
Synthesis of CuxNi(1−x)O coral-like nanostructures and their application in the design of a reusable toxic heavy metal ion sensor based on an adsorption-mediated electrochemical technique†
Sayan
Dey
a,
Sumita
Santra
b,
Anupam
Midya
c,
Prasanta Kumar
Guha
a and
Samit Kumar
Ray
*bc
aDepartment of Electronics & Electrical Communication Engineering, Indian Institute of Technology, Kharagpur, India
bDepartment of Physics, Indian Institute of Technology, Kharagpur 721302, India. E-mail: physkr@phy.iitkgp.ernet.in
cSchool of Nanoscience & Technology, Indian Institute of Technology, Kharagpur, India
First published on 10th November 2016
Abstract
Nickel oxide, and non-stoichiometric and Cu-doped variants (NiO, Ni2O3 and CuxNi(1−x)O) possessing porous coral-like nanostructures, were prepared by a facile, low temperature, hydrothermal approach. Structural and morphological characterization was performed by X-ray diffractometry, X-ray photoelectron spectroscopy and FESEM imaging. The Cu-doped oxide was found to possess lattice strain which resulted in the induction of surface defects. The prepared materials were used as sensing materials for toxic Cr(VI) ions in aqueous medium. A novel sensing technique was proposed based on adsorption, which was found to be more effective in terms of reproducibility and reusability of the sensor than the conventional electrochemical sensing technique. The sensing mechanism was explained based on the phenomenon of adsorption. This makes the efficiency of the sensor surface-dependent rather than chemical reactivity-dependent, thereby making it a non-destructive sensing technique. The Cu-doped NiO nanostructure (10% Cu doping) was found to show maximum sensitivity (252.62 at 1 ppm) and high selectivity, together with a low response time (∼2 s), towards Cr(VI) ions relative to Cd(II), As(V) and Pb(II) ions. This was due to its enhanced surface properties compared with its un-doped variants. The effect of Cu doping on the nanostructure morphology and consequently on sensor efficiency was also studied. The limit of detection was found to be 1 ppm (1 mg L−1) of Cr(VI) ions in aqueous solution. This material, along with the technique proposed, can thus be advantageous for the cost-efficient monitoring of water quality and drinking water standards.
Environmental significanceA CuxNi(1−x)O coral-like nanostructure was synthesized using a low-temperature, environmentally friendly hydrothermal technique and then used for toxic heavy metal (Cr(VI)) ion detection at ultra-low levels in water-quality monitoring. Introduction of a dopant (Cu) into the NiO lattice generated strain which enhanced the effective number of surface defects (adsorption sites). The novel adsorption–desorption-mediated sensing technique facilitates fabrication of ultra-fast (∼2 s) re-usable heavy metal sensors which, until now, have not been reported. The sensor is exceedingly selective towards Cr(VI) ions, due to the precise material engineering achieved through metal doping. This results in effective tuning of the electrical double layer formed in solution with Cr(VI), relative to other analytes. |
Introduction
Monitoring inorganic ions in water intended for consumption is an absolute necessity in order to maintain a healthy biota. With the increase in industrialization, the demand for water-contaminant monitoring systems to precisely detect toxic contaminants released as industrial effluents in natural consumable water sources has increased considerably. Inorganic water contaminants such as Cr(VI), Hg(II), As(III), Cd(II) etc. are common toxic metal ions introduced into the water system from industrial processes and they pose a serious threat to aquatic and human health.1 Among them, Cr(VI) ions are known to be carcinogenic to human beings if exposure exceeds a certain permissible limit.2 The median lethal dosage (LD50) of Cr(VI) is found to be approximately 500 ppb to 1.5 ppm.3 As a result, a high-precision sensor in terms of sensitivity, selectivity and fast response time is required to detect the presence of trace amounts of Cr(VI) in drinking water. In this context, transition metal oxide (TMO)-based nanostructures and their composites are of special interest, particularly due to their enhanced surface properties and ion adsorption capacity in aqueous solution.4,5 They are expected to produce excellent sensitivity, due to the presence of unsatisfied surface bonds that readily saturate when exposed to any ionic species. However, the sensitivity, selectivity and response time are highly dependent on the surface defects, surface potential and porosity of the synthesized nanomaterial.6 Among different transitional materials, developed so far, NiO has proven itself to be a promising nanomaterial that shows potential application in battery electrodes, super-capacitors, smart windows, catalysis, etc.7–11 Its mixed higher oxides are generally unstable at room temperature. Heavy metal removal by nanomaterials using different techniques is an important field of interest from the application perspective.12 However, Ni2O3 nanoparticles, reported to be synthesized at room temperature in stable form, show excellent surface properties due to their induced negative surface potential,13 and hence have been used for heavy metal ion removal in aqueous solution. Thus, oxides of nickel and their doped derivatives are expected to possess sensing properties for heavy metal ions. Synthetic procedures for TMO nanostructures and their metal-doped derivatives generally require high temperatures and so are not very energy-efficient. Consequently, there is a need for a synthetic procedure at a lower temperature to facilitate the energy-efficient synthesis of nanostructures. Inducing hierarchical morphologies enhances surface-to-volume ratio together with surface reactivity. Several hierarchical nanostructures of different oxides have been synthesized and applied in different electronic and environmental applications to achieve desirable results.14Conventional electrochemical sensing includes a redox reaction (either reduction or oxidation or both) at the electrodes (sensing material) in order to sense the presence of heavy metal contaminants in water. However, this leads to the destruction of the sensing material, thereby making it unusable for repeated use.15 Hence, reusability of such sensors is an issue. Non-destructive techniques have not yet been developed for heavy metal ion sensors.
To the best of our knowledge, metal-doped variants of NiO or its higher oxide (Ni2O3) coral-like hierarchical nanostructures have not yet been synthesized and used for heavy metal detection in aqueous solution. In this study, we synthesized NiO, Ni2O3 and Cu-doped NiO coral-like nanostructures by a low-temperature wet chemical synthetic route and tested their efficacy for Cr(VI) ion sensing in aqueous solution by non-destructive adsorption/desorption electrochemical techniques. We also compared the performance of the materials so prepared, and studied the effect of doping of metal ions in the NiO structure on its sensing properties. Moreover, extensive reproducibility and reusability tests of our sensors are presented, illustrating that the sensors can be used to develop a real-time flowing water monitoring system, which will in turn facilitate the development of a smart water remediation system.
Experimental section
Synthesis of coral-like hierarchical NiO, Ni2O3 and CuxNi(1−x)O nanostructures
All chemicals were purchased from Merck India Pvt. Ltd. and were used without further purification. All hierarchical oxides were prepared from a hydroxide precursor which was prepared by a hydrothermal approach. The hydroxide precursor as prepared by a hydrothermal approach was calcined at 400 °C for 2 h to synthesize hierarchical NiO. For the preparation of Ni2O3, the hydroxide precursor was treated with sodium hypochlorite (NaOCl) solution in alkaline medium. For the copper-doped oxide, the doping was done in situ hydrothermally by adding 10% by weight of copper precursor in the precursor medium. This results in the formation of a doped hydroxide. The so-prepared copper-doped hydroxide was further calcined 400 °C for 2 h to synthesize hierarchical NiO. The detailed synthetic procedures for all the three variants of nickel oxide are presented in ESI.†Material characterization and Cr(VI) ion sensing
The phase and possible doping of the oxide was predicted by X-ray diffraction (XRD, Ultima-III, Rigaku X-ray diffractometer (Cu Kα radiation, λ = 1.5404 Å)). Morphology studies of the synthesized samples were carried out using a field emission scanning electron microscope (FESEM, S-4800, Hitachi). A transmission electron microscope (JEM-2100, JEOL) was employed to investigate the microstructure and morphology of the synthesized samples. Investigation of possible copper doping was carried out by X-ray photoelectron spectroscopy (XPS, PHI 5000Versa Probe II (ULVAC-PHI Inc., Japan)) measurements. Spectroscopic investigations were carried out by Fourier transform infra-red spectroscopy (FTIR Nexus-870, USA) and Raman spectroscopy (MODEL T64000 (Jobin Yvon Horiba, France)) to investigate the type and nature of chemical bonds and differences between them in the three samples. A N2 adsorption–desorption experiment (BET/BJH) (Autosorb iQ2 automated gas sorption analyser, Quantachrome Instruments, Florida, USA) was carried out to study the porosity of the optimized sample (10% Cu-doped NiO).The sensing measurements were carried out using a Potentiostat Interface 1000 Gamry setup. Ag/AgCl and Pt were used as reference and counter electrodes, respectively, and the material was deposited on a 0.5 mm-thick aluminium sheet (3 cm × 3 cm) which was used as the working electrode for the half cell. 0.1 M NaCl solution was used as an electrolyte solution for the cyclic voltammetry experiment operated between −1 V to +1 V. Potassium dichromate solution at calculated concentrations was taken as the Cr(VI) ion source, which was dissolved in 0.1 M NaCl electrolyte solution (taken as the control for the experiment) for the subsequent measurements in 0.1 M NaCl electrolyte solution.
Results and discussion
The X-ray diffractograms taken for three dried powder samples of the oxide nanostructures, i.e. NiO, Ni2O3 and Cu(1−x)NixO nanostructures, are shown in Fig. 1(a). The blue spectrum with peaks at 34°, 31.66°, 45.41°, 56.38° and 66.12° may be readily indexed with (101), (002), (111), (202) and (004) planes of hexagonal Ni2O3 (JCPDS card number 14-0481, CAS number 1314-06-3), respectively. The red and black spectra with peaks at 37.25°, 43.30°, 62.92°, 75.45°, and 79.35° correspond to the (111), (200), (220), (311), and (222) planes of the fcc phase NiO (JCPDS card number 04-0835), respectively. No peak for crystalline copper or copper oxides or any other crystalline compound of copper is observed. However, in Fig. 1(b), a comparative study of the diffractograms obtained, respectively, for NiO (black) and CuxNi(1−x)O (red), is presented. The peaks in the red spectrum obtained for the doped oxide (i.e. CuxNi(1−x)O) appear to be broadened, with comparatively lower intensity (ΔI). This indicates the possible doping of copper in the NiO crystal lattice, which creates strain within the lattice, resulting in peak broadening. Moreover, a peak shift by ∼0.14° is observed in the case of the doped oxide with respect to the pure oxide sample. This shift is attributed to the possible Cu doping within the oxide lattice. The strain generated due to incorporation of a Cu dopant in the NiO lattice is estimated from Williamson–Hall and size-strain plot models.16,17 | ||
| Fig. 1 (a) X-Ray diffractograms for crystalline NiO, Cu(1−x)NixO and Ni2O3 hierarchical nanostructures. (b) Peak shift and intensity reduction for CuxNi(1−x)O in comparison with pure NiO. | ||
Table 1 presents the strain energy created within the NiO lattice when Cu atoms are doped. The strain energy in case of CuxNi(1−x)O is more than that in the pure NiO lattice. This may imply the possible replacement of Ni by Cu atoms within the crystal lattice, resulting in doping of NiO with Cu.
| Sample | Williamson–Hall plot | Size-strain plot | ||||
|---|---|---|---|---|---|---|
| Strain | Particle size (in nm) | r 2 | Strain | Particle size (in nm) | r 2 | |
| NiO | 9.42 × 10−2 | 31.80 | 0.927 | 3.16 × 10−1 | 36.09 | 0.998 |
| CuxNi(1−x)O | 2.2 × 10−1 | 15.72 | 0.856 | 4.24 × 10−1 | 21.45 | 0.993 |
The chemical composition of the doped oxide was extracted from XPS and estimated to have 42.3% nickel, 7.4% copper and 50.3% oxygen. The full spectrum and individual peaks of the respective elements are shown in the ESI† (Fig. S1). The analysis of the peaks confirms the copper doping in the NiO lattice from the obtained Cu peaks in the XPS spectra.
Analysis of the FTIR and Raman spectra reveals the bond nature and structure of the prepared materials. From the FTIR spectrum in Fig. 2(a), for all the three optimised samples, peaks are obtained at ∼450 cm−1 due to the presence of M–O (where M = Ni, Cu) bonds. Peaks between 1000 cm−1 and 1500 cm−1 are due to the –OH group bending vibrations, whereas peaks above 2000 cm−1 are due to water molecule (−OH) stretching vibrations. A peak is obtained at ∼1000 cm−1 in the case of Ni2O3 and is due to the formation of Ni–O–Ni bonds, which is different from the remaining two samples. The Raman spectra in Fig. 2(b) reveal two broad peaks at around 500 cm−1 and 1100 cm−1 corresponding to the coupling interaction of Ni–O oscillations. It can be seen that there are slight shifts in both peaks in the case of the CuxNi(1−x)O (10% doped) sample. This may be attributed to the possible doping of Cu ions in the NiO lattice resulting in the formation of strained hereto structures. A considerable amount of shift may have occurred due to the formation of the Ni–O–Ni bond.
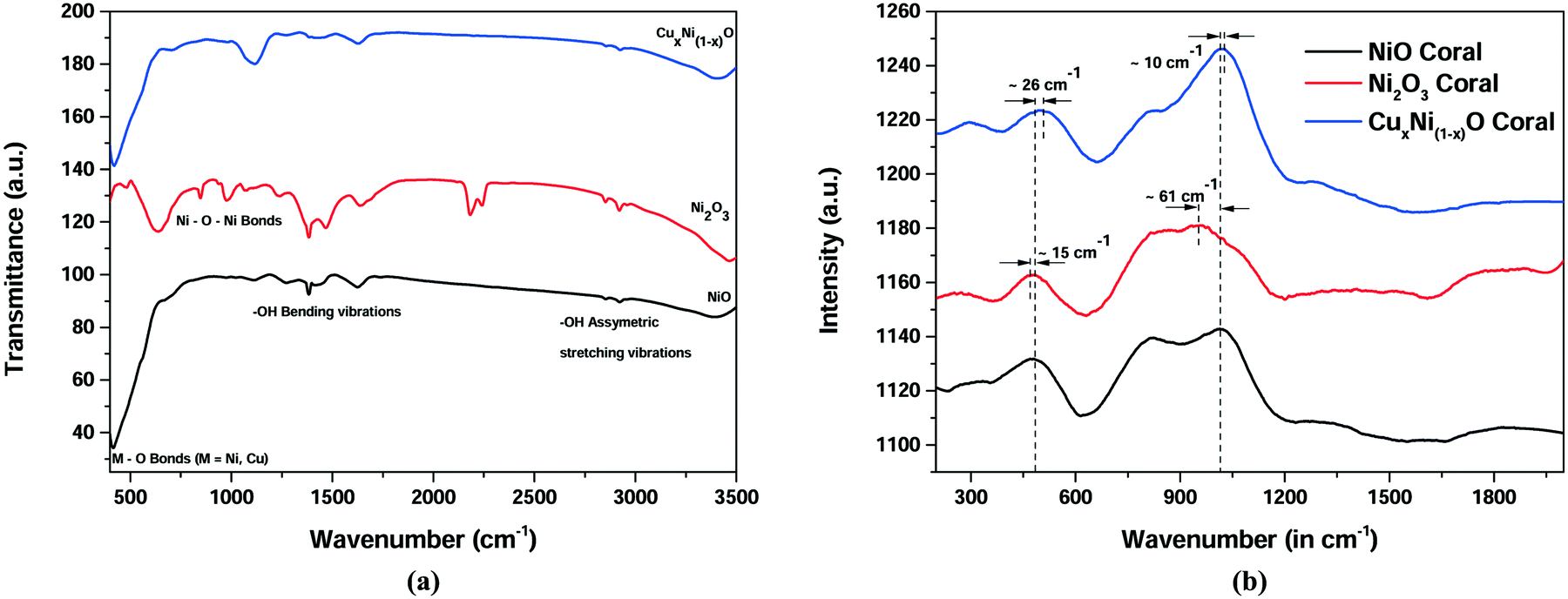 | ||
| Fig. 2 Spectroscopic investigation for NiO, Ni2O3 and CuxNi(1−x)O (10% Cu) (a) FTIR spectrum (b) Raman spectrum. | ||
FESEM images in Fig. 3(a)–(c) indicate the formation of a coral-like hierarchical morphology in the NiO, Ni2O3 and CuxNi(1−x)O samples, respectively. The coral-like hierarchical structures possess a higher density of defects, which may enhance the surface properties of the so-prepared oxides. It is also expected that the Cu-doped NiO sample should possess a higher surface activity, due to the strain generated by Cu atoms within the NiO crystal lattice. The formation of these coral-like structures is dependent on the basic structure forming the hierarchical structure. It can be observed from Fig. 3 that the pure oxides are formed from only a single type of structure, such as filaments or lengthened particles (in the case of NiO in Fig. 3(a)) and flakes (in the case of Ni2O3 in Fig. 3(b)). However, from Fig. 3(c) it can be seen that the hierarchical structure is a mixture of both filaments (and spherical particles) and flakes. This may be due to the possibility that the doping has controlled the nucleation rate and the orientation of growth, owing to the presence of two cations in the solution, thereby effectively increasing the surface area available for adsorption of chemical species (active adsorption sites). The EDAX spectrum and its analysis also support the XPS analysis in terms of chemical composition of the Cu-doped NiO sample (Fig. S2 in ESI†).
 | ||
| Fig. 3 FESEM images of (a) NiO (b) Ni2O3 and (c) Cu(1−x)NixO (10% Cu) hierarchical nanostructures. | ||
It is observed in Fig. 3 (nanostructures of each oxide type are magnified and presented) that, as the nickel oxide is varied from NiO to Ni2O3 and then to CuxNi(1−x)O, the surface morphology changes. From a visual perspective, it may be concluded that the doped variant has a much rougher surface as compared with the pure oxides of nickel. This can be attributed to the enhanced adsorption of Cr(VI) ions in aqueous solution, thereby leading to effective sensing of Cr(VI).
The microstructures so formed were studied by transmission electron microscopy (TEM) imaging. The lattice image and fringe pattern were also investigated. The doped oxide was expected to be larger in size compared with the pure variants. This may be attributed to the probable inflation of structure due to the incorporation of Cu (atomic number 29) having a fully filled d orbital (3d10 configuration) in place of Ni (atomic number 28) having a 3d9 configuration. Moreover, the surface roughness is greater in the case of CuxNi(1−x)O in comparison with the pure oxide variants, which provides more active adsorption sites. When comparing these three images, it can be established from Fig. 4(c) that the Cu-doped sample hierarchical structure is a mixture of flakes and filaments as primary growth units whereas, in the case of NiO (Fig. 4(a)), it is homogenous filaments, and in Ni2O3 (Fig. 4(b)), it is homogenous flakes. Thus, the presence of two basic units contributes a differential surface area for adsorption, which is expected to be greater than for those structures formed from a single basic unit (as in Fig. 3(a) and (b)). The lattice images and selected area electron diffraction (SAED) patterns of the three structures are provided in the ESI† (Fig. S4). The lattice spacing for all three samples was found to be ∼0.28 nm, confirming the formation of nickel oxides.
 | ||
| Fig. 4 TEM images of (a) NiO (b) Ni2O3 and (c) Cu(1−x)NixO hierarchical nanostructures. | ||
In order to study the effect of doping concentration on the structure, the doping amount was varied from 5% to 20% Cu. From the FESEM images shown in Fig. 5, it is seen that, on doping with Cu, the hierarchical structure is generated by spherical nanoparticles instead of flake-like nanostructures. As a result, the effective surface area increases and hence the number of effective adsorption sites also increases. However, on increasing the doping amount from 5% to 20%, the porosity of the surface of the nanostructure is seen to decrease (from Fig. 4(c)) and the spherical-like particle forms a coral-like nanostructure. As a result, surface porosity decreases, and hence the sensitivity and limit of detection are expected to decrease.
 | ||
| Fig. 5 Change in morphology of CuxNi(1−x)O based on percentage of Cu doping (% by weight) (a) 5% (b) 10% (c) 20%. | ||
The coral-like structure is selected because it is expected to provide more adsorption sites, owing to the induced surface roughness and intentionally generated surface defects. The surface morphology depends largely on the precursors used in the synthesis. In this regard, it should be mentioned that urea is used to produce an alkaline environment, which favours the heterogeneous nucleation responsible for the growth of coral-like mesoporous nanostructures.18 Hydrolysis of urea results in the formation of NH3 (NH4+ in solution) and CO32− at a temperature above 90 °C, which makes the solution alkaline and thus favourable for heterogeneous nucleation. Also, the bubble templating effect due to urea hydrolysis can be considered to be a synergistic cause of formation of hierarchical nanostructures. This takes place by the association of hydrogen bubbles in situ, thereby resulting in effective heteronucleation19 on PEG chains as templates to form directional Ni(OH)2 growth.
To investigate the sensing of Cr(VI) ions in aqueous solution by the prepared sensing materials, cyclic voltammetry was performed within the range −1 V to +1 V. It is observed from Fig. 6(a)–(c) that the current decreases with a decrease in the concentration of Cr(VI) in water. After a certain concentration of Cr(VI), it is observed that the current starts to increase and eventually the curve (i.e. current) becomes almost equal to the curve without an analyte (i.e. Cr(VI) ions in solution), which is shown in Fig. 6(a). Thus, the limit of detection can be considered to be the concentration at which a detectable reduction in current or an increase in resistance is maximum. The percentage increase in resistance is considered to be the sensitivity of the sensor. Moreover, the limit of detection is seen to depend on the electrode material, and this improved from NiO to Ni2O3 and then to CuxNi(1−x)O. The resistance is calculated from the measured current at 1 V. Table 2 represents the current (in mA), percentage change in current (from reference), resistance (at 1 V) and the sensitivity (percentage increase in resistance) for the three sensing oxide materials (NiO, Ni2O3 and CuxNi(1−x)O).
 | ||
| Fig. 6 C–V plots of (a) NiO (b) Ni2O3 and (c) Cu(1−x)NixO hierarchical nanostructures (−1 V to +1 V). | ||
| Concentration of Cr(VI) ions | NiO (coral-like nanostructure) | Ni2O3 (coral-like nanostructure) | CuxNi(1−x)O (coral-like nanostructure) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (ppm) | Sensed current (base current = 2.5 mA) | Increase in resistance (kΩ) | Response | Sensed current (base current = 1.3 mA) | Increase in resistance (kΩ) | Response | Sensed current (base current = 7 mA) | Increase in resistance (kΩ) | Response |
| 200 | 2.45 | 23.557 | 57.78 | 0.425 | 1.559 | 1.30 | 1.1 | 0.766 | 5.36 |
| 150 | 2.47 | 35.326 | 87.14 | 0.31 | 2.432 | 2.31 | 0.9 | 0.968 | 7.21 |
| 100 | 2.48 | 52.370 | 129.65 | 0.148 | 5.963 | 6.63 | 0.734 | 1.220 | 8.54 |
| 50 | — | — | — | 0.092 | 10.076 | 11.77 | 0.118 | 8.332 | 58.32 |
| 10 | — | — | — | 0.037 | 26.380 | 32.27 | 0.093 | 10.610 | 74.27 |
| 1 | — | — | — | — | — | — | 0.0276 | 36.089 | 252.62 |
Mechanism of Cr(VI) ion sensing in aqueous medium
Cr(VI) ion sensing by NiO and its variants can be explained by the mechanism of adsorption. In the nano regime, nickel oxides show excellent adsorption capacity of Cr(VI) ions, due to the formation of a porous structure, which aids the creation of a large number of surface defects.20–22 The probable sensing mechanism can be explained based on the adsorption capacity of Cr(VI) ions on the oxide surface. The schematic diagram for the sensing mechanism is predicted in Fig. 7. | ||
| Fig. 7 Schematic of a sensing mechanism by prepared oxide electrodes. | ||
The complete sensing mechanism by adsorption can be subdivided into the stages shown below.
Measurement of reference/base current
For any sensing activity, a reference must be set in order to measure the sensor parameters (i.e. sensitivity, resolution, limit of detection etc.) In this technique, a reference current is set by measuring the peak current of the electrode material in normal electrolyte (0.1 M NaCl solution) without any analyte. A certain current I0 is obtained that corresponds to the conductivity of the normal oxide–aluminium Schottky electrode. This current depends on the conductivity of the electrode material and hence should be different for different electrode materials (variants of NiO), as evident in Table 2.Adsorption of analyte onto the electrode surface Cr(VI) ions
When the analyte (i.e. Cr(VI) ions) is introduced into the solution, due to the porous surface morphology of the nanostructure, the Cr(VI) ions attach readily to the oxide surface. It is known from the literature that oxides of nickel can adsorb Cr(VI) ions23 very readily due to their negatively charged surface potential,13 surface morphology and surface porosity.25 The rate of adsorption increases with increase in concentration of Cr(VI) ions up to a certain concentration depending on the chemistry of the material. This may be attributed to the differences in defects and surface dangling bonds that act as adsorption sites in the three different materials. Doping of copper into the NiO crystal structure generates strain in the structure, which aids an increase in the number of adsorption sites. Also, with copper being more electron-rich, it supplies more electrons to the NiO structure, thereby increasing the adsorption efficiency relative to its un-doped counterparts. The fate and movement of Cr(VI) ions in aqueous solution in the presence of nickel oxide is due to the development of a partial positive charge over the Cr atom, which results in effective adsorption of the Cr(VI) species on the negatively charged adsorption sites.26 As the adsorption increases, the peak current in the solution decreases. This is due to the fact that the ionized electrolyte and dissolved ionic radicals aid the movement of current within an electrochemical cell. As the number of Cr(VI) ions decreases within the solution (due to their adsorption onto the electrode surface), the effective current also decreases. Thus, the resistivity at a particular voltage increases. Thus, it may be considered that the rate of adsorption (r) is directly proportional to the peak current change (ΔI), where ΔI is I0 − Isense.So, ΔI = I0 − Isense = K·r (where ‘Isense’ is the current after adding the analyte and ‘K’ is the constant of proportionality)
From Ohm's law,

The response of the sensor (S) can be expressed as a percentage by the following relation:
 | (1) |
i.e.
 (in times if more than 100%)
(in times if more than 100%)
The adsorption of Cr(VI) ions by NiO, Ni2O3 and Cu-doped NiO coral-like nanostructures has been tested by the batch adsorption technique. The details of the batch adsorption technique are explained in ESI.† The variation of adsorption rate with concentration is shown in Fig. 8.
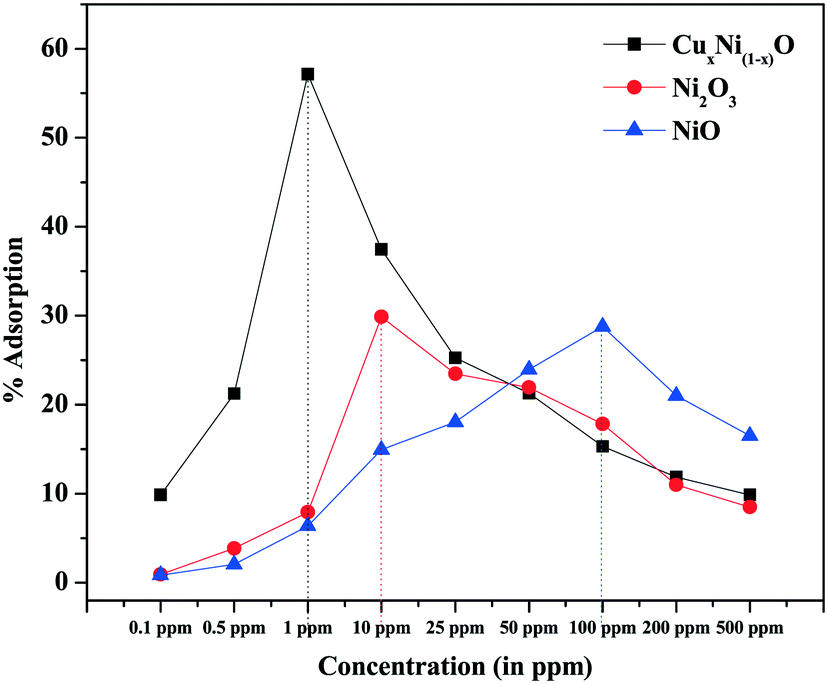 | ||
| Fig. 8 Variation of adsorption with concentration of Cr(VI) in ppm. | ||
From Fig. 8, it is observed that, as the concentration of Cr(VI) ions is increased, the adsorption rate is increased until a certain concentration depending on the chemistry of the adsorbent, which determines the number of adsorption sites and the surface potential (which are the determining factors for the adsorption of heavy metal ions). After this critical concentration, the rate of adsorption again decreases. It may be considered that, at very low concentrations, the number of heavy metal ions is less than the total number of available adsorption sites. The rate of adsorption is less and hence the current generated in the cathode by the electrochemical cell is very close to the base current (without analyte). After a critical concentration at which the adsorption is maximal, all the available adsorption sites are saturated. Hence, any concentration above the critical concentration would result in a decreased adsorption rate relative to the concentration. As the number of Cr(VI) ions in the electrolyte solution increases with increase in concentration, the current would saturate very close to the lowest current. Thus, for any concentration below the critical concentration of Cr(VI) ions Ccritical, if any current above the minimum current is observed then it can be due to the synergistic effect of excess Cr(VI) and Na+ ions in solution. However, this current is negligible and within the error bars for the experiments carried out. The possible reduction of current from the base current takes place because some of the sites having unsatisfied electrons (dangling bonds) act as adsorption sites and adsorb few Cr(VI) ions, and hence electrolyte discharge is not possible on those sites. When the concentration of Cr(VI) ions is greater than Ccritical, the total current contributed by the electrochemical cell has a synergistic effect on the excess un-adsorbed Cr(VI) and Na+ ions in solution. However, due to saturation of all the dangling bonds in the electrode, the current is still much lower than the base current and very close to the current corresponding to the concentration of maximum adsorption (though slightly higher than the minimum current). Hence, the concentration with the maximum adsorption (a Gaussian-type curve is expected for variation of adsorption rate) at which the minimum current (as well as the saturation current) is observed can be considered to be the limit of detection achieved by the sensor (Fig. 8).
Adsorption by a nanostructure is a surface phenomenon. Thus, it is expected to depend largely on the surface morphology of the prepared nanostructure. It is observed from Fig. 2 and 3 that, due to possible Cu doping, the coral-like structure so formed is composed of two types of basic units, i.e. filaments (and elongated particles) and flakes. When compared with the pure oxides, it is found that the coral-like structures are made of only one type of structure (either filament or flakes). Thus, the effective surface area of the surface available for adsorption is expected to increase due to the possible Cu doping when compared with pure oxides, as a result of which there is an increased adsorption of Cr(VI) ions. Doping with a more electron-rich species (i.e. Cu in comparison with Ni) is expected to increase the electron richness of the surface, thereby providing more negative centres for the effective adsorption of positive Cr(VI) ions (e.g. cationic centres of hydrated dichromate radicals at the mild ambient pH of waste water). This is evident from the zeta potential (surface charge) measurement of doped and pure NiO samples. The positivity of the surface is found to decrease ∼6.6 times in the doped sample in comparison with the pure NiO sample (5.51 mV for CuxNi(1−x)O and 36.3 mV for NiO coral-like nanostructures) (graphs provided in ESI,† Fig. S5 and S6).
To study the porosity of the optimized sample, a N2 adsorption–desorption experiment (BET/BJH) was performed for the CuxNi(1−x)O (10%-doped) sample (Fig. 9). The outgassing time was 180 min and the temperature was 150 °C. The outgassing time and temperature were recorded prior to exposure of the material to the adsorbent. Significant changes (if any) in the weight of the sample were recorded prior to the N2 adsorption–desorption experiment.27 It was observed that the surface area for adsorption for the sample was 44.543 m2 g−1 with a pore volume of 0.29 cc g−1 and an average pore radius of 15.718 Å (1.572 nm). Thus, from the BET study, it can be concluded that the prepared materials are very porous, with a higher surface area available for adsorption as compared with the pure NiO nanostructures (surface area ∼33 m2 g−1),27 and hence they show a comparably higher adsorption rate from their pure oxide counterparts. From the nature of the isotherm, it can be considered to be a typical mesoporous material. However, the pore size is slightly less as compared with those of mesoporous materials (∼2 nm to 50 nm). Thus, the material can be considered to be a pseudo-mesoporous material, which is microporous but tends to be mesoporous.
 | ||
| Fig. 9 N2 adsorption–desorption isotherm (BET) study of CuxNi(1−x)O (10% doped) sample. | ||
Desorption of the adsorbed Cr(VI) ions from the electrode material
As adsorption takes place, the sensing material can be revived by performing desorption of the adsorbed Cr(VI) ions. The electrode material carrying adsorbed Cr(VI) was washed with DI water or 0.1 M NaOH solution. When washed with NaOH, OH−, due to its more reactive affinity, readily forms Cr(OH)3 with the adsorbed Cr(VI), which is easily washed off in any liquid medium. The reactivity of the electrode materials (i.e. NiO, Ni2O3 and CuxNi(1−x)O) was studied and found to be stable (no reaction) in the presence of NaOH. Thus, the Cr(VI) desorbs from the electrode material such that it can be re-used several times.Effect of Cu doping on sensitivity to Cr(VI) ions in aqueous solution
To study the effect of Cu doping on NiO in sensing Cr(VI) ions in solution, three different doping concentrations, i.e. 5%, 10% and 20% of Cu doping, were prepared. The structure with 10% Cu doping was seen to display the maximum limit of detection and sensitivity, and the FESEM image shows it to be a combination of spherical nanoparticles and flake-like morphology to form a coral-like nanostructure. The formation of the maximum number of adsorption sites may be attributed to the formation of a mixed structured hierarchical morphology which is obtained with the 10% Cu-doped NiO sample. The sensing study for Cr(VI) ions was carried out with the three structures. It was observed that maximum sensitivity was shown by 10% Cu-doped NiO, as shown in Table 3.| Concentration of Cr(VI) ions | 5% Cu-doped NiO | 10% Cu-doped NiO | 20% Cu-doped NiO | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (in ppm) | Sensed current (base current = 34 mA) | Increase in resistance (Ω) | Response | Sensed current (base current = 7 mA) | Increase in resistance (kΩ) | Response | Sensed current (base current = 44 mA) | Increase in resistance (Ω) | Response |
| 200 | 30.8 | 3.06 | 0.104 | 1.1 | 0.766 | 5.36 | 41.6 | 1.287 | 0.05 |
| 100 | 28.7 | 5.43 | 0.18.46 | 0.9 | 0.968 | 8.54 | 31.5 | 8.996 | 0.396 |
| 50 | 24.3 | 11.74 | 0.3992 | 0.734 | 1.220 | 58.32 | 25.2 | 16.81 | 0.74 |
| 25 | 23.1 | 13.88 | 0.472 | 0.118 | 8.332 | 61.25 | — | — | — |
| 10 | 17.9 | 26.45 | 0.899 | 0.093 | 10.610 | 74.27 | — | — | — |
| 1 | — | — | — | 0.0276 | 36.089 | 252.62 | — | — | — |
From the results, it was observed that the sensitivity increased dramatically for 10% Cu-doped NiO in comparison with the other two variants. Also, the limit of detection was greater compared with the other two variants. This may be attributed to the effective adsorption of Cr(VI) on the Cu-doped NiO and the dependence of adsorption rate on the doping amount of Cu on the NiO. It also explained the morphological dependency of adsorption rate, which influenced the sensitivity of the sensing material. The response time (time required to detect a measurable change in Cr(VI) ion concentration in aqueous medium) for Cr(VI) ions also decreased with respect to the surface morphology, and was seen to be least for 10% Cu-doped NiO with respect to the other two variants (shown in Fig. S3 in ESI†).
Reusability studies on Cr(VI) ion sensors
Conventional heavy metal sensing by electrochemical techniques involves one-off use of the sensing material. This is due to the fact that the sensing material must react (either oxidation or reduction) in order to produce a detectable output. However, in our proposed technique involving adsorption, the sensing material can be re-used several times. Also, reproducing the result in the same range is an issue for conventional electrochemical sensors.23 To test the reproducibility, our sensor was tested over a period of 1 month to 6 months. The reusability study was performed by successive desorption from the same device of the adsorbed Cr(VI) ion immediately after sensing. The results show that the electrode revived completely after 2 cycles of washing. In a typical cycle, after sensing the Cr(VI) ion (50 ppm), the electrode was washed in 20 ml of DI water for 1 minute and then tested in normal electrolyte solution (without Cr(VI) ion analyte). The percentage recovery is shown in Table 4.| Desorption cycle | Initial current (before adsorption) (in amperes) | Final current (after desorption) (in amperes) | Percentage recovery |
|---|---|---|---|
| Cycle 1 | 0.0266 | 0.0259 | 97.3 |
| Cycle 2 | 0.0266 | 0.0264 | 99.24 |
| Cycle 3 | 0.0266 | 0.0264 | 99.24 |
From Table 4, it can be concluded that after two desorption cycles, complete recovery of the sensing material can be obtained with a percentage error of 0.75% and hence the material can be re-used for sensing.
To investigate reproducibility, Cr(VI) ion-sensing experiments were carried out periodically for up to 6 months and the percentage variation was analyzed. The reproducibility results are presented in Fig. 10. It is observed that the sensitivity error for the first month, for all concentrations, is within ∼0.5%. The error increases with time, showing a maximum of ∼2.5% at the end of 3 months (90 days) and ∼7.5% at the end of 6 months (180 days).
 | ||
| Fig. 10 Reproducibility of a Cu-doped NiO (10% Cu by weight) based Cr(VI) ion sensor. | ||
Selectivity of Cr(VI) ions over other heavy metal cations
The selectivity of the sensor material for Cr(VI) was tested relative to three heavy metal pollutants which are very common in drinking water: As(V), Pb(II) and Cd(II). It was observed that the prepared material is selective for Cr(VI) ions over other heavy metal cations, showing a high percentage sensitivity for the three concentrations, as shown in Fig. 11. | ||
| Fig. 11 Selectivity of a Cu doped NiO (10% Cu by weight) sensor for Cr(VI) over Cd(II), As(V) and Pb(II) ions. | ||
The effect of the K+ ion (sourced from K2Cr2O7) was also considered and the material was tested for any possible sensitivity to K+ ions that might affect the overall sensitivity. In a typical experiment, KCl (as source of K+) was used as an analyte and sensing measurements were performed. However, no significant change in base current was observed (less than 0.1% change). Thus, it may be considered that the sensitivity observed with Cr(VI) ions (sourced from K2Cr2O7) as an analyte may be solely due to Cr(VI) ions.
Negative adsorption sites are formed due to an excess electronic cloud (due to Cu doping) and cationic vacancies, whereas adsorption sites in pure oxides are due only to cationic vacancies. Thus, it should be capable of sharing 6 electrons to satisfy the Cr(VI) ion to its filled configuration. Moreover, the cations and the analyte (i.e. Cr(VI) ions) have comparable atomic radii. Hence, they are more accessible to the cationic defect sites acting as active adsorption sites. Therefore, due to the cumulative effect, the effective adsorption for Cr(VI) is much more than for other heavy metal cations. This results in the selective detection of Cr(VI) over other analytes.
Response time of a Cu doped NiO based Cr(VI) ions sensor
The response time of the sensor was tested using amperometric studies, whereby 1 ppm Cr(VI) ions were introduced in the electrolyte as the analyte and change in current was recorded against time and plotted. The Cr(VI) ion sensor was found to sense Cr(VI) ion at ∼2 s, as represented in Fig. 12. The response time was found to be much lower than that reported in the literature (9 s).24 The response time was found to be a minimum for the 10% doped sample (∼11 s for the 5% doped sample and ∼4.5 s for the 20% doped sample). This may be attributed to the possible defects formed due to the incorporation of Cu in the NiO lattice (amperometric plots for the 5% and 20% doped variants are provided in ESI† Fig. S3). | ||
| Fig. 12 (a) Response time of a Cu-doped NiO sensor for Cr(VI) ions. (b) Amperometric plot for 10% Cu-doped NiO. | ||
Conclusions
From the above results, it is concluded that an adsorption-based sensing mechanism can be used for sensing trace amounts of heavy metals in aqueous solution. Using this technique, sensing materials can be reused for sensing several times (tested for up to six months) without significant error (∼7.5% for 200 ppm), thereby making the technique exceptionally industry-friendly and cost-efficient. Moreover, the Cu-doped nickel oxide material shows excellent sensitivity, a low limit of detection (1 ppm) and selectivity towards Cr(VI) ions over As, Pb and Cd species in water, and with a very fast response of ∼2 seconds. In the proposed sensing technique, inert but high-adsorbing materials, such as mesoporous carbon, charcoal and their composites, can be used for sensing, thereby reducing the manufacturing cost relative to conventional materials. Conventional electrochemical sensing materials require specific synthetic techniques, thereby increasing the cost of production of the sensor. Thus, the proposed sensor may be used effectively to sense Cr(VI) ions in water, thereby aiding the efficient monitoring of water quality and drinking water standards.Acknowledgements
S. S. acknowledges the Department of Science and Technology (DST), India, for partial support of the work (Project SR/S2/RJN-104/2011). A. M. acknowledges the Department of Science and Technology (DST), India, for financial support (Project Number: IFA 13-MS-09). We acknowledge the DST FIST Laboratory (in the Department of Physics & Meteorology) for the use of the X-ray photoelectron spectrometer. We also acknowledge Dr. D. Pradhan for the BET N2 adsorption–desorption measurements.References
- R. Han, W. Zou, Z. Zhang, J. Shi and J. Yang, J. Hazard. Mater., 2006, 137, 384–395 CrossRef CAS PubMed.
- A. Zhitkovich, Chem. Res. Toxicol., 2011, 24, 1617–1629 Search PubMed.
- S. A. Katz and H. Salem, J. Appl. Toxicol., 1993, 13, 217–224 CrossRef CAS PubMed.
- L. Tan, J. Wang, Q. Liu, Y. Sun, X. Jing, L. Liu, J. Liu and D. Song, New J. Chem., 2015, 39, 868–876 RSC.
- M. E. Grass, Y. Yue, S. E. Habas, R. M. Rioux, C. I. Teall, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 4797–4804 CrossRef CAS.
- E. Comini, G. Faglia, G. Sberveglieri, Z. Pan and Z. L. Wang, Appl. Phys. Lett., 2002, 81, 1869–1871 CrossRef CAS.
- J. Zieliński, J. Mol. Catal., 1993, 83, 197–206 CrossRef.
- J. Zieliński, Appl. Catal., A, 1993, 94, 107–115 CrossRef.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- S. H. Park, S. H. Kang, C. S. Johnson, K. Amine and M. M. Thackeray, Electrochem. Commun., 2007, 9, 262–268 CrossRef CAS.
- J. Heffron, M. Marhefke and B. K. Mayer, Sci. Rep., 2016, 6, 28478 CrossRef CAS PubMed.
- S. Dey, S. Bhattacharjee, M. G. Chaudhuri, R. S. Bose, S. Halder and C. K. Ghosh, RSC Adv., 2015, 5, 54717–54726 RSC.
- T. N. Ravishankar, S. Muralikrishna, K. Suresh Kumar, G. Nagaraju and T. Ramakrishnappa, Anal. Methods, 2015, 7, 3493–3499 RSC.
- S. Sadeghi and A. Garmroodi, Mater. Sci. Eng., C, 2013, 33, 4972–4977 CrossRef CAS PubMed.
- V. D. Mote, Y. Purushotham and B. N. Dole, J. Theor. Appl. Phys., 2012, 6, 1–8 CrossRef.
- M. Jamal, S. Jalali Asadabadi, I. Ahmad and H. A. Rahnamaye Aliabad, Comput. Mater. Sci., 2014, 95, 592–599 CrossRef CAS.
- J. H. Pan, X. Zhang, A. J. Du, H. Bai, J. Ng and D. Sun, Phys. Chem. Chem. Phys., 2012, 14, 7481–7489 RSC.
- (a) W. Cao, J. Kang, G. Fan, L. Yang and F. Li, Ind. Eng. Chem. Res., 2015, 54, 12795–12804 CrossRef CAS; (b) L. Wei, G. Yang, R. Wang and W. Ma, J. Hazard. Mater., 2009, 164, 1159–1163 CrossRef CAS PubMed.
- C. Chen, J. Hu, D. Shao, J. Li and X. Wang, J. Hazard. Mater., 2009, 164, 923–928 CrossRef CAS PubMed.
- M. A. Behnajady and S. Bimeghdar, Chem. Eng. J., 2014, 239, 105–113 CrossRef CAS.
- E.-T. Liu, H. Zhao, H. Li, G. Li, Y. Liu and R. Chen, New J. Chem., 2014, 38, 2911–2916 RSC.
- S. Dey, S. Bhattacharjee, R. S. Bose and C. K. Ghosh, Appl. Phys. A: Mater. Sci. Process., 2015, 119, 1343–1354 CrossRef CAS.
- W. Jin and K. Yan, RSC Adv., 2015, 5, 37440–37450 RSC.
- T. A. Ali, A. L. Saber, G. G. Mohamed and T. M. Bawazeer, Int. J. Electrochem. Sci., 2014, 9, 4932–4943 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CAS.
- H. Qiao, Z. Wei, H. Yang, L. Zhu and X. Yan, J. Nanomater., 2009, 2009, 1–5 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6en00285d |
| This journal is © The Royal Society of Chemistry 2017 |