
Emerging investigator series: the effect of wildfire on streamwater mercury and organic carbon in a forested watershed in the southeastern United States†

Abstract
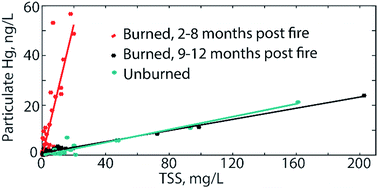
Wildfires alter forested ecosystems, which include large stores of mercury (Hg) and organic carbon, two compounds that are closely linked in vegetation, soils, and streamwater. Studies have shown that wildfires release elevated levels of mercury to the atmosphere which can be locally redeposited and leave charred organic material (vegetation and litter) on the soil surface. Both can contribute to the elevated mobilization of Hg into lakes and streams. However, no studies have conducted a detailed examination of hydrological transport of Hg following a wildfire. This study investigates the coupled transport of mercury and carbon at Twomile Run, a headwater stream located in the forested mountains of Shenandoah National Park, in the year following a low-severity wildfire. Weekly baseflow samples and bi-hourly high-flow storm samples were analyzed for dissolved and particulate mercury (HgD and HgP, respectively), dissolved organic carbon (DOC), UV absorbance at 254 nm (UV254, surrogate for DOC quantity and character), and total suspended solids (TSS), and were compared with identical measurements taken from a nearby unburned watershed. For all flow conditions sampled at the burned site (which did not include the 2 months following the fire), streamwater HgD and DOC concentrations, and corresponding UV254, were similar to the unburned system. TSS concentrations varied between sites but overall differences were relatively small in magnitude and likely attributable to site differences rather than fire effects. Notably, the HgP per unit of TSS at the burned site was an order of magnitude higher than the unburned site (2.66 and 0.13 ng HgP per mg TSS, respectively) for 8 months following the fire, resulting in elevated HgP concentrations for the range of flow conditions, after which there was a rapid return to non-disturbed conditions. Streamwater total Hg fluxes roughly doubled (0.55 to 1.04 μg m−2 yr−1) as a consequence of the fire, indicating that in addition to changing atmospheric and terrestrial Hg cycling, fires can rapidly and significantly alter the streamwater Hg which has implication for downstream ecosystems. These findings are particularly relevant as the occurrence and severity of wildfires are expected to increase in the mid-latitudes in response to climate change.
- This article is part of the themed collections: Emerging Investigator Series and Mercury Biogeochemistry, Exposure, and Impacts
 Please wait while we load your content...
Please wait while we load your content...

