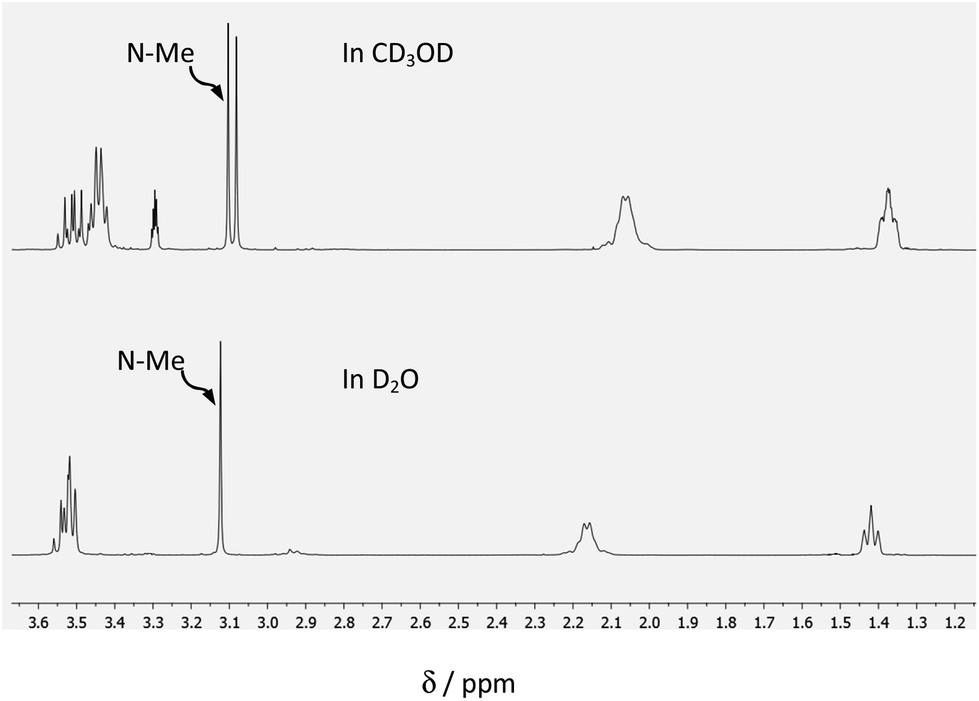 | ||
| Fig. 3 Partial 1H NMR (400 MHz; 25 °C) spectra of (1a; 15 mM) in: Top; CD3OD; and Bottom; D2O. | ||
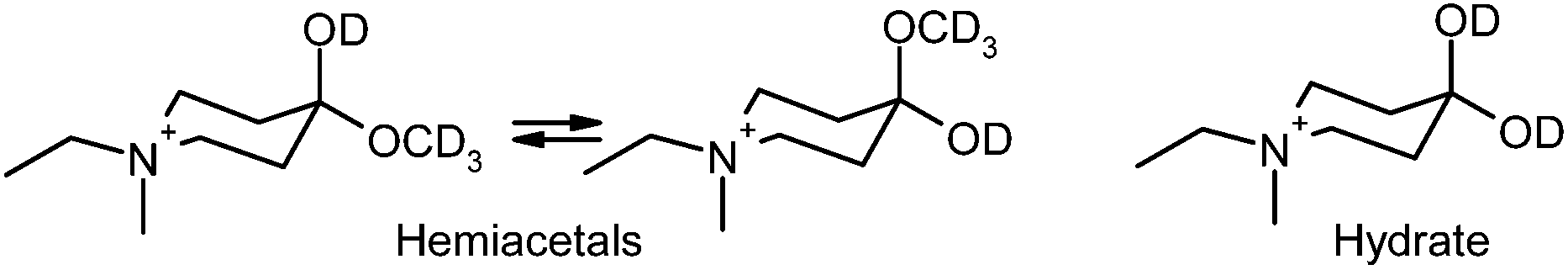 | ||
| Fig. 4 Hydrate and diastereoisomeric hemiacetals derived from (1a). | ||
Of interest, in CD3OD, formation of the hemiacetal creates a new chiral centre, generating two diastereoisomers in approximately equal proportions (clearly visible in the insets of Fig. 2(B)).
In contrast, the hydrate of (1a) is achiral, giving rise to a single isomer (see Fig. 4).
The 1H NMR spectra of (1a) in CD3OD exhibit two singlets of near equal intensity for the N-methyl group, whereas only one singlet is seen for the hydrate present in D2O. It is noteworthy that there is no evidence for the formation of a diacetal in the presence of the excess of CD3OD.
It was clearly of interest to determine the comparative reactivities of CD3OD and D2O towards (1a). Solutions of (1a) in mixtures of CD3OD and D2O with varying mole fractions were examined by 1H NMR spectroscopy (see Fig. 5). In an equimolar mixture of CD3OD and D2O, a significantly higher proportion of hemiacetal was present as compared to the hydrate (see Fig. 5). This could be attributed, in general, to the higher nucleophilicity of alcohols compared to water due to the electron-donating inductive effect of the alkyl group of alcohols.12 As the ratio of D2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3OD, is increased, the relative amount of hydrate formed also increased.
:CD3OD, is increased, the relative amount of hydrate formed also increased.
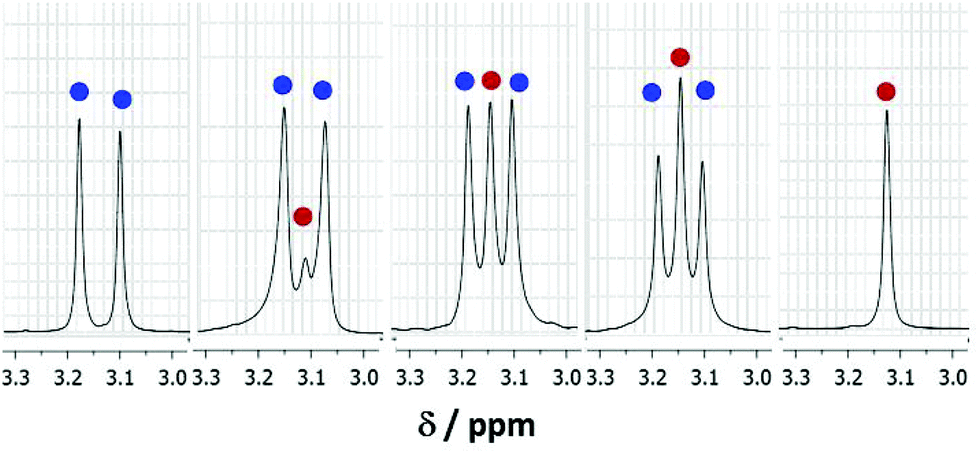 | ||
| Fig. 5 Partial 1H NMR spectra (δ 3.0–3.3) of (1a) showing the signal for N–Me group in: (from left to right) 100 mol% CD3OD; 67 mol% CD3OD; 50 mol% CD3OD; 33 mol% CD3OD and 100 mol% D2O. Blue and red dots represent the hemiacetal and the hydrate, respectively. | ||
This may have implications towards separating alcohols out of aqueous alcohol mixtures.
The presence of the carbonyl group in ionic liquid (1) ideally provides a probe for evaluating the magnitude of the interaction between (1) and a group of structurally diverse alcohols. Some of these alcohols belong to an important class of perfumes that could interact with (1) to differing extents, leading to controlled perfume delivery.13 FTIR spectroscopy was utilised to examine the stretching frequency of the liquid form of (1), viz. (1b). Eight diverse alcohols, including primary, secondary and tertiary, three of which are perfume raw materials (i.e. geraniol, 2-phenyethanol and linalool) were used for this study. One equivalent of (1b) was homogenised with 4 equivalents of the corresponding alcohol prior to running the FTIR spectra. Fig. 6 shows the carbonyl stretching frequency region of the FTIR spectra of the (1b)-alcohol mixtures. All primary alcohols interacted strongly with (1b), in many cases, resulting in complete disappearance of the CO stretching band. However, subtle variations can be seen even among the group of primary alcohols. In the case of 2-propanol, the interaction appears to be weak, with a slight shift in the intense band. Judging by these results, structurally diverse perfume alcohols would have interacted with (1b) to differing extent modulating their delivery. ES-MS spectra of mixtures of (1b + geraniol) or (1b + 2-phenyethanol) indicated the presence of two hemiacetals (cations) with masses 297 or 265, respectively. Higher masses corresponding to diacetals were not seen for these two alcohols.
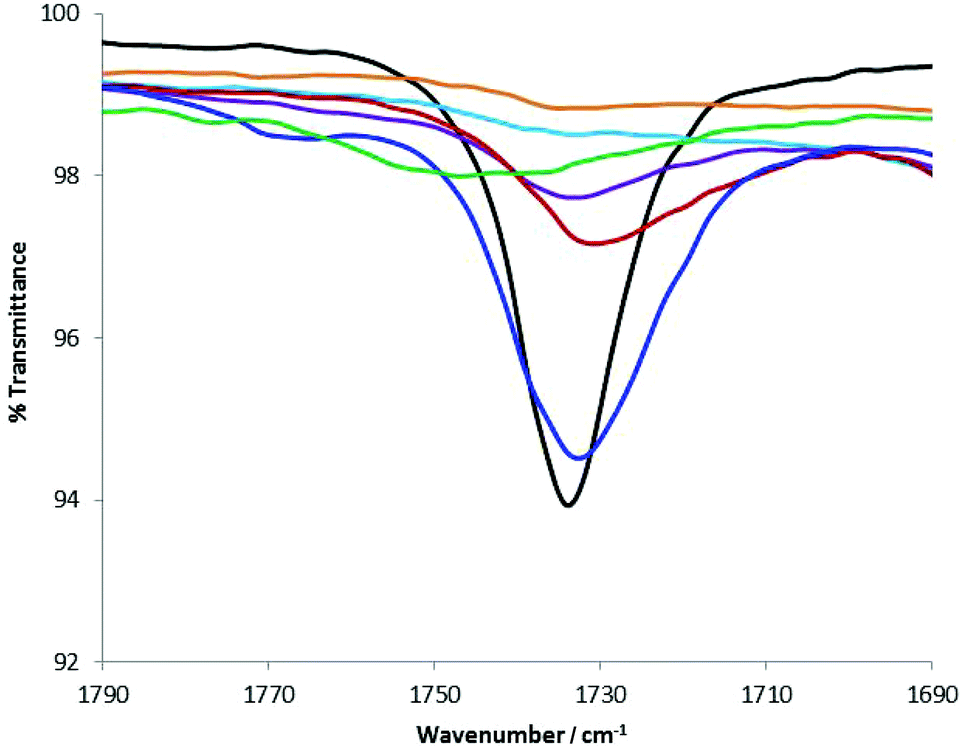 | ||
| Fig. 6 The carbonyl stretching frequency region of the FTIR spectra of (1b) and alcohol mixtures (in ratio of 1:4, respectively). Black (1b in CH3CN); turquoise (1b in MeOH); purple (1b in EtOH); red (1b in geraneol); blue (1b in 2-propanol); orange (1b in 1-butanol); green (1b in PhCH2CH2OH); it is noteworthy that (1b) was not soluble in either tBuOH or linalool, and are excluded from the figure. | ||
Finally we demonstrate here two applications of these systems in separating and modulating the vapour pressures14 of alcohol mixtures. When an equimolar mixture of geraniol (G) and linalool (L) was treated with (1b), at 30 °C for 1 h, phase separation occurred, and a sample of the top layer was analysed by gas chromatography (see Fig. 7). Linalool can be selectively extracted (88%) using a larger excess of (1b), and then by adding a large excess (>10 fold) of water to the ionic liquid layer released geraniol from (1b)-geraniol adduct. The ionic liquid was regenerated by removing all water under high vacuum.
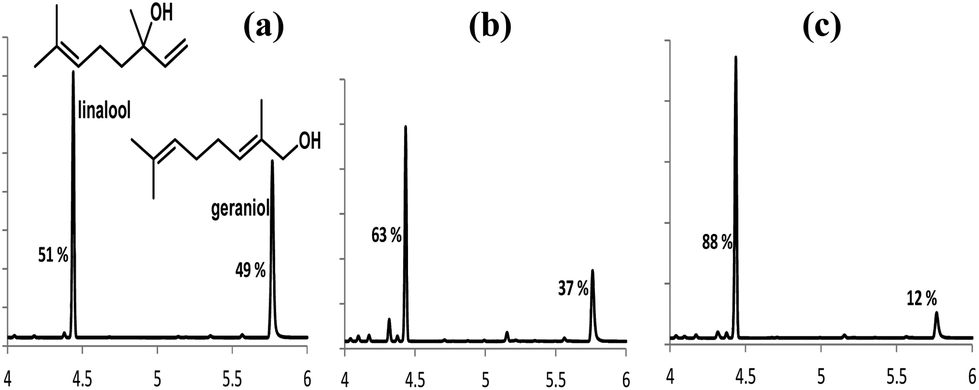 | ||
| Fig. 7 Gas chromatograms of diethyl ether extracts obtained after stirring an equimolar mixture of geraniol and linalool (G + L) with (1b) at different mole ratios. (a) G + L; (b) (G + L):(1b) = 1:2; (c) (G + L):(1b) = 1:4; the mole percentages of geraniol and linalool are shown next to each peak. | ||
Then, we examined how the vapour pressure of individual components of a mixture of alcohols (e.g. MeOH, Me2CHOH and Me3COH) could be modulated after interacting with these ionic liquids. An equimolar mixture of MeOH, Me2CHOH and Me3COH were treated with (1b) in a head-space GC vial followed by equilibration for 6 h at 30 °C. Head-space GC analysis was performed on this, together with a blank experiment using the mixture of alcohols without the ionic liquid. Results are shown in the Fig. 8. A dramatic reduction of the amount of the lowest boiling alcohol, MeOH, present in the vapour phase was seen. This demonstrates the potential of these materials for ‘perfume delivery’ type applications.
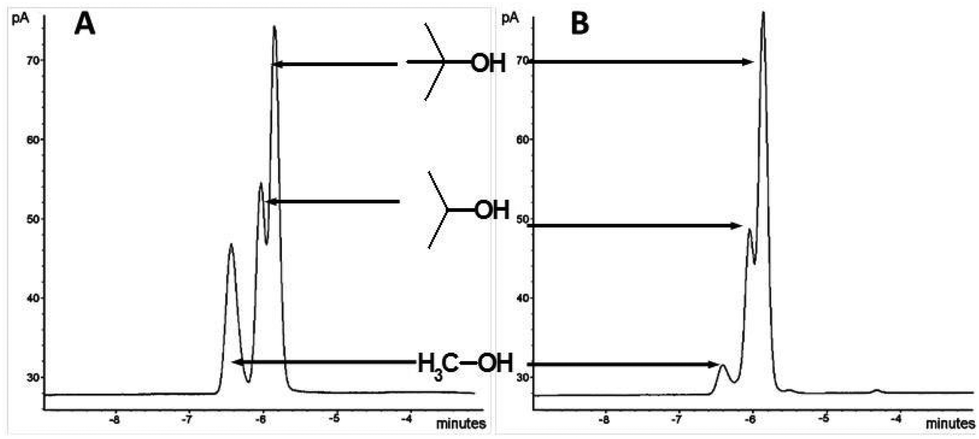 | ||
| Fig. 8 Head space GCs of equimolar mixture of MeOH–Me2CHOH–Me3COH: (A) without ionic liquids; (B) with (1b). Peaks corresponds to (from left to right) MeOH, iPrOH and tBuOH. | ||
There are reports, in the literature, of modulation of vapour pressures of organic solvents/liquids through interaction with ionic liquids15 albeit, the forces of interaction are weak in nature, i.e. dipolar, hydrogen-bond donor/acceptor and van der Waals forces. It must be stressed that the case demonstrated here, is distinctly different to such examples described before. The severe suppression of the vapour phase composition of the primary alcohol, methanol, compared to tBuOH and iPrOH, exemplifies the higher strength of the interaction of methanol by means of covalent bond formation with the ionic liquid.
In conclusion, we have designed, synthesised and studied the behaviour of a custom-designed class of ketone-functionalised ionic liquids that are capable of forming hemiacetals with alcohols, in the absence of any acid. Of particular interest and potential application, perfume raw materials containing an alcoholic function have also been shown to form hemiacetals with the same cations. Furthermore, the interaction of a mixture of alcohols with these ionic liquids was shown to drastically alter the vapour phase composition of each component. Moreover, the generated hemiacetal appear to be stable in the presence of a limited quantity of water. Our study on reactive ketone functionalised ionic liquids presents an opportunity for a novel separation of alcohol mixtures and for control of their relative vapour pressures. Thus, these reactive ionic liquids permits a novel ‘capture and release’ process for alcohols, which could find applications in their separation, or in fragrance delivery technologies.
Acknowledgements
We thank Drs Lalith Perera (NIH, USA), Peter Nockemann and Marijana Blesic for helpful discussions, Angela Brownlie for DSC and TGA, and Conor McGrann for ES-MS.Notes and references
- J. R. Hanson, Functional Group Chemistry, ed. A. G. Davies, Royal Society of Chemistry, 2001 Search PubMed; L. B. Clapp, The Chemistry of the OH Group, Foundations of Modern Chemistry, Series eds. R. W. Parry and H. Taube, New Jersey, 1967 Search PubMed; Patai's Chemistry of Functional Groups: The Chemistry of the Hydroxy Group, Part 1, J Wiley & Sons, Chichester Series Eds. S. Patai, 1971 Search PubMed.
- (a) J. F. Miller and A. Spaltenstein, Tetrahedron Lett., 1996, 37, 2521–2524 CrossRef CAS; P. M. Lukin, A. B. Zolotoi, S. V. Konovalikhin, S. P. Zil'berg, A. Kh. Bulai, O. A. D'yachenko, L. O. Atovmyan and O. E. Nasakin, Russ. Chem. Bull., 1986, 35, 1318–1319 Search PubMed; E. Neuzil, J. Josselin, J. C. Breton, J. L. Gaumet and B. Lemanceau, Bull. Soc. Pharm. Bordeaux, 1979, 118, 82–94 Search PubMed.
- Patai's Chemistry of Functional Groups: The Chemistry of the Amino Group, J Wiley & Sons, Series eds. S. Patai, 1968 Search PubMed; Patai's Chemistry of Functional Groups: The chemistry of the thiol group, Part 1, J Wiley & sons, Series eds. S. Patai, 1974 Search PubMed.
- M. Freemantle, An Introduction to Ionic Liquids, Royal Society of Chemistry, Cambridge, 2010 Search PubMed.
- H. Dreyfus, US Patent, US1991084A, 1935 Search PubMed; H. Dreyfus, US Patent, US2072101A, 1937 Search PubMed; W. Arlt, M. Seiler, C. Jork and T. Schneider, World Patent, 2002074718, 2002 Search PubMed; G. Ruffert, O. Pfohl and M. Grun, US Patent, US2008/0283383 A1, 2008 Search PubMed.
- J. Zhao, C.-C. Dong, C.-X. Li, H. Meng and Z.-H. Wang, Fluid Phase Equilib., 2006, 242, 147–153 CrossRef CAS; A. Westerholt, V. Liebert and J. Gmehling, Fluid Phase Equilib., 2009, 280, 56 CrossRef; V. K. Verma and T. Banerjee, J. Chem. Thermodyn., 2010, 42, 909–919 CrossRef; J. N. C. Lopes and L. P. N. Rebelo, Phys. Chem. Chem. Phys., 2010, 12, 1948–1952 RSC; F. S. Oliveira, R. Dohrn, A. B. Pereiro, J. M. M. Araújo, L. P. N. Rebelo and I. M. Marrucho, Fluid Phase Equilib., 2016, 419, 57–66 CrossRef.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed; T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef PubMed; N. D. Khupse and A. Kumar, J. Phys. Chem. B, 2011, 115, 711–718 CrossRef PubMed.
- H. Q. N. Gunaratne, P. Nockemann and K. R. Seddon, Green Chem., 2014, 16, 2411–2417 RSC; M. Blesic, H. Q. N. Gunaratne, J. Jacquemin, P. Nockemann, S. Olejarz, K. R. Seddon and C. R. Strauss, Green Chem., 2014, 16, 4115–4121 RSC; H. Q. N. Gunaratne, P. Nockemann and K. R. Seddon, Chem. Commun., 2015, 51, 4455–4457 RSC.
- M. Blesic, H. Q. N. Gunaratne, P. Nockemann, P. McCarron and K. R. Seddon, RSC Adv., 2013, 3, 329–333 RSC; J.-Y. Saint Laumer, E. Frerot and A. Herrmann, Helv. Chim. Acta, 2003, 86, 2871–2899 CrossRef CAS; S. Rochat, C. Minardi, J.-Y. Saint Laumer and A. Herrmann, Helv. Chim. Acta, 2000, 83, 1645–1671 CrossRef; B. Buchsa, W. Fiebera, D. Drahonovsky, J.-M. Lehn and A. Herrmann, Chem. Biodiversity, 2012, 9, 689–701 Search PubMed; A. Herrmann, Chem. – Eur. J., 2012, 18, 8568–8577 CrossRef PubMed; M. D. Soutullo, R. A. O'Brien, K. E. Gaines and J. H. Davis Jr., Chem. Commun., 2009, 2529–2531 RSC.
- A. Williams, Free Energy Relationships in Organic and Bio-Organic Chemistry, Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS; L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef; L. P. Hammett, Physical Organic Chemistry, McGraw-Hill Book Co. Inc., New York, NY, 1940, ch. III, IV, VII CrossRef; A. Streitwieser, R. W. Taft, P. R. Wells, S. Ehrenson and R. W. Taft, Progress in Physical Organic Chemistry, 2007, vol. 6 CrossRef; M. M. Kreevoy and R. W. Taft, J. Am. Chem. Soc., 1955, 77, 5590–5595 CrossRef; R. D. Topsom, in Progress in Physical Organic Chemistry, ed. R. W. Taft, Wiley, Hoboken, 1976, vol. 12, pp. 1–20 Search PubMed.
- N. E. Schore and P. C. Vollhardt, Organic Chemistry, Structure and Function, W.H. Freeman & Company, New York, 5th edn, 2007 CrossRef CAS; S. Marriott and R. D. Topsom, J. Am. Chem. Soc., 1984, 106, 7–10 CrossRef CAS; J. D. Roberts and W. T. Moreland, J. Am. Chem. Soc., 1953, 75, 2167 CrossRef; V. I. Galkin, J. Phys. Org. Chem., 1999, 12, 283–288 CrossRef.
- M. D. Soutullo, R. A. O'Brien, K. E. Gaines and J. H. Davis Jr., Chem. Commun., 2009, 2529–2531 RSC; J.-M. Lehn and A. Herrmann, Chem. Biodiversity, 2012, 9, 689–701 Search PubMed; A. Herrmann, Angew. Chem., Int. Ed., 2007, 46, 5836–5863 CrossRef CAS PubMed; L. Liu, G. Chen, M. L. Fishman and K. B. Hicks, Drug Delivery, 2005, 12, 149–157 CrossRef PubMed.
- K. Wong, J. Daugeron, S. Bonnus, R. Mounier, A. Furrer and D. Berthier, World Patent, WO2011/154926 A1, 2011 RSC; G. Mehlana, G. Ramon and S. A. Bourne, CrystEngComm, 2014, 16, 8160–8168 RSC; S. A. Ahmed, A. Chatterjee, B. Maity and D. Seth, J. Chem. Eng. Data, 2015, 60, 2301–2307 CrossRef CAS; P. Liu, M. Wang and Z.-M. Cheng, J. Chem. Eng. Data, 2015, 60, 836–844 CrossRef; N. Calvar, Á. Domínguez and E. A. Macedo, J. Chem. Thermodyn., 2014, 72, 9–15 CrossRef; M. García-Mardones, A. Barrós, I. Bandrés, H. Artigas and C. Lafuente, J. Chem. Thermodyn., 2014, 51, 17–24 CrossRef; N. Calvar, E. Gómez, Á. Domínguez and E. A. Macedo, Fluid Phase Equilib., 2012, 313, 38–45 CrossRef.
- K. Wysoczanska, N. Calvar and E. A. Macedo, J. Chem. Thermodyn., 2016, 97, 183–190 CrossRef CAS; N. Calvar, Á. Domínguez and E. A. Macedo, J. Chem. Thermodyn., 2014, 72, 9–15 CrossRef; N. Calvar, Á. Domínguez and E. A. Macedo, Thermochim. Acta, 2012, 549, 49–56 CrossRef; E. J. Gonzalez, L. Alonso and A. Domınguez, J. Chem. Eng. Data, 2006, 51, 1446–1452 CrossRef; F. M. Hinterholzinger, A. Ranft, J. M. Feckl, B. Ruhle, T. Bein and B. V. Lotsch, J. Mater. Chem., 2012, 22, 10356–10362 RSC; F. S. Emami, A. Vahid and J. R. Elliott, J. Chem. Thermodyn., 2009, 41, 530–537 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DSC, NMR, ESMS and synthetic procedures. See DOI: 10.1039/c6gc02708c |
| This journal is © The Royal Society of Chemistry 2017 |