
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward a benign strategy for the manufacturing of betulinic acid†
Anna K.
Ressmann
a,
Thomas
Kremsmayr
a,
Peter
Gaertner
a,
Ronald
Zirbs
b and
Katharina
Bica
 *a
*a
aInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: katharina.schroeder@tuwien.ac.at; katharina.bica@tuwien.ac.at; Fax: +43 1 58801 16360; Tel: +43 1 58801 163601
bInstitute for Biologically inspired materials, Department of Nanobiotechnology (DNBT), University of Natural Resources and Life Sciences, Muthgasse 11-II, A-1190 Vienna, Austria
First published on 12th December 2016
Abstract
We report a novel and efficient strategy for the preparation of the high-value triterpenoid betulinic acid based on extraction and streamlined oxidation of betulin from the industrial by-product birch bark. The initial extraction of betulin relies on a biphasic system and allowed extracting betulin in short times at room temperature. The crude extract could be directly oxidized, thereby providing a chromium-free, time- and energy saving strategy for the manufacturing of betulinic acid in high yield.
Introduction
Betulinic acid (1) is a naturally occurring pentacyclic triterpene which is widely found in the stem bark of a variety of tree species. Like many members of the lupane family, betulinic acid displays a number of biological and medicinal properties, and betulinic acid or its reduced species betulin (2) have been used in folk remedies since ancient times.1Recent studies have shown that betulinic acid exhibits potent cytotoxic activity specific for melanoma cells both in vitro and in vivo.2 Numerous papers over the past years aimed for the elucidation of the mode of action, as its anticancer activity is linked to its ability to induce apoptotic cell death in cancer cells.3 Betulinic acid and its derivatives have also been shown as potent inhibitors of the human immunodeficiency virus (HIV), while other sources report anti-bacterial, anti-malarial, anti-inflammatory, anthelmintic, antinociceptive and anti-HSV-1 activities.4
To date, two strategies exist for the manufacturing of betulinic acid that rely either on the direct extraction from plant sources or on a semi-synthetic approach based on its precursor triterpenoid betulin (2) (Fig. 1). While betulinic acid (1) may be directly isolated from a number of plants, its low content renders the extraction from most sources tedious and unsuitable for a reliable supply on industrial scale. The plane tree (platanus acerifola) is one of the most widely reported sources of betulinic acid with a comparably high content of 2.4–3.3 g per 100 g dry bark.5 However, its isolation suffers from the excessive use of mostly toxic solvents and time-consuming extraction or maceration strategies.6 Recent patent literature suggests the use of dichloromethane, methanol or toluene/2-pentanol/methanol mixtures as extractant.7–9 In any case, the obtained crude extract needs purification using chromatography techniques or multiple crystallization steps.
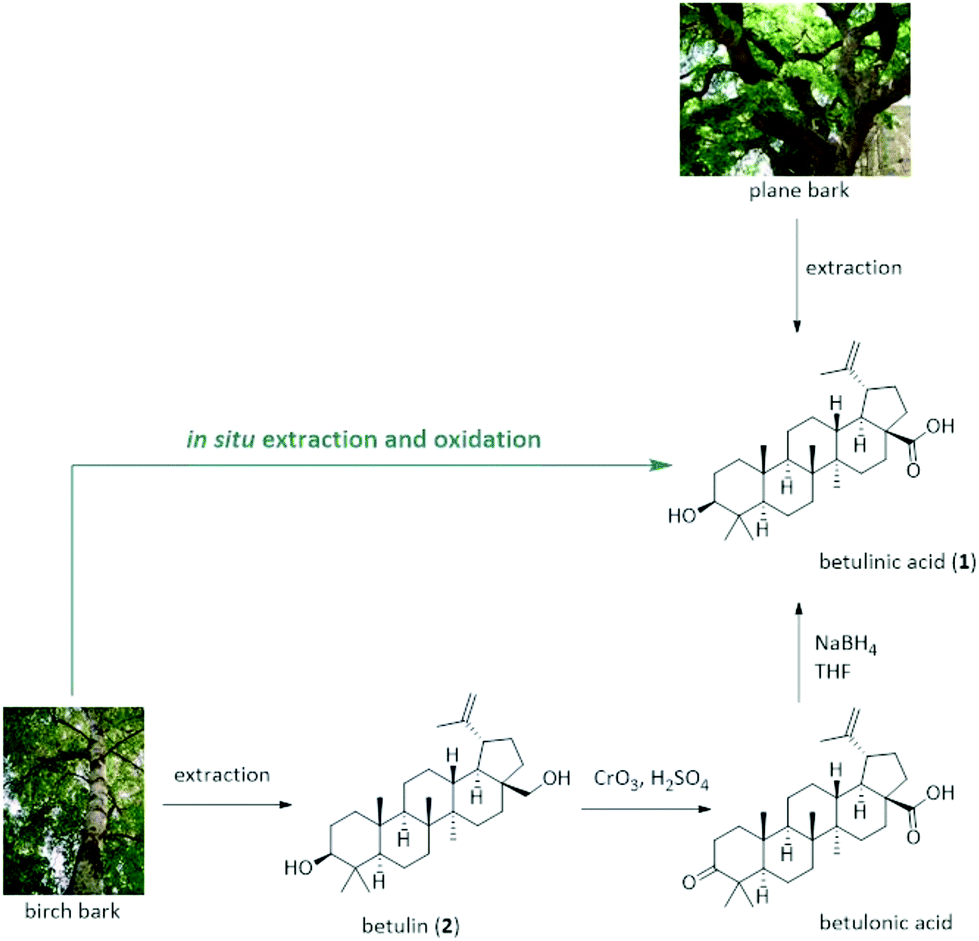 | ||
| Fig. 1 Conventional production of betulinic acid 1 relying either on extraction from plane bark (top) or semi-synthesis from betulin 2 (bottom). | ||
As alternative strategy, betulinic acid (1) can be prepared in a semi-synthetic pathway relying on its reduced species betulin (2). In contrast to betulinic acid, the triterpene betulin can be found in the white birch tree (betula alba) in significantly larger quantities of 10–35 wt% of the total dry weight of the outer bark.10–12 Due to the readily availability of birch bark as by-product and waste material from pulp and paper mills all over the world, this offers an attractive, cost-efficient and more reliable supply for the production of expensive triterpenes from an industrial waste stream that is usually burnt for energy production.
Yet, the consecutive conversion of betulin (2) to betulinic acid (1) is problematic and restricts its sustainable production: betulinic acid (1) is commonly synthesized via Jones oxidation of betulin (2) with CrO3/H2SO4 to betulonic acid followed by reduction to betulinic acid (1) using NaBH4.13,14 This is not only a two-step synthesis with only moderate yield, low atom efficiency and several purification steps but also relies on the use of stoichiometric Cr(VI) reagents that are extremely toxic and a serious safety and environmental concern. As a consequence, alternative strategies have been increasingly investigated, including a catalytic oxidation with (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) or its derivatives in BuOAc in combination with sub-stoichiometric amounts of NaOCl and NaClO2 in a buffered system.15,16 An oxidation method using [bis(acetoxy)iodo]benzene (BAIB) with TEMPO was recently disclosed.17 Chromic oxide in combination with potassium permanganate,18 as well as a ruthenium based oxidation using either a ruthenium catalyst e.g. RuCl2(PPh3)3 and TEMPO under O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 or TEMPO in combination with tetrapropylammonium perruthenat and N-methylmorpholin-N-oxide20,21 as well as an electrochemical22 and palladium based oxidation strategies23 have been also described. Despite this renewed interest in benign and chromium-free synthetic strategies for the oxidation of betulin, many of these procedures suffer from low yields and selectivities for betulinic acid, require multistep procedures, and rely typically on highly purified betulin (2) as starting material to obtain betulinic acid (1) in reasonable yield and purity.
19 or TEMPO in combination with tetrapropylammonium perruthenat and N-methylmorpholin-N-oxide20,21 as well as an electrochemical22 and palladium based oxidation strategies23 have been also described. Despite this renewed interest in benign and chromium-free synthetic strategies for the oxidation of betulin, many of these procedures suffer from low yields and selectivities for betulinic acid, require multistep procedures, and rely typically on highly purified betulin (2) as starting material to obtain betulinic acid (1) in reasonable yield and purity.
In here we describe our efforts towards a benign and scalable manufacturing of betulinic acid directly from birch bark, aiming to combine the extraction of betulin at room temperature with the direct oxidation towards betulinic acid while avoiding chromium reagents and the use of toxic solvents.
Materials and methods
General
All chemicals were purchased from commercial suppliers and used as received unless otherwise stated. White birch bark was collected in Vienna in summer 2012, frozen with liquid nitrogen and milled on a Retsch® CryoMill to a particle size of <1 mm.
1H, 13C and 31P NMR spectra were recorded from CDCl3, CD2Cl2, d4-MeOD or d6-DMSO solutions on a Bruker Advance UltraShield 400 (400 MHz) spectrometer. Chemical shifts (δ) are reported in ppm using tetramethylsilane as internal standard and coupling constants (J) are given in hertz (Hz). Melting points were measured on an automated melting point system OPTI MELT of Stanford ResearchSystems. Scanning electron microscopy (SEM) pictures were taken with a FEI Inspect F50 at 15 kV. All samples were coated with a 3.5 nm thick gold-layer using a Leica Cool Sputter Coater EM SCD005. HPLC analysis was performed on a Jasco HPLC unit equipped with a PDA detector. For the quantification of betulin (2) a Maisch ReproSil 100 C18 250 × 4.6, 5 μm was used with MeOH:H2O (0.1% TFA) = 87:13 as solvent and a flow of 1 ml min−1; detection was done at 210 nm, at 30 °C column oven temperature. Retention times were 7.3 min for the internal standard and 15.7 min for betulin (2). The same HPLC set-up used for the simultaneous quantification of betulin, betulinic aldehyde and betulinic acid in the oxidation process. Retention times were 7.3 min for the internal standard, 15.7 min for betulin (2), 16.5 min for betulinic acid (1) and 28.0 min for betulinic aldehyde. Microwave (MW) reactions were performed on a BIOTAGE Initiator™ sixty microwave unit. The reported times are hold times.
Screening of betulin extraction from birch bark
Isolation of betulin from birch bark
:EtOAc = 20:1–2:1) 230 mg betulin were obtained as colorless crystals. (23 wt% corresponding to birch bark) in >95% purity according to NMR.
Alternatively, betulin could be isolated via crystallization: the procedure was performed as described above using 300 mg birch bark. The crude product was crystallized from a mixture of methanol/water to yield betulin (62.5 mg, corresponding to 20.8 wt% yield) as colorless crystals with a purity of >90% according to NMR, containing traces of lupeol. M.p. 230.0–231.1 °C. 1H-NMR (600 MHz, CDCl3): δH = 0.67 (m, 1 H), 0.76 (s, 3H), 0.82 (s, 3H), 0.90 (s, 3H), 0.97 (s, 3H), 0.98 (s, 3H), 1.05 (m, 3H), 1.18–1.28 (m, 4H), 1.39 (m, 5H), 1.51–1.70 (m, 12H), 1.85 (m, 1H), 1.94 (m, 2H), 2.38 (m, 1H), 3.19 (dd, J = 11.5, 4.8, 1H), 3.33 (dd, J = 10.9, 1.3, 1H), 3.80 (dd, J = 10.8, 1.6, 1H), 4.58 (s, 1H), 4.68 (s, 1H). 13C-NMR (150 MHz, CDCl3): δC = 15.1, 15.7, 16.3, 16.5, 18.6, 19.4, 21.2, 25.5, 27.4, 27.7, 28.3, 29.5, 30.1, 34.3, 34.6, 37.5, 37.6, 39.0, 39.2, 41.3, 43.1, 48.1 (2C), 49.1, 50.7, 55.6, 60.9, 79.3, 110.1, 150.8.
Analytical data was in accordance with literature values.14
After purification via column chromatography (7 g SiO2, PE:EtOAc = 20:1–2:1) 25.4 mg betulin were obtained as colorless crystals in 25 wt% yield corresponding to birch bark.
Oxidation of betulin to betulinic acid
Betulin (2, 99.5 mg, 0.22 mmol), TEMPO (7.0 mg, 0.04 mmol, 0.2 eq.) and BAIB (217 mg, 0.67 mmol, 3 eq.) were dissolved in 10 ml n-BuOAc, 0.2 ml water and 42 μl t-BuOH. After 6 h stirring at room temperature TLC showed full conversion. To the clear yellow solution 0.2 ml water, 2-methyl-2-butene (72 μl, 1.03 mmol, 4.6 eq.), NaH2PO4 (70 mg, 0.58 mmol, 2.6 eq.) and NaClO2 (66 mg with 80% purity, 0.58 mmol, 2.6 eq.) were added and a colour change to brown was observed while a white solid was precipitating. The solution was quenched with saturated sodium thiosulfate solution and extracted with EtOAc three times, washed with saturated NaHCO3 solution and dried over Na2SO4. The solution was filtered and concentrated under reduced pressure. The off-white residue was crystallized from MeOH to obtain betulinic acid in 89% yield as colourless crystals.M.p.: 284.5–285.5 °C. 1H-NMR (400 MHz, CDCl3): δH = 0.67 (m, 1H), 0.75 (s, 3H), 0.8 (s, 3H), 0.93 (s, 3H), 0.96 (s, 3H), 0.97 (s, 3H), 1.18–1.69 (m, 15H), 1.69 (s, 3H), 1.97 (m, 2H), 2.19 (m, 1H), 1.27 (m, 5H), 2.26 (m, 1H), 3.00 (m, 1H), 3.19 (dd, J = 11.3, 5.0, 1H), 4.60 (s, 1H), 4.74 (s, 1H). 13C-NMR (100 MHz, CDCl3): δC = 15.1, 15.7, 16.4, 16.5, 18.6, 19.7, 21.2, 25.8, 27.8, 28.3, 30.1, 30.9, 32.5, 34.7, 37.4, 37.6, 38.7, 39.1, 39.2, 41.0, 42.8, 47.2, 49.6, 50.9, 55.7, 56.6, 79.4, 110.1, 150.8, 180.2.
Analytical data was in accordance with literature.14
Streamlined extraction and oxidation process
:1–2:1) to yield betulinic acid (1) in 37.5 mg corresponding to 18 wt% yield as colorless crystals with a purity of >95% according to 1H NMR.
Analytical data was in accordance with literature.14
Results and discussion
Biphasic extraction of betulin at room temperature
The extraction of betulin (2) from birch bark is typically performed using conventional Soxhlet extraction with a number of solvents such as dichloromethane, chloroform, ethanol or methanol.24–26 However, these conventional extraction strategies typically require large solvent volumes and suffer from long extraction times at elevated temperatures, resulting in high time and energy consumption. As for betulinic acid, the purity of the raw extracts is typically low, and additional purification steps are usually required.As a consequence, efforts to reduce the exhaustive use of undesirable solvents as extraction media for betulin have been made, and recent trends include the use of bio-derived solvents such as limonene,27 pressurized solvent10 and supercritical fluid extraction28 or ionic liquid-based extraction strategies.29 In a previous paper, we reported the microwave-assisted extraction of betulin (2) using several hydrophilic and hydrophobic ionic liquids. Ionic liquids as solvents for the extraction of value-added compounds may result in increased yields compared to conventional solvents through fractionation of the lignocellulosic matrix.30,31 In case of betulin, the best extraction yield was obtained after dissolution of birch bark in the ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2mim]OAc) at 100 °C for 15 min. An isolation strategy relying on the stepwise precipitation of biopolymers with ethanol followed by the addition of water to crystallize betulin was developed and allowed isolating betulin in good yields and purity.
However, aiming for the consecutive in situ oxidation of betulin (2) this strategy had to be modified in order to avoid ethanol as solvent and to remove any other biomass fragments that would interfere with the further conversion.
The search for an efficient extraction procedure of betulin that would allow the consecutive oxidation to betulinic acid lead us to the development of a biphasic extraction strategy relying on the dissolution or pretreatment of birch bark in ionic liquids or their aqueous solutions and an organic co-solvent, n-butyl acetate. This biphasic concept should enable not only a fast and efficient extraction of betulin from birch bark under mild conditions, but the simultaneous separation of the biopolymers or their fragments dissolved in the aqueous ionic liquid layer, while a relatively pure stream of the hydrophobic triterpene betulin (2) would be obtained in the organic phase (Fig. 2).
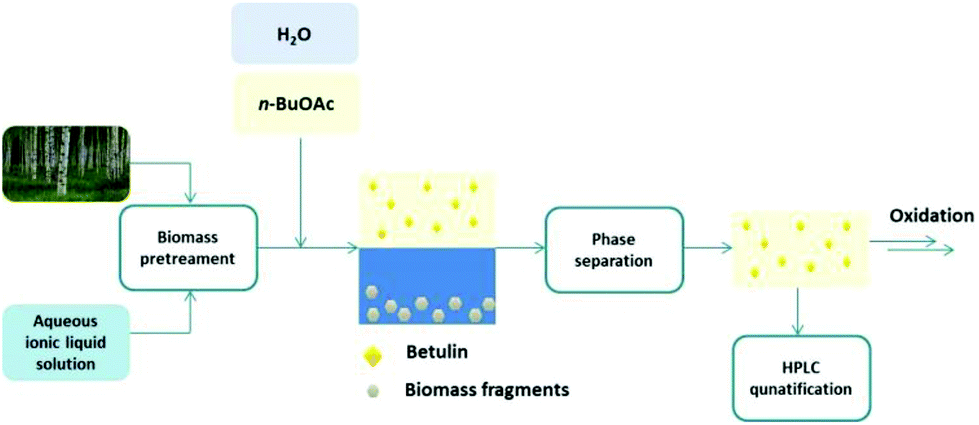 | ||
| Fig. 2 Biphasic system for the isolation of betulin (2) and the continued oxidation to betulinic acid (1). | ||
In 2013, Ohno et al. reported the efficient dissolution of woody biomass at 60 °C and of pine, cedar and poplar wood even at room temperature in aqueous solution of onium ionic liquids, e.g. in a 40% solution of tetrabutylammonium hydroxide ([P4444]OH) in water.32,33 The extraction degree of polysaccharides from poplar wood reached up to 37 wt% after 1 hour treatment with [P4444]OH solution at room temperature, suggesting that this phosphonium salt solution might also provide a powerful pretreatment medium for birch bark and could facilitate the extraction of betulin under mild conditions.
To evaluate the extraction performance birch bark was initially pretreated in the aqueous [P4444]OH solution for 15 min at 100 °C under microwave irradiation. Water and n-BuOAc were added to extract betulin in the organic phase. Several aspects contributed to the selection of n-butyl acetate as organic solvent at this stage. As key criteria, n-butyl acetate has been positively evaluated in the revised GSK solvent selection guide, suggesting that it might act as sustainable alternative to the more volatile ethyl acetate.34 Moreover, our studies showed that n-butyl acetate is a suitable solvent for the consecutive oxidation of betulin – even in the presence of water traces – which is of fundamental importance for the development of the envisaged streamlined extraction and oxidation process (vide infra, see also ESI Fig. S2 and Table S3†).
After these stepwise dissolution and extractions steps, the betulin (2) content in the upper organic phase was quantified via HPLC. Results showed that the phosphonium-based salt solution was indeed a suitable pretreatment media for birch bark, allowing a fast and efficient extraction of betulin with a high extraction yield of 29 wt% (Table 1, entry 1 and ESI Fig. S1†).‡ The influence of the concentration of tetrabutylammonium hydroxide in water was further investigated using more dilute solutions (Table 1, entries 2–4). A 20% solution of [P4444]OH in water still gave a good yield of 25%; however, if the concentration decreased to 5%, the yield dropped again until the same level as in the biphasic extraction with pure water/n-BuOAc was obtained (Table 1, entry 5).
| Entrya | Salt concentration in H2O | Conditions | Yield betulin (2)b,c [wt%] |
|---|---|---|---|
| a Performed using 100 mg ground birch bark and 900 mg aqueous [P4444]OH solution or H2O at the indicated conditions, followed by addition of 2 ml water and 3 ml BuOAc and stirring of 15 min, 100 °C under MW irradiation (entries 1–5) or 1 h at room temperature (entries 6–15). b Yields determined via HPLC using 1-methyl-1-cyclohexene as internal standard. c Mean ± STD, n = 3. | |||
| 1 | 40% [P4444]OH | 100 °C, MW, 15 min | 29.2 ± 0.6 |
| 2 | 20%[P4444]OH | 100 °C, MW, 15 min | 25.1 ± 1.8 |
| 3 | 10% [P4444]OH | 100 °C, MW, 15 min | 24.4 ± 0.9 |
| 4 | 5% [P4444]OH | 100 °C, MW, 15 min | 22.1 ± 1.0 |
| 5 | 0 (pure H2O) | 100 °C, MW, 15 min | 22.3 ± 1.1 |
| 6 | 0 (pure H2O) | r.t., 24 h | 15.0 ± 1.0 |
| 7 | 40% [P4444]OH | r.t., 24 h | 28.6 ± 1.3 |
| 8 | 40% [P4444]OH | r.t., 6 h | 27.0 ± 0.1 |
| 9 | 40% [P4444]OH | r.t.,3 h | 27.5 ± 1.2 |
| 10 | 40% [P4444]OH | r.t., 1 h | 26.5 ± 0.6 |
| 11 | 40% [P4444]OH | r.t., 30 min | 20.4 ± 0.8 |
| 12 | 20% [P4444]OH | r.t., 1 h | 24.5 ± 0.6 |
| 13 | 10% [P4444]OH | r.t., 1 h | 20.9 ± 0.8 |
| 14 | 8% NaOH | r.t., 1 h | 19.7 ± 1.5 |
| 15 | 6% NaCl | r.t., 1 h | 19.1 ± 0.9 |
In order to develop an energy saving process, the extraction efficiency of the salt solution/n-BuOAc system was further evaluated at room temperature, as it was previously shown that the phosphonium ionic liquid [P4444]OH in aqueous solution is able to dissolve wood even at room temperature.32 The effect of pretreatment with the aqueous solution is even more evident at room temperature. Only 15 wt% betulin could be extracted in biphasic extraction with pure water/n-BuOAc (Table 1, entry 6). In contrast, pretreatment with a 40% [P4444]OH solution in water followed by extraction with n-BuOAc dramatically increased the extraction yield to 28.6 wt%, indicating that the extraction yield can be doubled by the mere addition of phosphonium salt. Moreover, the extraction time could be dramatically reduced: while 30 min extraction time where not sufficient, 26.5 wt% betulin were already extracted after 1 hour extraction time, providing an ideal compromise between extraction yield and time consumption of the process (Table 1, entries 7–10). Further studies on the impact of phosphonium salt concentration at room temperature showed a similar effect as we observed in case of microwave treatment at 100 °C. With decreasing concentration of the phosphonium salt, the yield of betulin also decreased. A 20% solution of [P4444]OH in water resulted in a lower extraction yield of 24.5 wt% betulin. With a 10% solution the loss of extraction efficiency is even more pronounced and the extraction yield is reduced to 20.9 wt% betulin (Table 1, entries 12 and 13).
It is worthwhile noticing that this efficient pretreatment and strong increase in extraction yield obtained with a 40% solution of [P4444]OH in water is an unique effect arising from the combination of cation and anion in this system: when comparing the extraction yield with solutions of 40% [P4444]Br or 40% NaOH in water to mimic the impact of cation or anion separately, we did not observe the strong enhancement of extraction yield, and values remained drastically below those obtained with [P4444]OH solution (Fig. 3). Similarly, a saturated solution of sodium chloride did not result in a strong increase in comparison to pure water/n-BuOAc, thereby ruling out the presence of a pure salt effect on extraction and phase transfer. To exclude that the extraction of betulinic was inhibited by a kosmotropic effect of the 40% NaOH solution, experiments were also performed with a 8% solution NaOH that has the same molarity as 40% [P4444]OH in water (1.45 M). As it can be seen in Table 1, entries 14 and 15 yields remained significantly below the value obtained with the [P4444]OH solution. A solution of sodium chloride with the same molarity (1.45 M, corresponding to 6% NaCl in water) gave also only low yields (Table 1, entries 14–16).
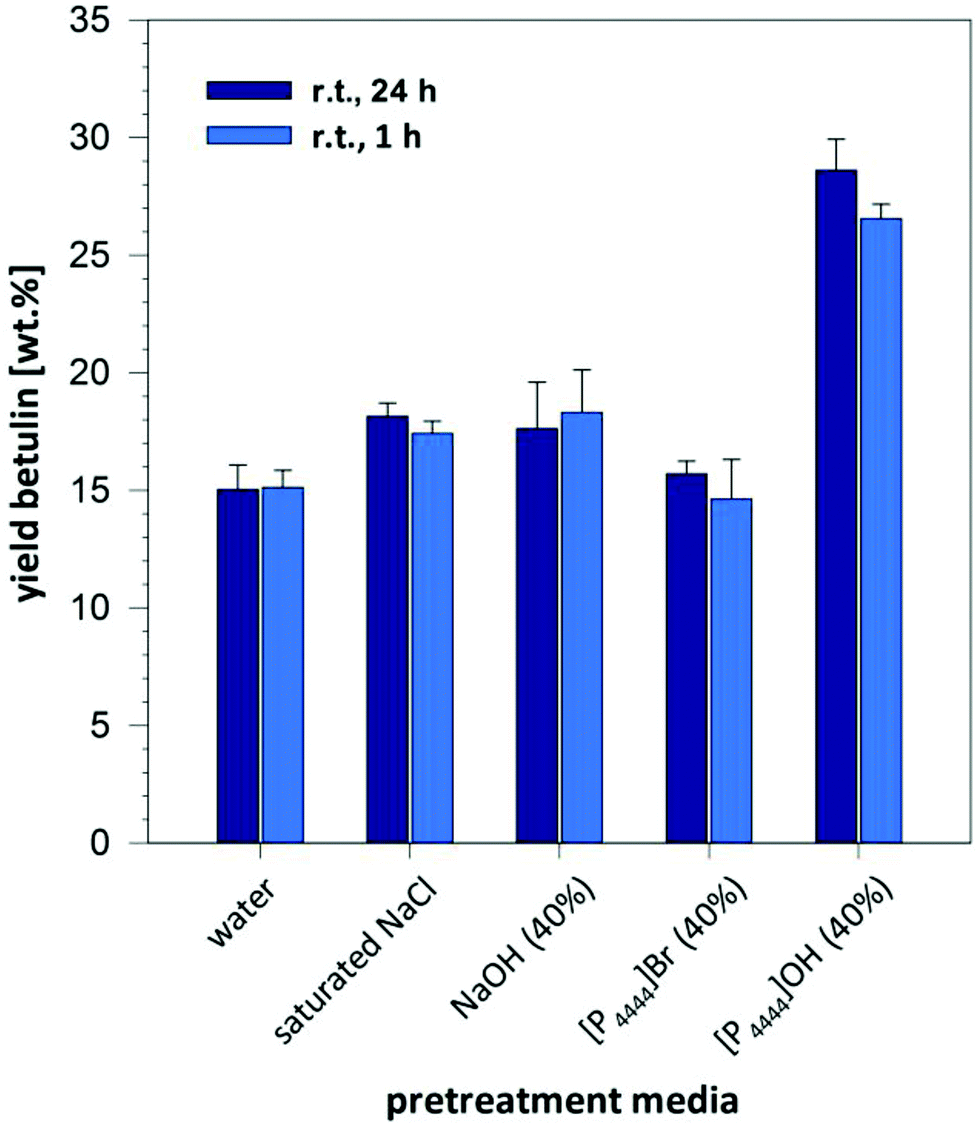 | ||
| Fig. 3 Impact of different salt solutions on the extraction of betulin from birch bark at room temperature. | ||
Additional studies showed the possiblility to recover and recycle the aqueous [P4444]OH solution after the extraction. In a straightforward process, betulin was extracted from the aqueous phase with butyl acetate without further dilution with water to maintain the concentration. Fresh biomass was added, and the extraction process was repeated. With this strategy, the aqueous [P4444]OH solution could be reused for three runs without any loss efficiency, and yields reached 29.5% and 32.5% betulin in the second and third run, respectively (see ESI Table S1†). The small increase in yield in the third run can be attributed to an incomplete phase separation. However, after the third addition of fresh biomass, extraction yields dropped drastically. Only 18.6% betulin were isolated in the 4th run, which might be related to the increasing viscosity of the aqueous [P4444]OH slurry that became increasingly difficult to stir. In order to avoid problems with viscosity due to the accumulation of biopolymers in the aqueous solution, we evaluated the possibility of their precipitation with ethanol as anti-solvent. This approach relied on a strategy that we previously used for the extraction of betulin from birch bark with the ionic liquid 1-ethyl-3-methylimidazolium acetate.29 After extraction of betulin with n-butyl acetate, the aqueous [P4444]OH solution was diluted with an excess ethanol. Precipitated biopolymers were removed via filtration and ethanol was removed via distillation before the aqueous [P4444]OH solution was subjected to another run. However, this strategy failed as the yield of betulin dropped already in the second run to 10.0%.
Eventually, the strong impact of lignocellulose pretreatment with a 40% [P4444]OH in water is also visible in electron microscopy. While preparing the biomass for the SEM-pictures it was already visible with the eye that pretreatment using [P4444]OH tremendously changed the biomass structure. As can be seen in Fig. 4 the morphology of the birch bark changed when treated with different solvents. The strongest effects were observed with a [P4444]OH solution, resulting in a perforated biopolymer matrix left after 1 h of pretreatment only.
 | ||
| Fig. 4 SEM pictures of untreated birch bark (top, left), birch bark treated with 40% solutions of NaOH (top, right), [P4444]Br (bottom, left) and [P4444]OH (bottom, right) in water. | ||
After the identification of suitable extraction conditions, it was possible to isolate betulin from the organic solution after phase separation. At this stage, purification via column chromatography gave betulin in 23 wt% yield and excellent purity after 1 h extraction time at room temperature only. Alternatively, betulin could be isolated via crystallization using water/methanol mixtures in 20.8 wt% yield. However, we were more interested in the in situ extraction and oxidation towards betulinic acid and therefore the oxidation reaction was investigated in a next step (see ESI Fig. S4–S7†).
Oxidation of betulin to betulinic acid
Once suitable extraction conditions were found, we focused on the oxidation of betulin (2) to betulin acid (1) that was initially optimized using pure betulin rather that the biomass-derived crude extraction stream (Fig. 5). Despite being one of the most frequently used reactions in organic synthesis, the oxidation of alcohols to carboxylic acids relied for a long time on the use of metal oxides or metal salts.35 | ||
| Fig. 5 Optimized oxidation of betulin (2) to betulinic acid (1). | ||
Apart from their outstanding toxicity, e.g. in case of notorious Cr(VI) salts, these methods suffer from the necessity of using stoichiometric or even higher amounts of the metal oxide, from the poor atom economy, and from the generation of heavy-metal waste.36 As a result, research has focused on the development of more benign oxidations protocols, particularly on catalytic versions using air or oxygen as oxidants.37,38 Yet a number of challenges exist for the oxidation of betulin to betulinic acid that limits the pool of available strategies for this particular reaction. Problems are mostly caused by the presence of a secondary alcohol group in position 3 that must not be oxidized, since otherwise a consecutive reduction step of betulonic acid would be required. Additionally, the presence of the electron-rich double bond in position 20 that is likely to be attacked under various conditions complicates the oxidation step.
Our search for a mild oxidation strategy that would fulfill the desired criteria led us to the evaluation of hypervalent iodine(III) reagents, and in particular to the catalytic oxidation using (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) in combination with [bis(acetoxy)iodo]benzene (BAIB). In general catalytic TEMPO mediated oxidations with secondary oxidants such as NaOCl,39 NaOCl/NaClO240 or BAIB41,42 are comparatively mild and known for the selective oxidation of primary alcohols in the presence of secondary alcohols due to the bulky oxoammonium salt derived from TEMPO.§ Moreover, the combination of BAIB with catalytic amounts of TEMPO was previously reported to be compatible with a number of functionalities, including double and triple bonds, esters, acetals, epoxides, amides, and azides as well as protection groups. Initial attempts on the oxidation of betulin using equimolar amount of TEMPO and 3 eq. BAIB in n-BuOAc showed complete conversion of betulin and a yield of 24% betulinic acid. Most importantly, we did not observe any attack of the reactive double bond or any over oxidation of the secondary alcohol group but detected betulinic acid and the intermittent betulinic aldehyde as sole products. The amount of TEMPO could be further reduced to 0.2 eq. TEMPO and 3 eq. BAIB. Further investigations into the reaction conditions revealed a strong influence of the concentration of betulin in n-BuOAc, as the highest yield and conversion was found at a concentration of 10 mg betulin per ml n-BuOAc (see ESI Tables S2 and S3†). After optimization of reaction conditions is was possible to obtain betulinic acid in 43% yield. However, at this stage, the yield of betulinic acid levelled, and a longer reaction time or the addition of fresh reagents did not shift the ratio between aldehyde and acid towards a higher yield of betulinic acid (Fig. 6).
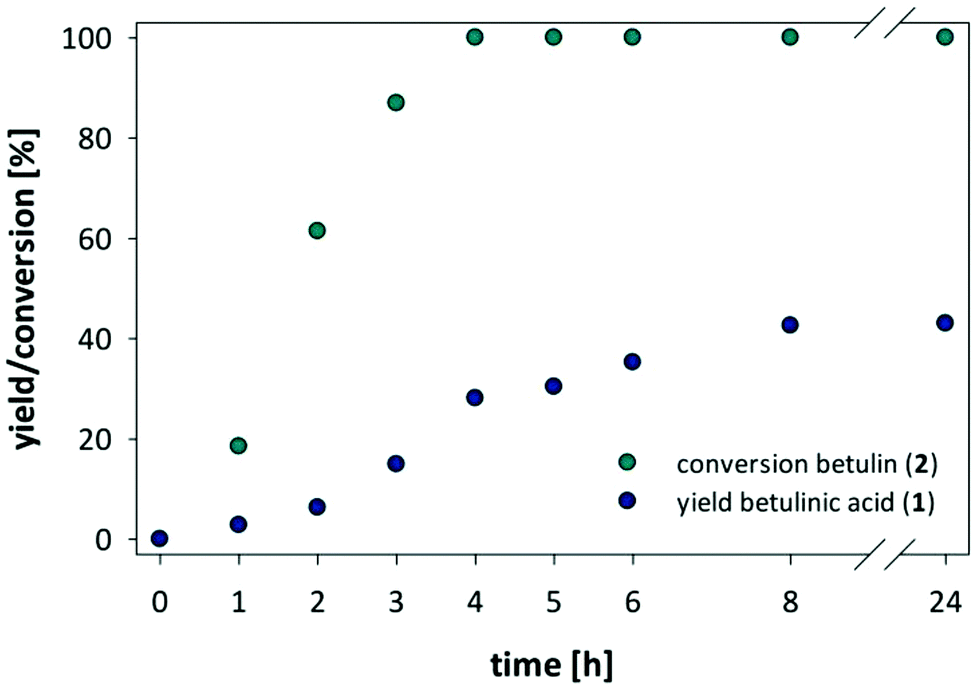 | ||
| Fig. 6 Conversion of betulin and yield of formed betulinic acid against time in the TEMPO/BAIB mediated oxidation of betulin (2) to betulinic acid (1). | ||
We found that the addition of acetic acid in excess can be beneficial for this oxidation and increases the yield of betulinic acid to 58%; however, it was still not possible to completely convert the intermittent aldehyde (see ESI Fig. S2†). In order for the further conversion of the crude mixture, we added a consecutive Pinnick oxidation step relying on NaClO2/NaH2PO4 with 2-methyl-2-butene as chlorine scavenger to convert the intermittent betulinic aldehyde to the acid.43 Eventually, after optimization of conditions the combination 0.2 eq. of TEMPO with 3 eq. BAIB followed by the addition of 2.6 eq. Pinnick reagents with a chlorine scavenger allowed to isolate betulinic acid in a reliable one-pot-two-step procedure in 89% yield without the use of chromium reagents (see ESI Fig. S8 and S9†).¶
Streamlined extraction and oxidation process
After finding suitable and compatible conditions for extraction and oxidation, we eventually merged the two individual processes, aiming for a one-pot extraction and in situ oxidation of the crude betulin extract. However, difficulties were faced when oxidizing the crude betulin (2) extract under the previously established conditions, and conversion and yield remained significantly below the values obtained for pure starting material. Part of these problems could be traced to the presence of small amounts of phosphonium salt in the organic extract after phase separation that inhibited the consecutive oxidation. When only 0.5 eq. of the phosphonium salt [P4444]OH were deliberately added with regard to betulin (2), conversion dropped from 100% to 88%, whereas no conversion was observed when 2 eq. of the phosphonium salt were present. A similar inhibition of the oxidation was observed in the presence of [P4444]Br. Attempts to remove undesired phosphonium salts via an additional extraction step of the crude betulin extract in n-BuOAc with water did not remove phosphonium contaminants, as 31P NMR still showed signals for the phosphonium cation in the n-BuOAc layer. It was however possible to remove phosphonium salts via a brief filtration over a batch of silica. Eventually, we were able to develop a streamlined process for the oxidation as shown in the flow scheme in Fig. 7: birch bark was pretreated in aqueous solution of [P4444]OH at room temperature for a short dissolution time of 1 h. After the addition of water and n-BuOAc the solution was stirred for another hour to extract betulin. After separation of the organic layer and filtration over SiO2 the solution was concentrated to obtain a 10 mg ml−1 solution of betulin (1) in n-BuOAc. The oxidation reagents were added, starting with TEMPO/BAIB reagents in combination with acetic acid. After 5 h reaction time Pinnick reagents were added and the solution was stirred for another 19 h.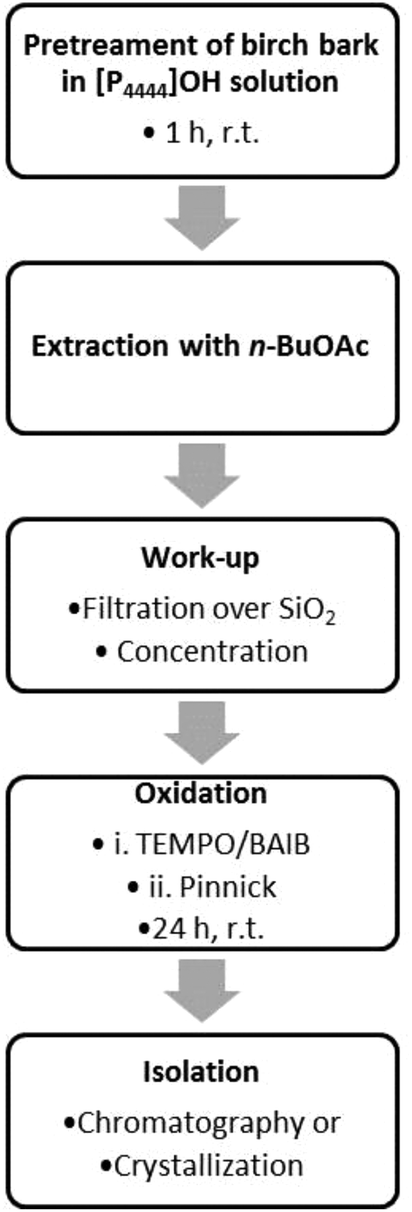 | ||
| Fig. 7 Flow scheme of the streamlined extraction and oxidation process for the direct preparation of betulinic acid (1) from birch bark. | ||
As a final challenge, the isolation of betulinic acid from the raw biomass-derived solution required a purification step to separate betulinic acid from lupeol, a triterpenoid whose content in birch bark varies between 1–4 wt%.44 Eventually, two options exist for the isolation of betulinic acid from the crude biomass-derived solution. The raw extract can be either directly subjected to flash column chromatography, allowing to separate the two triterpenoids and to isolate betulinic acid with high purity with a final yield of 18 wt% calculated from the amount of birch bark (see ESI Fig. S10 and S11†).
Alternatively, the final isolation and separation of lupeol and betulinic acid could be achieved via crystallization to avoid chromatography. Fractionated crystallization of the crude extract from water/methanol mixtures gave a purified fraction, and betulinic acid could be isolated in 22 wt% yield and good purity of >90% (see ESI Fig. S12 and S13†). However, this strategy could not separate betulinic acid and lupeol quantitatively, and betulinic acid was contaminated with 3 wt% lupeol. Additionally, a second fraction was obtained with 7 wt% yield betulinic acid and a purity <90%. Clearly, improvements in this final crystallization step for purification would add to the overall process efficiency and ease of manufacturing on large scale.
Conclusions
In this paper, we report a novel and efficient strategy for the synthesis of the high-value triterpenoid betulinic acid based on the extraction and streamlined oxidation of betulin (2).The initial extraction of betulin from the industrial by-product birch bark relies on a biphasic system using aqueous phosphonium hydroxide solution and n-butyl acetate. This strategy could extract betulin in high yields at room temperature after 1 hour only, thereby providing a cheap, benign and time and energy-saving strategy for the extraction of valuable ingredients from biomass compared to existing strategies (Fig. 8).
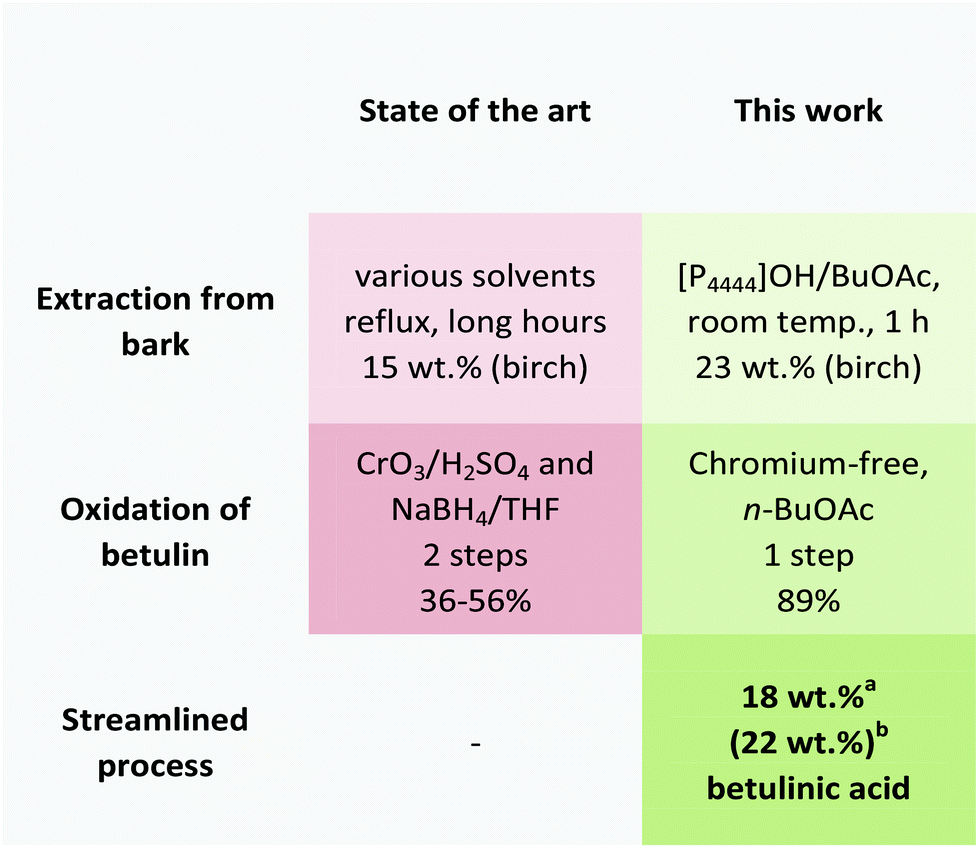 | ||
| Fig. 8 Comparison of conventional strategies and the novel approach featured in this paper for the synthesis of betulinic acid (1). aIsolated via column chromatography. bIsolated via recrystallization from water/methanol, 3 wt% lupeol present. | ||
Furthermore, this biphasic approach enables the further conversion of betulin (2) after simple phase separation toward the value-added triterpenoid betulinic acid (1). The crude extract could be directly oxidized using TEMPO and hypervalent iodine(III) reagents, thereby providing a reliable and high yielding strategy for the synthesis of betulinic acid.
In summary, we presented for the first time a streamlined extraction and oxidation strategy for the manufacturing of betulinic acid from birch bark that is running at room temperature, while simultaneously avoiding toxic chromium reagents or solvents (Fig. 8).
Notes and references
- R. H. Cichewicz and S. A. Kouzi, Med. Res. Rev., 2004, 24, 90–114 CrossRef CAS PubMed.
- R. Mukherjee, V. Kumar, S. K. Srivastava, S. K. Agarwal and A. C. Burman, Anticancer Agents Med. Chem., 2006, 6, 271–279 CrossRef CAS PubMed.
- S. Fulda, Int. J. Mol. Sci., 2008, 9, 1096–1107 CrossRef CAS PubMed.
- P. Yogeeswari and D. Sriram, Curr. Med. Chem., 2005, 12, 657–666 CrossRef CAS PubMed.
- S. Jäger, H. Trojan, T. Kopp, M. N. Laszczyk and A. Scheffler, Molecules, 2009, 14, 2016–2031 CrossRef PubMed.
- F. Chemat, M. Abert Vian and G. Cravotto, Int. J. Mol. Sci., 2012, 13, 8615–8627 CrossRef CAS PubMed.
- B. Draeger, R. Neubert, T. Galgon and W. Wohlrab, Herstellung von Betulinsäure, DE Pat19713768A1, 1997 Search PubMed.
- M. Sauter, Verbessertes Verfahren zur Gewinnung von Betulinsäure, WO Pat2003066659A3, 2003 Search PubMed.
- C. Puder, H. Graef, M. Thumerer and M. Heitzmann, Process for the extraction of betulinic acid, US Pat20070149490A1, 2007 Search PubMed.
- M. Co, P. Koskela, P. Eklund-Åkergren, K. Srinivas, J. W. King, P. J. R. Sjöberg and C. Turner, Green Chem., 2009, 11, 668–674 RSC.
- P. A. Krasutsky, Nat. Prod. Rep., 2006, 23, 919–942 RSC.
- G. Zha, W. Yan and D. Cao, J. Pharm. Biomed. Anal., 2007, 43, 959–962 CrossRef PubMed.
- D. S. H. L. Kim, Z. Chen, N. Van Tuyen, J. M. Pezzuto, S. Qiu and Z.-Z. Lu, Synth. Commun., 1997, 27, 1607–1612 CrossRef CAS.
- L. Pohjala, S. Alakurtti, T. Ahola, J. Yli-Kauhaluoma and P. Tammela, J. Nat. Prod., 2009, 72, 1917–1926 CrossRef CAS PubMed.
- R. Csuk, K. Schmuck and R. Schaefer, Tetrahedron Lett., 2006, 47, 8769–8770 CrossRef CAS.
- P. A. Krasutsky, A. Pushechnikov and T. Sergeeva, Selective oxidation of triterpenes employing tempo, WO Pat2006105354A1, 2006 Search PubMed.
- N. Wikhom, S. Alakurtti, J. Yli-Kauhaluoma and S. Koskimies, Method for preparation of betulinic acid, WO Pat2013038316A1, 2013 Search PubMed.
- A. Pichette, H. Liu, C. Roy, S. Tanguay, F. Simard and S. Lavoie, Synth. Commun., 2004, 34, 3925–3937 CrossRef CAS.
- J. Tulisalo, N. Wickholm, M. Pirttimaa, S. Alakurtti, J. Yli-Kauhauluoma and S. Koskimies, Method for preparation of betulinic acid from betulin, SE Pat.2011050818A1, 2013 Search PubMed.
- B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888–890 CrossRef CAS.
- J. Tulisalo, N. Wickhom, M. Pirtimaa, S. Alakurtti and J. Yli-Kauhaluoma, Method for preparation of betulinic acid from betulin, WO Pat2013038312A1, 2013 Search PubMed.
- H. Menard, C. M. Cirtiu, J.-M. Lalancette, L. Ruest and Z. Kaljaca, Process for preparing betulinic acid, WO Pat2006063464A1, 2006 Search PubMed.
- J. Tulisalo, M. Pirtimaa, S. Alakurtti, J. Yli-Kauhaluoma and S. Koskimies, Method for preparation of betulinic acid, WO Pat2013038314A1, 2013 Search PubMed.
- M. Drag, P. Surowiak, M. Drag-Zalesinska, M. Dietel, H. Lage and J. Oleksyszyn, Molecules, 2009, 14, 1639–1651 CrossRef CAS PubMed.
- A. Pichette, H. Liu, C. Roy, S. Tanguay, F. Simard and S. Lavoie, Synth. Commun., 2004, 34, 3925–3937 CrossRef CAS.
- C. Bender and M. Sauter, Verfahren zur Gewinnung von Betulin, DE Pat10204278C1, 2003 Search PubMed.
- R. Ferreira, H. Garcia, A. F. Sousa, C. S. R. Freire, A. J. D. Silvestre, W. Kunz, L. P. N. Rebelo and C. S. Pereira, RSC Adv., 2013, 3, 21285–21288 RSC.
- A. Felfoldi-Gava, B. Simandi, S. Plander, S. Szarka, E. Szoke and A. Kery, Acta Chromatogr., 2009, 21, 671–681 CrossRef CAS.
- A. K. Ressmann, K. Strassl, P. Gaertner, B. Zhao, L. Greiner and K. Bica, Green Chem., 2012, 14, 940–944 RSC.
- H. Passos, M. G. Freire and J. A. P. Coutinho, Green Chem., 2014, 16, 4786–4815 RSC.
- R. Zirbs, K. Strassl, P. Gaertner, C. Schröder and K. Bica, RSC Adv., 2013, 3, 26010–26016 RSC.
- M. Abe, S. Yamanaka, H. Yamada, T. Yamada and H. Ohno, Green Chem., 2015, 17, 4432–4438 RSC.
- M. Abe, T. Yamada and H. Ohno, RSC Adv., 2014, 4, 17136–17140 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC.
- R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 68, 381–388 CrossRef CAS.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- R. A. Sheldon, I. W. C. E. Arends, G. ten Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed.
- P. Lucio Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559–2562 CrossRef CAS.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564–2566 CrossRef CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef CAS.
- A. Dondoni, A. Massi, E. Minghini, S. Sabbatini and V. Bertolasi, J. Org. Chem., 2003, 68, 6172–6183 CrossRef CAS PubMed.
- B. S. Bal, W. E. Childers Jr. and H. W. Pinnick, Tetrahedron, 1981, 37, 2091–2096 CrossRef CAS.
- J. Rizhikovs, J. Zandersons, G. Dobele and A. Paze, Ind. Crops Prod., 2015, 76, 209–214 CrossRef CAS.
- M. M. Zhao, J. Li, E. Mano, Z. J. Song and D. M. Tschaen, Org. Synth., 2005, 81, 195–203 CrossRef CAS.
- A. Zanka, Chem. Pharm. Bull., 2003, 51, 888–889 CrossRef CAS PubMed.
- C. Noula, V. Loukas and G. Kokotos, Synthesis, 2002, 1735–1739 CAS.
- F. Hitzenhammer , Killer GmbH Co KG, personal communication Dec. 2016.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional graphs and data on extraction and oxidation experiments, copies of NMR spectra for isolated betulin and betulinic acid. See DOI: 10.1039/c6gc02641a |
| ‡ Several other hydrophilic ionic liquids, either in pure form or as aqueous solution, were also tested for the pretreatment of birch bark. Imidazolium derivatives [C2mim]OAc, [C2mim]Me2PO4, [C4mim]Cl, and [C4mim]MeSO3 as well as the low-prize ammonium protic ionic liquid [HNEt3]HSO4 were evaluated for the biphasic extraction of birch bark using the aqueous solution and n-BuOAc; however, extraction yields with these ionic liquids remained significantly below the values obtained with the phosphonium hydroxide solution. See ESI Fig. S1† for details. |
| § Although a TEMPO/metal catalyst/air-based oxidation would provide an ideal oxidation strategy in terms of sustainability and atom efficiency, this reaction is known to over-oxidise the secondary alcohol of betulin (2) towards betulonic acid.19 In case of TEMPO mediated oxidations with secondary oxidants such as NaOCl, NaOCl/NaClO2 we faced considerable problems when applying the protocols of Zhao,45 Csuk,15 Zanka46 or Noula47 to the oxidation of betulin and yields remained low, particularly when streams of crude betulin isolated from birch bark rather than highly purified starting materials were used. |
| ¶ Moreover, the change from stoichiometric chromium-based oxidations strategies towards the catalytic TEMPO/BAIB strategies also should also add to the financial impact of the process in terms of disposal costs. Although these savings might be relatively minor compared to the value of the product, the prevention and replacement of a mixed liquid waste containing halides and heavy metals, i.e. chromium with a dilute halide waste stream should result in a reduction in disposal cost by the factor 1.5.48 |
| This journal is © The Royal Society of Chemistry 2017 |