
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transaminase biocatalysis: optimization and application
Fei
Guo
and
Per
Berglund
 *
*
KTH Royal Institute of Technology, School of Biotechnology, Division of Industrial Biotechnology, AlbaNova University Centre, SE-106 91 Stockholm, Sweden. E-mail: per.berglund@biotech.kth.se
First published on 30th September 2016
Abstract
Transaminases (TAs) are one of the most promising biocatalysts in organic synthesis for the preparation of chiral amino compounds. The concise reaction, excellent enantioselectivity, environmental friendliness and compatibility with other enzymatic or chemical systems have brought TAs to the attention of scientists working in the area of biocatalysis. However, to utilize TAs in a more efficient and economical way, attempts have to be made to optimize their performance. The demand for various substrate specificities, stability under non-physiological conditions and higher conversions in reversible reactions have been targeted and investigated thoroughly. A number of both protein- and process-based strategies have been developed to improve TAs and systems involving TAs. Moreover, by combination with other enzymes in cascade reactions or even in more complex systems, so called synthetic biology and systems biocatalysis, TAs can be biocatalysts with immense potential in the industrial production of high-value chemical products. This review will highlight strategies for optimization of TAs and will discuss a number of elegant systems for improving their performance. Transaminase biocatalysis has been, and will continue to be, one of the most interesting topics in green organic synthesis.
 Fei Guo | Fei Guo was born in Baotou, China, in 1988. She obtained her Bachelor's degree in pharmaceutical engineering from Zhejiang University (Hangzhou, China) in 2010. Then she continued her study for a PhD in biological engineering there, supervised by Professor Hongwei Yu. After she obtained her PhD degree in 2015, she started a postdoctoral fellowship at KTH Royal Institute of Technology, Stockholm, Sweden, with Professor Per Berglund. Her research focuses on computer-guided engineering of biocatalysts in asymmetric synthesis. |
 Per Berglund | Per Berglund originates from Sundsvall, Sweden, where he studied Chemistry and obtained his PhD in Organic Chemistry from Mid Sweden University in 1995. He then moved to University of Toronto, Canada, for a postdoc with Prof. J. Bryan Jones. Per returned to Sweden in 1997 and started at KTH Royal Institute of Technology, Stockholm, where he became Associate Professor and Docent in Organic Chemistry and finally Full Professor in Biochemistry in 2009. His research interests include stereoselective biocatalysis and enzyme engineering and his group now focuses on transaminases in cascade reactions for industrial applications. In 2010, Prof. Berglund was elected Vice Dean of KTH and has been responsible for the educational structure and content at KTH since then. |
Introduction
Transaminases (TAs), or aminotransferases, are enzymes that mediate the transfer of an amino group from the amino donor to the acceptor for chiral amino acid or amine synthesis, providing a more concise route for reductive amination compared to its chemical counterparts.1 This process consists of two steps (Scheme 1): (i) deamination of an amino acid or amine (amino donor, a) releasing amino donor product (b), and (ii) amination of a keto acid, ketone or aldehyde (amino acceptor, c) producing a new amino acid or amine (d).2 A vitamin B6-based cofactor, pyridoxal 5′-phosphate (PLP), bound to a catalytic lysine of the enzyme via an imine bond, assists the reaction by acting as a transient “custodian” of the amino group.3,4 By the end of the transamination, the cofactor releases the amino group and returns to its initial state. This automatic cofactor recycling is advantageous in enzymatic syntheses since no additional cofactor regeneration is required as is often the case for many other enzymes.5,6 Another appealing property of TAs is their extraordinary stereoselectivity.7 With the almost exclusive substrate-binding mode constrained by the relative position of PLP and two substrate-binding pockets of different sizes, the enantiomeric excess of the chiral products is remarkably high. These distinguished natural properties have brought TAs to the attention of the biocatalysis community.8–10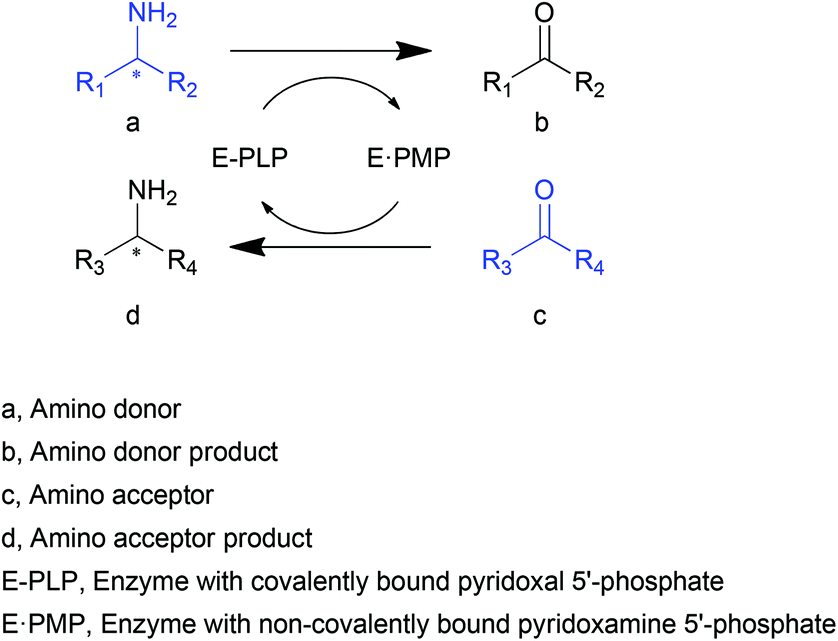 | ||
| Scheme 1 TA-mediated transamination. | ||
Since their first applications in industry and academia at the end of the last century,11,12 an increasing number of attempts have been made to utilize TAs in a more efficient and economical fashion. The primary demand for the application of TAs is the practicability for various substrates other than α-amino acids. Of special interest are the transaminases that accept substrates with an amino group distant from the carboxylate (β- or other amino acids), or even lacking a carboxyl group (amines). The favourable substituent groups include not only small or medium aliphatic chains but also larger aromatic moieties. The development of biotechnology and protein engineering has facilitated the search for and generation of new TAs with diverse substrate specificities.13–15
Even with appropriate substrate specificity, the TA stability may become an obstacle. Maintaining the quaternary and tertiary structures under non-physiological conditions (such as high concentrations of substrate and product, organic solvent, mechanical force, etc.) for a long period of time (10–100 h working time and an even longer storage time) is a huge task for all biocatalysts. Attempts have been made by strategies of protein engineering, biocatalyst formulation and reaction medium engineering to solve the stability issue.16–18 These long-lived and even recyclable TAs are able to reduce the cost of enzymes prominently.
Nevertheless, organic synthesis by TAs suffer from the unfavourable reaction equilibrium, especially in the asymmetric synthesis of amines from an amino acid as amino donor.19 To maximize the productivity of TAs, shifting the equilibrium to the side of product is extremely important.20,21 Process control strategies have been developed in order to remove product in situ, among which cascade reactions employing additional biochemical reactions of the side products have gained much attention.22
Afterwards, the optimized enzyme and process can be applied in the production of amino compounds, which are in most cases important synthons in pharmaceutical, food additive, cosmetic, agrochemical and material industries.8–10 Combining TAs with other enzymatic or chemical routes, even metabolic pathways in a one-pot fashion, has been demonstrated to be a smart method for practical applications, particularly in regard to shortening the reaction routes, avoiding protecting steps, reducing chemical waste and achieving a high atom-efficiency.22–24 In this process development, intelligent synthesis route design is the most critical and appreciable step. Besides this, metabolic flux control and systematic regulation are also able to improve the final production.
After nearly 20 years of study, TA has been proven to be a powerful biocatalyst in organic synthesis. A number of elaborate reviews have been published on this topic.2,7–10,20,22,25–28 In the current review, we focus on attempts to optimize the TA-mediated reactions in order to propose methodological options for the problems that may be encountered in the application of TAs. The elegant ways of using TAs in cascade reactions and in even more complex multi-step systems, so called synthetic biology and systems biocatalysis, are also presented to illustrate the immense potential of TAs in industrial applications.
Exploring and generating novel enzymes
The specificity for the chemicals of interest is the primary property to be considered and optimized when an enzyme is applied in organic synthesis. Basically, two strategies are practical in generating biocatalysts with the expected properties: gene mining to discover new enzymes and protein engineering to tailor-make enzymes. Gene mining is advantageous due to the vast database in nature to be searched in; however, the searching itself is a time-consuming process. Protein engineering, especially the rational strategy based on biochemical mechanisms and biological structures, is more efficient with a reduced number of designed mutants, although the efficiency of this process could be restricted by inappropriate starting-point proteins. To accelerate the process of both these methods, high throughput screening strategies have been developed on the basis of diverse smart biochemical reactions. Combination of these techniques will provide effective and rapid methods for generating TAs with the expected substrate specificities.Gene mining
As a tremendous library of proteins, nature has been employed for mining a large number of biocatalysis genes for enzymatic organic synthesis.29,30 With the development of biotechnology, the dominant strategy to discover new biocatalysts has varied from straightforward experimental assays for certain functions to deduction of protein functions from bioinformation databases. In this section, we will discuss the mining of TA genes with different strategies (Table 1 and Fig. 1).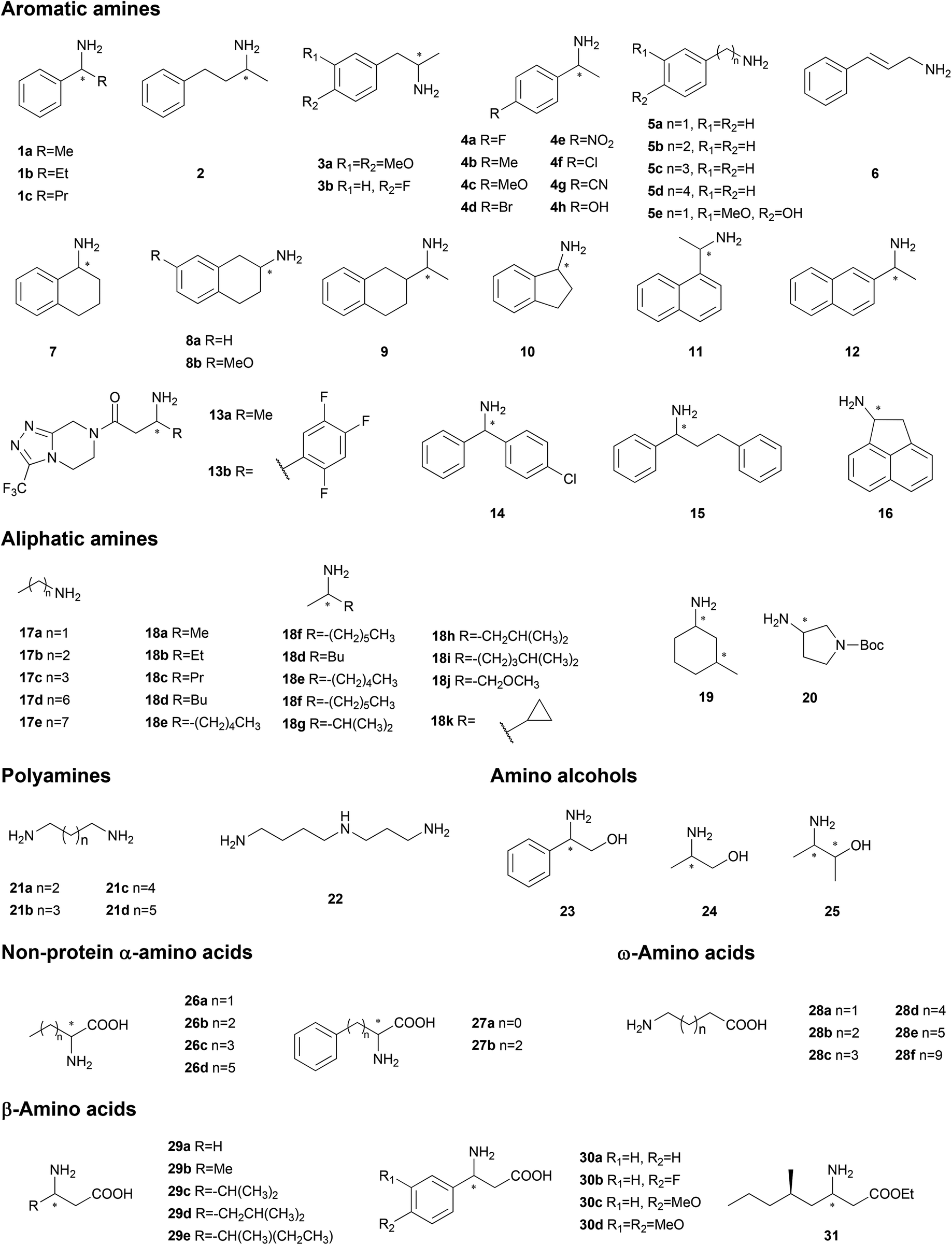 | ||
| Fig. 1 Amines as model substrates in gene mining and protein engineering. | ||
| Strategy | Organism | Commenta | Ref. |
|---|---|---|---|
| a SNS, sole nitrogen source. | |||
| Enrichment cultivation | Klebsiella pneumoniae JS2F | SNS, 1a | 32 |
| Bacillus thuringiensis JS64 | SNS, 1a | ||
| Vibrio fluvialis JS17 | SNS, 1a | 13 and 33 | |
| Arthrobacter sp. KNK168 | SNS, 3a; R-selective | 37 | |
| Pseudomonas sp. KNK425 | SNS, 3a | ||
| Bacillus megaterium SC6394 | SNS, 18k | 38 | |
| Moraxella lacunata WZ34 | SNS, 24 | 39 | |
| Pseudomonas fluorescens KNK08-18 | SNS, 8b | 40 | |
| Alcaligenes denitrificans Y2k-2 | SNS, 29b; β-TA | 41 | |
| Mesorhizobium sp. LUK | SNS, 30c; β-TA | 42 | |
| Pseudomonas sp. AAC | SNS, 28f; ω-TA | 43 and 44 | |
| Metabolic function analysis | Capsicum | Vanillin TA for 3c | 45 and 46 |
| GC-based assay | Janibacter terrae DSM13953 | TA for S-1a | 47 |
| Pseudomonas cichorii DSM 50259 | TA for S-1a | ||
| Pseudomonas fluorescens ATCC49838 | TA for S-1a | ||
| Homologous sequence searching | Chromobacterium violaceum DSM30191 | 38% identity to V. fluvialis TA | 52 |
| Paracoccus denitrificans | 94% identity to V. fluvialis TA | 57 | |
| Ochrobactrum anthropi | 42% identity to P. denitrificans TA | 58 | |
| Acinetobacter baumannii | 17% identity to P. denitrificans TA | ||
| Acetobacter pasteurianus | 21% identity to P. denitrificans TA | ||
| Caulobacter crescentus | 65% identity to A. denitrificans TA; β-TA | 59 | |
| Polaromonas sp. JS666 | 54% identity to Mesorhizobium TA; β-TA | 60 | |
| Halomonas elongata | 38% identity to V. fluvialis TA; halo-adapted | 104 | |
| Sphaerobacter thermophilus | 41% identity to Polaromonas TA; thermophile | 18 | |
| Geobacillus thermodenitrificans | 51% identity to V. fluvialis TA; thermophile | 103 | |
| Motif-based sequence searching | Aspergillus terreus | R-Selective | 14 and 61 |
| Penicillium chrysogenum | R-Selective | ||
| Aspergillus oryzae | R-Selective | ||
| Aspergillus fumigatus | R-Selective | ||
| Neosartorya fischeri | R-Selective | ||
| Gibberella zeae | R-Selective | ||
| Hyphomonas neptunium | R-Selective | ||
| Mycobacterium vanbaalenii | R-Selective | ||
| Mesorhizobium loti | R-Selective | ||
| Mesorhizobium loti MAFF303099 | 10 putative TAs from protein family profile | 63 | |
| Nectria haematococca | R-TA from fungal source | 62 | |
| Escherichia coli K12 | ygjG gene, TA for 21 | 64 | |
| Pseudomonas putida | spuC gene, TA for 21 | 65 | |
| Pseudomonas chlororaphis | spuC gene, TA for 21 | ||
| Pseudomonas fluorescens | spuC gene, TA for 21 | ||
| Sequence of known function | Pseudomonas aeruginosa | β-TA | 66 |
| Arthrobacter citreus | Aromatic amine TA | 16 | |
| Rhodobacter sphaeroides | Aromatic amine TA | 67 | |
| Burkholderia graminis | β-TA | 68 | |
| Protein structure database | Silicibacter pomeroyi | PDB ID, 3HMU | 69 |
| Rhodobacter sphaeroides KD131 | PDB ID, 3I5T | ||
| Ruegeria sp. TM1040 | PDB ID, 3FCR | ||
| Mesorhizobium loti MAFF30399 | PDB ID, 3GJU | ||
| Bacillus anthracis | PDB ID, 3N5M | 70 | |
Experimental analysis of biosynthetic pathways for secondary metabolites may also provide TA candidates for biocatalysis. For example, by the investigation of the biosynthetic pathway of capsaicinoid in Capsicum annuum CH-19 sweet, a non-pungent pepper mutant derived from a pungent pepper strain, a T insertion in a putative vanillin TA gene catalysing the synthesis of vanillylamine (5e) was identified as the main reason for the phenotype alteration of this pepper.45 Further in vitro investigation revealed that this vanillin TA can be employed in the biocatalysis of chiral amines due to its high stereoselectivity and activity.46
Recently, another example of experimental screening was reported with a GC based transamination activity determination of more than 100 lyophilized cell preparations from diverse microorganisms.47 Among them, Janibacter terrae DSM 13953, Pseudomonas cichorii DSM 50259 and Pseudomonas fluorescens ATCC 49838 were identified as highly enantioselective biocatalysts for the kinetic resolution of amine 1a, albeit all strains showed S-enantiomer preference. The best strain J. terrae was further applied in a kinetic resolution of 143 mg rac-1a, resulting in 51% of conversion and product of >99% ee (E > 200) after 24 h of reaction. Some disadvantages of whole cell biocatalysis, indicated in this work, are that the side-products from the microorganism could disturb the results of screening; meanwhile, the strategies of equilibrium shifting may also be limited by other enzymes in the system.
With elegant substrate promiscuity, the TAs developed may exhibit novel activities towards new substrates. For the scientists in organic chemistry and biocatalysis, commercial collections available as “ready to use” screening toolkits could be another practical option. For example, TAs from Codexis, including ATA-103, ATA-113, ATA-114, ATA-117 and ATA-256, has been applied in a number of investigations.15,48–51
Taking the two β-TAs explored through microorganism screening as a template, Caulobacter crescentus TA59 (65% identity to A. denitrificans TA) and Polaromonas sp. JS666 TA60 (54% identity to Mesorhizobium TA) were identified by homologous searching, exhibiting high activity towards β-amino acids.
Nevertheless, the straightforward sequence alignment is only able to identify enzymes with similar specificities to the template enzymes. One severe issue of amine TAs has, until recently, been the fact that although S-selective enzymes have been largely explored and investigated, R-selective ones were barely reported. Bornscheuer et al., at that time with no R-selective amine TA sequence reported, developed a motif-based algorithm for enzyme function prediction and screening. PLP-dependent enzymes in fold class IV56 was chosen as the protein database to be searched according to a hypothesized evolution routine of R-selective amine TA. The sequences and structures of D-amino acid aminotransferases, L-branched chain amino acid aminotransferases and 4-amino-4-deoxychorismate lyase in this fold class were investigated and a set of key motifs that are possible to use as markers for enzyme function and enantioselectivity were proposed. The algorithm was performed in the exclusion of template specificities and led to the subsequent identification of desired enzymes. As a result, seventeen R-selective TAs were identified by this strategy,14 seven of which were further characterized and proven to be efficient biocatalysts with excellent R-enantioselectivity.61 With a similar strategy, a novel R-selective TA was also identified from a fungal source, Nectria haematococca.62
For a more complex motif analysis, a sequence set of proteins with certain functions is constructed with a seed enzyme in the protein family profile strategy. By this method, the genome of Mesorhizobium loti MAFF303099, a nitrogen fixation bacterium with more TAs than a normal strain, was analysed, from which 10 putative TAs were identified and characterized.63
Multi-sequence alignment and functional motif analysis were also employed in the gene mining for TAs catalysing reactions with polyamines. Putrescine TA ygjG from Escherichia coli K12 was firstly identified as a potential biocatalyst for terminal diamines (21), although it showed a strict preference for pyruvate over ketones.64 Further alignments among amine TAs, gene cluster ygjG and gene cluster supC (putrescine TA gene in Pseudomonas species) revealed a higher similarity between gene cluster supC and amine TA, indicating that putrescine TAs encoded by gene cluster supC show high potential in accepting both diamines and ketones as substrates.65 Three supC genes were identified and characterized from P. putida, P. chlororaphis and P. fluorescens with obvious activity towards diamines (21a, 21b and 22) with ketones as amino acceptors.
Furthermore, although the strategy for bioinformation acquisition has not been reported, TAs from Pseudomonas aeruginosa,66Arthrobacter citreus,16Rhodobacter sphaeroides67 and Burkholderia graminis68 were also cloned and applied in organic synthesis of amino compounds.
Protein engineering
Protein engineering, including directed evolution based on random mutagenesis and rational design based on sequence and structure analysis, has been demonstrated extensively as a practical and efficient strategy for the optimization of biocatalysts.71,72 This strategy has also been employed in the improvement of TA. In this section, these studies will be discussed and their details are listed in Table 2 and the relevant compounds are shown in Fig. 1.| TA | Strategya | Mutation | Result | Ref. |
|---|---|---|---|---|
| a Protein engineering strategies. Random: random mutagenesis based directed evolution; structure: structure analysis based rational design; sequence: multi-sequence alignment based rational design. | ||||
| V. fluvialis TA | Random | — | 3-Fold improvement of specificity towards 30a | 73 |
| TAmla (P233L, V297A) | Decrease the inhibition from product of 2-butylamine (18b, 6-fold) and 2-aminoheptane (18e, 4-fold) | 74 | ||
| 2-Fold higher activities towards long chain aliphatic amines (18(e, f and j)) | ||||
| Structure | W57G | Up to 30-fold improvement of activity for aromatic amines (1a, 2, 4(b, d and e), 5(a–d), 7, 8a and 10) and aliphatic amines (18(a, b, c, e, g and j)) | 75 | |
| F85L, V153A | 26-Fold improvement of specificity towards 1c | 36 | ||
| Y150 M, V153A | 53-Fold improvement of specificity towards 23 | |||
| L56V | Improvement of enantioselectivity towards 19 from 14% de (1R,3S) to 66% de (1R,3S) | 85 | ||
| L56I | Reversion of enantioselectivity towards 19 from 14% de (1R,3S) to 70% de (1S,3S) | |||
| M3 (W57F, R415L, L417 V) | 3.4-Fold improvement of specificity of pentanal and loss of activity towards pyruvate | 87 | ||
| Structure & sequence | r414 (F19 W, W57F, F85A, R88 K, V153A, K163F, I259 V, R415F) | 60-Fold improvement of the specificity towards pre-31 ketone | 35 | |
| A. citreus TA | Random | Mut6 (M46 T, D48G, Y60C, Y164F, Y185C, N186S, P195S, M197 T, C205Y, A242 V, A245 T, I252 V, F255I, N268S, T409R, K424E, C436A) | 260-Fold improvement of activity towards substituted S-aminotetralin (8a) | 16 |
| Optimal temperature from 30 °C to 55 °C | ||||
| Structure | CNB05-01-V328A | Reversion of enantioselectivity towards 3b from 98% ee (S) to 58% ee (R) | 82 | |
| C. crescentus TA | Structure | N285A | 3-Fold improvement of specificity towards 30a | 59 |
| V227G | 2-Fold improvement of specificity towards 30a | |||
| ATA-117 (Arthrobacter sp.) | Structure | Mut1a (S223P) | 11-Fold improvement of activity towards pre-13a ketone, but non-active for 13b | 15 |
| Mut1b (adding V69G, F122I, A284G to Mut1a) | Margin activity towards pre-13b ketone | |||
| Mut2 (adding H62 T, G136Y, E137I, V199I, A209L, T282S to Mut1b) | 75-Fold improvement of activity to produce 13b compare to Mut1b | |||
| Mut11 (adding S8P, Y60F, L61Y, G69 T, D81G, M94 T, I96L, I122 M, S124 T, S126 T, Y136F, I137E, Y150S, V152C, A169L, G215C, G217N, L269P, L273Y, P297S, S321P to Mut2) | Improvement of reaction conditions: substrate concentration 50 g l−1, amino donor concentration 1 M, pH 8.5, temperature 45 °C and cosolvent DMSO concentration 50% | |||
| C. violaceum TA | Structure | W60C | Improvement of enantioselectivity of 7 and 8a | 83 and 84 |
| 29-Fold improvement of specificity towards 1a and about 5-fold improvement of specificity towards 4(b, c, e, g and h) | ||||
| A231F, F88A | Reversion of enantioselectivity of 7 and 8a | 83 | ||
| Sequence | Y153 M | 4-Fold improvement of specificity towards serine | 88 | |
| W60Q | 4-Fold improvement of specificity towards serine | |||
| F88R | 3-Fold improvement of specificity towards serine | |||
| P. denitrificans TA | Structure | V153A | 4-Fold improvement of specificity towards 26d | 76 |
| O. anthropi TA | Structure | L57A | Up to 150-fold improvement of specificity towards aromatic amines (1(a–c), 2, 4(a–c), 5a, 7, 9 and 10) | 77 and 78 |
| Up to 5-fold improvement of specificity towards aliphatic amines (18(a–d, f–i and k)) | ||||
| Up to 48-fold improvement of specificity towards α-keto acids of corresponding amino acids (26(a–d)) | ||||
| W58L | At least 86-fold improvement of specificity towards aromatic ketones from corresponding amines (1(a–c), 2, 4(a–c), 7, 9 and 10) | 79 | ||
| At least 40-fold improvement of specificity towards aliphatic ketones from corresponding amines (18(c, d, f, g, h and k)) | ||||
| Ruegeria TA | Structure | Y59W | 22-Fold improvement of specificity towards 1a | 81 |
| T231A | 4.3-Fold improvement of specificity towards 1a | |||
| Structure | Y59 W, Y87F, Y152F, T231A, P423H | Up to 8900-fold improvement of specificity towards bulky amines 14, 15 and 16 | 80 | |
Mutation of the residues in the small pocket was also investigated in the engineering of substrate specificity of other TAs. For instance, V153A in the small pocket of P. denitrificans TA was identified as a beneficial mutant for α-keto acid amino acceptors with a long linear alkyl side chain (26d for example).76 The enzymatic activity in the transamination of 26d and 1a was improved 4 times, although the mutant did not exhibit enhanced specificities towards α-keto acids with shorter side chains (26(a–c)) that were readily accepted by the wild type. In another example, the structure of a β-TA from C. crescentus was investigated and the two-pocket model was also demonstrated in its active site.59 To broaden its substrate specificity towards aromatic β-amines with a bulky phenyl ring supposed to bind in the small pocket, two residues were mutated into smaller ones to create a larger space in this pocket. As a result, the mutants N285A and V227G improved the specificity towards 30a by 3- and 2-fold, respectively. Recently, Shin et al. investigated an amine TA from O. anthropi and expanded its substrate specificity to bulky substrates by engineering the residues in both the small and the large pocket. L57 has been identified as a residue which exhibits steric interference with the substrate substituent bound in the small pocket.58,59 An alanine mutation at this site removed this hindrance and resulted in a mutant with improved specificity towards a large amount of bulky substrates, including up to 150-fold improvement for aromatic amines (1(a–c), 2, 4(a–c), 5a, 7, 9 and 10),77 up to 5-fold improvement for aliphatic amines (18(a–d, f–i and k))77 and up to 48-fold improvement for α-keto acids of the corresponding amino acids (26(a–d)).78 Besides, W58 in the large pocket was also mutated into a leucine with a smaller volume in order to reduce the steric barrier.79 W58L has been proven to be a superb biocatalyst for amination of bulky acetones from the corresponding amines (1(a–c), 2, 4(a–c), 7, 9, 10 and 18(c, d, f, g, h and k)) with isopropylamine (18a) as the amino donor.
As shown above, structure-based protein engineering has been utilized successfully to expand the TA substrate spectrum to substrates with bulky side chains. Among these compounds, sitagliptin (13b) is the most bulky one with large groups at both sides of the amino group. Its corresponding ketone is totally inert to the wild type ATA-117.15 Nevertheless, by structure-based engineering of both substrate-binding pockets, the specificity towards prositagliptin ketone was established and improved significantly. Twelve residues in the large pocket were firstly mutated and screened resulting in a variant (Mut1a) with a single mutation S223P, which improved the activity of the simplified product analogue (13a) with only one bulky side chain binding into the large pocket, although no sitagliptin activity was detected. Then the small pocket was opened by mutagenesis at four sites to accommodate the large trifluorophenyl group, establishing the activity to give 13b. A mutant (Mut1b) with three mutations along with S223P was determined as a TA with marginal, but far from sufficient, activity. To enhance this activity, all the beneficial mutations from the two previous rounds of engineering were combined and tested. Finally, Mut2 with 6 added mutations was obtained with a 75-fold improvement of the activity to produce 13b. In this research, the substrate binding pocket of ATA-117 was successfully opened up by structure-based protein engineering for a bulky substrate which was not accepted at all by the wild type enzyme.
Recently, another successful engineered S-selective TA for bulky substrates was also reported.80Ruegeria TA was chosen as the initial scaffold for engineering due to its potential evolvability, which was based on a previous investigation, in which single-point mutations caused an improvement of the specificity towards 1a (22-fold improvement by Y59W and 4.3-fold improvement by T231A).81 By combining these two sites with three more sites revealed from a structural analysis of the intermediate model, the mutant Y59W/Y87F/Y152F/T231A/P423H was constructed and determined to be a superb biocatalyst with up to 8900-fold improvement of specificity towards bulky amines (14–16). This motif of mutations was also validated in other protein scaffolds from in silico screening, resulting in more candidates for the biocatalysis of bulky amines.
High throughput screening
Identifying the positive enzymes or variants in libraries from gene mining or protein engineering is a crucial step and the analytical methods used determine the efficiency of the whole process. The conventional analytical methods including GC, HPLC or MS are disadvantageous with low throughput, consequently consuming time and labour resources. Therefore, novel high throughput screening methods are of great interest to accelerate the identification of optimized proteins. A number of strategies have been developed and reported for transaminases, including enrichment cultivation, detection of the common substrates and employment of smart substrates. In this section, these approaches and their characteristics will be discussed. Among them, the screening assays based on common and smart substrates are summarized in Table 3 and Scheme 2 as AI–AVII and BI–BIV. | ||
| Scheme 2 High throughput screening strategies of TA. | ||
| Substrate dependence | Additional reagent | Detection and equipment | Sensitivity | Background | Enantioselectivity | Continuous or end point | Liquid or solid | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| n.r., not reported. | |||||||||
| AI | α-Amino acid | CuSO4/MeOH | Blue colour | ε = 10 M−1 cm−1 linear in 0.1 mM–20 mM | Amino acids in cell extract | Y | E | L | 73 |
| AII | Alanine and glycine | Oxidase, HRP, colour indicators | Colour change | 1% of initial activity, nonlinear | False positive (<0.5 mU) | Y | C | Both | 89–92 |
| AIII | Alanine and glutamate | Oxidase, tetrazolium | Colour change | Linear in low product concentration (10–100 μM) | Amino acids in cell extract, PLP | Y | C | L | 65 and 93 |
| AIV | Alanine | Oxidase, pH indicator | Colour change | n.r. | From pH disturbing substance | Y | C | L | 94 and 95 |
| AV | Complex of Cu(II)–Ala instead of alanine | Cu(II) and BSA | Fluorescence quenching with a fluorescence spectrometer | Linear, high sensitivity, LOD 3 U L−1, 5–400 U L−1 | From Cu(II)–Ala complex degradation | N | C | L | 96 |
| AVI | 2-Hydroxy ketone | Tetrazolium | Colour change | Linear in low activity, dependent on substrates | Amino acids in cell extract | Y | E | L | 97 and 98 |
| AVII | 1-Phenylethylamine (1-PEA, 1a) | — | Absorption at 245 nm with a UV spectrometer | ε = 12 mM−1 cm−1 | From protein | Y | C | L | 67 and 99 |
| BI | Substituted aminotetraline | — | Coloured product exposed to air | n.r. | n.r. | Y | C | L | 16 |
| BII | ortho-Xylylenediamine | — | Dark colour | High, nonlinear | Strong | N | C | Both | 100 |
| BIII | EWG-substituted aromatic amines | — | Coloured product | High quantitative LOD 1.2–30% | Low | N | C | Both | 101 |
| BIV | 1-(6-Methoxynaphth-2yl) alkylamines | — | Fluorescence at 450 nm with a fluorescence spectrometer | High LOD 0.35 nmol min−1 LOQ 1.2 nmol min−1 | Low | Y | C | L | 102 |
As the natural substrates of TA, α-amino acids and the corresponding keto acids are the most commonly employed amino donors and acceptors. Especially in kinetic resolution of racemic chiral amines, using α-keto acid as the amino acceptor is advantageous due to the favourable reaction equilibrium towards the product side. Kim et al. reported a colorimetric high throughput screening method taking advantage of the blue colour generated from amino acids with CuSO4 and MeOH, which could be detected at 595 nm (Method AI in Table 3 and Scheme 2).73 The absorbance is linearly related to the concentration of amino acid generated in the reaction in 0.1 mM–20 mM with a sensitivity of 10 M−1 cm−1. The main drawback of this strategy is the strong background colour from other amino acids from the cell extract. Moreover, the chromogenic reagent (CuSO4 and MeOH) is detrimental to the enzymes, which limits the application of this strategy in a continuous fashion.
Another approach is a colorimetric detection of alanine production by a combination of amino acid oxidase (AAO) and horseradish peroxidase (HRP) in a cascade reaction system (Method AII).89–91 In this system, alanine is deaminated by amino acid oxidase to produce hydrogen peroxide (H2O2), which can be detected by using horseradish peroxidase and some colorimetric indicators. Moreover, glycine is also able to be detected by deamination of glycine oxidase and the following H2O2 detection.92 This strategy can be used as solid phase selection and the co-product was recycled, which is beneficial to the reaction equilibrium. However, multiple enzymes involved in the system may cause operational problems and H2O2 may be detrimental to proteins. Another disadvantage is that this strategy can only be employed as a qualitative method due to the nonlinear correlation between the enzyme activity and the colour intensity as well as the obvious false positive results.
Similarly, another strategy based on AAO was employed for fast detection of TA activity (Method AIII).65,93 Produced in a TA-mediated reaction, alanine65 or glutamate93 can be oxidized by the specific oxidases AlaDH or GDH, resulting in a conversion from NAD+ to NADH. Simultaneously, tetrazolium salts in the system can be reduced into a coloured product, the amount of which was correlated to the concentration of NADH. The strategy with GDH was determined to be sensitive and linear with low glutamate concentrations (10–100 μM). Purified enzymes were required since amino acids and proteins in the cell extract resulted in a strong background colour. Moreover, PLP for the transamination was determined to be detrimental to the GDH activity.
In asymmetric synthesis of chiral amines, alanine is one of the most commonly used amino donors. Therefore, the efficient detection of the amino donor product pyruvate is of great interest. A pH change-based method has been developed where the pyruvate produced is reduced by lactate dehydrogenase (LDH) accompanied by the oxidation of glucose to gluconic acid by glucose dehydrogenase (GDH), which causes a colour change of the pH indicator (Method AIV).94,95 The results of this method could be affected by other pH disturbing substances in the reaction system.
Recently, a method for detecting transamination from alanine to pyruvate was developed based on fluorescence quenching of bovine serum albumin (BSA) in a reaction system containing the metal coordinate complex Cu(II)–Ala (Method AV).96 Coordination of Cu(II) with L-alanine will be destroyed by conversion of L-alanine into pyruvate. Then the free Cu(II) ion will combine with BSA and cause the detectable fluorescence quenching. This method is highly sensitive with a limit of detection (LOD) of 3 U L−1 and linear in 5–400 U L−1. However, it also suffers from drawbacks such as the background from the degradation of the Cu(II)–Ala complex and the requirement of additional equipment for fluorescence detection.
Tetrazolium salts have been demonstrated to be efficient reagents in the detection of 2-hydroxyl ketone (Method AVI). Colourless 2,3,5-triphenyltetrazolium chloride (TTC) can be reduced into a red-coloured formazan precipitate in the presence of 2-hydroxyl ketone, which has been employed as a strategy for detection of the TA activity for amino alcohols.97 Recently, the application of a newer generation of tetrazolium reagents (WST-1) was proposed to improve the sensitivity of the alkaline 2-hydroxy ketone assay by 2 orders of magnitude, with the linear range changed from 1–10 mM (TTC) to 1–100 μM (WST-1).98 Moreover, the coloured formazan product remains fully soluble, with increased dynamic range and reproducibility.
Another commonly used amino donor is 1-PEA (1a), used as a model structure of aromatic amines and a highly active amino donor in transaminations. A high throughput screening strategy was developed for the TA reaction of this compound due to the UV absorbance (245 nm) of its corresponding product acetophenone (Method AVII).67 The significant (ε = 12 mM−1 cm−1) and exclusive absorbance (other components in the reaction system do not show a comparable absorbance at 245 nm) promotes this strategy to be an efficient high throughput screening method for quantitative analysis.99 Moreover, this approach has other advantages including no need for additional reagents, the capability to determine the enantioselectivity and the capability to work in a continuous fashion. The main drawback aside from the substrate dependence is the requirement for a UV-detection spectrophotometer.
Martin and co-workers reported the first example in a directed evolution study of TA for activity and thermostability. Substituted aminotetraline (8) gives the product ketone 32, which turns into a coloured product after exposure to air (Method BI).16 In another report, ortho-xylylenediamine (33) was employed as the smart amino donor. After its deamination spontaneous polymerization of the isoindole by-product (34) generates dark-coloured derivatives (Method BII).100 This strategy is sensitive but nonlinear, with a strong background colour observed. However, the advantage of the method is the capability to be applied in solid screening. Similarly, another smart amino donor 2-(4-nitrophenyl)ethan-1-amine (35) and several other 4-nitroaryl amines were used in a HTS method for both solid and liquid screening (Method BIII). The 4-nitroaryl electron withdrawing group (EWG) in the smart co-product derivatives enhances its tendency to form an enamine which showed different colours.101 The variation of smart co-substrates provided a wider generality of this method. Moreover, the sensitivity was reported to be high with a LOD of 1.2–30% of the initial enzyme activity. This method is also applicable in both liquid and solid screening like Method BIII, although with the background colour much lower than the previous one.
Recently, our group in collaboration with Fessner developed a fluorescence-based method involving 1-(6-methoxynaphth-2-yl)alkylamines (37) as the amino donor (Method BIV). The produced acetonaphthone (38) can be monitored using its bright fluorescence at 450 nm with very high sensitivity (LOD 0.35 nmol min−1) and selectivity (weak background response).102 The limitations are obviously the substrate dependence and the equipment requirement.
Constructing stable biocatalysts
In organic synthesis, TAs are practical and economical biocatalysts only if their stability under non-physiological conditions can be improved to a sufficient level. The stability includes enzyme operational stability, such as thermostability, organic solvent tolerance, chemical tolerance, and storage stability. A number of strategies have been employed to enhance the stability of TAs including gene mining, protein engineering, biocatalyst formulation/immobilization and medium engineering. In this section, these methods and their applications will be discussed.Gene mining
Thermophilic strains provide a natural library of proteins with high thermostability due to their unique living environments. Therefore, gene mining of these microorganisms will result in thermostable proteins with optimal temperature over 60 °C. For example, an S-selective TA from Sphaerobacter thermophilus was identified by homologous sequence searching with Polaromonas TA as the template (Table 1).18 The optimal temperature of this TA was determined to be 60 °C, and after 1 h of incubation at this temperature, no loss of activity was observed. Similarly, TAs from the thermophilic Geobacillus thermodenitrificans was also identified as thermostable biocatalysts with an optimal temperature determined to be 65 °C (Table 1).103 Moreover, an increase of activity was observed after the incubation in high temperature: a 2.5-fold increase after 8 h in 50 °C or 30 min in 60 °C, which was ascribed to temperature-related folding improving the enzyme stability.Another microorganism which grows in special environments is the halophilic strain, consequently containing a library of halophilic proteins. These proteins are highly tolerant of high salt concentration and have potential to tolerate high substrate concentration. Recently, a TA was identified from Halomonas elongate, a moderate halophilic bacterium, with a sequence identity of 38% to V. fluvialis TA and 55% to C. violaceum TA (Table 1).104 The stability of this enzyme was not decreased on incubation in a high concentration of salt (KCl and NaCl) buffer (concentration from 1 M to 3 M) for 24 h, although the reactivity was repressed to some extent. More interestingly, the maintained stabilities were also observed in the presence of organic co-solvents (MeOH, DMSO, EtOH and tBuOH) up to 20%. With these stabilities, H. elongate TA was reported as a promising candidate for organic synthesis.
Protein engineering
Protein engineering has also been employed to improve the stability of TA. For example, by error-prone PCR-based directed evolution, Martin et al. engineered a mesophilic A. citreus TA into a thermostable mutant with 17 mutations after 5 rounds of screening (Table 2).16 The best mutant exhibited good activity at an optimal temperature of 55 °C, much higher than that of the wild type protein (30 °C).A similar strategy has been employed in the protein engineering of TA for the production of sitagliptin (Table 2).15 To generate a practical and economical biocatalyst for industrial application, the TA-mediated reaction conditions were optimized, including substrate concentration, pH, temperature and organic co-solvent concentration. After 11 rounds of evolution, the optimized biocatalyst showed remarkable activity in 50 g l−1 pro-sitagliptin ketone, 1 M amino donor IPA, pH 8.5, 45 °C and 50% co-solvent DMSO. This extraordinary success set up a milestone for protein engineering of TA.
Aside from the conventional protein engineering strategy, non-canonical amino acid substitution has also been demonstrated as an efficient method for stability improvement of TA. Global substitution by fluorotyrosine (FY) was conducted to V. fluvialis TA, resulting in a superior mutant TA[FY] with higher thermostability and more tolerance towards DMSO.105 The half-life at 50 °C was prolonged from 3 h (WT) to 7 h (TA[FY]); the half-life at 50 °C with 20% DMSO was also prolonged from 7 h (WT) to 48 h (TA[FY]). This significant improvement of both thermostability and organic solvent tolerance was ascribed to the stronger interactions between two protein monomers provided by the fluorotyrosine on the dimer interface.
Biocatalyst formulation
One enzyme in different biocatalyst formulations may exhibit diverse performance in the same reaction. Common biocatalyst formulations are summarized in Table 4 and compared in the biocatalyst production process, specificity to the target reaction, biocatalyst activity, stability, cost and application area. Purified enzyme is the most specific, which is required in most biochemical research projects. However, the production process and cost prevent it from large-scale usage. A crude cell extract may show comparable specificity and higher activity than purified enzyme only if the target protein is over-expressed in a sufficient amount. This formulation is the most widely used for its easy production and low cost. Nevertheless, neither purified enzyme nor crude cell extract is a stable formulation for long-time processing and storage. In this case, other biocatalyst formulations are needed.| Production | Specificity | Activity | Stability | Cost | Application | |
|---|---|---|---|---|---|---|
| a Specificity of this formulation depends on whether the favourable protein is over-expressed or not. b Specificity of this formulation depends on whether the enzyme is purified or not. c Some method of immobilization is detrimental for the enzyme activity. d The cost will be largely reduced if the enzyme could be well-recycled. | ||||||
| Purified enzyme | +++ | +++ | ++ | + | +++ | Biochemical |
| Crude cell extract | + | ++/+a | +++ | + | + | Biochemical |
| Chemical | ||||||
| Industrial | ||||||
| Lyophilized enzyme | ++ | +++/++b | ++ | ++ | ++ | Chemical |
| Industrial | ||||||
| Immobilized enzyme | ++ | +++/++b | +c | +++ | +d | Chemical |
| Industrial | ||||||
| Whole-cell biocatalyst | + | ++/+a | +++ | ++ | + | Chemical |
| Industrial | ||||||
For the application of TAs in organic solvents, lyophilization is an option that is commonly employed due to the easy production, superior organic solvent tolerance as well as long possible storage time.54,106,107 The process of lyophilization basically includes rapid freezing and water removal, which may maintain the protein conformation while preventing its degradation in the solvent.
Immobilization is the most widely used strategy for practical and economical biocatalysts in industry.108–110 Although additional immobilization reactions are required and in some methods the enzyme activity may be damaged to some extent, immobilized enzyme is still favoured due to its high operational and storage stability. In most cases, immobilized enzyme can be recycled by some physical separation from the reaction mixture, which largely reduces the cost of the biocatalyst. For instance, TA-117 was immobilized in a sol–gel/celite matrix resulting in the optimal temperature rising from 30 °C to 40–50 °C and almost no loss of activity after 5 reaction cycles.111 In another example, cellulose-immobilized V. fluvialis TA showed a broad temperature range of 30–50 °C and maintained its activity after 4 cycles.112 Besides, TAs have also been immobilized on other materials including resin,17 chitosan,113 Fe3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 114 and glass beads,115 resulting in stable and recyclable biocatalysts.
114 and glass beads,115 resulting in stable and recyclable biocatalysts.
Furthermore, the use of whole-cell biocatalysts may also improve TA stability. For instance, the chemical tolerance of V. fluvialis TA was remarkably enhanced by this strategy. Used as crude cell extract, V. fluvialis TA was inactivated thoroughly by 80 mM of the substrate 2-aminoheptane, whereas the whole cell biocatalysts tolerated up to 200 mM of the same substrate.74 Due to the short production process (fermentation only), this formulation is also favourable in industrial applications. However, the separation issue that is caused by the cell lysis in the reaction medium limits its application in organic synthesis.
Medium engineering
Due to its significant effects on protein structures, the reaction or storage medium is crucial for the biocatalyst stability. Organic solvents are generally regarded as detrimental for proteins; however some solvents have been reported to show positive effects on maintaining the enzyme structure. For instance, 20% DMSO prolonged the half-life of V. fluvialis TA at 50 °C from 3 h to 7 h and the half-life of the mutant TA[FY] even longer (from 7 h to 48 h).105 In another example, addition of 20–50% glycerol, 10–20% methanol or 10–50% DMSO in the storage buffer improved the storage stability of C. violaceum TA.106Aside from the solvents, additives may also affect protein stability. As the cofactor of TAs, PLP is regarded as an efficient stabilizer for TA. The addition of 0.1 mM PLP improved the residual activity of M. vanbaalenii TA at 60 °C from 33% to 87%.116 Aside from the thermostability, the tolerance of chemicals may also be enhanced by adding PLP. With 80 mM of the substrate 2-aminoheptane, V. fluvialis TA lost all of its activity. However, only 5% of the activity was lost in the presence of 1 mM PLP.74 The stabilization of TA may be ascribed to the important role of PLP in the dimerization of TA.106 Release of PLP may accelerate the dissociation of the TA dimer and consequently destabilize the enzyme.
Shifting the reaction equilibrium
As reversible reactions, TA-mediated transaminations suffer from equilibrium issues. Especially in some asymmetric amine syntheses, the equilibrium can be far on the side of the substrates. To optimize TA performance in organic synthesis, a number of physical and chemical strategies have been developed and described to displace the reaction equilibrium to the product side,10,20–22 which will be discussed in this section (Scheme 3).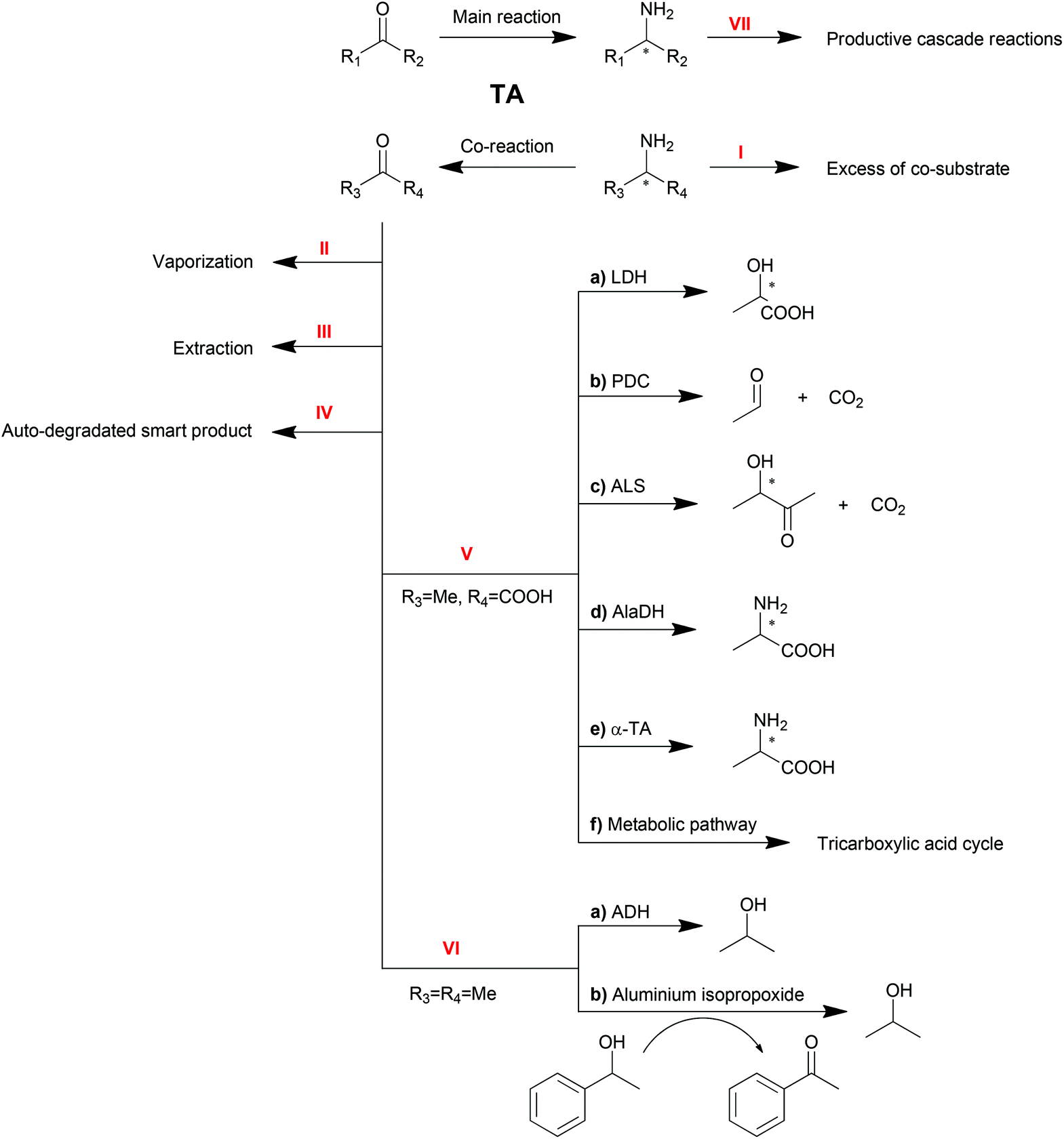 | ||
| Scheme 3 Strategies for TA reaction equilibrium shifting. | ||
Excess of co-substrate
One of the conventional strategies is providing an excess amount of co-substrate (Scheme 3, Method I). Nearly 50-fold excess of the amino donor isopropylamine (1 M) was applied in the transamination of acetophenone (20 mM) to 1-PEA (1a) in order to push transamination reactions towards complete conversion.94 This strategy is straightforward and easy to be conducted albeit its applicability is limited by the initial equilibrium of the reaction, the substrate solubility and the enzyme inhibition.Removal of product or co-product
Removal of the products is another straightforward method to shift the equilibrium of reversible reactions. Multiple strategies are available to remove the products, including physical vaporization/extraction methods and chemical reactions.Because of the different polarities of amines and ketones, the extraction of ketone product by organic solvent is another option for product removal (Method III). Bi-phasic reaction systems, either liquid–solid120,121 or liquid–liquid,34,122 have been proven to be practical for ketone product extraction. Nevertheless, the application of this strategy is limited due to drawbacks including the poor distinction between substrate and product or the destabilization of proteins by organic solvents.
For the elimination of the amine product which is hydrophilic, a tri-phasic system has been developed consisting of an organic solvent bridge between two aqueous phases: an alkaline reaction phase and an acidic extraction phase.123 With isooctane as the organic solvent, the inhibitive product 1-PEA (1a) moved from the reaction phase (pH = 8.0) to the extraction phase (pH = 3.0) leaving alanine and pyruvate in the reaction phase due to their ionization state. To further improve this tri-phasic system, a supported liquid membrane (SLM) contactor with a packed bed reactor was recently proposed in the production of 1-PEA (1a) with a yield of 98% compared to 50% with no product extraction.124 This system avoids the contact of the biocatalyst with the organic solvent and has promising potential for continuous operation. Moreover, pH control and SLM regeneration can also be employed to optimize this system.125 Besides, alanine was proposed as a better amino donor in this system because of its non-extractability.126
Furthermore, spontaneous tautomerization of an unstable co-product was utilized when a transamination system with 3-aminocyclohexa-1,5-dienecarboxylic acid as the amino donor was developed by our group.129 The unstable ketone product from the amino donor spontaneously turned into its stable isomer 3-hydroxybenzoic acid, pushing the equilibrium to the product side with a theoretical yield of 100%.
A recent update of an AlaDH involved process is the combination with an alanine racemase.137 For R-selective TA-mediated transaminations, the more expensive D-alanine is required. Combining AlaDH and alanine racemase, the produced pyruvate was transformed firstly into L-alanine and then racemized. Therefore, the readily available L-alanine can be used as an amino donor, which significantly reduced the cost.
Due to its role in the carbon metabolic pathway, pyruvate can be removed by the innate pathway, acting as a strategy for shifting transamination equilibria in whole cell biocatalysts.138 The production of S-sec-butylamine by O. anthropi TA was improved by carbon-starved cultivation without glucose. The cells were instead taking the produced pyruvate as the carbon source. However, given the complexity of the cell metabolic pathway, a practical application of this strategy is yet to be developed.
Another amino donor product, acetone, can also be removed by cascade reactions (Method VI). Alcohol dehydrogenase (ADH) can be one option to convert ketones or aldehydes into their corresponding alcohols, which will not be substrates for TAs.139 Recently, a chemical catalyst mediated Oppenauer oxidation was introduced into the transamination system, which converted the ketone product back to the amino acceptor.140 In the presence of aluminium isopropoxide, the catalyst of the Oppenauer oxidation, and 1-phenylethanol, the produced acetone is able to be transformed into isopropanol and concomitantly generate acetophenone, the substrate of the transamination. Both reactors are interconnected through a semi-permeable membrane, where multicomponent intra-membrane transport takes place by diffusion and viscous flow.
It is worth noting that the productive linear cascade reactions of the major product (Method VII) are also able to shift the equilibrium, which will be discussed in the next sections.
Substrate engineering
Despite a number of successful cases reported, the applicability of using an excess of substrate or removal of product has been restricted by the intrinsic equilibrium constant of the transaminase reactions. This constant depends on the physical conditions, initial component concentrations as well as the substrates pair: the amino donor and acceptor.19 In the reaction of α-TA, the amino acids and keto acids have almost the same ability for deamination and amination, which results in equilibrium constants approximately equal to 1. However, the situation is significantly different in the reaction of amine TA. For instance, the amino group transfer between alanine/pyruvate and 1-PEA/acetophenone performs quite differently depending on which is the amino donor. Alanine has great propensity to hold an amino group while 1-PEA (1a) is a superb amino donor instead. Therefore, the matching pair of substrates is of great significance for the reaction equilibrium.141,142 Consequently, the theoretical and experimental calculation of the equilibrium constant is also of interest in the substrate engineering of TAs.143Three pairs of common substrates are listed and compared in Table 5. The difference in deamination free energy between alanine/pyruvate, which is the hardest to be deaminated, and 1-PEA/acetophenone, which is the easiest to be deaminated, is approximately 18 kJ mol−1.141 In the purpose of shifting the transamination reaction to the product side, the amino donor should be easier to be deaminated than the amino acceptor product, for example, transamination from 1-PEA (1a) to pyruvate. Therefore, alanine is not a good amino donor in this aspect. However, being an easy and cheap source, acceptance by most TAs and hardly any inhibition to enzymes are the reasons why alanine has been employed anyway in many cases. Moreover, as previously discussed, the development of strategies for pyruvate elimination and alanine recycling further promotes the application of alanine.
| Substrate pair | Chirality | Deamination | Model substrate | Inhibition | Detection | Elimination |
|---|---|---|---|---|---|---|
| Alanine | Chiral | Hard | α-Amino acid | — | — | Cascade reactions |
| Pyruvate | — | — | — | — | ||
| IPA | Achiral | Easy | — | — | — | Vaporization |
| Acetone | — | — | Severe | — | Cascade reactions | |
| 1-PEA | Chiral | Very easy | Aromatic amine | Observed | — | Extraction |
| Acetophenone | — | — | Severe | UV | Cascade reactions |
IPA and acetone are a promising pair of substrates in the application of TAs in organic synthesis, although a limited number of TAs can accept them as efficient substrates,22,141 including A. citreus TA mutant,139 ATA117 mutant15 and TAs from P. aeruginosa, P. denitrificans and A. terreus.142 The advantages include the achiral nature of IPA, which works with either S- or R-selective TAs, and the easy elimination of acetone by physical or chemical strategies.
1-PEA (1a) is widely used as the model substrate of aromatic amines. The great propensity to lose the amino group and the easy detection of its product acetophenone by UV absorbance increase the practicability of this pair of substrates. Although acetophenone shows severe inhibition of enzymes, a number of elimination approaches including extraction and chemical reactions have been developed.32,144
Eliminating the substrate and product inhibition
TA suffers the inhibition from both substrates and products. An overdosed substrate may inhibit the enzyme by binding to alternative sites and destabilizing the protein. Product inhibition, as a strategy in most metabolic regulations, is ascribed to not only the effects on the reaction equilibrium but also the destabilization of the enzyme. To eliminate these inhibitions, both process and protein perspectives should be taken into consideration.Process engineering
Removal of product, as indicated above, is a practical method for product inhibition elimination as well as shifting the equilibrium.For the TAs suffering the substrate inhibition, the fed-batch addition can be an option not only to ensure the total amount of substrate but also to prevent the detrimental effects of them. Employing a fed-batch strategy with a 300 mM 1-PEA (1a) added after 180 min in a constant velocity resulted in a final conversion yield of 62%, which was 25% higher than the corresponding yield using the one shot addition.145 An alternative solution is to search for a less inhibitive substrate for the enzyme in use.
Protein engineering
With the aid of gene mining and protein engineering, TAs with decreased or eliminated inhibition are available to be further explored.TAs from O. anthropi146 and M. vanbaalenii116 were reported with low product inhibition (Table 1). The tolerance of acetophenone in both cases was reported up to 20 mM while O. anthropi TA can also tolerate 500 mM of 1-PEA (1a).
Moreover, by error-prone PCR mutagenesis and enrichment culture selection, the aliphatic ketone inhibition of V. fluvialis TA was reduced significantly (Table 2).74 The mechanism of product inhibition of TAs has not yet been fully uncovered, so the analysis of these advanced proteins with low inhibition is of great interest for future work.
Enzymatic cascade reactions
Enzymatic cascade can be used as a smart synthesis strategy in which two or even more biocatalysts are combined for the production of a target compound. This approach has been demonstrated to be advantageous in shortening reaction routes, avoiding unstable or toxic intermediates, increasing the atom efficiency and reducing the amount of waste.147–150 Taking advantage of this method, TAs could be applied in organic synthesis in a more efficient fashion. A few elaborate reviews have been published on the topic of cascade reactions involving TAs, which provide a great amount of details.10,22,26 Therefore, in this section we will only discuss some possible cascade reaction routes and representative reactions (Scheme 4). | ||
| Scheme 4 Representative cascade reactions involving TA. | ||
Unnatural amino acids
Unnatural amino acids have been utilized as building blocks in the synthesis of a number of chiral drugs. Unlike natural amino acids, fermentation and extraction strategies are not practical in the production of unnatural amino acids. That is why TA is a promising biocatalyst in the synthesis of unnatural amino acids. Moreover, in combination with other enzymes, TA is also able to catalyse the synthesis of unnatural amino acids from accessible and cheap chemicals.Unnatural L-homoalanine (L-26a) has been synthesized from natural L-threonine instead of from expensive keto acid by a cascade reaction of threonine deaminase (TD) and TA (Scheme 4a).57E. coli TD catalysed the dehydration and deamination of L-threonine, generating 2-oxobutyrate, which was then stereoselectively converted to L-homoalanine (L-26a) by P. denitrificans TA with amine 5a as the amino donor. This reaction was conducted in a one-pot fashion with a 5-fold excess of TD to avoid inhibition from the final product. With this method, 91% conversion of L-threonine to L-homoalanine (L-26a) was achieved. A significant advantage in the application of amine TA to produce an amino acid is the fact that the reaction equilibrium lies on the side of amino acid product, which results in 100% theoretical conversion in the transamination step.
Another strategy for unnatural amino acid production is the combination of α-TA and amine TA.151 With the advantages found in synthesis of amino acids as described above, amine TA was employed as an “equilibrium shifter” for the transamination mediated by α-TA. The amino group was transformed from the amine into the keto acid by the sequential transamination of amine and α-TA with a pair of amino acid and keto acid as intermediates, which needs to be accepted by both of the two TAs. For example, after a screening of substrates, L-homoalanine (L-26a) and its keto acid product were chosen as the intermediate substrates for E. coli branched-chain transaminase and O. anthropi TA. Employing IPA as amino donor, 99% conversion of 40 resulted in L-26c (ee >99%) after 9 h (Scheme 4b). With this strategy, both enantioselective TAs were investigated, resulting in a number of successful examples of production of unnatural amino acids with different side chains.
Aside from the unnatural α-amino acids, β-amino acids may also be important synthesis intermediates of chiral drugs. The asymmetric synthesis of β-amino acids was hampered by the unstable β-keto acid amino acceptor as well as the low β-activity of TAs.42 By gene mining and protein engineering, more TAs with β-activity have been explored (Tables 1 and 2).42,68 The challenge of the unstable β-keto acid has been overcome by cascade reactions of TAs combined with lipases. The accumulation of the unstable β-keto acid can be avoided by a one-pot reaction of lipase-mediated hydrolysis of a β-keto ester and the sequential TA-catalysed transamination.42S-30a was produced through a cascade reaction of Candida rugosa lipase and Mesorhizobium TA in 20% yield and >99% ee (Scheme 4c). Further optimization can be conducted by manipulating the concentration of lipase.152 The total activity of TA should be slightly higher than that of the lipase in order to minimize the accumulation of intermediate β-keto acid.
Deracemization of amines
Deracemization has been shown to be an efficient strategy for the production of optically pure amines from their racemates. A straightforward route is the combination of two TAs with complementary enantioselectivity, turning one enantiomer into ketone and then switching this ketone into the other enantiomer.111,153–155 This method has been limited by the substrate compatibility of these two TAs, whereas the cascade reaction needs to be conducted in a stepwise fashion with the elimination of the first enzyme. For example, deracemization of rac-18c by a combination of ATA-113 (S-selective) and ATA-117 (R-selective) resulted in only 75% ee (S) or 29% ee (R), depending on the enzyme addition order (Scheme 4d).153 However, if an additional heat inactivation step was conducted before addition of the second enzyme, the final ee of each enantiomer could be achieved over 99%. Further optimization of this strategy was proposed using a smart amino acceptor in the first step which will not be accepted by the second TA so as to drive the reaction to the product side.156 The inactivated step was also avoided with this smart substrate.In another method avoiding the interference of two TAs, oxidases have been employed in the first step of deamination. Like TAs, enantioselective oxidases also transform one of the amine enantiomers into its keto product, followed by amination by a TA with complementary enantioselectivity. D-Amino acid oxidase157 and monoamine oxidase158 have been demonstrated to be promising candidates for this strategy. The disadvantage of this approach is the consumption of oxidant in the first step, which could be compensated for by additional recycling of these compounds.
Imines, secondary amines and amides
Primary amines are the only products that can be directly obtained in the straightforward transamination catalysed by TAs due to the enzyme mechanism. However, with the aid of other reactions, for instance, spontaneous dehydration or enzyme-mediated condensation, imines, secondary amines or amides are possible to be produced.Kroutil et al. reported a regio- and stereoselective monoamination of 2,6-diketones (44) by both S- and R-selective TAs (Scheme 4e).55 With five tested TAs, the ω-1 carbonyl moiety was exclusively aminated leading to a cyclic imine Δ1-piperideine (46) with either S- or R-product in an optically pure form (ee >99%). The generality of this strategy was also demonstrated by regio- and stereoselective synthesis of 2,6-diketones with a number of other substituents. Further reduction of the double bond provides chiral piperidines (47); all of the four diastereomers were obtained by utilizing R- and S-selective TAs with different chemical chiral reductants.159 With an alternative strategy in the reduction step, monoamine oxidase (MAO-N) was employed in a one-pot cascade reaction system producing enantiomerically pure 2,5-disubstituted pyrrolidines.160 Furthermore, this strategy may also be applied in the stereoselective monoamination of triketone compounds giving a chiral pyrrolizidine product.161 In a recent example, by combination with a carboxylic acid reductase at the beginning of the cascade reaction, piperidines and pyrrolidines can be synthesized from keto acids.162
A novel triangular cascade reaction for cyclic amines was reported consisting of TA and norcoclaurine synthase (NCS) taking both the substrate and product of TA as substrates (Scheme 4f).163 With a second Pictet–Spengler cyclisation by sequential addition of formaldehyde, pharmaceutically active tetrahydroprotoberberine alkaloids (S-51) can be produced with high enantiomeric purity (>95% ee). Another similar cascade route for cyclic amines was proposed as the combination of TA and strictosidine synthase for the synthesis of C3-methylated strictosidine derivatives from indolylketone and secologanin.164
Aside from amines, cyclic amides can also be prepared by cascade reactions involving TA and spontaneous dehydration. Keto acids or keto esters were firstly aminated by TA and then turned into cyclic amides by spontaneous cyclization and dehydration. For example, a dynamic kinetic resolution route was developed with amination of a racemic 4-oxo-3-phenylbutyric acid ethyl ester (52) followed by spontaneous racemization into 4-phenylpyrrolidin-2-one (54), which after subsequent chemical reactions gives 3-phenyl-GABA (Scheme 4g).165 A similar strategy has also been applied in the production of Niraparib, an orally active poly(ADP-ribose)polymerase inhibitor, by amination of a 5-carbon aldehyde ester.49 Another example is the chemoenzymatic synthesis of the dual orexin receptor antagonist MK-6096. Amination of a keto ester followed by a cyclization provided a chiral piperidone.166
Biocatalytic amidation may be achieved by a hydrolase only in organic solvents or by an acyl transferase in aqueous solutions. Recently, our group proposed a one-pot one-step cascade reaction combining TA and acyl transferase for the amidation of an aldehyde (Scheme 4h).167 After optimization of pH, acyl donor and enzyme concentration, a conversion of 97% was reached in the synthesis of N-benzylacetamide (56).
Amination of –OH or –CH3
Alcohols have gained a great amount of attention in the production of amines due to their oxidation state, which is the same as that of amines. Alcohols are firstly oxidized into ketones or aldehydes and then reductively aminated by TAs into amines (Scheme 4i). The combination of TAs with oxidants or oxidases has provided a number of successful examples in asymmetric amination of primary,168–170 secondary alcohols,171,172 diols169 and saccharides with multiple hydroxyl groups.173 One of the elegant strategies for the amination of alcohols is recycling of the cofactor NAD/NADH, which is consumed in the oxidation step, regenerating the cofactor in an AlaDH-mediated alanine recycling employed in the transamination step.169,171,174A terminal methyl group can also be aminated in a cascade reaction. This inert group in dodecanoic acid methyl ester (57) was oxidized into an aldehyde (59) in two steps by an alkane monooxygenase (AlkBGT), and then transformed into a terminal amine (60) by TA (Scheme 4j).175 This cascade reaction was achieved in a single cell biocatalyst with the two involved enzyme genes recombined.
Amines with two stereo-centres
Working with other stereoselective enzymes, TAs are able to be applied in the synthesis of amines with two stereo-centres. The advantage of these routes is the circumvention of the protection step of the first stereo-centre before the production of the second stereo-centre.Chiral amino alcohols have been produced in a cascade reactions together with transketolase (TK)/TA, acetohydroxyacid synthase I (AHAS-I)/TA, TA/ADH and keto reductase (KRED)/TA. Among them, the TK/TA cascade reaction is the most studied system. TK, important in the pentose phosphate pathway, mediates the C–C bond formation between aldehydes and keto-alcohols. The produced ketose containing one chiral hydroxyl group in the vicinity of a carbonyl group (63) is further catalysed by TA to produce chiral amino alcohols (64) with two stereo-centres (Scheme 4k).66 An engineered variant of E. coli transketolase (D469 T) and C. violaceum TA were employed in this system with an isolated yield of 26% in the 100 ml reaction scale.176 Recently, the routine was further optimized by adjusting the expression of the enzymes and reducing the substrate inhibition providing a 6-fold greater yield.145 Moreover a deamination of serine was introduced in the reaction system for producing hydroxypyruvate from a common source.24
Like TK, thiamine diphosphate (ThDP)-dependent acetohydroxyacid synthase I (AHAS-I) from E. coli can catalyse C–C bond formation providing a keto alcohol to TA for further amination.177 Pyruvate was reported to be decarboxylated and subsequently ligated to benzaldehyde yielding R-phenylacetylcarbinol, which is a substrate for TAs. Two diastereomers were produced by employing R- or S-selective TA. However, limited by the enantioselectivity of the first step, only the 1R-product can be obtained. All four diastereomers were produced by involving another S-selective AHAS-I.178 Meanwhile, starting from 1-phenylpropane-1,2-dione (65), another cascade reaction with amination and reduction turned two carbonyl groups into an amine and a hydroxyl group respectively (Scheme 4l). Similarly, all four diastereomers of 1,3-amino alcohol were accessed by the combination of a ketoreductase and TA.179 The regio-selectivity in both cases is crucial for the purity of products.
Recently, other cascade reactions to obtain chemicals with two stereo-centres have been developed by the combination of TAs and other enzymes. Ene-reductase, catalysing the reduction of olefins, is one of the candidates. Both β-substituted cyclic enone85 (Scheme 4m, 68) and open-chain α-alkyl-β-arylenones50 were turned into amines with two stereo-centres. By employing enantio-complementary reductases and TAs, all four diastereomers can be obtained.
Transaminase in synthetic biology and systems biocatalysis
As important biocatalysts in nitrogen metabolism, TAs have been found to be involved in amino acid synthesis and degradation as well as in the biosynthesis of some secondary metabolites.180,181 Integration of cascade reactions involving TAs into a single microbial cell could be a new proposal in synthetic biology and the strategies of metabolic engineering may also be applicable in these cases. Alternatively, complex multistep synthesis pathways with multiple enzymes in vitro provide the examples of systems biocatalysis, which is a promising strategy in organic synthesis.182Natural biosynthetic pathway
The innate amino acid biosynthesis pathways in microorganisms can be regulated for the industrial production of these amino acids by manipulating the TAs involved. For instance, the production of L-tyrosine was improved significantly by combinatorial overexpression of aromatic amino acid biosynthesis genes in E. coli.181 Besides ydiB and aroK, coding for a shikimate dehydrogenase and a shikimate kinase, overexpression of tyrB encoding the last enzyme of the AAAB pathway, an aromatic amino acid transaminase, improved L-tyrosine production significantly when co-overexpressed together with ydiB or aroK. Similarly, overexpression of a putative class II aminotransferase increased the L-ornithine production in Corynebacterium glutamicum from 8.42 g l−1 for the control to 12.48 g l−1.183Aside from natural amino acids, unnatural ones have also been produced by construction of artificial biosynthetic pathways based on the innate ones. L-Phenylglycine, non-proteinogenic but important for the preparation of several antibiotics and taxol, was biosynthesized from glucose with an engineered E. coli containing an L-phenylglycine synthetic pathway from Amycolatopsis orientalis and Streptomyces coelicolor.184 The production of L-phenylglycine was increased by 224-fold compared with the original strain by optimization of the heterologous pathway and attenuation of native L-phenylalanine transamination.
The control of the degradation of amino acids has also been proposed as an efficient strategy for production of other organic chemicals. The yeast Ehrlich pathway (Scheme 5a) has been investigated and employed as the biosynthesis route for 2-phenylethanol. This pathway including an aromatic transaminase was constructed in either Saccharomyces cerevisiae185 or E. coli,186 both of which achieved an improvement in 2-phenylethanol production.
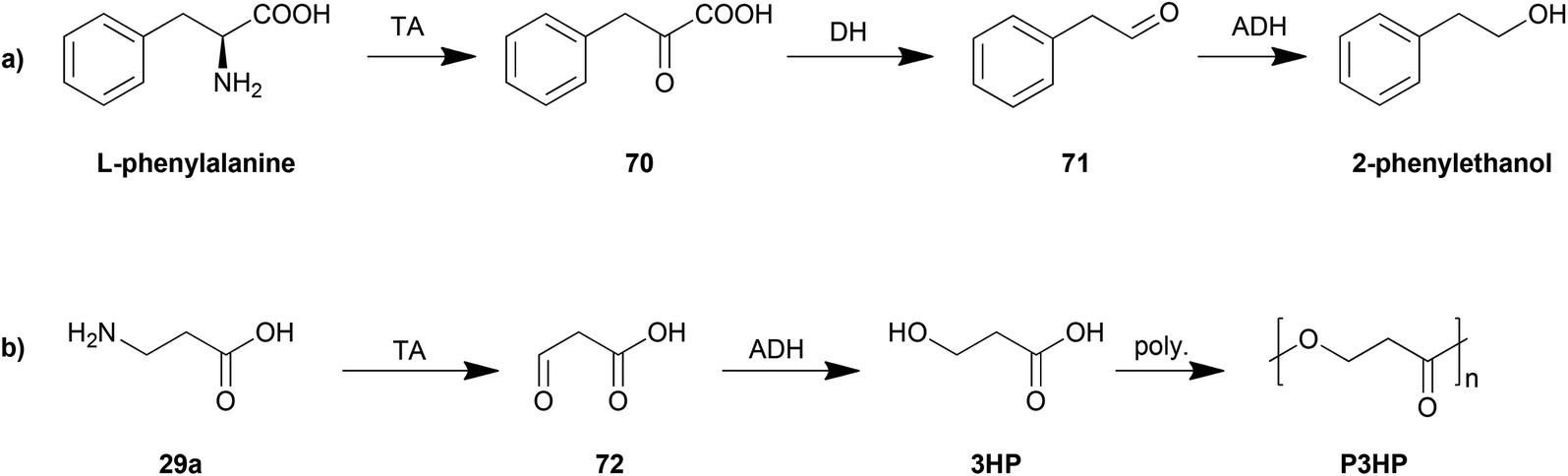 | ||
| Scheme 5 Biosynthesis based on degradation of amino acids. | ||
Another example is the biosynthesis of 3-hydroxypropionic acid (3HP), a potential chemical building block for sustainable production of superabsorbent polymers and acrylic plastics, from the degradation of β-alanine (Scheme 5b). The pathway was engineered and optimized by using a β-alanine-pyruvate aminotransferase to convert β-alanine into 3HP.187 By further employing polymerization steps, poly(3-hydroxypropionate) (P3HP) can be obtained.188
Besides the pathways of amino acids, the biosynthesis of some secondary metabolites may also be achieved and optimized by engineering of TA-mediated processes. For instance, in the biotin biosynthesis in B. subtilis, the bottleneck of the process was identified as the TA which catalyses the formation of the intermediate 7,8-diaminopelargonic acid (DAPA) from 7-keto-8-aminopelargonic acid (KAPA).180 This limitation of this TA was ascribed to the unfavourable amino donor S-adenosylmethionine. As a consequence, substitution of this compound by a favourable L-lysine dramatically stimulated the conversion of KAPA to DAPA by the B. subtilis DAPA TA.
Artificial biosynthetic pathway
Cascade reactions involving TAs have been thoroughly investigated for organic synthesis, which has been discussed in the previous section. Based on these routes, expressing all of the needed enzymes in one whole cell generates a cell factory for the target product. An example of this cell factory is the construction of a whole-cell biocatalyst catalysing the terminal amino-functionalization of fatty acid methyl esters (e.g., dodecanoic acid methyl ester) and alkanes (e.g., octane) by heterologous pathway engineering (Scheme 4j).175 Coupling oxygenase and transaminase catalysis in vivo, both substrates are converted with absolute regiospecificity to the terminal amine via two sequential oxidation reactions followed by an amination step. This route was further tuned by metabolic engineering methods recently.189 Hydrophobic outer membrane porin AlkL was employed to improve the substrate uptake, consequently increasing the oxygenation and transamination activities by 8.3- and 7.6-fold, respectively. Moreover, a heterologous alanine dehydrogenase was introduced to construct an alanine self-sufficient whole-cell transamination. Finally, the introduction of an alcohol dehydrogenase enabled an increase in pathway flux and minimized the undesired over oxidation.Similarly, another biosynthesis pathway was also monitored using strategies of metabolic engineering. To biosynthesize (2S,3R)-2-amino-1,3,4-butanetriol (ABT), Ward et al. established an artificial pathway containing E. coli transketolase (TK) and the C. violaceum TA in E. coli (Scheme 4k). The kinetic models and the analysis of parameters of each enzymatic node identified that TA is the bottleneck with a 25 times slower conversion rate than the upstream enzyme TK.145 Increasing the expression amount of TA and decreasing the substrate inhibition of TA successfully scaled up the production of ABT to preparative scales. Furthermore, a modification of the pathway by adding a production of hydroxypyruvate (HPA) from serine by TAs made the biosynthetic pathway more economical.24 This system may be further improved by constructing a recycling cascade where serine is used as the donor to aminate erythrulose (catalysed by a transketolase) for the simultaneous synthesis of ABT and HPA.
An increasing amount of interest has been attracted by this promising biocatalyst even though the application of TA is limited by some inherent problems as discussed above. The whole cell containing all the cascades reaction nodes can be a promising strategy in TA catalysis which may degrade the co-products, can improve the stability and even perform the synthesis of cofactors. With versatile enzymes from either gene mining or protein engineering, the current long organic synthesis routes have possibilities to be transformed into artificial biosynthesis pathways and integrated into whole-cell factories.
Systems biocatalysis
Working in a cell-free fashion, systems biocatalysis employs in vitro “artificial metabolisms” as pathways for the preparation of valuable chemicals.182 Even though it is a much smaller system than the total metabolisms of any host cell, it is still highly important that every reaction step and their compatibility should be carefully designed. Regardless of the complexity, systems biocatalysis has a great number of advantages including avoiding competing metabolic pathways, avoiding physical barriers and toxicity problems and, the most significant advantage, construction of novel systems for non-natural substrates.One example is the total synthesis of capsaicin analogues from lignin-derived compounds explored by our group involving heterogeneous metal, organocatalytic and enzymatic cascade routes (Scheme 6).174 This synthesis process consisted of three steps: the production of vanillin (74), the amination of vanillin (74) and the production of amide 78, whereas each of them can be achieved by either chemo- or bio-catalysis. Vanillin (74) can be synthesized by either the oxidation of vanillyl alcohol (73) or the retro-aldol transformation of aldol compound 75. Each method is compatible with the TA mediated transamination afterwards. Especially, the ADH-catalysed oxidation of vanillyl alcohol (73) contributed to the regeneration of NAD/NADH in the transamination process, which resulted in an elegant one-pot one-step cascade reaction. As the last step, the amidation of amine 5e was also performed with either chemo- or bio-catalysts. Lipase-mediated amidation is a greener strategy avoiding the employment of halogenated organics, although the chemical method catalysed by SOCl2 gave a higher conversion. This last step is connected to the previous reactions in a one-pot fashion without any purification steps, although the removal of water as solvent was required. In this total synthesis of capsaicin analogues, multiple steps with various catalysts were integrated in a one-pot fashion without any protection and purification steps, which resulted in a more efficient and economical route.
 | ||
| Scheme 6 Total synthesis of capsaicin analogues. | ||
Another example is the biosynthesis of the nylon-6 monomer 6-aminohexanoic acid (28c) from cyclohexanol (79) (Scheme 7).23 This synthesis route consisted of two cofactor self-sufficient cascade modules, the production of ε-caprolactone (81) from cyclohexanol (79) and subsequent production of 6-aminohexanoic acid (28c). In the first module, a NADP/NADPH-dependent alcohol dehydrogenase (ADH) and a Baeyer–Villiger monooxygenase (BVMO) were combined in a cascade reaction, resulting in a recycling of the cofactor. The reaction route in the second module is longer, with four enzymes combined in four steps: the ring-opening reaction by an esterase, the oxidation of the terminal hydroxyl group by ADH, the subsequent amination by TA together with AlaDH and the hydrolysis into the final product 28c. The oxidation and amination steps contributed to the regeneration of cofactor NAD/NADH, which is an elegant strategy for the amination of alcohols. Moreover, the methanol added in the ring-opening step resulted in the methyl ester product (82) instead of its acid analogue, which is a dead end molecule for the following oxidation by ADH. Besides, methanol was more practical than ethanol due to its low activity for ADH afterwards. To conduct these two modules simultaneously, more details have been taken into consideration. Employed in both modules, the ADHs need to be highly specific towards the substrates, sec-alcohol in the first module while prim-alcohol in the other, to avoid the interferences. Similarly, the cofactors used were also different in the two modules. The concentration of methanol was also adjusted since it improved the production in the second module while it significantly destabilized BVMO in the first module. Therefore, further optimization of this route may be achieved by enhancing the MeOH tolerance of BVMO or improving the specificity of the esterase towards methyl ester product in the ring-opening reaction.
 | ||
| Scheme 7 Production of 6-aminohexanoic acid. | ||
Conclusions
TAs have been proven to be promising biocatalysts in organic synthesis due to their extraordinary nature of producing highly enantiomerically enriched amino acids and amines. Various strategies developed at every step from enzyme discovery to product elimination have played an increasingly significant part in the optimization of TAs and of processes involving TAs. These exquisite processes are practical and efficient in the production of chiral pharmaceuticals, giving a strong competition to conventional chemical routes.190,191 The greenness of these outstanding biocatalysts, including aspects such as shorter routes without protecting steps, lower cost of equipment and energy, less environment-detrimental waste, easier procedures for work-up and even lower cost of catalysts considering the recycling of enzymes, has made them one of the top options for industrial organic synthesis.192Acknowledgements
This work was funded by KTH Royal Institute of Technology. Henrik Land, KTH, is gratefully acknowledged for critical comments on the text.References
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC.
- P. Berglund, M. S. Humble and C. Branneby, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, vol. 7, pp. 390–401 Search PubMed.
- A. C. Eliot and J. F. Kirsch, Annu. Rev. Biochem., 2004, 73, 383–415 CrossRef CAS PubMed.
- F. Steffen-Munsberg, C. Vickers, H. Kohls, H. Land, H. Mallin, A. Nobili, L. Skalden, T. van den Bergh, H. J. Joosten, P. Berglund, M. Höhne and U. T. Bornscheuer, Biotechnol. Adv., 2015, 33, 566–604 CrossRef CAS PubMed.
- S. Kara, J. H. Schrittwieser, F. Hollmann and M. B. Ansorge-Schumacher, Appl. Microbiol. Biotechnol., 2014, 98, 1517–1529 CrossRef CAS PubMed.
- W. F. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS PubMed.
- D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS PubMed.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- H. Kohls, F. Steffen-Munsberg and M. Höhne, Curr. Opin. Chem. Biol., 2014, 19, 180–192 CrossRef CAS PubMed.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 CrossRef CAS PubMed.
- G. Matcham, M. Bhatia, W. Lang, C. Lewis, R. Nelson, A. Wang and W. Wu, Chimia, 1999, 53, 584–589 CAS.
- J. S. Shin and B. G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS PubMed.
- J.-S. Shin and B.-G. Kim, Biosci., Biotechnol., Biochem., 2001, 65, 1782–1788 CrossRef CAS PubMed.
- M. Höhne, S. Schatzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- A. R. Martin, R. DiSanto, I. Plotnikov, S. Kamat, D. Shonnard and S. Pannuri, Biochem. Eng. J., 2007, 37, 246–255 CrossRef CAS.
- M. D. Truppo, H. Strotman and G. Hughes, ChemCatChem, 2012, 4, 1071–1074 CrossRef CAS.
- S. Mathew, S. P. Nadarajan, T. Chung, H. H. Park and H. Yun, Enzyme Microb. Technol., 2016, 87–88, 52–60 CrossRef CAS PubMed.
- J. H. Seo, D. Kyung, K. Joo, J. Lee and B. G. Kim, Biotechnol. Bioeng., 2011, 108, 253–263 CrossRef CAS PubMed.
- P. Tufvesson, J. Lima-Ramos, J. S. Jensen, N. Al-Haque, W. Neto and J. M. Woodley, Biotechnol. Bioeng., 2011, 108, 1479–1493 CrossRef CAS PubMed.
- R. Abu and J. M. Woodley, ChemCatChem, 2015, 7, 3094–3105 CrossRef CAS.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- J. H. Sattler, M. Fuchs, F. G. Mutti, B. Grischek, P. Engel, J. Pfeffer, J. M. Woodley and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 14153–14157 CrossRef CAS PubMed.
- M. F. Villegas-Torres, R. J. Martinez-Torres, A. Cázares-Körner, H. Hailes, F. Baganz and J. Ward, Enzyme Microb. Technol., 2015, 81, 23–30 CrossRef CAS PubMed.
- M. S. Malik, E.-S. Park and J.-S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- S. Mathew, G. Shin, M. Shon and H. Yun, Biotechnol. Bioprocess Eng., 2013, 18, 1–7 CrossRef CAS.
- W. John and W. Roland, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef.
- J. S. Gong, Z. M. Lu, H. Li, Z. M. Zhou, J. S. Shi and Z. H. Xu, Appl. Microbiol. Biotechnol., 2013, 97, 6603–6611 CrossRef CAS PubMed.
- L. P. Wackett, Curr. Opin. Biotechnol., 2004, 15, 280–284 CrossRef CAS PubMed.
- S. Dover and Y. S. Halpern, J. Bacteriol., 1972, 109, 835–843 CAS.
- J.-S. Shin and B.-G. Kim, Biotechnol. Bioeng., 1997, 55, 348–358 CrossRef CAS PubMed.
- J. S. Shin, H. Yun, J. W. Jang, I. Park and B. G. Kim, Appl. Microbiol. Biotechnol., 2003, 61, 463–471 CrossRef CAS PubMed.
- R. E. Meadows, K. R. Mulholland, M. Schürmann, M. Golden, H. Kierkels, E. Meulenbroeks, D. Mink, O. May, C. Squire, H. Straatman and A. S. Wells, Org. Process Res. Dev., 2013, 17, 1117–1122 CrossRef CAS.
- K. S. Midelfort, R. Kumar, S. Han, M. J. Karmilowicz, K. McConnell, D. K. Gehlhaar, A. Mistry, J. S. Chang, M. Anderson, A. Villalobos, J. Minshull, S. Govindarajan and J. W. Wong, Protein Eng., Des. Sel., 2013, 26, 25–33 CrossRef CAS PubMed.
- A. Nobili, F. Steffen-Munsberg, H. Kohls, I. Trentin, C. Schulzke, M. Höhne and U. T. Bornscheuer, ChemCatChem, 2015, 7, 757–760 CrossRef CAS.
- A. Iwasaki, K. Matsumoto, J. Hasegawa and Y. Yasohara, Appl. Microbiol. Biotechnol., 2011, 93, 1563–1573 CrossRef PubMed.
- R. L. Hanson, B. L. Davis, Y. Chen, S. L. Goldberg, W. L. Parker, T. P. Tully, M. A. Montana and R. N. Patel, Adv. Synth. Catal., 2008, 350, 1367–1375 CrossRef CAS.
- D. Chen, Z. Wang, Y. Zhang, Z. Sun and Q. Zhu, Bioprocess Biosyst. Eng., 2007, 31, 283–289 CrossRef PubMed.
- N. Ito, S. Kawano, J. Hasegawa and Y. Yasohara, Biosci., Biotechnol., Biochem., 2014, 75, 2093–2098 CrossRef PubMed.
- H. Yun, S. Lim, B. K. Cho and B. G. Kim, Appl. Environ. Microbiol., 2004, 70, 2529–2534 CrossRef CAS PubMed.
- J. Kim, D. Kyung, H. Yun, B. K. Cho, J. H. Seo, M. Cha and B. G. Kim, Appl. Environ. Microbiol., 2007, 73, 1772–1782 CrossRef CAS PubMed.
- M. Wilding, E. F. Walsh, S. J. Dorrian and C. Scott, Microb. Biotechnol., 2015, 8, 665–672 CrossRef CAS PubMed.
- M. Wilding, T. S. Peat, J. Newman and C. Scott, Appl. Environ. Microbiol., 2016, 82, 3846–3856 CrossRef PubMed.
- Y. Lang, H. Kisaka, R. Sugiyama, K. Nomura, A. Morita, T. Watanabe, Y. Tanaka, S. Yazawa and T. Miwa, Plant J., 2009, 59, 953–961 CrossRef CAS PubMed.
- N. Weber, A. Ismail, M. Gorwa-Grauslund and M. Carlquist, BMC Biotechnol., 2014, 14, 1–6 CrossRef PubMed.
- D. Clay, D. Koszelewski, B. Grischek, J. Gross, I. Lavandera and W. Kroutil, Tetrahedron: Asymmetry, 2010, 21, 2005–2009 CrossRef CAS.
- D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell and W. Kroutil, Adv. Synth. Catal., 2008, 350, 2761–2766 CrossRef CAS.
- C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215–227 CrossRef CAS.
- D. Monti, M. C. Forchin, M. Crotti, F. Parmeggiani, F. G. Gatti, E. Brenna and S. Riva, ChemCatChem, 2015, 7, 3106–3109 CrossRef CAS.
- A. Gomm, W. Lewis, A. P. Green and E. O'Reilly, Chem. – Eur. J., 2016, 22, 1–5 CrossRef PubMed.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- M. S. Humble, K. E. Cassimjee, M. Hakansson, Y. R. Kimbung, B. Walse, V. Abedi, H. J. Federsel, P. Berglund and D. T. Logan, FEBS J., 2012, 279, 779–792 CrossRef CAS PubMed.
- F. G. Mutti and W. Kroutil, Adv. Synth. Catal., 2012, 354, 3409–3413 CrossRef CAS.
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS PubMed.
- R. D. Finn, A. Bateman, J. Clements, P. Coggill, R. Y. Eberhardt, S. R. Eddy, A. Heger, K. Hetherington, L. Holm, J. Mistry, E. L. L. Sonnhammer, J. Tate and M. Punta, Nucleic Acids Res., 2013, 1–9 Search PubMed.
- E. Park, M. Kim and J.-S. Shin, Adv. Synth. Catal., 2010, 352, 3391–3398 CrossRef CAS.
- E.-S. Park, M. Kim and J.-S. Shin, Appl. Microbiol. Biotechnol., 2011, 93, 2425–2435 CrossRef PubMed.
- B.-Y. Hwang, S.-H. Ko, H.-Y. Park, J.-H. Seo, B.-S. Lee and B.-G. Kim, J. Microbiol. Biotechnol., 2008, 18, 48–54 CAS.
- H.-S. Bea, H.-J. Park, S.-H. Lee and H. Yun, Chem. Commun., 2011, 47, 5894–5896 RSC.
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef.
- C. Sayer, R. J. Martinez-Torres, N. Richter, M. N. Isupov, H. C. Hailes, J. A. Littlechild and J. M. Ward, FEBS J., 2014, 281, 2240–2253 CrossRef CAS PubMed.
- J.-H. Seo, J.-Y. Hwang, S.-H. Seo, H. Kang, B.-Y. Hwang and B.-G. Kim, Biosci., Biotechnol., Biochem., 2014, 76, 1308–1314 CrossRef PubMed.
- I. Slabu, J. L. Galman, N. J. Weise, R. C. Lloyd and N. J. Turner, ChemCatChem, 2016, 8, 1038–1042 CrossRef CAS.
- J. L. Galman, I. Slabu, N. J. Weise, C. Iglesias, F. Parmeggiani, R. C. Lloyd and N. J. Turner, Green Chem., 2016 10.1039/c6gc02102f.
- C. U. Ingram, M. Bommer, M. E. Smith, P. A. Dalby, J. M. Ward, H. C. Hailes and G. J. Lye, Biotechnol. Bioeng., 2007, 96, 559–569 CrossRef CAS PubMed.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81, 8244–8248 CrossRef PubMed.
- S. Mathew, H. Bea, S. P. Nadarajan, T. Chung and H. Yun, J. Biotechnol., 2015, 196–197, 1–8 CrossRef CAS PubMed.
- F. Steffen-Munsberg, C. Vickers, A. Thontowi, S. Schätzle, T. Tumlirsch, M. Svedendahl Humble, H. Land, P. Berglund, U. T. Bornscheuer and M. Höhne, ChemCatChem, 2013, 5, 150–153 CrossRef CAS.
- F. Steffen-Munsberg, P. Matzel, M. A. Sowa, P. Berglund, U. T. Bornscheuer and M. Höhne, Appl. Microbiol. Biotechnol., 2016, 100, 4511–4521 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- J. M. Woodley, Curr. Opin. Chem. Biol., 2013, 17, 310–316 CrossRef CAS PubMed.
- B.-Y. Hwang and B.-G. Kim, Enzyme Microb. Technol., 2004, 34, 429–436 CrossRef CAS.
- H. Yun, B. Y. Hwang, J. H. Lee and B. G. Kim, Appl. Environ. Microbiol., 2005, 71, 4220–4224 CrossRef CAS PubMed.
- B.-K. Cho, H.-Y. Park, J.-H. Seo, J. Kim, T.-J. Kang, B.-S. Lee and B.-G. Kim, Biotechnol. Bioeng., 2008, 99, 275–284 CrossRef CAS PubMed.
- E.-S. Park, S.-R. Park, S.-W. Han, J.-Y. Dong and J.-S. Shin, Adv. Synth. Catal., 2014, 356, 212–220 CrossRef CAS.
- S.-W. Han, E.-S. Park, J.-Y. Dong and J.-S. Shin, Adv. Synth. Catal., 2015, 357, 2712–2720 CrossRef CAS.
- S.-W. Han, E.-S. Park, J.-Y. Dong, J.-S. Shin and S. J. Liu, Appl. Environ. Microbiol., 2015, 81, 6994–7002 CrossRef CAS PubMed.
- S.-W. Han, E.-S. Park, J.-Y. Dong and J.-S. Shin, Adv. Synth. Catal., 2015, 357, 1732–1740 CrossRef CAS.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016 DOI:10.1038/nchem.2578.
- F. Steffen-Munsberg, C. Vickers, A. Thontowi, S. Schätzle, T. Meinhardt, M. Svedendahl Humble, H. Land, P. Berglund, U. T. Bornscheuer and M. Höhne, ChemCatChem, 2013, 5, 154–157 CrossRef CAS.
- M. Svedendahl, C. Branneby, L. Lindberg and P. Berglund, ChemCatChem, 2010, 2, 976–980 CrossRef CAS.
- M. S. Humble, K. E. Cassimjee, V. Abedi, H.-J. Federsel and P. Berglund, ChemCatChem, 2012, 4, 1167–1172 CrossRef.
- K. E. Cassimjee, M. S. Humble, H. Land, V. Abedi and P. Berglund, Org. Biomol. Chem., 2012, 10, 5466–5470 CAS.
- L. Skalden, C. Peters, J. Dickerhoff, A. Nobili, H. J. Joosten, K. Weisz, M. Höhne and U. T. Bornscheuer, ChemBioChem, 2015, 16, 1041–1045 CrossRef CAS PubMed.
- M. S. Humble and P. Berglund, Eur. J. Org. Chem., 2011, 3391–3401 CrossRef CAS.
- M. Genz, C. Vickers, T. van den Bergh, H.-J. Joosten, M. Dörr, M. Höhne and U. Bornscheuer, Int. J. Mol. Sci., 2015, 16, 26953–26963 CrossRef CAS PubMed.
- D. Deszcz, P. Affaticati, N. Ladkau, A. Gegel, J. M. Ward, H. C. Hailes and P. A. Dalby, FEBS J., 2015, 282, 2512–2526 CrossRef CAS PubMed.
- J. Hopwood, M. D. Truppo, N. J. Turner and R. C. Lloyd, Chem. Commun., 2011, 47, 773–775 RSC.
- J. E. B. Barber, A. M. Damry, G. F. Calderini, C. J. W. Walton and R. A. Chica, Anal. Biochem., 2014, 463, 23–30 CrossRef CAS PubMed.
- S. C. Willies, J. L. Galman, I. Slabu and N. J. Turner, Philos. Trans. R. Soc., A, 2016, 374, 20150084 CrossRef PubMed.
- M. S. Weiss, I. V. Pavlidis, C. Vickers, M. Höhne and U. T. Bornscheuer, Anal. Chem., 2014, 86, 11847–11853 CrossRef CAS PubMed.
- M. Bommer and J. M. Ward, Enzyme Microb. Technol., 2013, 52, 218–225 CrossRef CAS PubMed.
- M. D. Truppo, J. D. Rozzell, J. C. Moore and N. J. Turner, Org. Biomol. Chem., 2009, 7, 395–398 CAS.
- M. D. Truppo and N. J. Turner, Org. Biomol. Chem., 2010, 8, 1280–1283 CAS.
- M. Chen, L. Rong and X. Chen, RSC Adv., 2015, 5, 103557–103562 RSC.
- T. Sehl, R. C. Simon, H. C. Hailes, J. M. Ward, U. Schell, M. Pohl and D. Rother, J. Biotechnol., 2012, 159, 188–194 CrossRef CAS PubMed.
- M. Bommer and J. M. Ward, Anal. Biochem., 2016, 493, 8–10 CrossRef CAS PubMed.
- K. E. Cassimjee, M. S. Humble, V. Miceli, C. G. Colomina and P. Berglund, ACS Catal., 2011, 1, 1051–1055 CrossRef CAS.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- D. Baud, N. Ladkau, T. S. Moody, J. M. Ward and H. C. Hailes, Chem. Commun., 2015, 51, 17225–17228 RSC.
- T. Scheidt, H. Land, M. Anderson, Y. Chen, P. Berglund, D. Yi and W.-D. Fessner, Adv. Synth. Catal., 2015, 357, 1721–1731 CrossRef CAS.
- Y. Chen, D. Yi, S. Jiang and D. Wei, Appl. Microbiol. Biotechnol., 2016, 100, 3101–3111 CrossRef CAS PubMed.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B: Enzym., 2015, 120, 141–150 CrossRef CAS.
- K. Deepankumar, M. Shon, S. P. Nadarajan, G. Shin, S. Mathew, N. Ayyadurai, B.-G. Kim, S.-H. Choi, S.-H. Lee and H. Yun, Adv. Synth. Catal., 2014, 356, 993–998 CrossRef CAS.
- S. Chen, H. Land, P. Berglund and M. S. Humble, J. Mol. Catal. B: Enzym., 2016, 124, 20–28 CrossRef CAS.
- H. Yun, J. Kim, K. Kinnera and B.-G. Kim, Biotechnol. Bioeng., 2006, 93, 391–395 CrossRef CAS PubMed.
- R. Bahulekar, N. R. Ayyangar and S. Ponrathnam, Enzyme Microb. Technol., 1991, 13, 858–868 CrossRef CAS PubMed.
- Y. J. Lu, P. J. Li, Y. L. Guo, Y. Q. Wang and G. Z. Lu, Prog. Chem., 2008, 20, 1172–1179 CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- D. Koszelewski, N. Müller, J. H. Schrittwieser, K. Faber and W. Kroutil, J. Mol. Catal. B: Enzym., 2010, 63, 39–44 CrossRef CAS.
- S. P. de Souza, I. I. Junior, G. M. A. Silva, L. S. M. Miranda, M. F. Santiago, F. Leung-Yuk Lam, A. Dawood, U. T. Bornscheuer and R. O. M. A. de Souza, RSC Adv., 2016, 6, 6665–6671 RSC.
- H. Mallin, M. Höhne and U. T. Bornscheuer, J. Biotechnol., 2014, 191, 32–37 CrossRef CAS PubMed.
- J. Yang, K. Ni, D. Wei and Y. Ren, Biotechnol. Bioprocess Eng., 2015, 20, 901–907 CrossRef CAS.
- K. Engelmark Cassimjee, M. Kadow, Y. Wikmark, M. Svedendahl Humble, M. L. Rothstein, D. M. Rothstein and J. E. Bäckvall, Chem. Commun., 2014, 50, 9134–9137 RSC.
- G. Shin, S. Mathew and H. Yun, J. Ind. Eng. Chem., 2015, 23, 128–133 CrossRef CAS.
- E.-S. Park, J.-Y. Dong and J.-S. Shin, Org. Biomol. Chem., 2013, 11, 6929–6933 CAS.
- H. Yun, B.-K. Cho and B.-G. Kim, Biotechnol. Bioeng., 2004, 87, 772–778 CrossRef CAS PubMed.
- P. Tufvesson, C. Bach and J. M. Woodley, Biotechnol. Bioeng., 2014, 111, 309–319 CrossRef CAS PubMed.
- J.-S. Shin, B.-G. Kim, A. Liese and C. Wandrey, Biotechnol. Bioeng., 2001, 73, 179–187 CrossRef CAS PubMed.
- J.-S. Shin, B.-G. Kim and D.-H. Shin, Enzyme Microb. Technol., 2001, 29, 232–239 CrossRef CAS.
- B.-K. Cho, H. J. Cho, H. Yun and B.-G. Kim, J. Mol. Catal. B: Enzym., 2003, 26, 273–285 CrossRef CAS.
- H. Yun and B.-G. Kim, Biosci., Biotechnol., Biochem., 2008, 72, 3030–3033 CrossRef CAS PubMed.
- G. Rehn, P. Adlercreutz and C. Grey, J. Biotechnol., 2014, 179, 50–55 CrossRef CAS PubMed.
- G. Rehn, B. Ayres, P. Adlercreutz and C. Grey, J. Mol. Catal. B: Enzym., 2016, 123, 1–7 CrossRef CAS.
- T. Börner, G. Rehn, C. Grey and P. Adlercreutz, Org. Process Res. Dev., 2015, 19, 793–799 CrossRef.
- H.-H. Lo, S.-K. Hsu, W.-D. Lin, N.-L. Chan and W.-H. Hsu, Biotechnol. Prog., 2005, 21, 411–415 CrossRef PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Adv. Synth. Catal., 2016, 358, 1618–1624 CrossRef.
- B. Wang, H. Land and P. Berglund, Chem. Commun., 2013, 49, 161–163 RSC.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, N. G. Turrini, J. H. Sattler, A. Lerchner, A. Skerra and W. Kroutil, Eur. J. Org. Chem., 2012, 1003–1007 CrossRef CAS.
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS.
- M. Höhne, S. Kuhl, K. Robins and U. T. Bornscheuer, ChemBioChem, 2008, 9, 363–365 CrossRef PubMed.
- D. Koszelewski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozzell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 9337–9340 CrossRef CAS PubMed.
- M. D. Truppo, N. J. Turner and J. D. Rozzell, Chem. Commun., 2009, 2127–2129 RSC.
- D. Koszelewski, M. Göritzer, D. Clay, B. Seisser and W. Kroutil, ChemCatChem, 2010, 2, 73–77 CrossRef CAS.
- N. Richter, J. E. Farnberger, D. Pressnitz, H. Lechner, F. Zepeck and W. Kroutil, Green Chem., 2015, 17, 2952–2958 RSC.
- S.-W. Han and J.-S. Shin, Biosci., Biotechnol., Biochem., 2014, 78, 1788–1790 CrossRef CAS PubMed.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- T. Esparza-Isunza, M. González-Brambila, R. Gani, J. M. Woodley and F. López-Isunza, Chem. Eng. J., 2015, 259, 221–231 CrossRef CAS.
- E.-S. Park, M. S. Malik, J.-Y. Dong and J.-S. Shin, ChemCatChem, 2013, 5, 1734–1738 CrossRef CAS.
- K. Fesko, K. Steiner, R. Breinbauer, H. Schwab, M. Schürmann and G. A. Strohmeier, J. Mol. Catal. B: Enzym., 2013, 96, 103–110 CrossRef CAS.
- P. Tufvesson, J. S. Jensen, W. Kroutil and J. M. Woodley, Biotechnol. Bioeng., 2012, 109, 2159–2162 CrossRef CAS PubMed.
- H. Yun, Y.-H. Yang, B.-K. Cho, B.-Y. Hwang and B.-G. Kim, Biotechnol. Lett., 2003, 25, 809–814 CrossRef CAS PubMed.
- L. Rios-Solis, P. Morris, C. Grant, A. O. O. Odeleye, H. C. Hailes, J. M. Ward, P. A. Dalby, F. Baganz and G. J. Lye, Chem. Eng. Sci., 2015, 122, 360–372 CrossRef CAS.
- E.-S. Park and J.-S. Shin, Appl. Environ. Microbiol., 2013, 79, 4141–4144 CrossRef CAS PubMed.
- P. A. Santacoloma, G. R. Sin, K. V. Gernaey and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 203–212 CrossRef.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- I. Oroz-Guinea and E. Garcia-Junceda, Curr. Opin. Chem. Biol., 2013, 17, 236–249 CrossRef CAS PubMed.
- R. Sigrist, B. Z. D. Costa, A. J. Marsaioli and L. G. de Oliveira, Biotechnol. Adv., 2015, 33, 394–411 CrossRef CAS PubMed.
- E.-S. Park, J.-Y. Dong and J.-S. Shin, ChemCatChem, 2013, 5, 3538–3542 CrossRef CAS.
- S. Mathew, S.-S. Jeong, T. Chung, S.-H. Lee and H. Yun, Biotechnol. J., 2016, 11, 185–190 CrossRef CAS PubMed.
- D. Koszelewski, D. Clay, D. Rozzell and W. Kroutil, Eur. J. Org. Chem., 2009, 2289–2292 CrossRef CAS.
- D. Koszelewski, D. Pressnitz, D. Clay and W. Kroutil, Org. Lett., 2009, 11, 4810–4812 CrossRef CAS PubMed.
- D. Koszelewski, B. Grischek, S. M. Glueck, W. Kroutil and K. Faber, Chem. – Eur. J., 2011, 17, 378–383 CrossRef CAS PubMed.
- G. Shin, S. Mathew, M. Shon, B. G. Kim and H. Yun, Chem. Commun., 2013, 49, 8629–8631 RSC.
- Y.-M. Seo, S. Mathew, H.-S. Bea, Y.-H. Khang, S.-H. Lee, B.-G. Kim and H. Yun, Org. Biomol. Chem., 2012, 10, 2482–2485 CAS.
- E. O'Reilly, C. Iglesias and N. J. Turner, ChemCatChem, 2014, 6, 992–995 CrossRef.
- R. C. Simon, F. Zepeck and W. Kroutil, Chem. – Eur. J., 2013, 19, 2859–2865 CrossRef CAS PubMed.
- E. O'Reilly, C. Iglesias, D. Ghislieri, J. Hopwood, J. L. Galman, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 2447–2450 CrossRef PubMed.
- S. E. Payer, J. H. Schrittwieser, B. Grischek, R. C. Simon and W. Kroutil, Adv. Synth. Catal., 2016, 358, 444–451 CrossRef CAS.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852–855 RSC.
- E.-M. Fischereder, D. Pressnitz and W. Kroutil, ACS Catal., 2016, 6, 23–30 CrossRef CAS.
- D. Koszelewski, D. Clay, K. Faber and W. Kroutil, J. Mol. Catal. B: Enzym., 2009, 60, 191–194 CrossRef CAS.
- M. Girardin, S. G. Ouellet, D. Gauvreau, J. C. Moore, G. Hughes, P. N. Devine, P. D. O'Shea and L.-C. Campeau, Org. Process Res. Dev., 2013, 17, 61–68 CrossRef CAS.
- H. Land, P. Hendil-Forssell, M. Martinelle and P. Berglund, Catal. Sci. Technol., 2016, 6, 2897–2900 CAS.
- M. Fuchs, K. Tauber, J. Sattler, H. Lechner, J. Pfeffer, W. Kroutil and K. Faber, RSC Adv., 2012, 2, 6262–6265 RSC.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- M. Pickl, M. Fuchs, S. M. Glueck and K. Faber, ChemCatChem, 2015, 7, 3121–3124 CrossRef CAS PubMed.
- K. Tauber, M. Fuchs, J. H. Sattler, J. Pitzer, D. Pressnitz, D. Koszelewski, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Chem. – Eur. J., 2013, 19, 4030–4035 CrossRef CAS PubMed.
- C. Palo-Nieto, S. Afewerki, M. Anderson, C.-W. Tai, P. Berglund and A. Córdova, ACS Catal., 2016, 6, 3932–3940 CrossRef CAS.
- A. Lerchner, S. Achatz, C. Rausch, T. Haas and A. Skerra, ChemCatChem, 2013, 5, 3374–3383 CrossRef CAS.
- M. Anderson, S. Afewerki, P. Berglund and A. Córdova, Adv. Synth. Catal., 2014, 356, 2113–2118 CrossRef CAS.
- M. Schrewe, N. Ladkau, B. Bühler and A. Schmid, Adv. Synth. Catal., 2013, 355, 1693–1697 CrossRef CAS.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC.
- H. Kohls, M. Anderson, J. Dickerhoff, K. Weisz, A. Córdova, P. Berglund, H. Brundiek, U. T. Bornscheuer and M. Höhne, Adv. Synth. Catal., 2015, 357, 1808–1814 CrossRef CAS.
- S. W. Van Arsdell, J. B. Perkins, R. R. Yocum, L. Luan, C. L. Howitt, N. Prasad Chatterjee and J. G. Pero, Biotechnol. Bioeng., 2005, 91, 75–83 CrossRef CAS PubMed.
- T. Lütke-Eversloh and G. Stephanopoulos, Metab. Eng., 2008, 10, 69–77 CrossRef PubMed.
- W.-D. Fessner, Nat. Biotechnol., 2015, 32, 658–664 CAS.
- D.-J. Kim, G.-H. Hwang, J.-N. Um and J.-Y. Cho, J. Mol. Microbiol. Biotechnol., 2015, 25, 45–50 CrossRef CAS PubMed.
- S. P. Liu, R. X. Liu, A. A. M. M. El-Rotail, Z. Y. Ding, Z. H. Gu, L. Zhang and G. Y. Shi, J. Biotechnol., 2014, 186, 91–97 CrossRef CAS PubMed.
- S. Yin, H. Zhou, X. Xiao, T. Lang, J. Liang and C. Wang, Curr. Microbiol., 2015, 70, 762–767 CrossRef CAS PubMed.
- J.-Y. Hwang, J. Park, J.-H. Seo, M. Cha, B.-K. Cho, J. Kim and B.-G. Kim, Biotechnol. Bioeng., 2009, 102, 1323–1329 CrossRef CAS PubMed.
- I. Borodina, K. R. Kildegaard, N. B. Jensen, T. H. Blicher, J. Maury, S. Sherstyk, K. Schneider, P. Lamosa, M. J. Herrgård, I. Rosenstand, F. Öberg, J. Forster and J. Nielsen, Metab. Eng., 2015, 27, 57–64 CrossRef CAS PubMed.
- Q. Wang, P. Yang, M. Xian, L. Feng, J. Wang and G. Zhao, Biotechnol. Lett., 2014, 36, 2257–2262 CrossRef CAS PubMed.
- N. Ladkau, M. Assmann, M. Schrewe, M. K. Julsing, A. Schmid and B. Buhler, Metab. Eng., 2016, 36, 1–9 CrossRef CAS PubMed.
- A. A. Desai, Angew. Chem., Int. Ed., 2011, 50, 1974–1976 CrossRef CAS PubMed.
- L. Frodsham, M. Golden, S. Hard, M. N. Kenworthy, D. J. Klauber, K. Leslie, C. Macleod, R. E. Meadows, K. R. Mulholland, J. Reilly, C. Squire, S. Tomasi, D. Watt and A. S. Wells, Org. Process Res. Dev., 2013, 17, 1123–1130 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
| This journal is © The Royal Society of Chemistry 2017 |