
An elegant method for large scale synthesis of metal oxide–carbon nanotube nanohybrids for nano-environmental application and implication studies†
Dipesh
Das
a,
Jaime
Plazas-Tuttle
a,
Indu Venu
Sabaraya
a,
Sneha S.
Jain
b,
Tara
Sabo-Attwood
c and
Navid B.
Saleh
*a
aDepartment of Civil, Architectural and Environmental Engineering, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: navid.saleh@utexas.edu; Tel: +1 (512) 471 9175
bDepartment of Chemical Engineering, The University of Texas at Austin, Austin, TX 78712, USA
cDepartment of Environment and Global Health, University of Florida, Gainesville, FL 32610, USA
First published on 17th October 2016
Abstract
Nanohybrids (NHs) with synergistic and emergent properties are used as electrocatalysts, photocatalysts, and antimicrobial agents in numerous applications. Carbon nanotube (CNT)–metal oxide NHs are one of the most commercialized heterostructures because of their advantages as catalyst supports in the fuel cell industry. To date, there has been little understanding of their environmental behavior, primarily due to the lack of a robust yet facile synthesis technique. This study presents an elegant synthesis method, which varies the reagent composition and ratio to grow metal oxides of choice onto multiwalled carbon nanotube (MWNT) surfaces. This technique can synthesize heterostructures with elements ranging from transition (Ti and Zn) to lanthanide series (Er and Pr) metals. This modified sol–gel method can provide large material yields (100s of mg) with a high degree of overall homogeneity between synthesized batches. Such a method for preparing complex NHs from component materials can be extremely useful to perform systematic environmental analyses.
Environmental significanceThis study presents a facile multiwalled carbon nanotube–metal oxide nanohybrid (NH) synthesis technique, where simple variation in reagent composition and ratio allows the metal oxide type and loading on nanotube surfaces to be controlled. Carbon–metal oxide nanohybrids with on-demand metal oxide composition can be synthesized using this sol–gel method. The large material yield makes this method particularly useful for nano-environmental studies. |
Introduction
Materials engineering has moved on from passive nanostructures to hierarchical nanohybrids1,2 (NHs) in quest of achieving multifunctionality and improved efficiency. One of the major classes of such complex heterostructures is carbon nanotube–metal oxide (CNT–MO) NHs. These heterostructures have shown improved performance compared to their component materials when integrated as electro- and photo-catalysts,3 and into electronics,4 gas sensing,5 biosensing,6 and laser technology.7 With such a wide range of application possibilities, commercialization of these heterostructures is on the rise, especially with an incredible growth in the fuel cell industry in recent years.8 CNT–MOs, such as CNT–TiO2 and CNT–ZnO are mostly used as anodes as well as catalyst supports in fuel cells and are likely to result in environmental release during the end of life processing for resource recovery (e.g., Pt recovery).9 Once released into the environment, the environmental health and safety (EHS) of these multicomponent heterostructures with emergent properties will likely differ from those of their component materials, resulting in new unpredictable environmental risks.10–13 To determine if linear superposition of the environmental behavior of the components will adequately capture the heterostructure EHS, gaining control over the synthesis process from their component materials becomes necessary.Electronic band engineering of MO nanocrystals allows their efficient use in microelectronic circuits, sensors, piezoelectric devices, and fuel cells, and as corrosion resistant coating and catalysts.14 With recent advances in nanotechnology and a constant need for improvement in the energy sector, researchers have focused on hybridizing MOs with a secondary element or nanostructure in order to achieve synergistic advantages.15 CNTs with unique mechanical, thermal, physicochemical, and optoelectronic properties have been used as nano-scale supports for MOs.15 The ballistic electron conductivity and high surface area of the CNTs also enhance the catalytic and optoelectronic efficiency of the MOs upon hybridization.16 We posit that these advantageous NHs with novel properties will manifest new and unknown environmental risks.
Multiwalled carbon nanotubes (MWNTs) and several MO nanocrystals have displayed toxicity when introduced to a wide range of microorganisms and aquatic species.17–19 MWNTs have been reported to generate reactive oxygen species (ROS), exhibit physical perturbation to cell walls,21 and even compromise membrane integrity.20,21 Nano ZnO and TiO2 are examples of nano-MOs that have shown antimicrobial properties by ROS generation.22 ZnO NPs have also demonstrated dissolution mediated toxicity.23 The toxicological manifestation of the nano-MOs is not only a function of the chemical composition and band architecture of the materials24 but is also strongly dependent on their size,25 shape and morphology,26 surface chemistry,27 and crystallinity.28 As hybridization or chemical attachment of MO nanocrystals to CNT surfaces develops novel heterostructures, the following is the critical question for the nano-EHS community: will the hybrid CNT–MO attain new properties and exhibit environmental behavior unique to its components? The answer likely lies in potentially emergent material characteristics of these NHs.
Hybridization of CNTs with nano-MOs has been known to modulate their physicochemical properties.1,2 For example, the photocatalytic activity of TiO2 (visible photoactivation) is enhanced when TiO2 is hybridized onto CNTs; lowering of the band gap is known to be behind such an enhancement.29 CNTs hybridized with ZnO show enhanced photocatalytic activity compared to their component materials (when UV irradiated).30 Hybridization is also known to alter van der Waals interaction energy,31 likely resulting in unknown interaction of the NHs with environmental interfaces (i.e., aggregation and deposition in porous media). Determination of property changes in a systematic way from components to nano-heterostructures is key to better predict the EHS of the NHs, where controlled synthesis in a large enough quantity becomes essential.
Several methods have been employed to synthesize CNT–MO NHs including sol–gel,3,32–39 hydrothermal,40–43 solvothermal,44 thermal decomposition,45 direct deposition,46,47 direct mixing,48 microwave irradiation,49 and atomic layer deposition,50 among others. Some of these techniques are fairly simple and can be used to achieve on-demand MO composition and loading. However, a simple technique, where varying the reagent composition and/or amount yields a large amount of hybrid materials with on-demand hybrid composition, is yet to be developed. Such a technique will not only strengthen the synthesis capability by widening the heterostructure chemical composition space but will also facilitate performing comparable EHS studies on a large set of CNT–MOs.
The objective of this study is to develop an elegant method to synthesize CNT–MOs in large quantities (100s of mg), where MOs of different chemical compositions can be conjugated with MWNTs by merely changing the precursor type. A modified sol–gel method8 has been employed to grow MO nanocrystals of two transitional metals (Ti and Zn) and two lanthanide series metals (Er and Pr) in situ onto acid-etched MWNTs. The oxides of Ti and Zn are chosen for their excellent antimicrobial and catalytic properties,51 while those of Er and Pr for their superior optical properties.52 Precursors with relevant chemical compositions are utilized to form these MO nanocrystals. The physical morphology and elemental distribution of these NHs are characterized by employing electron microscopy techniques. To identify if the grown MOs on MWNT surfaces are crystalline, X-ray diffraction (XRD) is performed. To ensure that reproducibility has been achieved between batches, X-ray photoelectron spectroscopy (XPS) is used to determine the material composition. One of the key questions in hybridization is whether these MO nanocrystals are chemically bound to the MWNTs or are physically associated with the tubules. The peak oxidation temperature, which is a strong function of the MWNT chemical bond structure, is determined by thermal gravimetric analysis (TGA). Furthermore, a synthesized NP is only useful for environmental studies if it possesses appreciable colloidal stability in water. A common suspension technique has been employed for all the NHs and their stability is determined by dynamic light scattering (DLS). The elegance of this synthesis technique lies in the simplicity of changing the MO type by merely changing the precursor composition as well as in the large yield of the synthesized materials. Such a large material yield is essential to perform detailed nano-EHS studies of any synthesized NP.
Materials and methods
Chemicals and reagents
Pristine MWNTs (O.D. 8–15 nm) were procured from Cheap Tubes Inc. (Brattleboro, VT). Concentrated nitric acid, sulfuric acid, titanium(IV) isopropoxide (TTIP), erbium(III) nitrate pentahydrate, and praseodymium(III) nitrate hexahydrate were purchased from Sigma Aldrich (St. Louis, MO). Isopropanol was obtained from Fisher Scientific (Pittsburgh, PA) while zinc(II) nitrate hexahydrate was purchased from J.T. Baker (Center Valley, PA). For preparing all aqueous suspensions and solutions, 18.2 mΩ (Milli-Q) water was used unless otherwise stated.Synthesis of CNT–MO NHs
MWNTs (1 g) were acid-etched by ultrasonication (Qsonica LLC, Newtown, CT) in 300 mL of concentrated nitric and sulfuric acid mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume basis). Upon sonication, the mixture was refluxed at 100 °C for 3 h under continuous stirring. The oxidized MWNTs were subsequently filtered until the pH of the filtrate reached >5.5 and then were dried for 48 h in a desiccator. Once dried, 50 mg of the oxidized MWNTs was re-suspended in 100 mL of isopropanol with an ultrasonic dismembrator (Qsonica, Newtown, CT) and transferred into a round bottom flask. Appropriate amounts of precursors (10:1 carbon to precursor molar ratio for MWNT–TiO2 and MWNT–ZnO and 16:1 carbon to precursor molar ratio for MWNT–Er2O3 and MWNT–Pr6O11 synthesis) were added to 10 mL of isopropanol and introduced dropwise to the MWNT–isopropanol suspension at 0.301 mL min−1 with a peristaltic pump (Ismatec, Wertheim, Germany). The slow rate of precursor addition was maintained to provide sufficient mixing time. The entire suspension was refluxed at 80 °C for 3 h in a nitrogen environment. For MWNT–TiO2 NHs, 5 mL of water was added dropwise into the reaction vessel to promote TiO2 crystal formation on the MWNT backbone via a hydrolysis process over 1 h. Hydrolysis was necessary for the MWNT–TiO2 NHs only; the precursors used for the other NHs were already in the hydrated form. Afterwards, the refluxed mixture was washed 4 times with isopropanol (as a purification step), which removed any unreacted reagent. Finally, isopropanol was evaporated, the dry materials were powdered using a mortar and pestle, and the resultant materials were calcined at 400 °C for 3 h under nitrogen to facilitate crystal formation. Synthesis of all four NHs was performed in triplicate using the same method to ensure reproducibility. The synthesis setup is shown in Fig. S1.†
:1 volume basis). Upon sonication, the mixture was refluxed at 100 °C for 3 h under continuous stirring. The oxidized MWNTs were subsequently filtered until the pH of the filtrate reached >5.5 and then were dried for 48 h in a desiccator. Once dried, 50 mg of the oxidized MWNTs was re-suspended in 100 mL of isopropanol with an ultrasonic dismembrator (Qsonica, Newtown, CT) and transferred into a round bottom flask. Appropriate amounts of precursors (10:1 carbon to precursor molar ratio for MWNT–TiO2 and MWNT–ZnO and 16:1 carbon to precursor molar ratio for MWNT–Er2O3 and MWNT–Pr6O11 synthesis) were added to 10 mL of isopropanol and introduced dropwise to the MWNT–isopropanol suspension at 0.301 mL min−1 with a peristaltic pump (Ismatec, Wertheim, Germany). The slow rate of precursor addition was maintained to provide sufficient mixing time. The entire suspension was refluxed at 80 °C for 3 h in a nitrogen environment. For MWNT–TiO2 NHs, 5 mL of water was added dropwise into the reaction vessel to promote TiO2 crystal formation on the MWNT backbone via a hydrolysis process over 1 h. Hydrolysis was necessary for the MWNT–TiO2 NHs only; the precursors used for the other NHs were already in the hydrated form. Afterwards, the refluxed mixture was washed 4 times with isopropanol (as a purification step), which removed any unreacted reagent. Finally, isopropanol was evaporated, the dry materials were powdered using a mortar and pestle, and the resultant materials were calcined at 400 °C for 3 h under nitrogen to facilitate crystal formation. Synthesis of all four NHs was performed in triplicate using the same method to ensure reproducibility. The synthesis setup is shown in Fig. S1.†
Characterization
To determine the elemental composition of the dry MWNT and NH samples, a Kratos X-ray photoelectron spectrometer-Axis Ultra DLD, equipped with a monochromated Al Kα X-ray source (1.486 keV) and a concentric hemispherical analyzer was employed. A thin layer of the powdered sample was placed on a double-sided copper taped stainless steel bar. The bar was then placed in the analysis chamber and degassed for at least 3 h. X-ray photoelectron spectroscopy (XPS) analysis was then performed to obtain the survey spectra as well as the spatial high-resolution spectra and the data was analyzed by fitting the high-resolution element specific peaks with CasaXPS (Casa Software Ltd., Japan). To ensure reproducibility and overall homogeneity, a total of 9 samples for each material (MWNT and four NHs) were analyzed (3 samples each in triplicate batches for all NHs).
:25 sensitivity. The experimental details of size measurement have been described elsewhere.54,56,58,59,61,62 In brief, 2 mL of the 2 mg L−1 of suspensions was injected into pre-cleaned borosilicate glass vials54,56,58,59,61,62 and inserted into the toluene-filled goniometer sample housing. The scattered laser light was collected every 15 s for at least 25 min at 90°. A cumulant fit was used to analyze the collected data and the average hydrodynamic radii (HDR) were obtained for every 15 s segment during the entire duration of the experiments.
Results and discussion
Physical morphology
Representative TEM (Fig. S2†) micrographs of the MWNTs show that the tubes are mostly debundled with an average shell thickness of 21.3 ± 2.6 nm. The HRTEM images illustrate the presence of catalyst-metal free MWNTs. TEM and STEM micrographs with elemental mapping of the NHs (Fig. 1) confirm the presence of the metal atoms (Ti, Zn, Er and Pr), which are distributed throughout the MWNT backbone. These micrographs and the elemental mapping of the MWNT–TiO2 NHs suggest that TiO2 nanocrystals (HRTEMs show lattice fringes) have successfully been grown throughout the MWNT surfaces. Similarly, the TEM images of the other NHs (i.e., MWNT–ZnO, MWNT–Er2O3, and MWNT–Pr6O11) exhibit distinct nanocrystalline MO features on the MWNTs. It should also be noted that though most of MONPs are observed to be at the exterior of the MWNTs, some of the nanocrystals might have been encapsulated within the tube interior. Further experimentation is necessary to determine and analyze the encapsulation ratio. It was also observed that the spatial distribution of the MOs was not uniform throughout the MWNT surfaces for all NHs. This most likely has occurred due to the lack of homogeneity in distribution of the oxygen-containing surface binding sites on the MWNT backbone in the first place. The presence of lattice fringes, as observed in the HRTEM images, confirms the crystalline growth of TiO2, ZnO, Er2O3, and Pr6O11. | ||
| Fig. 1 Representative TEM micrographs of (a) MWNT–TiO2, (c) MWNT–ZnO, (e) MWNT–Er2O3, and (g) MWNT–Pr6O11. HRTEM images with lattice fringes of MOs are shown in the insets. Representative STEM micrographs and elemental mapping of (b) MWNT–TiO2, (d) MWNT–ZnO, (f) MWNT–Er2O3, and (h) MWNT–Pr6O11. | ||
Crystallinity and chemical composition
The XRD patterns of the MWNTs and the NHs are presented in Fig. 2. The lattice planes corresponding to the grown MO nanocrystals possess signature XRD peaks and are in excellent agreement with previously reported XRD patterns of oxidized MWNTs,63 MWNT–TiO263 and MWNT–ZnO NHs.30 Since there are no literature report on XRD patterns of MWNT–Er2O3 and MWNT–Pr6O11 NHs, spectral patterns for these materials were compared to those of Er2O364 and Pr6O1165 nanocrystals (Fig. S3†).
 | ||
| Fig. 2 Representative XRD spectra of (a) oxidized MWNTs, (b) MWNT–TiO2, (c) MWNT–ZnO, (d) MWNT–Er2O3, and (e) MWNT–Pr6O11 NHs. Numbers in parentheses indicate the lattice planes of CNTs (a) and MOs in the respective NHs (b–e). | ||
MWNTs and the NHs display a strong graphitic carbon peak at 25.7°. The MWNT–TiO2 NHs exhibited a major peak for the lattice plane (101), typical of the anatase crystalline phase. Although weaker, the peak occurrences at 37.5°, 48°, and 55.5° correspond to the (004), (200), and (211) crystalline planes, respectively,63 which further confirm the dominance of the anatase phase in the grown TiO2. MWNT–ZnO NHs showed strong peaks corresponding to the (100), (002), (101), (102), and (110) planes, which are consistent with the ZnO nanocrystal structure.30 The XRD spectrum of MWNT–Er2O3 NHs (Fig. 2 and S3a†) displays a highly crystalline Er2O3 signature with a sharp peak at 29.4°, corresponding to the (222) diffraction planes. Other diffraction planes analyzed are (211), (431), (440), and (622), which match with literature reports.64 The MWNT–Pr6O11 NHs show peaks (Fig. 2 and S3b†) with broader ‘full width at half maximum’ (FWHM). When compared to the peaks of Pr6O11,65 the lattice planes (111), (200), and (220) were identified. The peaks of MWNT–Pr6O11 with FWHM features suggest that the Pr6O11 particles are likely smaller than the MOs in the other NHs.66 The TEM, HRTEM and STEM images of MWNT–Pr6O11 (Fig. 1) confirm this observation, displaying ∼5 nm Pr6O11 nanocrystalline features on the MWNTs.
The chemical composition of the NHs is further analyzed by XPS. Fig. S4† presents the XPS spectra for C 1s in the oxidized MWNTs. The peak positions of the deconvoluted spectrum indicate the presence of multiple oxygen containing moieties on the MWNT surfaces, which is in agreement with previously reported XPS data on acid-treated MWNTs.67 The percent oxidation of the MWNTs was determined to be 10.8 ± 1.5%. The survey spectra of the oxidized MWNTs (Fig. 3) show that these are free from any catalyst metal, while those of the NHs (Fig. 3) confirm the presence of the respective metals with no traces of other metal impurities. XPS peaks, corresponding to different orbitals of the metal atoms as well as the Auger peaks, are identified (Fig. 3) from the CasaXPS library and the National Institute of Standards and Technology X-ray photoelectron spectroscopy database.68 The high-resolution XPS spectra of the metal atoms in all the NHs are shown in Fig. S4.† The MWNT–TiO2 NHs show peaks at 458.3 and 464.3 eV, which are characteristic of Ti 2p3/2 and Ti 2p1/2 on anatase phase TiO2.69 For the other NHs, the characteristic ZnO peak for Zn 2p3/2 at 1022.3 eV,30 the Er2O3 peak for Er 4d at 168.8 eV,68 and the Pr6O11 peak for Pr 3d at 933.3 eV68 are located on MWNT–ZnO, MWNT–Er2O3 and MWNT–Pr6O11, respectively. The deconvoluted spectrum for Pr3d (Fig. S4e†) confirms the presence of multiple oxidation states (i.e., III and V) of Pr.65
 | ||
| Fig. 3 Representative XPS survey spectra of (a) oxidized MWNTs, (b) MWNT–TiO2, (c) MWNT–ZnO, (d) MWNT–Er2O3, and (e) MWNT–Pr6O11 NHs. The peak labels indicate the atomic orbitals associated with the binding energies at the respective peak positions. | ||
Table 1 presents the carbon and metal percentages on the NHs. The % MOs are calculated using the % metal values and metal equivalence (i.e., 1 eq. of Ti on TiO2, 1 eq. of Zn in ZnO, 2 eq. of Er in Er2O3, and 6 eq. of Pr in Pr6O11). Table 1 shows that MWNT–TiO2 NHs contain the maximum MO percentage (∼8.5%), while MWNT–Pr6O11 NHs have the lowest (∼0.7%) among all NH types. The XPS results showed a ∼6.2% molar ratio of zinc in the MWNT–ZnO NHs. This value appeared to be lower than that of the MWNT–TiO2 NHs (∼8.5%), though a similar molar ratio of carbon to precursor (10:1) was used for both the NHs. Similarly, the metal content in MWNT–Pr6O11 and MWNT–Er2O3 was similar to each other (∼4.7% for MWNT–Er2O3 and ∼4.2% for MWNT–Pr6O11) but varied with other NHs, in spite of using a carbon to precursor molar ratio of 16:1 for synthesizing these lanthanide oxide NHs. It should be noted that XPS is a highly surface sensitive method with a maximum X-ray penetration depth of 10 nm. This might have caused the observed differences in the metal content of the NHs with similar carbon to precursor molar ratios. The highlight of the XPS results is the low standard deviation (<10%) of the mean values for all the NHs, which confirms the achievement of reproducibility between different batches of the synthesized NHs. Fig. S5† further demonstrates the reproducibility of different NHs among triplicates for each of the materials.
:metal estimated from the XPS spectra using CasaXPS software
| Sample | % C | % metal | % metal oxide in the NHs |
|---|---|---|---|
| MWNT–TiO2 | 63.9 ± 0.7 | 8.5 ± 0.6 | ∼8.5 |
| MWNT–ZnO | 78.5 ± 1.0 | 6.2 ± 0.5 | ∼6.2 |
| MWNT–Er2O3 | 81.4 ± 0.9 | 4.7 ± 0.4 | ∼2.35 |
| MWNT–Pr6O11 | 79.5 ± 1.2 | 4.2 ± 0.4 | ∼0.7 |
Evidence of chemical hybridization
Thermal oxidation of materials, i.e., TGA (Fig. 4 and Table S1†) assesses the nature of hybridization by providing insight into the oxidation of carbonaceous materials.70 The peak oxidation temperature for MWNTs is determined to be 635.4 ± 0.6 °C by plotting the rate of mass loss over the temperature range. Oxidation temperatures for the NHs show a significant leftward shift for all NHs, when compared to the oxidized MWNTs; i.e., the peak oxidation temperatures are 74.9, 48.3, 161.4 and 197.9 °C lower for TiO2–MWNT, ZnO–MWNT, Er2O3–MWNT, and Pr2O3–MWNT, respectively. Lowering in the peak oxidation temperature for hybridized CNTs has been reported earlier, where MOs were found to have caused an enhancement in oxidation reaction.70,71 The change in the oxidation temperature is also reported to vary based on the degree of MO crystallinity and concentration of defects on the MWNT surfaces.70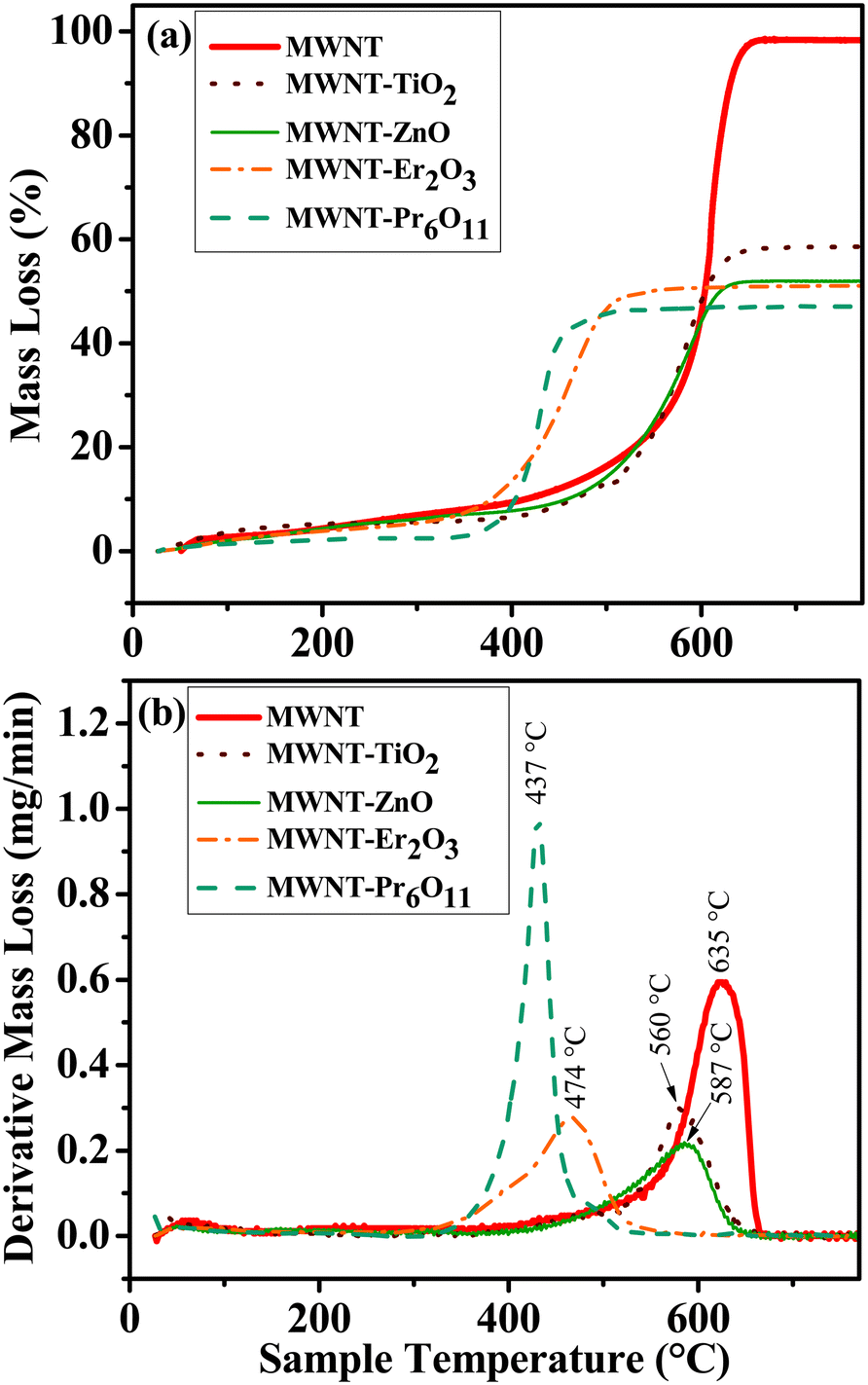 | ||
| Fig. 4 TGA mass loss profiles as a function of oxidation temperature: (a) percent mass loss and (b) derivative mass loss. The peak oxidation temperatures of the oxidized MWNTs and the NHs are labeled. | ||
The residual mass for the oxidized MWNTs is determined to be 1.4 ± 0.2%, which can be attributed to any metal impurities in the oxidized MWNTs. However the other characterization techniques (i.e., TEM, HRTEM, XRD, and XPS) did not find any trace impurities in the oxidized MWNTs. Other likely sources of the residual mass are possible incomplete combustion of MWNTs or measurement sensitivity issues. The residual mass for the MWNT–TiO2 NHs and MWNT–ZnO NHs is ∼40.3% and ∼46.1%, respectively, while that for MWNT–Er2O3 and MWNT–Pr6O11 is found to be ∼47.3% and 51.8%, respectively. The maximum difference in the aforementioned residual masses among the NHs is ∼11.5%. This difference can be lowered by changing the carbon to precursor ratio at the beginning of the synthesis process. It is important to note that the differences in determined MO mass using TGA and XPS cannot be equated, since the former is a mass-based bulk technique while the latter is a surface technique. However, similar to the XPS results, the low standard deviation for the percent mass loss (Table S1†) and similar oxidation temperatures among triplicates for each of the NHs (Fig. S6†) further highlight the reproducibility between synthesized batches.
The residual MOs remaining upon complete oxidation of the NHs with TGA were further analyzed by TEM to assess their physical morphology. The TEM micrographs (Fig. S7†) show similar sized MO nanocrystals with individual particle diameters ranging from 5–12 nm, sintered to form larger aggregates. All the MO nanocrystals show distinct lattice fringes, which confirm the ordered crystallinity in the MOs. This analysis presents valuable insight into the MO nanocrystal morphology, which were grown onto the MWNT surfaces.
Suspension stability
The oxidized MWNTs and the NHs can be suspended in water with a high degree of stability (Fig. S8†). The hydrodynamic radius (HDR) of the oxidized MWNT clusters is determined to be 75 ± 2.4 nm (Fig. 5a), which increases upon hybridization; i.e. 101.5 ± 2.3, 128.5 ± 8.2, 185.5 ± 11.8, and 150.7 ± 16.8 for MWNT–TiO2, MWNT–ZnO, MWNT–Er2O3 and MWNT–Pr6O11, respectively (Fig. 5a). The higher HDR of the NHs is likely due to altered physical properties of the MWNTs, which has influenced the packing of the clusters. No significant settling of the aqueous suspensions of oxidized MWNTs and the MWNT–TiO2 NHs is observed for more than 30 d at a concentration of 10 mg L−1, while the other NHs (i.e., MWNT–ZnO, MWNT–Er2O3 and MWNT–Pr6O11) at the same concentration started to settle after 48 h from the time of suspension preparation. The difference is likely caused by variation in van der Waals attractive interaction among ZnO, Er2O3 and Pr6O11 nanocrystals or due to altered electrokinetic behavior. To achieve higher colloidal stability of the MWNT–ZnO, MWNT–Er2O3 and MWNT–Pr6O11 NHs, surface functionalization (with polymers/surfactants) may be applied.72,73 | ||
| Fig. 5 (a) Average hydrodynamic radii and (b) electrophoretic mobility values of MWNTs and the NHs. All measurements were carried out at pH of 6.9 and at 20 °C. | ||
Electrokinetic properties
The oxidized MWNTs as well as all the NHs (Fig. 5b) exhibit negative surface potential. The EPM values are found to be −3.15 ± 0.1, −2.94 ± 0.3, −2.27 ± 0.3, −2.24 ± 0.1, and −2.33 ± 0.2 m2 V−1 S−1 for oxidized MWNTs, MWNT–TiO2 NHs, MWNT–ZnO NHs, MWNT–Er2O3 NHs, and MWNT–Pr6O11 NHs, respectively. The measured EPM values of the MWNTs agree with previously reported EPMs for oxidized MWNTs (−3.5 to −2.4 m2 V−1 S−1 with high and low oxygen content, respectively, at low ionic strength),74 which likely originated from the oxygen containing moieties on the MWNT surfaces via acid-etching.74–76 Other carbon allotropes such as single-walled carbon nanotubes (−2.8 m2 V−1 S−1 to −1.25 m2 V−1 S−1)56,77,78 and graphene79,80 have shown similar surface potential upon oxidation. The EPM values of the MWNT–ZnO, MWNT–Er2O3, and MWNT–Pr6O11 NHs are significantly lower than those of the oxidized MWNTs and the MWNT–TiO2 NHs. Hence the relative instability of the MWNT–ZnO, MWNT–Er2O3, and MWNT–Pr6O11 NHs, as discussed earlier, may have been dominated by the electrokinetics of these materials.Conclusion
A large-scale, elegant, and facile synthesis process has been developed for four different CNT–MO NHs. The novelty of this study lies in this synthesis technique being a singular standardized method synthesizing carbonaceous–metallic nanohybrids with a wide range of metal/metal oxides including new nanohybrids that involve lanthanide series metals, and in the fact that this method has allowed at least 100s of mg of nanohybrids to be synthesized in a single synthesis cycle while achieving a high degree of reproducibility. The synthesized NHs were characterized to identify their physical morphology, crystallinity, and chemical composition as well as to demonstrate proper hybridization and reproducibility of the NHs. The characterization results demonstrate the successful preparation of CNT–MO NHs with a wide range of MO compositions, which are highly reproducible between batches. One of the limitations of this method is the limited homogeneity in the spatial distribution of the MONPs on the MWNT surfaces. However, it is important to note that these NHs will interact with other environmental entities as a cluster rather than as individual tubules. Hence, the overall homogeneity in MO distribution may not play a significant role in their manifested EHS. The strength of this synthesis technique lies in the large material yield with high reproducibility between synthesized batches. The simplicity in tuning MO composition by merely changing the reagent type widens the choice of materials, which can be extremely useful to perform nano-EHS studies on carbon–metal oxide heterostructures.Acknowledgements
This work is supported by an NSF award, bearing award#1602273. The authors also thank Dr. Karalee Jarvis and Dr. Hugo Celio at the Texas Materials Institute for their assistance in elemental mapping and XPS analysis of the NHs.References
- N. Aich, J. Plazas-Tuttle, J. Lead and N. Saleh, Environ. Chem., 2014, 11, 609–623 CrossRef CAS.
- N. B. Saleh, N. Aich, J. Lead, J. Plazas-Tuttle and G. V. Lowry, Environ. Sci.: Nano, 2015, 2, 11–18 RSC.
- D. Eder and A. H. Windle, Adv. Mater., 2008, 20, 1787–1793 CrossRef CAS.
- N. J. Alley, K. S. Liao, E. Andreoli, S. Dias, E. P. Dillon, A. W. Orbaek, A. R. Barron, H. J. Byrne and S. A. Curran, Synth. Met., 2012, 162, 95–101 CrossRef CAS.
- E. Llobet, E. Espinosa, E. Sotter, R. Ionescu, X. Vilanova, J. Torres, A. Felten, J.-J. Pireaux, X. Ke and G. Van Tendeloo, Nanotechnology, 2008, 19, 375501 CrossRef CAS PubMed.
- Z. Liu, J. Wang, D. Xie and G. Chen, Small, 2008, 4, 462–466 CrossRef CAS PubMed.
- Y. Zhu, H. I. Elim, Y. L. Foo, T. Yu, Y. Liu, W. Ji, J. Y. Lee, Z. Shen, A. T.-S. Wee and J. T.-L. Thong, Adv. Mater., 2006, 18, 587–592 CrossRef CAS.
- W. A. Rigdon and X. Huang, J. Power Sources, 2014, 272, 845–859 CrossRef CAS.
- M. Pehnt, Handbook of Fuel Cells, 2003 Search PubMed.
- J. Plazas-Tuttle, L. Rowles, H. Chen, J. Bisesi, T. Sabo-Attwood and N. Saleh, Nanomaterials, 2015, 5, 1102 CrossRef CAS.
- N. B. Saleh, A. Afrooz, J. H. Bisesi Jr, N. Aich, J. Plazas-Tuttle and T. Sabo-Attwood, Nanomaterials, 2014, 4, 372–407 CrossRef.
- N. B. Saleh, N. Aich, J. Plazas-Tuttle, J. R. Lead and G. V. Lowry, Environ. Sci.: Nano, 2015, 2, 11–18 RSC.
- N. Aich, J. Plazas-Tuttle, J. R. Lead and N. B. Saleh, Environ. Chem., 2014, 11, 609–623 CrossRef CAS.
- M. Fernández-García and J. A. Rodriguez, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2011 Search PubMed.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS PubMed.
- S. H. Lee, D. H. Lee, W. J. Lee and S. O. Kim, Adv. Funct. Mater., 2011, 21, 1338–1354 CrossRef CAS.
- I. L. Bergin and F. A. Witzmann, Int. J. Biomed. Nanosci. Nanotechnol., 2013, 3, 163–210 CrossRef CAS PubMed.
- J. Cheng and S. H. Cheng, Int. J. Nanomed., 2012, 7, 3731–3739 CrossRef PubMed.
- M. S. Diniz, A. P. de Matos, J. Lourenço, L. Castro, I. Peres, E. Mendonça and A. Picado, Microsc. Microanal., 2013, 19, 1131–1140 CrossRef CAS PubMed.
- H. Chen, B. Wang, D. Gao, M. Guan, L. Zheng, H. Ouyang, Z. Chai, Y. Zhao and W. Feng, Small, 2013, 9, 2735–2746 CrossRef CAS PubMed.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409–6413 CrossRef CAS PubMed.
- L. K. Adams, D. Y. Lyon and P. J. Alvarez, Water Res., 2006, 40, 3527–3532 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- N. B. Saleh, D. J. Milliron, N. Aich, L. E. Katz, H. M. Liljestrand and M. J. Kirisits, Sci. Total Environ., 2016, 568, 926–932 CrossRef CAS PubMed.
- D. Xiong, T. Fang, L. Yu, X. Sima and W. Zhu, Sci. Total Environ., 2011, 409, 1444–1452 CrossRef CAS PubMed.
- N. Balázs, K. Mogyorósi, D. F. Srankó, A. Pallagi, T. Alapi, A. Oszkó, A. Dombi and P. Sipos, Appl. Catal., B, 2008, 84, 356–362 CrossRef.
- K. Mogyorosi, N. Balazs, D. F. Sranko, E. Tombacz, I. Dekany, A. Oszko, P. Sipos and A. Dombi, Appl. Catal., B, 2010, 96, 577–585 CrossRef CAS.
- C. M. Sayes, R. Wahi, P. A. Kurian, Y. Liu, J. L. West, K. D. Ausman, D. B. Warheit and V. L. Colvin, Toxicol. Sci., 2006, 92, 174–185 CrossRef CAS PubMed.
- K.-h. Jung, J. S. Hong, R. Vittal and K.-J. Kim, Chem. Lett., 2002, 864–865 CrossRef CAS.
- K. Dai, G. Dawson, S. Yang, Z. Chen and L. Lu, Chem. Eng. J., 2012, 191, 571–578 CrossRef CAS.
- Z. Hua, J. Zhang, X. Bai, Z. Ye, Z. Tang, L. Liang and Y. Liu, Sci. Total Environ., 2016, 539, 196–205 CrossRef CAS PubMed.
- M. T. Nguyen, C. K. Nguyen, T. M. P. Vu, Q. Van Duong, T. L. Pham and T. C. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2014, 5, 045018 CrossRef.
- C. F. Yang, W. C. Hsu, S. M. Wu and C. C. Su, J. Nanomater., 2015 DOI:10.1155/2015/952797.
- Y. Haldorai, A. Rengaraj, J.-B. Lee, Y. S. Huh and Y.-K. Han, Synth. Met., 2015, 199, 345–352 CrossRef CAS.
- A. Jitianu, T. Cacciaguerra, M.-H. Berger, R. Benoit, F. Béguin and S. Bonnamy, J. Non-Cryst. Solids, 2004, 345, 596–600 CrossRef.
- M. Ding, D. C. Sorescu and A. Star, J. Am. Chem. Soc., 2013, 135, 9015–9022 CrossRef CAS PubMed.
- M. Zarezade, S. Ghasemi and M. R. Gholami, Catal. Sci. Technol., 2011, 1, 279–284 CAS.
- J. Khanderi, R. C. Hoffmann, A. Gurlo and J. J. Schneider, J. Mater. Chem., 2009, 19, 5039–5046 RSC.
- W.-L. Song, M.-S. Cao, B. Wen, Z.-L. Hou, J. Cheng and J. Yuan, Mater. Res. Bull., 2012, 47, 1747–1754 CrossRef CAS.
- K. Byrappa, A. Dayananda, C. Sajan, B. Basavalingu, M. Shayan, K. Soga and M. Yoshimura, J. Mater. Sci., 2008, 43, 2348–2355 CrossRef CAS.
- K. Dai, X. Zhang, K. Fan, T. Peng and B. Wei, Appl. Surf. Sci., 2013, 270, 238–244 CrossRef CAS.
- C. Liu, H. Chen, K. Dai, A. Xue, H. Chen and Q. Huang, Mater. Res. Bull., 2013, 48, 1499–1505 CrossRef CAS.
- T. A. Saleh, M. Gondal and Q. Drmosh, Nanotechnology, 2010, 21, 495705 CrossRef PubMed.
- L. H. Tian, L. Q. Ye, K. J. Deng and L. Zan, J. Solid State Chem., 2011, 184, 1465–1471 CrossRef CAS.
- G. Guo, J. Guo, D. Tao, W. Choy, L. Zhao, W. Qian and Z. Wang, Appl. Phys. A: Mater. Sci. Process., 2007, 89, 525–528 CrossRef CAS.
- B. C. Kim, S. Kim, J. Chung, J. Chen, S. Park and G. G. Wallace, Synth. Met., 2012, 162, 650–654 CrossRef CAS.
- X. M. Yan, D. Y. Pan, Z. Li, B. Zhao, J. C. Zhang and M. H. Wu, Mater. Lett., 2010, 64, 1694–1697 CrossRef CAS.
- T. Ueda, K. Takahashi, F. Mitsugi and T. Ikegami, Diamond Relat. Mater., 2009, 18, 493–496 CrossRef CAS.
- Y. Du, C. Hao and G. Wang, Mater. Lett., 2008, 62, 30–32 CrossRef CAS.
- J. M. Green, L. Dong, T. Gutu, J. Jiao, J. F. Conley Jr and Y. Ono, J. Appl. Phys., 2006, 99, 094308 CrossRef.
- D. Jassby, J. Farner Budarz and M. Wiesner, Environ. Sci. Technol., 2012, 46, 6934–6941 CrossRef CAS PubMed.
- L. L. Wilkinson, M.S. Thesis, Ball State University, Department of Physics and Astronomy, 2012, http://liblink.bsu.edu/catkey/1678999#holdings Search PubMed.
- A. N. Afrooz, D. Das, C. J. Murphy, P. Vikesland and N. B. Saleh, Water Res., 2016, 99, 7–15 CrossRef CAS PubMed.
- N. Aich, L. K. Boateng, I. V. Sabaraya, D. Das, J. R. Flora and N. B. Saleh, Environ. Sci. Technol., 2016, 50, 3562–3571 CrossRef CAS PubMed.
- A. N. Afrooz, I. A. Khan, S. M. Hussain and N. B. Saleh, Environ. Sci. Technol., 2013, 47, 1853–1860 CrossRef CAS PubMed.
- I. A. Khan, A. R. M. N. Afrooz, J. R. V. Flora, P. A. Schierz, P. L. Ferguson, T. Sabo-Attwood and N. B. Saleh, Environ. Sci. Technol., 2013, 47, 1844–1852 CrossRef CAS PubMed.
- I. A. Khan, N. Aich, A. R. M. N. Afrooz, J. R. V. Flora, L. Ferguson, T. Sabo-Attwood and N. B. Saleh, Chemosphere, 2013, 93, 1997–2003 CrossRef CAS PubMed.
- I. A. Khan, J. R. V. Flora, A. R. M. N. Afrooz, N. Aich, P. A. Schierz, P. L. Ferguson, T. Sabo-Attwood and N. B. Saleh, Environ. Chem., 2015, 12, 652–661 CrossRef CAS PubMed.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7963–7969 CrossRef CAS PubMed.
- E. Mansfield, A. Kar and S. A. Hooker, Anal. Bioanal. Chem., 2010, 396, 1071–1077 CrossRef CAS PubMed.
- A. R. M. N. Afrooz, I. A. Khan, S. M. Hussain and N. B. Saleh, Environ. Sci. Technol., 2013, 47, 1853–1860 CrossRef CAS PubMed.
- A. R. M. N. Afrooz, S. T. Sivalapalan, C. J. Murphy, S. M. Hussain, J. J. Schlager and N. B. Saleh, Chemosphere, 2013, 91, 93–98 CrossRef CAS PubMed.
- W. A. Rigdon and X. Huang, J. Power Sources, 2014, 272, 845–859 CrossRef CAS.
- M. Miritello, R. L. Savio, A. Piro, G. Franzo, F. Priolo, F. Iacona and C. Bongiorno, J. Appl. Phys., 2006, 100, 013502 CrossRef.
- M. S. Hassan, M. S. Akhtar, K.-B. Shim and O.-B. Yang, Nanoscale Res. Lett., 2010, 5, 735 CrossRef CAS PubMed.
- C. Baker, A. Pradhan, L. Pakstis, D. J. Pochan and S. I. Shah, J. Nanosci. Nanotechnol., 2005, 5, 244–249 CrossRef CAS PubMed.
- K. A. Wepasnick, B. A. Smith, J. L. Bitter and D. H. Fairbrother, Anal. Bioanal. Chem., 2010, 396, 1003–1014 CrossRef CAS PubMed.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST Standard Reference Database 20, Version 4.1, 2012 Search PubMed.
- K. H. Leong, H. Y. Chu, S. Ibrahim and P. Saravanan, Beilstein J. Nanotechnol., 2015, 6, 428–437 CrossRef CAS PubMed.
- S. Aksel and D. Eder, J. Mater. Chem., 2010, 20, 9149–9154 RSC.
- D. Eder and A. H. Windle, J. Mater. Chem., 2008, 18, 2036–2043 RSC.
- N. Aich, L. K. Boateng, J. R. Flora and N. B. Saleh, Nanotechnology, 2013, 24, 395602 CrossRef PubMed.
- I. A. Khan, J. R. Flora, A. N. Afrooz, N. Aich, P. A. Schierz, P. L. Ferguson, T. Sabo-Attwood and N. B. Saleh, Environ. Chem., 2015, 12, 652–661 CrossRef CAS PubMed.
- P. Yi and K. L. Chen, Langmuir, 2011, 27, 3588–3599 CrossRef CAS PubMed.
- B. Smith, K. Wepasnick, K. E. Schrote, H.-H. Cho, W. P. Ball and D. H. Fairbrother, Langmuir, 2009, 25, 9767–9776 CrossRef CAS PubMed.
- B. Smith, K. Wepasnick, K. E. Schrote, A. R. Bertele, W. P. Ball, C. O'Melia and D. H. Fairbrother, Environ. Sci. Technol., 2009, 43, 819–825 CrossRef CAS PubMed.
- N. B. Saleh, L. D. Pfefferle and M. Elimelech, Environ. Sci. Technol., 2010, 44, 2412–2418 CrossRef CAS PubMed.
- M. Sano, J. Okamura and S. Shinkai, Langmuir, 2001, 17, 7172–7173 CrossRef CAS.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- J. D. Lanphere, C. J. Luth and S. L. Walker, Environ. Sci. Technol., 2013, 47, 4255–4261 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6en00294c |
| This journal is © The Royal Society of Chemistry 2017 |