
A metabolite of nobiletin, 4′-demethylnobiletin and atorvastatin synergistically inhibits human colon cancer cell growth by inducing G0/G1 cell cycle arrest and apoptosis
Xian
Wu†
*a,
Mingyue
Song†
ab,
Peiju
Qiu
ac,
Fang
Li
a,
Minqi
Wang
a,
Jinkai
Zheng
ad,
Qi
Wang
a,
Fei
Xu
a and
Hang
Xiao
 *a
*a
aDepartment of Food Science, University of Massachusetts, Amherst, MA, USA. E-mail: hangxiao@foodsci.umass.edu; xianwu@alumni.umass.edu
bCollege of Food Science, South China Agricultural University, Guangzhou, China
cSchool of Pharmacy, Ocean University of China, Qingdao, Shandong, P. R. China
dInstitute of Agro-Products Processing Science and Technology, Chinese Academy of Agricultural Sciences, Beijing, China
First published on 2nd October 2017
Abstract
Combining different chemopreventive agents is a promising strategy to reduce cancer incidence and mortality due to potential synergistic interactions between these agents. Previously, we demonstrated that oral administration of nobiletin (NBT, a citrus flavonoid) at 0.05% (w/w, in diet) together with atorvastatin (ATST, a lipid-lowering drug) at 0.02% (w/w, in diet) produced much stronger inhibition against colon carcinogenesis in rats in comparison with that produced by NBT (at 0.1% w/w in diet) or ATST (at 0.04% w/w in diet) alone at higher doses. To further elucidate the mechanism of this promising synergy between NBT and ATST, herein, we measured the levels of NBT, its major metabolites and ATST in the colonic tissue of rats fed NBT (0.05% w/w, in diet) + ATST (0.02% w/w, in diet), and determined the mode of interaction between the major NBT metabolite and ATST in inhibiting colon cancer cell growth. HPLC-MS analysis showed that 4′-demethylnobiletin (4DN) is the most abundant metabolite of NBT with a level about 5-fold as high as that of NBT in the colonic tissue, which indicated the potential significance of 4DN in mediating the biological effects of NBT in the colon. We found that co-treatments of 4DN/ATST at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentration ratio produced much stronger growth inhibitory effects on human colon cancer HT-29 cells than 4DN or ATST alone, and isobologram analysis confirmed that this enhanced inhibitory effect by the 4DN/ATST combination was highly synergistic. The co-treatment of 4DN/ATST led to G0/G1 cell cycle arrest and induced extensive apoptosis in HT-29 cells. Furthermore, the 4DN/ATST co-treatment profoundly modulated key signaling proteins related to the regulation of the cell cycle and apoptosis. Our results demonstrated a strong synergy produced by the 4DN/ATST co-treatment in inhibiting colon cancer cell growth, which provided a novel mechanism by which NBT/ATST in combination synergistically inhibit colon carcinogenesis.
:1 concentration ratio produced much stronger growth inhibitory effects on human colon cancer HT-29 cells than 4DN or ATST alone, and isobologram analysis confirmed that this enhanced inhibitory effect by the 4DN/ATST combination was highly synergistic. The co-treatment of 4DN/ATST led to G0/G1 cell cycle arrest and induced extensive apoptosis in HT-29 cells. Furthermore, the 4DN/ATST co-treatment profoundly modulated key signaling proteins related to the regulation of the cell cycle and apoptosis. Our results demonstrated a strong synergy produced by the 4DN/ATST co-treatment in inhibiting colon cancer cell growth, which provided a novel mechanism by which NBT/ATST in combination synergistically inhibit colon carcinogenesis.
1 Introduction
At present, colon cancer is one of the leading causes of human death in the United States.3 Many of the current treatment modalities for colon cancer are limited due to undesirable side effects and complications associated with their long-term use. Thus, this underscores the need for novel strategies to prevent and treat colon cancer. Recently, combination regimens of using food bioactive components with pharmacological agents have gained growing attention. Accumulating evidence suggests that the combination of different chemopreventive agents that have complementary mechanisms of action may produce a synergistic interaction, thus an enhanced inhibition against carcinogenesis. Moreover, the enhanced efficacy can reduce the dosage required for each agent in the combination, which may lower potential adverse effects associated with long-term high-dose administration of a certain agent.4–6Polymethoxyflavones (PMFs) are a unique group of flavonoids found in citrus fruit. There have been more than 20 PMFs being isolated and identified from citrus fruit, such as sinensetin (5,6,7,3′,4′-pentamethoxyflavone), nobiletin (5,6,7,8,3′,4′-hexamethoxyflavone, NBT), and tangeretin (5,6,7,8,4′-pentamethoxyflavone). PMFs are the secondary compounds located in the peel, especially in the flavedo that act as antifungal agents.7 NBT has been shown to be a potent anti-carcinogenic agent against different cancers in animal models, including colon,8–11 prostate12 and lung13 cancers. Previously, we found that oral administration of NBT in combination with ATST synergistically suppressed colon carcinogenesis in azoxymethane (AOM)-treated rats via modulating multiple pathways associated with cell cycle progress, apoptosis, inflammation, angiogenesis and metastasis in colon cancer.11
ATST (commercially sold as Lipitor® or Torvast®) is an important member of statin drugs that are mainly used to lower blood cholesterol and prevent cardiovascular disease. Besides their lipid-lowering effects, statins have been reported to reduce the risk of colon cancer in observational, preclinical and clinical studies.14 The chemopreventive property of statins is mainly due to their inhibitory activity on HMG-CoA reductase, which thereby reduces the levels of mevalonate and its associated products. It in turn inhibits the membrane localization and activation of some small G-proteins, such as RhoA and RhoC, and suppresses the epidermal growth factor-induced invasion of cancer cells. RhoA and RhoC have been found to be overexpressed in many human cancers, and they are related to uncontrolled cell cycle regulation and increased immigration and metastasis of cancer cells.4
Biotransformation plays essential roles in the bioactivities of dietary compounds. The metabolites generated after biotransformation in the body may have different biochemical and pharmacological properties. Oral administration of a dietary compound may result in higher levels of metabolites than the parent compound in certain tissues. NBT undergoes extensive biotransformation leading to its structural modifications after oral consumption.9,11,15 We previously reported that NBT was transformed into its demethylated metabolites such as 4′-demethylnobiletin (4DN), 3′-demethylnobiletin (3DN) and 3′,4′-didemethylnobiletin (34DN) in the colon of mice after the long-term feeding of NBT.9 Interestingly, it was found that 4DN was the most abundant colonic metabolite of NBT. The colonic level of 4DN was about 12-fold higher than that of NBT and 2-fold higher than that of 34DN, the second most abundant metabolite. More importantly, 4DN exhibited stronger bioactivities than NBT, such as anti-cancer and anti-inflammatory effects.9,16,17 Under this circumstance, 4DN, rather than the ingested parent compound NBT, may exert more important roles in the anti-carcinogenic activities of NBT in the colon.
Previously, the synergistic colon cancer preventive effects of NBT and ATST in combination observed in an animal model were ascribed to NBT itself.11 There is no detailed information available about the role of the biotransformation of NBT in the chemopreventive effect of the NBT/ATST co-treatment in the colon. To better understand the possible mechanisms involved in the synergistic interaction between NBT and ATST, in this study, we determined the colonic levels of ATST, NBT, 4DN, 3DN and 34DN in the rats given NBT/ATST co-treatment. Moreover, we determined the growth inhibitory effects of the combination of 4DN and ATST in HT-29 human colon cancer cells, and elucidated the underlying molecular mechanisms.
2 Materials and methods
2.1 Animals, diets, and experimental procedure
NBT and ATST were purchased from Bepharm Ltd (Shanghai, China) with a purity greater than 98%. 4DN, 3DN and 34DN were chemically synthesized as we previously described.18,19 The chemical structures of ATST, NBT and its metabolites are illustrated in Fig. 1. The protocol (#2011-0066) for the animal experiment was approved by the Institutional Animal Care and Use Committee of the University of Massachusetts. | ||
| Fig. 1 Chemical structures of ATST, NBT, 4DN, 3DN and 34DN. | ||
Twelve male F344 rats (5 weeks of age) were obtained from Charles River Laboratories (Wilmington, MA, USA). Upon arrival, the rats were kept in a temperature-controlled animal room (23 °C), with humidity at 65–70% under a day/night cycle (12 h light, 12 h dark) with free access to water and a modified high-fat AIN-76A diet20 for one week for acclimation. Rats were then randomly divided into two groups (6 animals each). The rats in the control group were fed a basal diet (control group), while the NBT/ATST-treated group was fed with a modified AIN-76A diet containing 0.05% NBT plus 0.02% ATST (w/w, in diet) (combination group). Both groups were allowed to eat and drink ad libitum during the whole experiment period. After ten weeks of treatment, all rats were sacrificed by CO2 asphyxiation. Colonic mucosa was collected and stored at −80 °C for LC-MS analysis.
2.2 Determination of colonic levels of ATST, NBT and NBT metabolites
Colonic mucosa samples from the rats fed with a basal diet (control group) and a basal diet containing NBT/ATST (combination group) were homogenized with 50% methanol using a Bead Ruptor homogenizer (Omni International, Kennesaw, GA, USA). Samples were then extracted with an equal volume of ethyl acetate three times. The combined ethyl acetate extracts were dried under vacuum, and then dissolved in 50% methanol for LC-MS (Model 2020, Shimadzu, Kyoto, Japan) analysis. The identification and quantification of ATST, NBT and NBT metabolites (4DN, 3DN and 34DN) were carried out using a previously published method.21 ATST, NBT, 4DN, 3DN and 34DN, with a purity greater than 98%, were used as external standards to identify and quantify their levels in the colonic mucosa of the rats.2.3 Cell cultures and treatments
The human colorectal cancer cell line HT-29 was obtained from American Type Cell Collection (ATCC, Manassas, VA, USA). HT-29 cells were maintained in RPMI 1640 media (Mediatech, Herndon, VA, USA) supplemented with 5% heat-inactivated fetal bovine serum and 100 U mL−1 of penicillin and 0.1 mg mL−1 of streptomycin at 37 °C with 5% CO2 and 95% air. The cells were maintained subconfluent and the media were changed every 3–4 days. All cells used in experiments were between 4 and 25 passages. Dimethylsulfoxide (DMSO) was used as a vehicle to deliver 4DN and ATST to the cells. The final concentration of DMSO in all experiments was 0.1% v/v in cell culture media.2.4 Cell viability assay and analyses of synergy
HT-29 (2000 cells per well) cells were seeded in 96-well plates. After 24 h, the cells were treated with serial concentrations of 4DN and ATST and their half dose combinations in 200 μL of serum complete media. After 72 h, the cells were subjected to the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The media were replaced by 100 μL of fresh media containing 0.5 mg mL−1 of MTT (Sigma-Aldrich, St Louis, MO, USA). After 1 h incubation at 37 °C, MTT-containing media were removed and the reduced formazan dye was solubilized by adding 100 μL of DMSO to each well. After gentle mixing, the absorbance was monitored at 570 nm using a plate reader (Elx800TM absorbance microplate reader, BioTek Instrument, Winooski, VT).The analyses were based on Chou and Talalay's empirical method1 with modifications as described previously.22–24 It was assumed that the dose response model follows log[E/(1 − E)] = α(logd − logDm), which is a linear regression model with the response log[E/(1 − E)] and the regressor log(d). This model is used for 4DN and ATST and the co-treatment of 4DN/ATST with the fixed ratio of the doses of two agents. E is the fraction of cells survived, d is the dose applied, Dm is the median effective dose of a drug (IC50), and α is a slope parameter. On the basis of the regression model, median effect plots were constructed using the data from the MTT assay. Suppose that the combination (d1, d2) elicits the same effect x as 4DN alone at the dose level Dx,1, and ATST alone at the dose level Dx,2, then the interaction index of 4DN/ATST = (d1/Dx,1) + (d2/Dx,2) (Dx,1 and Dx,2 were calculated from median effect models). The combination index was used to determine the additivity, antagonism, and synergy of the combination treatment of 4DN and ATST at different dose pairs depending on the combination index = 1, >1, and <1, respectively. The delta method2 was used to calculate the variance of the interaction index, which is given by var(combination index) = var(Dx,1)(d12/Dx,14) + var(Dx,2)(d22/Dx,24). Data were further analyzed by using R program software.
2.5 Cell cycle analysis
HT-29 (7 × 104 cells per well) cells were seeded in 6-well plates. After 24 h of incubation for cell attachment, the cells were treated with serial concentrations of 4DN, ATST, and their half dose co-treatment in 2 mL of serum complete media. After 24 h of the treatment, media containing any floating cells were harvested and combined with adherent cells that were detached by brief trypsinization (0.25% trypsin-EDTA; Mediatech). Cell pellets were washed with 1 mL of ice-cold PBS and then resuspended in 1 mL of 70% ethanol at −20 °C overnight. After centrifugation (1600g, 1 min), the supernatant was removed and the cells were incubated with 0.5 mL of PBS containing 50 mg RNAse (Sigma-Aldrich) and 5 mg propidium iodine (PI, Sigma-Aldrich) for 30 min at room temperature. Single-cell suspension was generated by gentle pipetting. The cell cycle was analyzed using a BD LSR II cell analyzer at the analytical cytometry facility (University of Massachusetts Amherst), and data were processed using Modifit software.2.6 Detection of apoptosis
HT29 cells were seeded and treated exactly the same as in the cell cycle analysis described above. Apoptotic cells were quantified by Annexin V/PI double staining assay. Annexin V/PI staining was performed using an apoptotic detection kit (BioVision, Mountain View, CA, USA) following the manufacturer's instruction. After 48 h of the treatment, the cells were gently detached by brief trypsinization (any floating cells were also collected), and then washed with ice cold PBS. After another wash with binding buffer, the cells were suspended in 300 mL binding buffer containing annexin V and PI, and incubated for 5 min at room temperature. Early apoptotic cells were identified as annexin V-positive/PI-negative cells, while late apoptotic/necrotic cells were identified as annexin V-positive/PI-positive cells using a BD LSR II cell analyzer.2.7 Immunoblot analysis
Whole-cell lysates were prepared as previously described.22,25 For immunoblot analysis, equal amounts of proteins (50 μg) were resolved over 12% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes containing the transferred protein were blocked in blocking buffer (5% nonfat dry milk, 1% Tween-20 in 20 mM Tris-buffered saline, pH 7.6) for 2 h at room temperature, and then incubated with the appropriate monoclonal primary antibody in blocking buffer overnight at 4 °C. After incubation with appropriate secondary antibodies, the membranes were washed three times with Tris buffer with Tween-20, and then visualized using enhanced chemiluminescence (Boston Bio Products, Ashland, MA, USA). Antibodies for CDK4, cleaved caspase 3 (Asp175), p53 and p21Cip1/Waf1 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody for β-actin was from Sigma-Aldrich.2.8 Statistical analysis
All data were presented as mean ± standard deviation (SD) or standard error (SE). Student's t test was used to test the mean difference between two groups. An analysis of variance (ANOVA) model was used for the comparison of differences among more than two groups. A P-value <0.05 was considered statistically significant. The effect of the interaction between 4DN and ATST (synergistic, additive, or antagonistic) on cell cycle distribution and apoptosis in HT29 cells was analyzed based on the “simple designs and model free tests for synergy” by Laska et al.263 Results
3.1 4DN was a major metabolite of NBT in the colonic tissue after oral administration of NBT/ATST in rats
In this study, we fed the F344 rats for ten weeks with the same doses of NBT/ATST as we used in the previous rat study (0.05% NBT + 0.02% ATST w/w in diet)11 to generate colonic mucosa samples for LC-MS analysis. We found that 4DN, 3DN and 34DN were present in the colonic mucosa of the rats fed with NBT/ATST. It was observed that ATST, NBT and three colonic metabolites of NBT showed identical molecular weights and retention times as chemical standards of these compounds. As shown in Fig. 2, the concentrations of ATST, NBT, 4DN, 3DN and 34DN in the rat colonic mucosa were 1.48 ± 0.34, 0.59 ± 0.13, 2.95 ± 0.38, 1.82 ± 0.55, and 2.06 ± 0.89 nmol g−1 of tissue, respectively. It is noteworthy that the level of 4DN was the most abundant among three major NBT metabolites, and was 4.95-fold as high as that of NBT. The high abundance of 4DN in the colonic tissue may lead to a significant role of 4DN in the anti-carcinogenic effects of NBT/ATST combination in rats.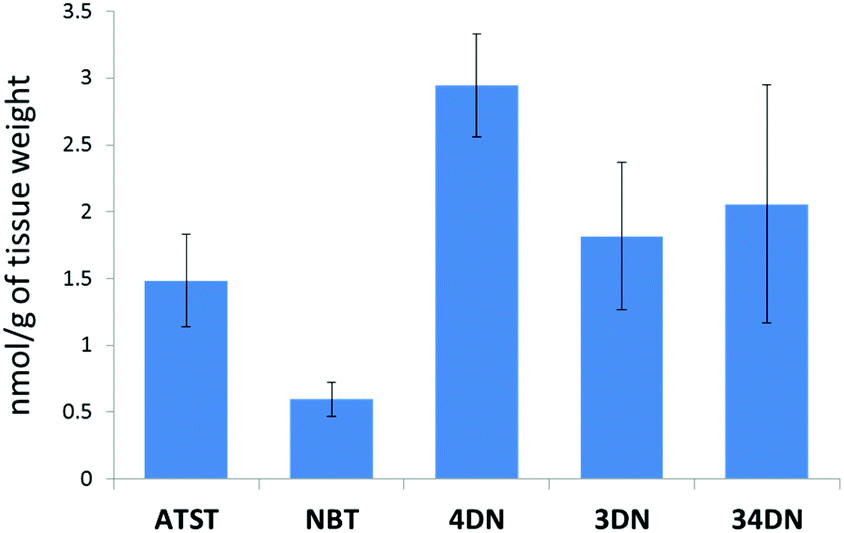 | ||
| Fig. 2 Quantification of ATST, NBT, 4DN, 3DN and 34DN in the colonic mucosa of rats given the NBT/ATST co-treatment by LC/ESI-MS analysis. Data were shown as mean ± SE (n = 6). | ||
3.2 4DN/ATST co-treatment synergistically inhibited the growth of human colon cancer cells
To establish the potential significance of 4DN in mediating synergistic inhibition in colon carcinogenesis in rats by NBT/ATST combination as we described previously,11 the inhibitory effects of 4DN, ATST and 4DN/ATST combination on human colon cancer HT29 cells were determined. HT-29 human colon cancer cells were treated with the serial concentrations of 4DN (at 7.2, 14.4, 21.6, 28.8, or 36 μM), ATST (at 3.6, 7.2, 10.8, 14.4, or 18 μM), and 4DN/ATST combinations at a 2:1 ratio (3.6/1.8, 7.2/3.6, 10.8/5.4, 14.4/7.2, or 18/9 μM) for 72 h. The 2:1 ratio between 4DN and ATST used in the cell culture experiment was based on the fact that the colonic level of 4DN was about 2-fold that of ATST. As shown in Fig. 3, single treatment with 4DN or ATST resulted in dose-dependent growth inhibition in HT-29 cells. However, at the highest concentration tested (36 μM), 4DN only modestly inhibited cell growth by 25.89%. Similarly, at the highest concentration tested (18 μM), the ATST treatment showed a moderate 20.89% growth inhibition in HT-29 cells. In contrast, the half dose combinations of 4DN and ATST led to more potent growth inhibitory effects on cancer cells than the individual treatment with NBT or ATST at their full doses. For example, a combination of 14.4 μM 4DN with 7.2 μM ATST caused a 53.84% inhibition, while 28.8 μM 4DN alone and 14.4 μM ATST alone led to only 11.65 and 11.15% inhibition, respectively. Similarly, 18 μM 4DN combined with 9 μM ATST caused a 68.89% inhibition, while 36 μM 4DN alone and 18 μM ATST alone led to only 25.89 and 20.89% inhibition, respectively. It is noteworthy that even 10.8 μM 4DN plus 5.4 μM ATST co-treatment produced slightly stronger inhibition than that produced by 36 μM 4DN alone or 18 μM ATST alone. These results demonstrated that 4DN/ATST combination led to much stronger inhibitory effects on human colon cancer cells in comparison with 4DN or ATST alone.
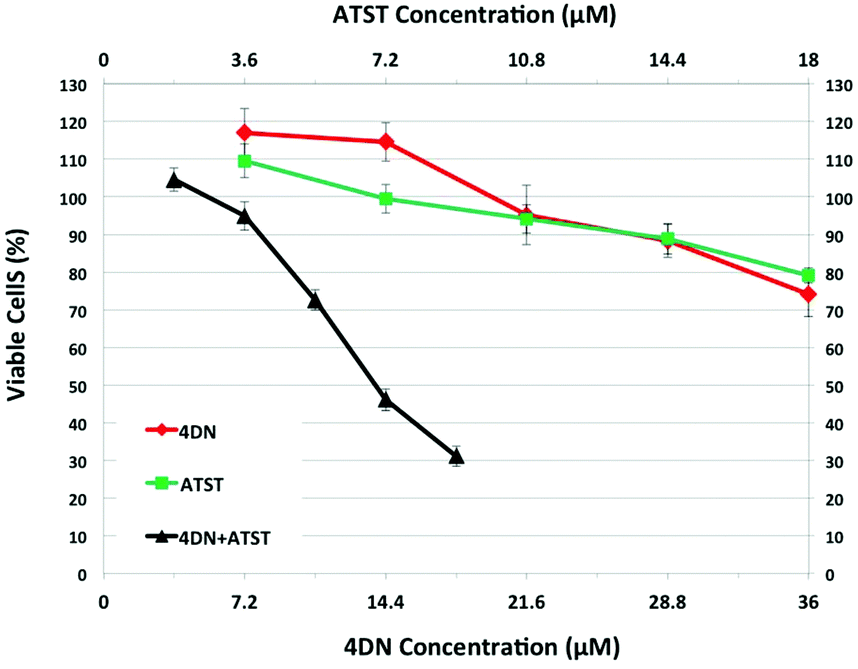 | ||
| Fig. 3 Growth inhibitory effects of 4DN, ATST and their half dose combination on HT-29 human colon cancer cells. HT-29 cells were treated with serial concentrations of 4DN, ATST, and their combinations for 72 h. Viable cells were measured by a cell viability assay as described in Materials and methods. The half-dose combination produced much stronger growth inhibition on HT-29 cells than those caused by the full-dose individual treatments of 4DN and ATST. Data were shown as mean ± SD (n = 6), and viable cells of controls were used as 100%. | ||
To determine whether the enhanced inhibitory effects on cancer cells from 4DN/ATST co-treatments were statistically synergistic, we next performed an isobologram analysis as we described previously.22–24 As shown in Fig. 4A, the median effect plot verified that the linear regression model used in the isobologram analysis was well-fitted to the dose–response relationship of 4DN, ATST and their combinations within the concentration ranges exploited herein. The interaction index was used to determine the additivity, synergy, or antagonism of the combination at different dose pairs depending on the interaction index = 1, <1, or >1, respectively. An interaction index between 0.3 and 0.8 indicates moderate synergism, whereas an interaction index smaller than 0.3 indicates a strong synergism between the two tested compounds. The interaction index of each co-treatment dose pair of 4DN/ATST was determined on the basis of the median effect plot. As shown in Fig. 4B, all of the 4DN/ATST co-treatment dose pairs resulted in interaction indexes smaller than 0.7, suggesting synergistic interactions between 4DN and ATST in suppressing HT-29 cell growth within the concentration ranges tested. Furthermore, the interaction indexes of 4DN/ATST combination treatments at the two higher dosed pairs (14.4 μM 4DN + 7.2 μM ATST, and 18 μM 4DN + 9 μM ATST) were 0.22 and 0.14, respectively, suggesting a strong synergistic effect between 4DN and ATST. Altogether, our results demonstrated a significant synergistic effect between 4DN and ATST in inhibiting the growth of human colon cancer cells.
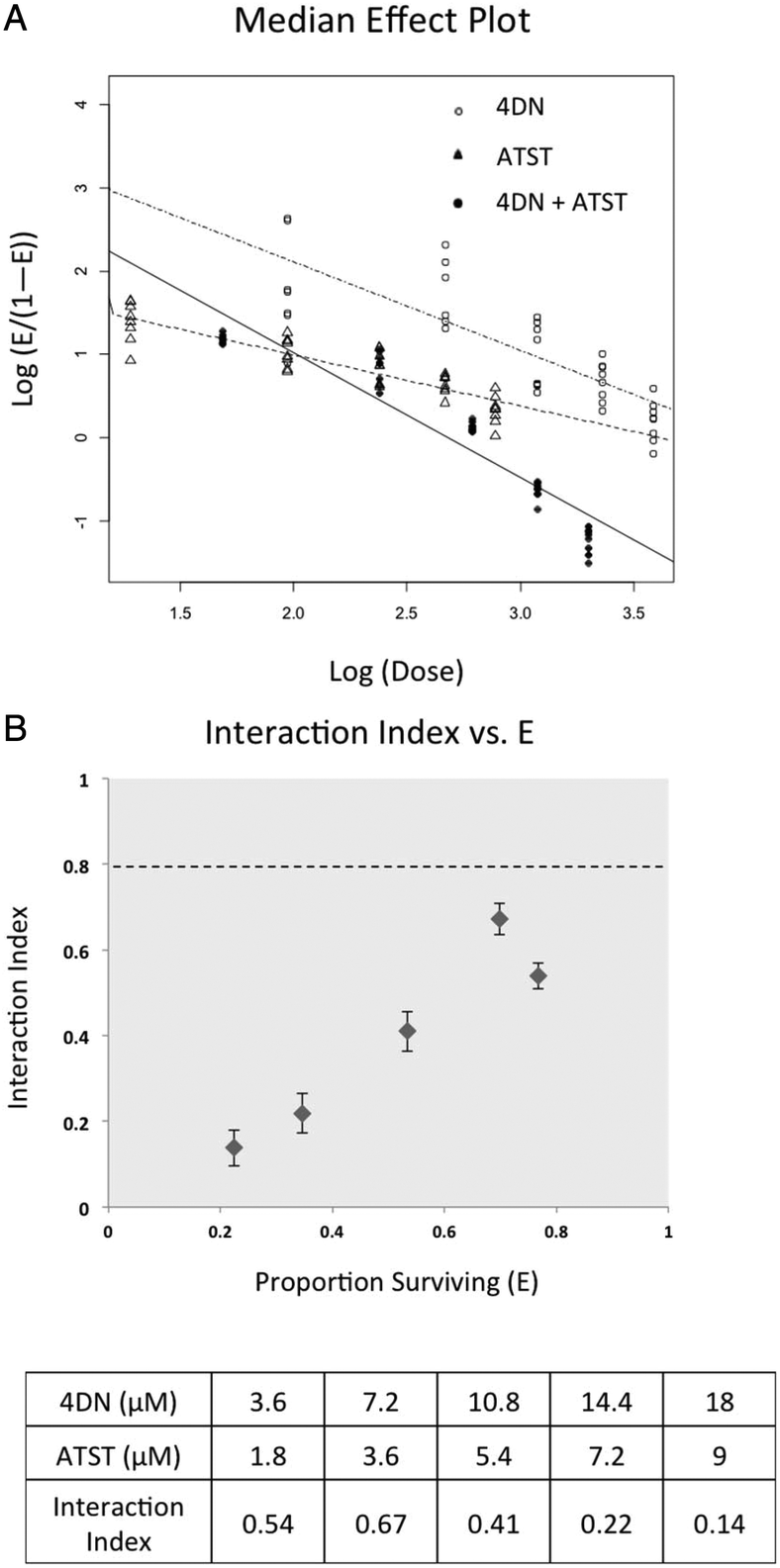 | ||
| Fig. 4 Median effect (A) and interaction index (B) of 4DN, ATST, and their combination in HT-29 human colon cancer cells. Cells were treated with 4DN or ATST alone or in combination at serial concentrations for 72 h in 96-well plates. Cell viability was determined by the MTT assay as described in Materials and methods. The median effect plot and interaction index plot were constructed by using modified Chou and Talalay's method1 as described in Materials and methods. The synergistic effect of two agents was defined as an interaction index lower than 1.0. Error bars in interaction index plots represented 95% confidence interval and were calculated using a delta method.2 | ||
3.3 4DN/ATST co-treatment synergistically induced G0/G1 cell cycle arrest in human colon cancer cells
Uncontrolled and dysregulated cell proliferation is one of the most common features of cancer. To characterize the molecular mechanism underlying the synergistic anti-cancer effects of 4DN and ATST, we determined their effects on cell cycle progression by flow cytometric analysis. As shown in Fig. 5, individual treatment with 4DN (36 μM) or ATST (18 μM) for 24 h significantly increased HT-29 cell accumulation in the G0/G1 phase by 23.1 and 12.7%, respectively, compared to the untreated control cells. However, their half dose co-treatment (4DN at 18 μM + ATST at 9 μM) resulted in 29.5%, in comparison with the control cells, which was stronger than the effects of 4DN or ATST alone at their full doses. Using “simple designs and model free tests for synergy” established by Laska et al.,26 we analysed the cell cycle data and it was confirmed that the stronger effects produced by the 4DN/ATST co-treatment was synergistic in comparison with those of 4DN or ATST alone. This finding suggested that the induction of cell cycle arrest might contribute to the enhanced growth inhibitory effects of the 4DN/ATST co-treatment on cancer cells.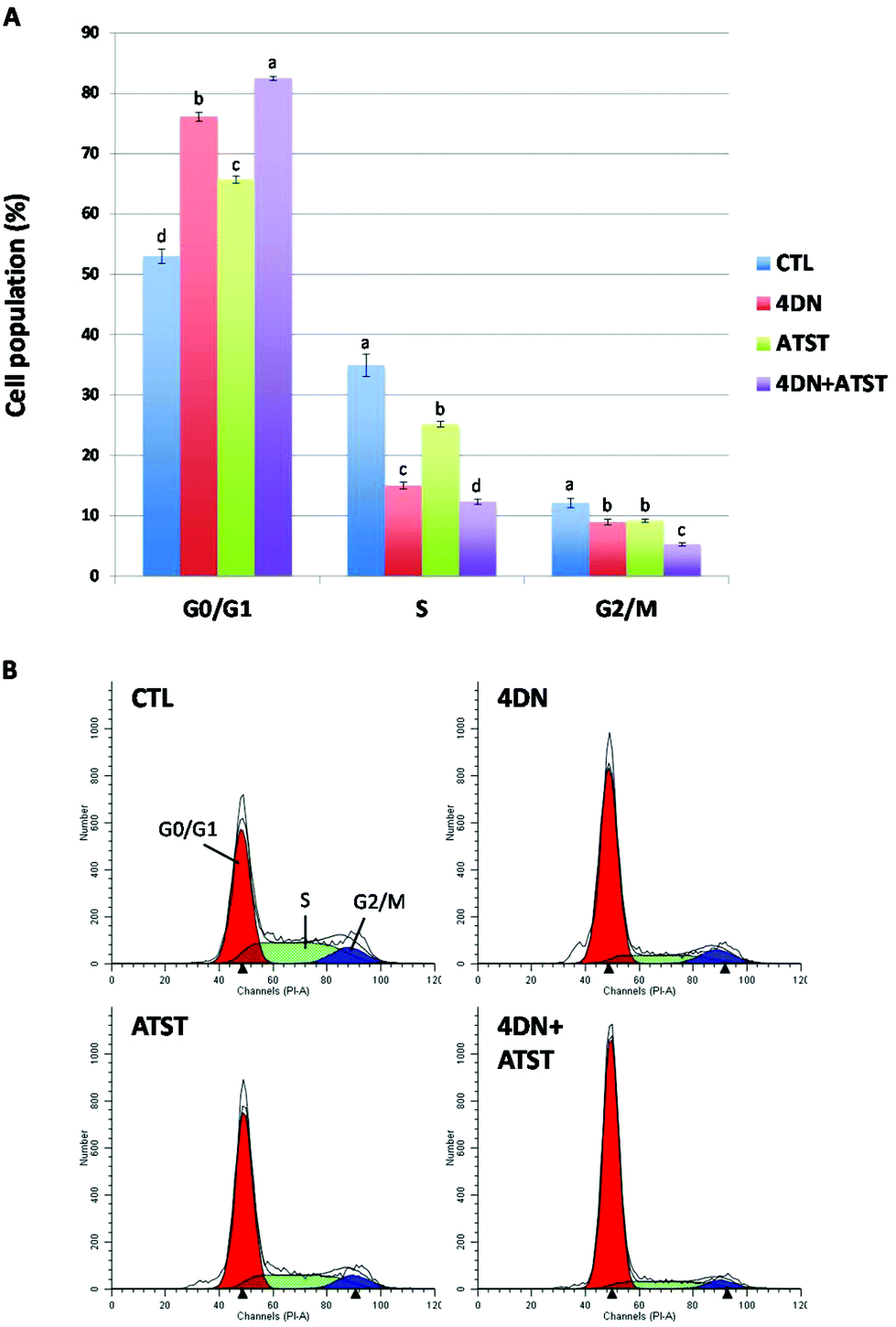 | ||
| Fig. 5 (A) Effects of 4DN (36 μM), ATST (18 μM), and their half dose combination (4DN at 18 μM + ATST at 9 μM) on cell cycle progression of HT-29 human colon cancer cells. Cells were seeded in 6-well plates for 24 h, and then treated with 4DN, ATST, and their half dose combination. After 24 h of treatments, the cells were harvested and subjected to cell cycle analysis, respectively. Data are shown as mean ± SD. Different notations in the bar charts indicate statistical significance (p < 0.05, n = 3) by ANOVA. (B) DNA histogram of cell cycle distribution of HT-29 cells. | ||
3.4 4DN/ATST co-treatment synergistically triggered cellular apoptosis in human colon cancer cells
Similar to abnormal cell cycle progression, evasion of apoptosis is another common feature of cancer. Thus, the induction of apoptosis in cancer cells is an essential approach to prevent and treat cancer.27 Annexin-V/propidium iodine double staining assay was used to determine the pro-apoptotic activities of the 4DN/ATST co-treatment. As shown in Fig. 6, after 48 h of the treatment, 4DN at 36 μM did not cause a significant change in the apoptotic cell population, compared to the control cells. ATST at 18 μM led to a 2.3-fold increase in the late apoptotic cell population, while it showed no effect on early apoptosis. On the other hand, the half dose co-treatment of 4DN/ATST (4DN at 18 μM + ATST at 9 μM) exerted a much stronger induction of early apoptosis of HT-29 cells. The co-treatment greatly increased the early apoptotic cell population by 7.9-fold, in comparison with the control cells. Although the 4DN/ATST co-treatment did not induce the late apoptosis of HT-29 cells, compared to the control cells, it elevated the total apoptotic cell population by 4.1-fold. Based on the statistical method established by Laska et al.,26 the apoptosis-inducing efficacy by the 4DN/ATST co-treatment was verified to be synergistic. Therefore, these findings indicated that the growth inhibitory effects of the 4DN/ATST co-treatment in human colon cancer HT-29 cells were associated with its ability to trigger cellular apoptosis synergistically.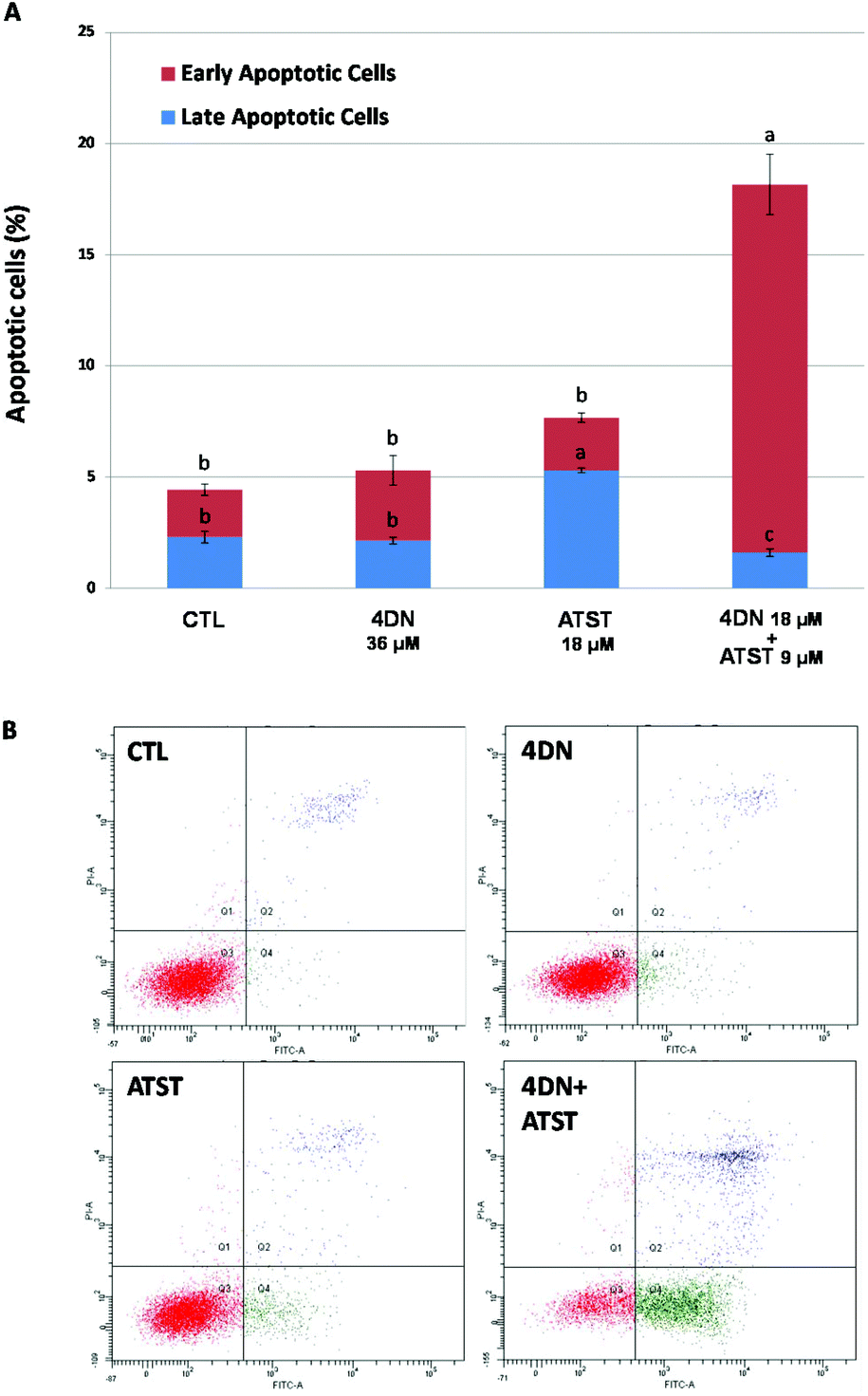 | ||
| Fig. 6 (A) Effects of 4DN (36 μM), ATST (18 μM), and their half dose combination (4DN at 18 μM + ATST at 9 μM) on apoptosis of HT-29 human colon cancer cells. Cells were seeded in 6-well plates for 24 h, and then treated with 4DN, ATST, and their half dose combination. After 48 h of treatments, the cells were harvested and subjected to apoptosis analysis. Data are shown as mean ± SD. Different notations in the bar charts indicate statistical significance (p < 0.05, n = 3) by ANOVA. (B) Dot plots of apoptotic cells of HT-29 cells. | ||
3.5 4DN/ATST co-treatment modulated the expression levels of key proteins related to cell cycle progression and apoptosis
To further understand the molecular mechanisms involved in the synergistic chemopreventive activities of the 4DN/ATST co-treatment, western blotting analysis was performed to evaluate the effects of this combination on the expression levels of some key signalling proteins related to the cell cycle and apoptosis in HT-29 cells. As shown in Fig. 7, 4DN at 36 μM enhanced the protein levels of p21 by 1.53-fold and showed no effect on CDK4. ATST at 18 μM even decreased the level of p21 and did not significantly change the level of CDK4. In contrast, the 4DN/ATST co-treatment (4DN at 18 μM + ATST at 9 μM) showed stronger effects on p21 and CDK4 than 4DN or ATST alone. It resulted in a 1.84-fold increase in the p21 level and 32% decrease in the CDK4 level, compared to the control cells. All treatment groups increased the expression levels of tumor suppressor p53 and cleaved caspase-3. However, the half dose co-treatment of 4DN/ATST led to a much stronger increase than 4DN or ATST alone at full dose. It significantly elevated the protein levels of p53 and cleaved caspase-3 by 3.97- and 6.71-fold, respectively. This may explain why the co-treatment showed greater apoptosis-inducing and cell cycle arresting effects on colon cancer cells than 4DN or ATST alone.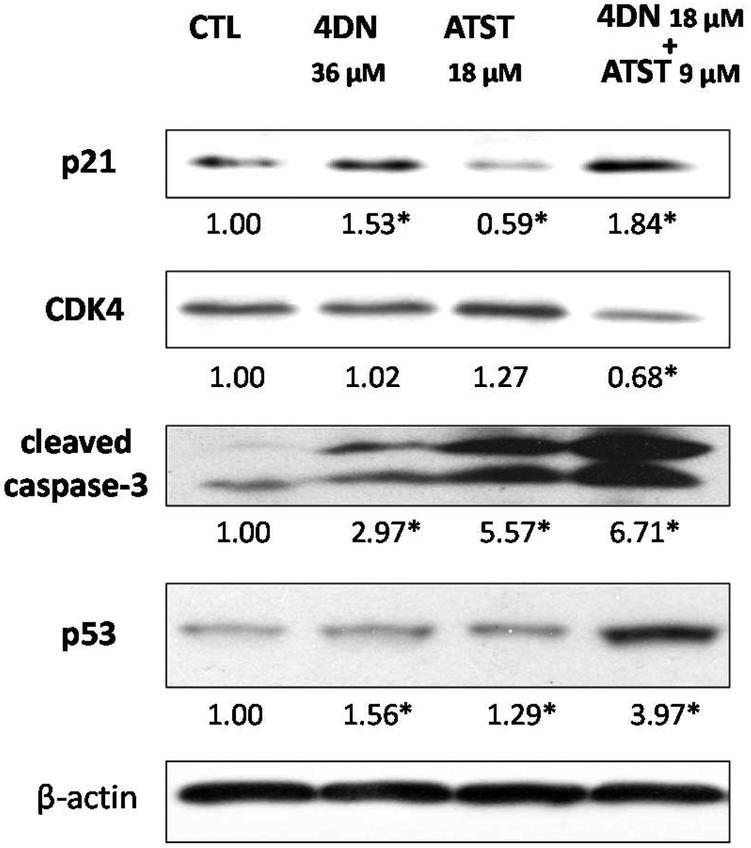 | ||
| Fig. 7 Effects of 4DN (36 μM), ATST (18 μM), and their half dose combination (4DN at 18 μM + ATST at 9 μM) on the cell cycle and apoptosis-related signaling proteins in HT-29 human colon cancer cells. HT-29 cells were seeded in 10 cm dishes for 24 h, and then the cells were treated with 4DN, ATST, and their half dose combination. After another 24 or 48 h of incubation, the cells were collected for western blotting as described. The numbers underneath the blots represent band intensity (normalized to β-actin, means of three independent experiments) measured by Image J software. The SDs (all within ± 20% of the means) were not shown. β-Actin served as an equal loading control. * indicates statistical significance in comparison with the control (p < 0.05, n = 3). | ||
4 Discussion
Combination regimens of multiple chemopreventive agents that show a minimal risk of side effects during long-term treatment are of great interest to all in search of a solution to reduce the incidence and mortality of cancer. Numerous dietary bioactive compounds have been found to have cancer preventive potential but cause little or no adverse effects. A growing number of studies have suggested that combining chemopreventive dietary components with commonly used pharmaceutical drugs is a promising strategy for cancer prevention and treatment.4,5 Previously, we have demonstrated the potential synergistic anti-cancer activities of NBT and ATST combination in colon carcinogenesis both in vitro and in vivo.11 Accumulating evidence has indicated that biotransformation dictates the biological fate and functions of PMFs including NBT.9,18,28,29 We previously reported that 4DN was the most abundant metabolite of NBT in mouse colonic mucosa after the long-term oral administration of NBT, and its colonic concentration was much higher than that of NBT.9 Therefore, 4DN might play important roles in the biological activities of orally administered NBT in the colon. Based on our previous finding that the oral administration of NBT and ATST resulted in a strong synergy in inhibiting colon carcinogenesis in rats,11 we hypothesize that as a major NBT metabolite, 4DN may synergistically interact with ATST in the colon to produce enhanced anti-cancer effects. If this hypothesis is true, it will provide a novel mechanism for the synergy between NBT and ATST in inhibiting colon carcinogenesis.In this study, we fed rats with identical doses of NBT and ATST to those we used in the previous study11 to generate colonic mucosa samples for LC-MS analysis to determine the levels of ATST, NBT, and major metabolites of NBT. Our results confirmed that 4DN was the major metabolite of NBT in the rat colon, and its level in colonic mucosa was 4.95-fold as high as that of NBT. This finding supported the hypothesis that 4DN may significantly contribute to the synergistic anti-carcinogenic effects of the NBT/ATST co-treatment. To test our hypothesis, herein, the inhibitory effects of the combination of 4DN and ATST were determined in human colon cancer HT-29 cells, and the underlying mechanisms were elucidated. It is noteworthy that the colonic level of ATST in the rats fed with 4DN and ATST was about half of that of 4DN; therefore, the concentrations of 4DN and ATST used in the cell culture study were in the ratio of 2:1.
Using a cell viability assay, we found that the half dose combinations of 4DN/ATST produced enhanced growth inhibitory effects on HT-29 cells in comparison with 4DN or ATST alone at their full doses. Moreover, the isobologram analysis verified that the enhanced bioactivity by the combination of 4DN and ATST was synergistic. All 4DN/ATST combination dose pairs resulted in interaction indexes lower than 0.7, and the two highest dose pairs tested even led to the interaction indexes lower than 0.3, indicating a strong synergy. This result demonstrated that there was a significant synergism between 4DN and ATST in inhibiting cancer cell growth. Considering the relatively high abundance of 4DN available in rat colon and the potent synergistic interactions produced by 4DN in combination with ATST in the cell culture, it is reasonable to state that the in vivo synergistic chemopreventive effects by NBT/ATST combination observed in the AOM-treated rats might be strongly dependent on the synergistic interactions between 4DN and ATST.
Uncontrollable cell cycle progression and evasion of programed cell death (apoptosis) are common hallmarks of colon cancer. These molecular events leading to colon cancer are important targets for chemopreventive agents.30 To further reveal the underlying molecular mechanisms, we utilized flow cytometric and western blotting analyses to determine the effects of the 4DN/ATST co-treatment on the cell cycle progression and apoptosis of HT-29 cells. Our results showed that the 4DN/ATST half dose co-treatment markedly arrested HT-29 cell division in the G0/G1 phase during cell cycle progression, and this effect was significantly stronger than those produced by 4DN or ATST alone at their full doses. Additionally, we investigated the effects of 4DN/ATST on two key proteins associated with the cell cycle: p21 and CDK4. Critical signaling proteins related to cell cycle control are often deregulated during the promotion stage of carcinogenesis. Cyclin–cyclin-dependent kinase (CDK) complexes and checkpoints tightly regulate the cell cycle progression. P21 is an essential CDK inhibitor that can bind to multiple cyclin–CDK complexes, inhibit their activities, and in turn arrest the cell cycle in the G1 and S phase. In addition to the inhibition of Ras farnesylation in cancer cells, it has been suggested that the anti-proliferative effects of statins are also the consequence of p21 and p27 (another critical CDK inhibitor) stabilization.31 The transition of the cell cycle from the G1 phase to the S phase is precisely controlled by cyclinD–CDK4/6 complexes. Increased levels of p21 can lead to the inactivation and/or degradation of these complexes, thus resulting in G0/G1 phase cell cycle arrest.32,33 As a major regulator of G1 phase progression, CDK4 is also an important target for anti-cancer drug research. Palbociclib, a recently FDA approved drug for the treatment of breast cancer, is a selective inhibitor of CDK4.34 Our results suggested that both the individual treatment with 4DN and the co-treatment of 4DN/ATST increased the expression level of p21 by 1.53- and 1.84-fold, respectively, while only the co-treatment significantly decreased the levels of CDK4 in colon cancer cells. These results were consistent with the results from cell cycle analysis.
Our results showed that the prolonged co-treatment with 4DN/ATST greatly elevated the levels of cellular apoptosis of colon cancer cells. In contrast, the 4DN treatment alone did not cause any significant change in the apoptotic cell population, whereas the ATST treatment alone only resulted in a modest induction in late apoptotic cells. We determined their effects on important proteins that have been found to play critical roles in the regulation of apoptosis, which are tumor suppressor p53 and cleaved caspase-3. p53 is one of the most potent suppressors of various types of cancer. Loss of p53 function can result in evasion of apoptosis, genomic instability, and promotion of angiogenesis. The expression of p21 is also mediated by p53, which can trigger G0/G1 phase cell cycle arrest in response to a range of stimuli.33,35 Down-regulation of caspase-3 has been found in cancer patients and animal models. Caspase-3 is a member of the caspase protein family. Activation (cleavage) of caspase-3 and its downstream target PARP play key roles in initiating cellular apoptosis.36 The mechanism of statin-induced apoptosis has been suggested to be mediated predominantly via the depletion of geranylgeranylated proteins. Statins are also capable of activating caspase pathways, such as caspases-3, -8 and -9.31 Western blotting analysis revealed that all of the treatments induced the expression of p53 and cleaved capase-3, while the 4DN/ATST co-treatment exhibited the strongest effects. The enhanced effects of 4DN/ATST combination were confirmed to be synergistic. HT-29 cells carry a mutant form of the p53 gene (R273H), which has been shown to increase cancer cell proliferation and migration, propagate the cell cycle, and contribute to cancer cell survival.37 Nevertheless, our result suggests that the co-treatment of 4DN/ATST has the potential to up-regulate p53 gene expression in cancer cells, which in turn may contribute to its apoptosis-inducing effect. Future investigations using colon cancer cells and animal models with wild type p53 will provide definitive proof of this mechanistic scheme. Interestingly, 4DN seemed to play a more important role in arresting cell cycle progression than did ATST, while ATST exerted a stronger effect in inducing apoptosis than did 4DN, while they were used individually on cancer cells. The results suggested that 4DN and ATST had a distinct molecular target in colon cancer cells, and may partially explain why these two compounds could act synergistically in inhibiting colon cancer.
5 Conclusions
This study demonstrated for the first time the synergistic inhibitory effects on human colon cancer cells by the combination of 4DN and ATST. These effects were associated with the synergistic induction of cell cycle arrest in the G0/G1 stage and cellular apoptosis of colon cancer cells by the 4DN/ATST co-treatment. Further studies on (i) the interaction between other abundant metabolites of NBT (e.g. 3DN and 34DN) and ATST as well as its metabolites, and (ii) the combination of NBT, ATST and their major metabolites as a whole are important to provide mechanistic insights into the synergism between NBT and ATST in inhibiting colon carcinogenesis. Information obtained in these studies will provide a solid scientific basis for using NBT and ATST in combination to prevent colon cancer in humans.Abbreviations
| PMF | Polymethoxyflavone |
| NBT | Nobiletin |
| 4DN | 4′-Demethylnobiletin |
| 3DN | 3′-Demethylnobiletin |
| 34DN | 3′,4′-Didemethylnobiletin |
| ATST | Atorvastatin |
Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
This work was partially supported by a NIH grant (CA139174), an AICR grant (10A044), and USDA Grants on bioactive food components (MAS00450 and MAS00492). Jinkai Zheng was partially supported by China Natural Science Foundation project 31428017.References
- T. C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- G. W. Oehlert, Am. Stat., 1992, 46, 27–29 Search PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- H. Xiao and C. S. Yang, Int. J. Cancer, 2008, 123, 983–990 CrossRef CAS PubMed.
- C. DiMarco-Crook and H. Xiao, Annu. Rev. Food Sci. Technol., 2015, 6, 505–526 CrossRef CAS PubMed.
- X. Wu, A. C. Pfalzer, G. Y. Koh, S. Tang, J. W. Crott, M. J. Thomas, M. Meydani and J. B. Mason, J. Agric. Food Chem., 2017, 65, 7200–7209 CrossRef CAS PubMed.
- J. A. del Rio, P. Gomez, A. G. Baidez, M. C. Arcas, J. M. Botia and A. Ortuno, J. Agric. Food Chem., 2004, 52, 1913–1917 CrossRef CAS PubMed.
- S. Miyamoto, Y. Yasui, T. Tanaka, H. Ohigashi and A. Murakami, Carcinogenesis, 2008, 29, 1057–1063 CrossRef CAS PubMed.
- X. Wu, M. Song, M. Wang, J. Zheng, Z. Gao, F. Xu, G. Zhang and H. Xiao, Mol. Nutr. Food Res., 2015, 59, 2383–2394 CAS.
- X. Wu, M. Song, Z. Gao, Y. Sun, M. Wang, F. Li, J. Zheng and H. Xiao, J. Nutr. Biochem., 2017, 42, 17–25 CrossRef CAS PubMed.
- X. Wu, M. Song, P. Qiu, K. Rakariyatham, F. Li, Z. Gao, X. Cai, M. Wang, F. Xu, J. Zheng and H. Xiao, Carcinogenesis, 2017, 38, 455–464 CrossRef PubMed.
- M. X. Tang, K. Ogawa, M. Asamoto, T. Chewonarin, S. Suzuki, T. Tanaka and T. Shirai, Nutr. Cancer, 2011, 63, 227–233 CrossRef CAS PubMed.
- G. Luo, X. Guan and L. Zhou, Cancer Biol. Ther., 2008, 7, 966–973 CrossRef CAS PubMed.
- M. F. Demierre, P. D. Higgins, S. B. Gruber, E. Hawk and S. M. Lippman, Nat. Rev. Cancer, 2005, 5, 930–942 CrossRef CAS PubMed.
- S. Li, Z. Wang, S. Sang, M. T. Huang and C. T. Ho, Mol. Nutr. Food Res., 2006, 50, 291–299 CAS.
- X. Wu, M. Song, K. Rakariyatham, J. Zheng, M. Wang, F. Xu, Z. Gao and H. Xiao, J. Agric. Food Chem., 2015, 63, 10921–10927 CrossRef CAS PubMed.
- X. Wu, M. Song, K. Rakariyatham, J. Zheng, S. Guo, Z. Tang, S. Zhou and H. Xiao, J. Funct. Foods, 2015, 19, 278–287 CrossRef CAS PubMed.
- J. Zheng, M. Song, P. Dong, P. Qiu, S. Guo, Z. Zhong, S. Li, C. T. Ho and H. Xiao, Mol. Nutr. Food Res., 2013, 57, 1999–2007 CAS.
- J. Zheng, J. Bi, D. Johnson, Y. Sun, M. Song, P. Qiu, P. Dong, E. Decker and H. Xiao, J. Agric. Food Chem., 2015, 63, 509–516 CrossRef CAS PubMed.
- C. V. Rao, Y. Hirose, C. Indranie and B. S. Reddy, Cancer Res., 2001, 61, 1927–1933 CAS.
- Y. Sun, X. Wu, X. Cai, M. Song, J. Zheng, C. Pan, P. Qiu, L. Zhang, S. Zhou, Z. Tang and H. Xiao, Mol. Nutr. Food Res., 2016, 60, 1924–1932 CAS.
- H. Xiao, Q. Zhang, Y. Lin, B. S. Reddy and C. S. Yang, Int. J. Cancer, 2008, 122, 2115–2124 CrossRef CAS PubMed.
- S. Guo, P. Qiu, G. Xu, X. Wu, P. Dong, G. Yang, J. Zheng, D. J. McClements and H. Xiao, J. Agric. Food Chem., 2012, 60, 2157–2164 CrossRef CAS PubMed.
- A. Funaro, X. Wu, M. Song, J. Zheng, S. Guo, K. Rakariyatham, M. T. Rodriguez-Estrada and H. Xiao, J. Food Sci., 2016, 81, H1320–H1327 CrossRef CAS PubMed.
- X. X. Chen, X. Wu, W. Ouyang, M. Gu, Z. L. Gao, M. Y. Song, Y. J. Chen, Y. Y. Lin, Y. Cao and H. Xiao, J. Agric. Food Chem., 2017, 65, 1566–1573 CrossRef CAS PubMed.
- E. M. Laska, M. Meisner and C. Siegel, Biometrics, 1994, 50, 834–841 CrossRef CAS PubMed.
- L. Ouyang, Z. Shi, S. Zhao, F. T. Wang, T. T. Zhou, B. Liu and J. K. Bao, Cell Proliferation, 2012, 45, 487–498 CrossRef CAS PubMed.
- M. Song, X. Wu, N. Charoensinphon, M. Wang, J. Zheng, Z. Gao, F. Xu, Z. Li, F. Li, J. Zhou and H. Xiao, Food Funct., 2017, 8, 954–963 CAS.
- M. Song, N. Charoensinphon, X. Wu, J. Zheng, Z. Gao, F. Xu, M. Wang and H. Xiao, J. Agric. Food Chem., 2016, 64, 4943–4949 CrossRef CAS PubMed.
- R. G. Mehta, G. Murillo, R. Naithani and X. Peng, Pharm. Res., 2010, 27, 950–961 CrossRef CAS PubMed.
- P. Kubatka, P. Kruzliak, V. Rotrekl, S. Jelinkova and B. Mladosievicova, Crit. Rev. Oncol. Hematol., 2014, 92, 296–311 CrossRef PubMed.
- M. Malumbres and M. Barbacid, Nat. Rev. Cancer, 2009, 9, 153–166 CrossRef CAS PubMed.
- T. Waldman, K. W. Kinzler and B. Vogelstein, Cancer Res., 1995, 55, 5187–5190 CAS.
- N. S. Mangini, R. Wesolowski, B. Ramaswamy, M. B. Lustberg and M. J. Berger, Ann. Pharmacother., 2015, 49, 1252–1260 CrossRef CAS PubMed.
- N. S. Chari, N. L. Pinaire, L. Thorpe, L. J. Medeiros, M. J. Routbort and T. J. McDonnell, Apoptosis, 2009, 14, 336–347 CrossRef CAS PubMed.
- R. U. Janicke, M. L. Sprengart, M. R. Wati and A. G. Porter, J. Biol. Chem., 1998, 273, 9357–9360 CrossRef CAS PubMed.
- P. A. J. Muller and K. H. Vousden, Cancer Cell, 2014, 25, 304–317 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |