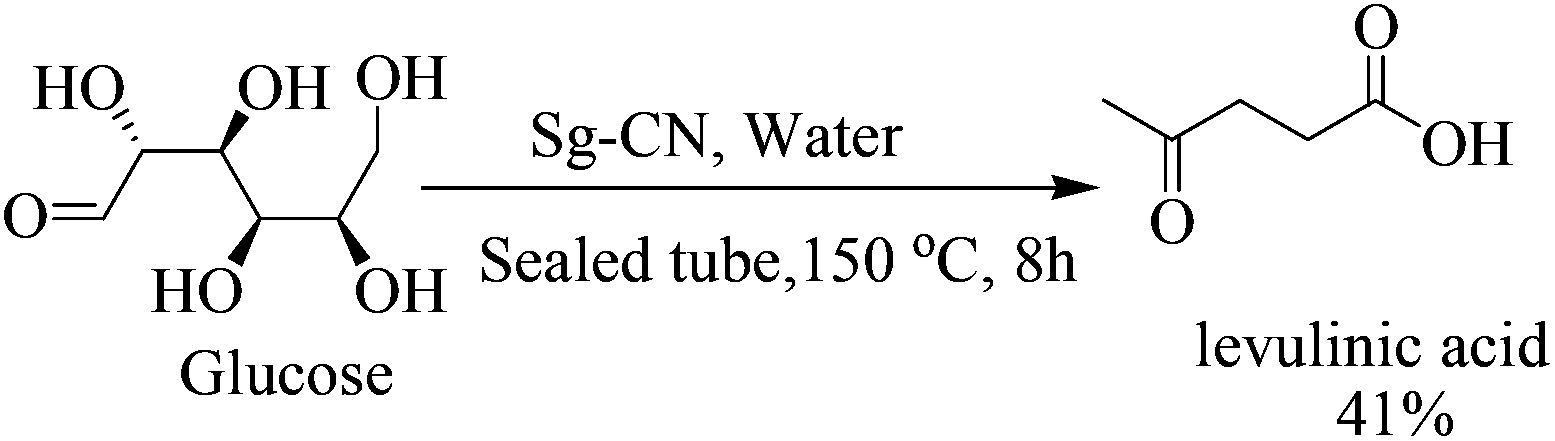DOI:
10.1039/C6GC02551J
(Paper)
Green Chem., 2017,
19, 164-168
Sustainable pathway to furanics from biomass via heterogeneous organo-catalysis†
Received
11th September 2016
, Accepted 31st October 2016
First published on 31st October 2016
Abstract
An organic sulfonated graphitic carbon nitride is synthesized and its application has been demonstrated in the conversion of carbohydrates into furanics and related value-added products. The most important feature of the material is the stability and acidity, which could be utilized at elevated temperatures for cleaving carbohydrates and converting them into biologically important scaffolds and platform chemicals.
Introduction
The threat of climate change lies at the doorstep of our planet1 and its symptoms are increasingly visible every passing year in terms of unpredicted weather conditions. Although technological advancements are catering to the need of increased energy demand, their environmental consequences are rampant. In near future, we have to substitute petroleum-based feedstocks with bio-derived secondary sources to minimize the ongoing environmental changes and their effects on living species.2 The scientific community has started seeking sustainable solutions for the conversion of biomass to value-added platform chemicals, which can be used as fuel substitutes and feedstocks for industrial applications.3 Plants are a bio-renewable source of energy, which ultimately can generate most of the chemicals while maintaining an ecological balance. The popular and general consensus is that carbohydrates obtained from plants could be benign substitutes, which can meet the requirements of human beings without compromising the environment.4
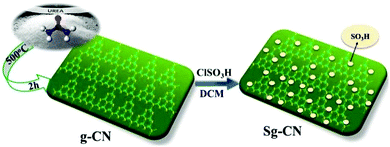
Glucose and fructose are among the most abundant plant-derived carbohydrates.5 They have been converted into building blocks for the synthesis of value-added chemicals used in drug discovery and in the synthesis of polymers among other industrial applications.6 In general, these carbohydrates are heated with mineral acids to convert them into 5-hydroxymethylfurfural (HMF) and levulinic acid notwithstanding some limitations in terms of product yields and experimental conditons.7 Researchers are incessantly investigating pathways to achieve higher yields by reconnoitring a wide range of homogenous and heterogeneous catalysts8 using ionic liquids9 and biphasic systems.10 Heterogeneous and homogeneous catalysis8 have shown limited success in terms of productivity, yield and cost-efficiency. The quest for identification of a highly acidic and relatively benign material that could efficiently accomplish the conversion of carbohydrates to furanics with a recyclability attribute has been a challenge.11 Heterogeneous organo-catalysis is probably an ideal choice as it does not involve the use of any metals.12 Cao et al. reported the use of ammonium resin to convert glucose in to HMF.13 However, this reaction required high temperature (140 °C) along with an aqueous–organic solvent (70% DMSO in water) with a maximum yield of 56%. Bhaumik et al. used sulfonated triphenylaniline polymer as an acidic organo-catalyst14 wherein the catalyst preparation required the use of toxic triphenylaniline, polymerization under anhydrous conditions followed by a sulfonation step. Furthermore, the high temperature and requirement of anhydrous DMSO have limited the wider scope of this reaction. In continuation of our focus on the development of sustainable methods,15 herein, we report a heterogeneous organo-acid catalyst derived from the electron-rich graphitic carbon nitride for the conversion of carbohydrates into value-added platform chemicals (Scheme 1).
 |
| | Scheme 1 Synthesis of sulfonated graphitic carbon nitride (Sg-CN). | |
Synthesis and characterization
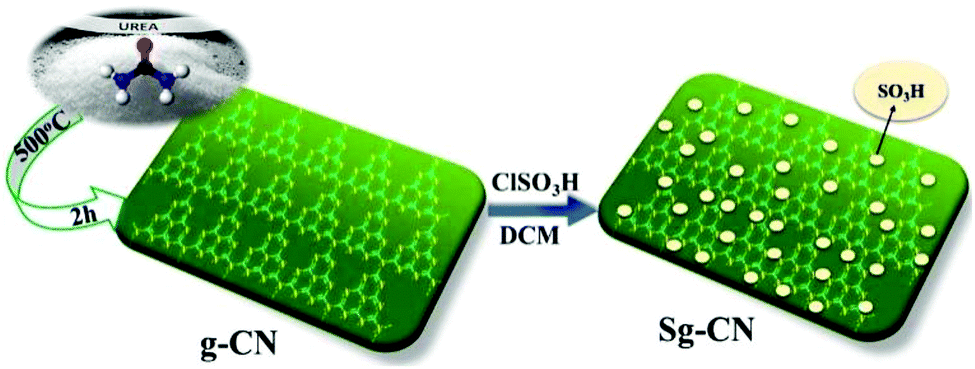
Invariably, carbohydrates can be converted into furanics under acidic conditions.16 The scarcity of an active heterogeneous acidic organic framework obtainable from readily available starting materials enthused us to design and develop an inexpensive solid acid organo-catalyst. Accordingly, we designed and synthesized sulfonated graphitic carbon nitride (Scheme 1). Synthesis of the catalyst involves calcination of urea at 500 °C for 2 hours,17 followed by stirring with chlorosulfonic acid leading to the formation of sulfonated graphitic carbon nitride (Sg-CN).
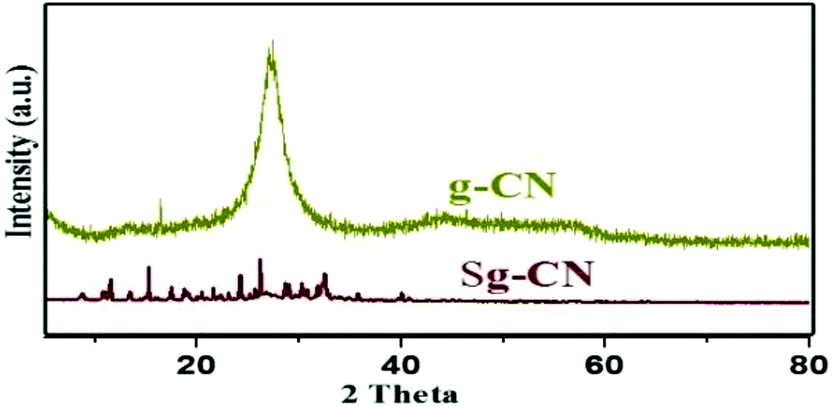
The synthesized catalyst was characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR) and solid state 13C NMR. The SEM (Fig. 1a and b), TEM (Fig. 1c and d) and XRD (Fig. 2) of graphitic carbon nitride (g-CN) and sulfonated graphitic carbon nitride (Sg-CN) show a visible difference in morphology and crystalline nature. The SO3H-immobilized Sg-CN becomes crystalline, whereas before immobilization, g-CN was amorphous in nature.
 |
| | Fig. 1 SEM images of (a) g-CN and (b) Sg-CN; TEM images of (c) g-CN and (d) Sg-CN. | |
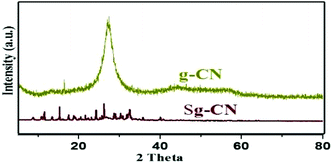 |
| | Fig. 2 XRD spectra of g-CN and Sg-CN. | |
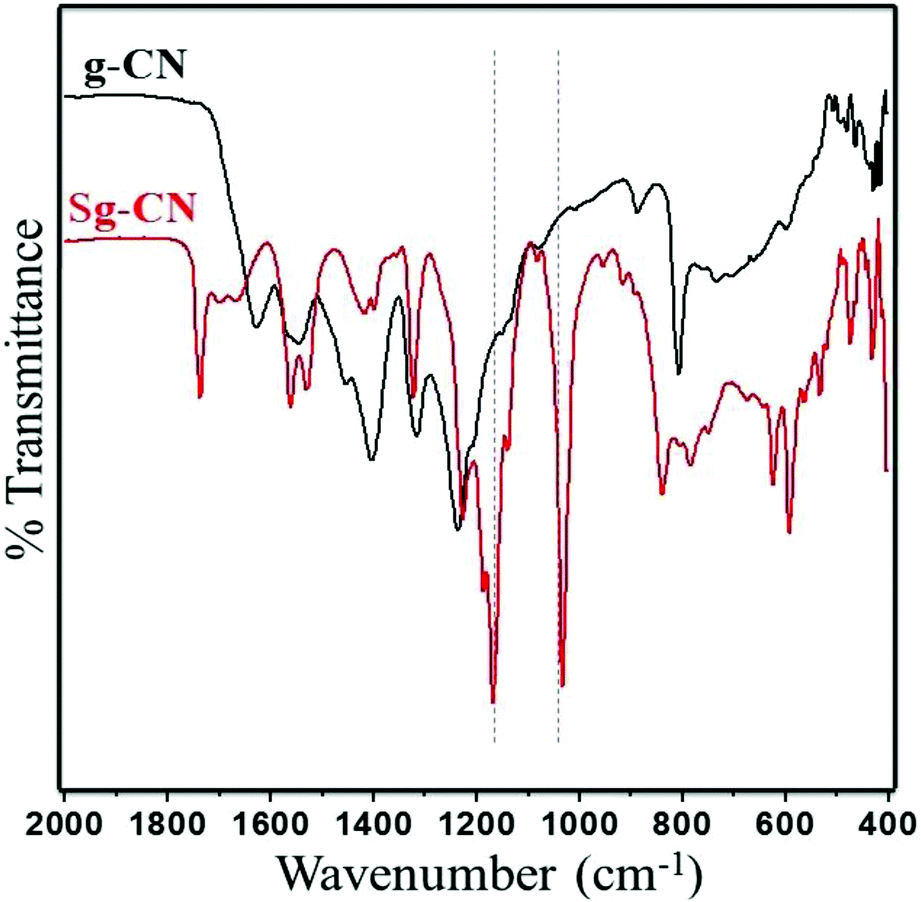
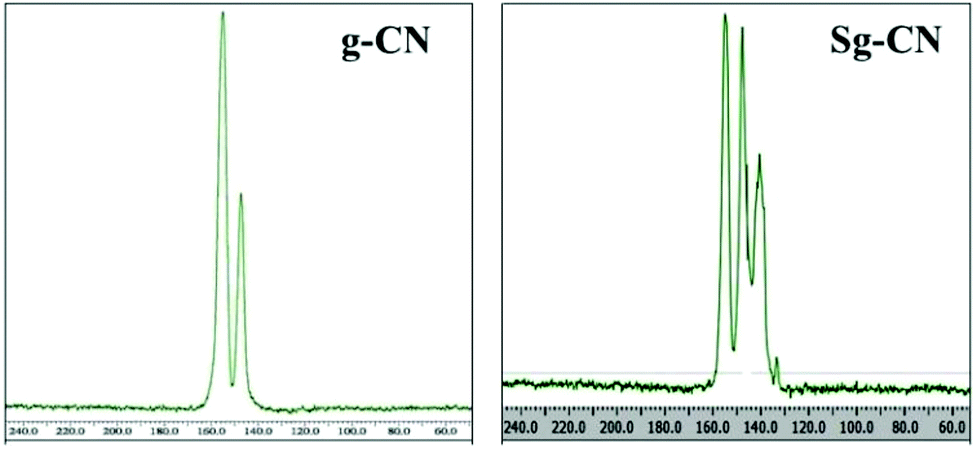
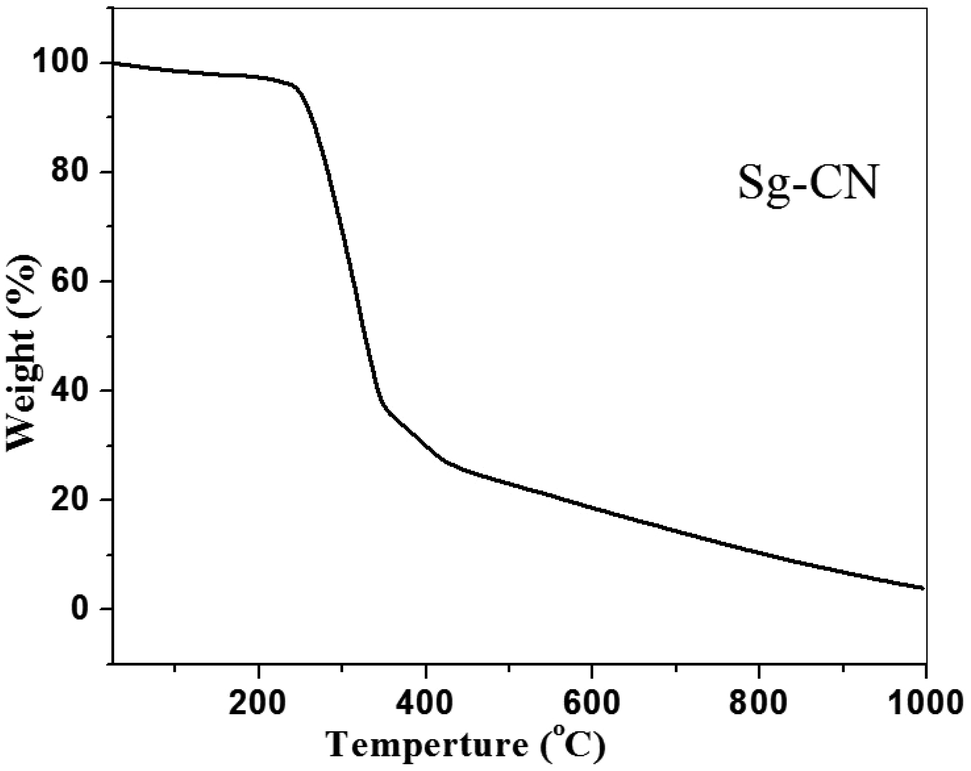
The FTIR (Fig. 3) and solid state 13C NMR (Fig. 4) confirm the functionalization of graphitic carbon nitride surface. The presence of strong absorption between 1190 and 1170 cm−1 and 1020 and 1040 cm−1 confirms the presence of the –SO3H moiety in Sg-CN. The different pattern is due to a change in the electronic environment after sulfonation. For the 13C NMR spectra of graphitic carbon nitride and sulfonated graphitic carbon nitride, the peak at 140 ppm is the characteristic signal for sulfonated graphitic carbon nitride (Sg-CN), which is affirmed by FTIR of g-CN and Sg-CN. The stability of Sg-CN was examined using thermogravimetric analysis (TGA). The TGA analysis curve of sulfonated graphitic carbon nitride (Sg-CN) confirmed that the catalyst is stable up to ∼250 °C as negligible weight loss (<3%) was observed in the TGA (Fig. 5). The immobilization of the sulfonic group was further confirmed by BET surface analysis of the sulfonated graphitic carbon nitride (10.04 m2 g−1, ESI†) and graphitic carbon nitride (35.42 m2 g−1, ESI†). After the immobilization, –SO3H ion goes and sits inside the pores via ionic interaction with the nitrogen lone pairs. A comparison of adsorption and desorption curves for g-CN (ESI, Fig. S7 and S8†) and Sg-CN (ESI, Fig. S5 and S6†) clearly indicates that the SO3H ions are sitting in the pores and binding with the nitrogen in Sg-CN. Due to this, the nature of g-CN changes from amorphous to crystalline in Sg-CN, which results in better packing and consequently a decrease in the total surface area although the pore distribution remains uneven.
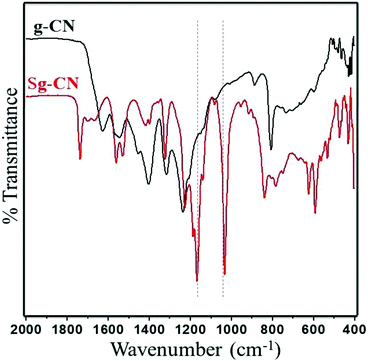 |
| | Fig. 3 FTIR spectra of g-CN and Sg-CN. | |
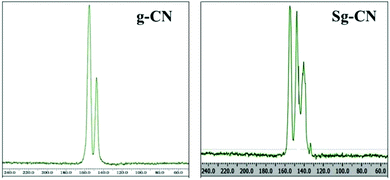 |
| | Fig. 4 Solid state 13C NMR spectra of g-CN and Sg-CN. | |
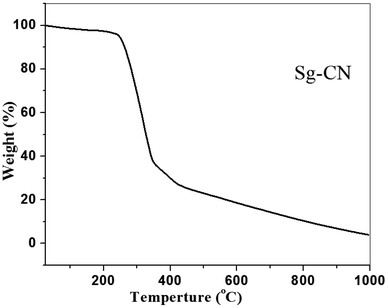 |
| | Fig. 5 TGA trace of Sg-CN. | |
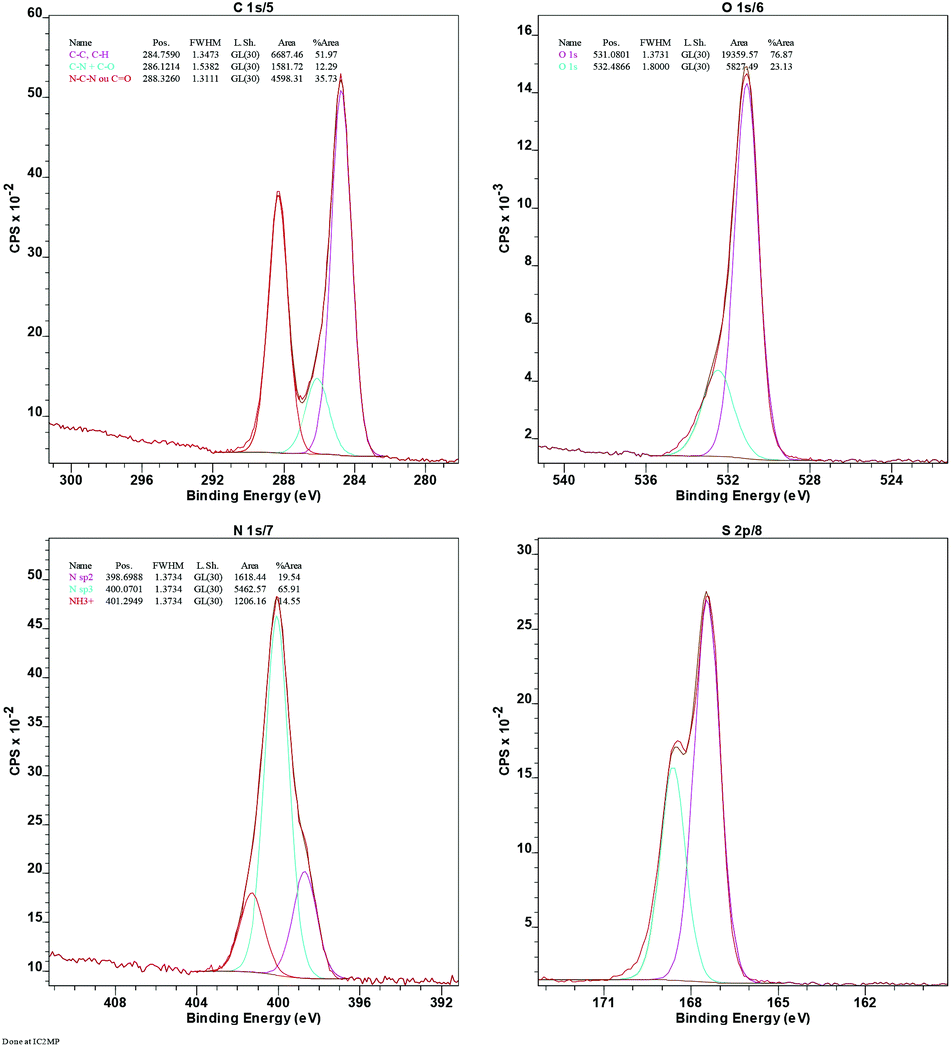
The immobilization of –SO3H is confirmed by the presence of sulfur in the energy-dispersive X-ray (EDX) spectrum (ESI, Fig. S9†) and further supported by the X-ray photoelectron spectrum (XPS) of Sg-CN (Fig. 6). The exact concentration of sulfur was determined using elemental analysis and found to be 5.47 mmol g−1. The higher acidity strength of the Sg-CN catalyst was anticipated due to the positive charge developed on the nitrogenous framework after the –SO3H immobilization.
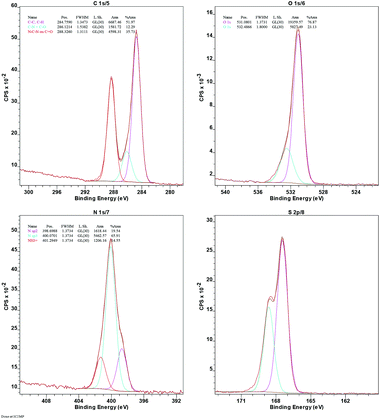 |
| | Fig. 6 XPS analysis of Sg-CN. | |
Results and discussion
After synthesizing the sulfonated graphitic carbon nitride (Sg-CN), it was imperative to find ideal conditions for the conversion of carbohydrates into furanics. Accordingly, xylose was treated with Sg-CN in DMSO under ambient conditions and the reaction was monitored at regular time intervals. DMSO was chosen as a solvent as it was the most effective media known for the reaction.14 Apparently, ∼3% formation of furfural was isolated after 24 hours stirring at room temperature (Table 1, entry 1). Increasing the reaction temperature to 50 °C to accelerate the rate of reaction resulted in a 60% yield of furfural after 8 hours (Table 1, entry 2). Increasing the reaction temperature to 100 °C afforded a quantitative yield of the desired aldehyde within 25 min (Table 1, entry 3). After finding the ideal conditions, we screened low-boiling solvents to accomplish the transformation. Consequently, the reaction was run in dimethylformamide (DMF), dioxane, acetonitrile, ethanol, methanol and water (Table 1, entries 4–11). We observed selective conversion of xylose into furfural in pure water, which was not demonstrated earlier. Other solvents were not as effective as water and DMSO (Table 1). The appropriate weight percentage of the catalyst requirement was evaluated to accomplish the speedy conversion of xylose to furfural. With this goal in mind, the reactions were performed using 50 mg, 25 mg and 12.5 mg of the catalyst for 2.0 mmol of the substrate (Table 1, entries 9–11). The reactions with 50 mg and 25 mg of the Sg-CN were completed within 30 min, whereas the reaction with 12.5 mg of Sg-CN required a longer reaction time of 4 hours. Finally, studies were conducted with 25 mg of the catalyst to maintain the expeditious conversion of carbohydrates to the corresponding furan derivatives.
Table 1 Reaction optimization for the conversion to furfural from xylosea
|
|
| Entry |
Solvent |
Temperture (°C) |
Time |
Yieldb |
| Reaction conditions: Xylose (2 mmol), Sg-CN (50 mg). Isolated yield. 25 mg of Sg-CN used. 12.5 mg of Sg-CN used. |
| 1 |
DMSO |
25 |
24 h |
<3% |
| 2 |
DMSO |
50 |
8 h |
60% |
| 3 |
DMSO |
100 |
25 min |
95% |
| 4 |
DMF |
100 |
1 h |
30% |
| 5 |
Dioxane |
100 |
1 h |
10% |
| 6 |
Acetonitrile |
82 |
1 h |
trace |
| 7 |
Ethanol |
78 |
1 h |
9% |
| 8 |
Methanol |
65 |
1 h |
10% |
| 9 |
Water |
100 |
30 min |
95% |
| 10c |
Water |
100 |
30 min |
96% |
| 11d |
Water |
100 |
4 h |
86% |
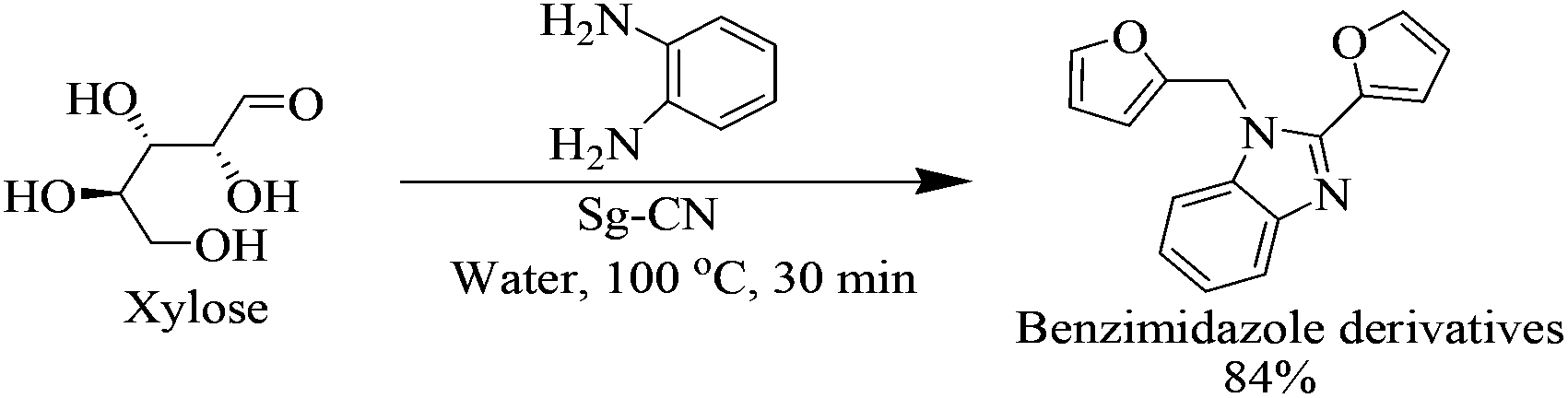
After optimizing the reaction conditions, we embarked on the applications of sulfonated graphitic carbon nitride (Sg-CN). In order to make broader use of this methodology, we added 1,2-phenylenediamine to the reaction mixture, which directly gave the pharmaceutically important benzimidazole derivative in 84% yield (Scheme 2).
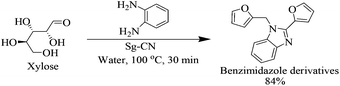 |
| | Scheme 2 Conversion of xylose to benzimidazoles. | |
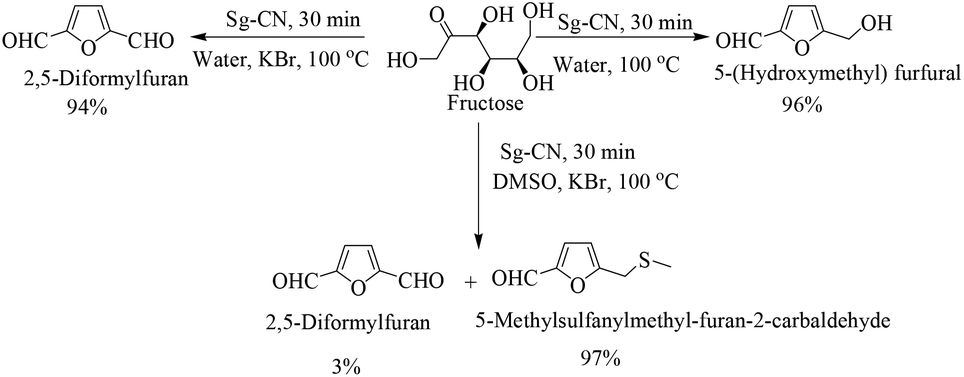
The success of Sg-CN-catalyzed conversions of xylose into value-added platform chemicals motivated us to expand further the prospects and applications of this catalyst. Consequently, we treated fructose with sulfonated graphitic carbon nitride under similar conditions, leading to the formation of the corresponding 5-hydroxymethylfurfural (HMF) in very good yield (Scheme 3). Utilizing a one-pot protocol, KBr, Sg-CN catalyst and fructose afford a 94% yield of 2,5-diformylfuran (DFF) via in situ oxidation of HMF (Scheme 4). KBr serves as an internal oxidant that oxidizes HMF in situ, leading to the selective formation of DFF. Replacement of KBr oxidant with DMSO, which may also serve as an oxidant,13 resulted in no formation of desired product. Interestingly, the use of KBr in conjunction with DMSO afforded predominantly 5-methylsulfanylmethyl-furan-2-carbaldehyde with a minor amount of DFF (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio; Scheme 3), the thiolated compound being the major product. In most of the documented results under similar conditions, DFF was the sole product described with occasional traces of 5-methylsulfanylmethyl-furan-2-carbaldehyde.13,18 However, herein, the reaction of fructose with Sg-CN afforded the selective formation of 5-methylsulfanylmethyl-furan-2-carbaldehyde with insertion of methylsulfide (CH3S–) in very high yields (Scheme 3). This unprecedented formation of 5-methylsulfanylmethyl-furan-2-carbaldehyde is an important finding, missing in earlier explorations. Interestingly, heating the aqueous solution of fructose/glucose with Sg-CN in a sealed tube over a longer period of time (8 h) at 150 °C provided levulinic acid (Scheme 4). Levulinic acid is an important intermediate of use in the synthesis of basic building blocks such as γ-valerolactone and many natural products.19
:5 ratio; Scheme 3), the thiolated compound being the major product. In most of the documented results under similar conditions, DFF was the sole product described with occasional traces of 5-methylsulfanylmethyl-furan-2-carbaldehyde.13,18 However, herein, the reaction of fructose with Sg-CN afforded the selective formation of 5-methylsulfanylmethyl-furan-2-carbaldehyde with insertion of methylsulfide (CH3S–) in very high yields (Scheme 3). This unprecedented formation of 5-methylsulfanylmethyl-furan-2-carbaldehyde is an important finding, missing in earlier explorations. Interestingly, heating the aqueous solution of fructose/glucose with Sg-CN in a sealed tube over a longer period of time (8 h) at 150 °C provided levulinic acid (Scheme 4). Levulinic acid is an important intermediate of use in the synthesis of basic building blocks such as γ-valerolactone and many natural products.19
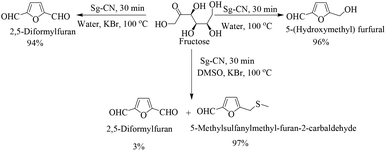 |
| | Scheme 3 Sg-CN catalysed conversion of fructose to HMF, DFF and thiolated furfural derivatives. | |
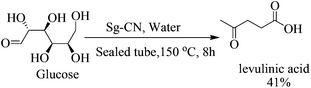 |
| | Scheme 4 Sg-CN catalysed conversion of glucose to levulinic acid. | |
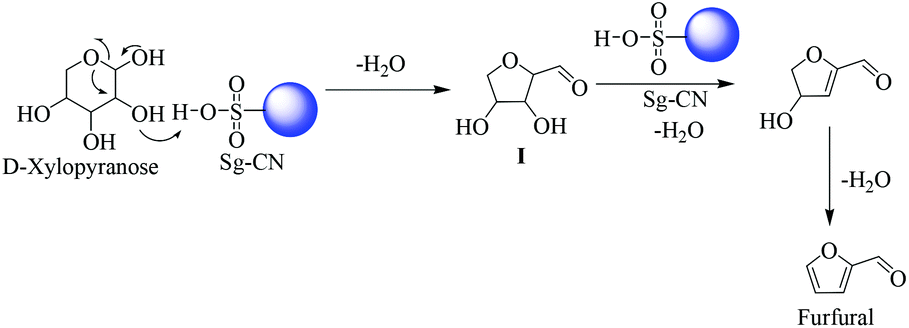
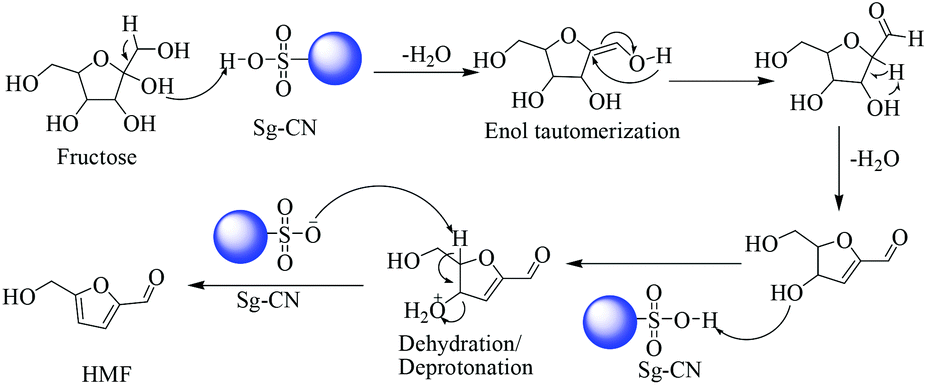
The plausible mechanisms for the dehydration of xylose and fructose have been proposed in Schemes 5 and 6, respectively. The dehydration of xylose to furfural takes place via the formation of 2,5-anhydroxylose furanose cyclic intermediate I20 and dehydration of fructose to HMF via an enol tautomerization intermediate.21
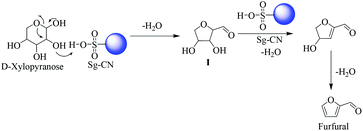 |
| | Scheme 5 Xylose dehydration mechanism via cyclic intermediates. | |
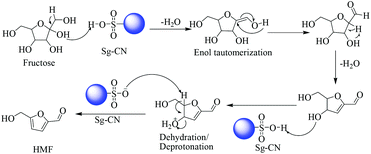 |
| | Scheme 6 Fructose dehydration mechanism via enol tautomerization intermediate. | |
It was imperative to evaluate the stability of the catalyst in recycling experiments using fructose as the model substrate. The reaction of fructose in water at 100 °C gave the formation of HMF. After the reaction, the catalyst was separated by filtration/centrifugation, washed with acetone and re-used for the next cycle with the fresh reactants. The catalyst was found to be active even after three cycles, clearly indicating that it does not lose its activity during the course of reaction (ESI, Fig. S1†). The SEM, TEM and XRD of the recycled catalyst showed that it retained its morphology and crystalline nature (ESI†), which is in corroboration with the outcome of the recycling experiments.
Conclusions
Heterogenized sulfonated graphitic carbon nitride (Sg-CN) has been synthesized, which showed its ability as a highly acidic solid organo-catalyst. Several applications have been demonstrated in the conversion of carbohydrates to value-added furan derivatives. The generated furanics could be used as starting materials in the syntheses of natural products and polymers. We have also shown that the furan derivatives could be used in situ for the synthesis of biological important heterocycles in cascade reactions. The most important feature of the catalyst is its accessibility from benign material, recyclability and stability.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Any mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Acknowledgements
S. V. and N. B. R. B. were supported by the Postgraduate Research Program at the National Risk Management Research Laboratory administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Environmental Protection Agency.
Notes and references
- C. D. Thomas, A. Cameron, R. E. Green, M. Bakkenes, L. J. Beaumont, Y. C. Collingham, B. F. Erasmus, M. F. De Siqueira, A. Grainger, L. Hannah and L. Hughes, Nature, 2004, 427, 145–148 CrossRef CAS PubMed.
-
(a) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC;
(b) R. Parajuli, T. Dalgaard, U. Jørgensen, A. P. S. Adamsen, M. T. Knudsen, M. Birkved, M. Gylling and J. K. Schjørring, Renewable Sustainable Energy Rev., 2015, 43, 244–263 CrossRef CAS;
(c) R. Schlçgl, ChemSusChem, 2010, 3, 209–222 CrossRef PubMed.
- N. Lucas, N. R. Kanna, A. S. Nagpure, G. Kokate and S. Chilukuri, J. Chem. Sci., 2014, 126, 403–413 CrossRef CAS.
-
(a) M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91–109 CrossRef CAS PubMed;
(b) A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS PubMed.
- N. J. Kruger, Plant: Physiol. Biochem. Mol. Biol., 1990, 1, 59–76 Search PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
-
(a) R. J. van Putten, J. C. V. D. Waal, E. D. Jong, C. B. Rasrendra, H. J. Heeres and J. G. D. Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed;
(b) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 (
Angew. Chem.
, 2007
, 119
, 7298–7318
) CrossRef CAS PubMed;
(c) E. I. Grbz, D. M. Alonso, J. Q. Bond and J. A. Dumesic, ChemSusChem, 2011, 4, 357–361 CrossRef PubMed.
-
(a) P. Srinivasu, D. Venkanna, M. L. Kantam, J. Tang, S. K. Bhargava, A. Aldalbahi, K. C. W. Wu and Y. Yamauchi, ChemCatChem, 2015, 7, 747–751 CrossRef CAS;
(b) H. L. Wang, Q. Q. Kong, Y. X. Wang, T. S. Deng, C. M. Chen, X. L. Hou and Y. L. Zhu, ChemCatChem, 2014, 6, 728–732 CrossRef CAS;
(c) Y. Romn-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed;
(d) D. H. K. Jackson, D. Wang, J. M. R. Gallo, A. J. Crisci, S. L. Scott, J. A. Dumesic and T. F. Kuech, Chem. Mater., 2013, 25, 3844–3851 CrossRef CAS;
(e) J. Liu, Y. Tang, K. Wu, C. Bi and Q. Cui, Carbohydr. Res., 2012, 350, 20–24 CrossRef CAS PubMed;
(f) L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. T. Horvath, ACS Catal., 2014, 4, 1470–1477 CrossRef CAS.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489–2499 RSC.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
-
(a) M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima and H. Kato, Langmuir, 2009, 25, 5068–5075 CrossRef CAS PubMed;
(b) X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2009, 10, 1771–1775 CrossRef CAS.
- S. K. Kundu and A. Bhaumik, ACS Sustainable Chem. Eng., 2015, 3, 1715–1723 CrossRef CAS.
- X. Cao, S. P. Teong, D. Wu, G. Yi, H. Su and Y. Zhang, Green Chem., 2015, 17, 2348–2352 RSC.
- S. Mondal, J. Mondal and A. Bhaumik, ChemCatChem, 2015, 7, 3570–3578 CrossRef CAS.
-
(a) R. S. Varma, Green Chem., 2014, 16, 2027–2041 RSC;
(b) R. S. Varma, ACS Sustainable Chem. Eng., 2016, 4, 5866–5878 CrossRef CAS;
(c) R. B. Nasir Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2014, 2, 2155–2158 CrossRef CAS;
(d) S. Verma, R. B. Nasir Baig, C. Han, M. N. Nadagouda and R. S. Varma, Green Chem., 2016, 18, 251–254 RSC;
(e) R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 4333–4338 RSC.
- B. R. Caes, R. E. Teixeira, K. G. Knapp and R. T. Raines, ACS Sustainable Chem. Eng., 2015, 3, 2591–2605 CrossRef CAS.
-
(a) J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed;
(b) S. Verma, R. B. Nasir Baig, C. Han, M. N. Nadagouda and R. S. Varma, Chem. Commun., 2015, 51, 15554–15557 RSC.
- X. Hu, L. Wu, Y. Wang, D. Mourant, C. Lievens, R. Gunawan and C. Z. Li, Green Chem., 2012, 14, 3087–3098 RSC.
- S. Verma, R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, ChemCatChem, 2016, 8, 690–693 CrossRef CAS.
- B. Danon, G. Marcotullio and W. de Jong, Green Chem., 2014, 16, 39–54 RSC.
- F. C. de Melo, R. F. de Souza, P. L. A. Coutinho and M. O. de Souza, J. Braz. Chem. Soc., 2014, 25, 2378–2384 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02551j |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a