
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indole-based conjugated macromolecules as a redox-mediated electrolyte for an ultrahigh power supercapacitor†
Ting
Xiong‡
ab,
Wee Siang Vincent
Lee‡
 a,
Li
Chen
c,
Teck Leong
Tan
d,
Xiaolei
Huang
*a and
Junmin
Xue
*a
a,
Li
Chen
c,
Teck Leong
Tan
d,
Xiaolei
Huang
*a and
Junmin
Xue
*a
aNational University of Singapore, Department of Materials Science and Engineering, Singapore 117573, Singapore. E-mail: msexuejm@nus.edu.sg; msehx@nus.edu.sg; Fax: +65 65164655; Tel: +65 65164655
bCentre for Advanced 2D Materials and Graphene Research Centre, National University of Singapore, Singapore 117546, Singapore
cDepartment of Chemistry, Tianjin University, Tianjin 300072, People's Republic of China
dInstitute of High Performance Computing, A*STAR, 1 Fusionopolis Way, #16-16 Connexis, Singapore 138632, Singapore
First published on 6th October 2017
Abstract
Balancing energy density and power density has been a critical challenge since the inception of supercapacitors. Introducing redox-active additives in the supporting electrolyte has been shown to increase the energy density, however the power density and cycling stability are severely hampered in the process. Herein, an extensively conjugated indole-based macromolecule consisting of 5,6-dihydroxyindole/5,6-quinoneindole motifs, prepared by electrochemical polymerization of dopamine under acidic conditions, was employed as a redox-active additive. By utilizing the conjugation effect, the HOMO–LUMO gap (HLG) of the extensively conjugated indole-based macromolecule was reduced to ca. 2.08 eV, which enhanced the electronic transfer kinetics, in turn improving the power density and reversibility of redox reactions. When coupled with a porous honeycomb-like carbon (PHC) electrode, the assembled supercapacitor delivered an excellent rate performance with a high specific capacitance of 205 F g−1 at 1000 A g−1. This work reports one of the highest power densities recorded at 153 kW kg−1 for redox-mediated electrolyte systems with a respectable energy density of 8.8 W h kg−1. In addition to an excellent cycling stability of 97.1% capacitance retention after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge/discharge cycles, the conjugation degree has to be considered when engineering the redox-active electrolyte so as to improve the power density and stability.
000 charge/discharge cycles, the conjugation degree has to be considered when engineering the redox-active electrolyte so as to improve the power density and stability.
Broader contextPortable and reliable energy storage devices (ESDs) have become gradually integrated into technological advancements. Supercapacitors have gained significant recognition as important ESDs due to their high power density arising from fast physical ion adsorption based on the electric double layer (EDL) principle. However, the fast EDL principle is dependent on the electrode surface area which ironically is responsible for poor energy density. Thus, such a limitation has resulted in a research interest shift towards pseudocapacitive capacitors as additional pseudocapacitance can be achieved from the Faradaic reaction. One strategy to incorporate pseudocapacitance is via the addition of small redox species into the supporting electrolyte (resulting in 5–7 times energy density enhancement). However, the use of small redox species typically leads to a poor power density and poor cycling stability which renders such energy density enhancement insignificant. In this work, a supercapacitor based on a novel indole-based conjugated macromolecule redox-mediated electrolyte is reported to achieve one of the highest power densities in redox mediated electrolyte supercapacitor research, while excellent cycling stability was simultaneously attained in this system. |
1. Introduction
Electric double layer capacitors (EDLCs) are widely favoured in high power applications such as regenerative braking due to their high power density of more than 10 kW kg−1.1–4 Despite the fast physical adsorption/desorption mechanism present in the electric double layer principle, EDLCs are fundamentally dependent on the surface area which ultimately creates a capacitance bottleneck (energy density of 5–10 W h kg−1)5–7 that needs to be urgently addressed. As such, research into pseudocapacitive systems has gained increasing traction in recent decades,8,9 whereby incorporating transition metal oxides,10,11 conducting polymers,12 and grafting functional groups onto carbon electrode materials13 are considered to be a few of the most commonly favoured synthesis techniques. However these strategies not only increase the synthesis complexity, but the electrode stability and power density can also be significantly reduced which poses some key challenges for pseudocapacitors.10–13 Hence, alternative means of incorporating pseudocapacitance have to be explored in greater detail so as to achieve a high energy density without sacrificing power density greatly.Recently, redox-mediated electrolytes have gained increasing attention due to their efficiency in enhancing the energy storage of traditional EDLCs. Such a method is attractive as a simple and reliable carbon-based electrode material can be used which greatly reduces the fabrication cost and complexity. Furthermore, the preparation process for the redox-mediated electrolyte is rather straightforward as controlled amounts of redox-active species are added directly into the supporting electrolyte.14–17 To date, small organic redox-active species such as hydroquinone,18p-phenylenediamine,19p-nitroaniline,20 methylene blue,21 indigo carmine22etc. have been reported to show remarkable improvements in capacitance of nearly 5–7 fold as compared to that in a pure supporting electrolyte. However, one major flaw of using small organic molecules lies with the large HOMO–LUMO gap (HLG) which reduces the ease of electron transfer from the donor groups to the acceptor groups.23,24 Due to the large HOMO–LUMO gap, i.e. lower electronic conductivity, electrons have to overcome a higher energy barrier in order for the electron transfer to take place, which inevitably leads to a lower power density. Hence, most supercapacitors that employ small organic molecules as redox-active additives do not emphasize power densities as an electrochemical highlight due to this limitation. Thus, a more efficient and highly conductive redox active species system is highly desirable so as to harmonize the improved energy density with enhanced power density.
To enhance the electron transfer kinetics, reducing the HOMO–LUMO gap (HLG) of the organic molecule is one established method25 which can be achieved via extending conjugation.26 In addition to improving the conductivity, i.e. reduced HLG, due to the delocalized π-electron in conjugated systems, extra stability in terms of delocalization energy is conferred to the conjugated molecule which translates to higher structural stability.27 Furthermore, multiple redox centres are available in the conjugated structure which will aid in further enhancing the capacitance.28 Therefore, engineering an extensively conjugated macromolecule as a redox-active additive should address the problems brought forward by the small redox-additive molecules.
Irrefutably, engineering a redox-active conjugated macromolecule electrolyte is important towards achieving high energy and power density, however compatible electrode–electrolyte interaction (e.g. ease of electrolyte accessibility, large surface area, electrolyte wettability etc.) is also necessary to further enhance its performance.1 Hence, designing an electrode material to complement the redox-active electrolyte is considered to be an essential step. Activated carbon is regarded as the holy-grail in ELDC materials, but the limitation still lies in its pore structure and morphology.29 Therefore to improve the electrode–electrolyte interaction, a specialized carbon design has to be considered such as (1) a highly interconnected carbon network to facilitate rapid ion/electron transport,30 (2) a high surface area with nano-pores of ≤2 nm which is crucial for high energy density,31 and (3) nitrogen doping on carbon to firstly impart pseudocapacitance and secondly increase the wettability towards aqueous electrolytes,32 so as to improve on the traditional activated carbon.
Herein, an indole-based conjugated macromolecule consisting of a 5,6 dihydroxyindole/5,6-qunioneindole motif, to the best of our knowledge, is proposed for the first time as a redox-active additive. The system can be prepared via the simple electrochemical polymerization of dopamine in an acidic supporting medium. The formation of the extensively conjugated structure firstly enhanced the electronic transfer kinetics due to lower HLG, and secondly increased the stability of the structure due to higher delocalization energy. When coupled with the interconnected porous, honeycomb-like carbon, the assembled supercapacitor delivered an excellent rate performance with a high specific capacitance of 205 F g−1 at an ultra-high current density of 1000 A g−1. A maximum power density of 153 kW kg−1 was recorded as one of the highest reported power densities in a redox-active electrolyte system, with a respectable energy density of 8.8 W h kg−1, suggesting a successful harmonious blend between energy and power density. On top of the ultra-high power density, the system demonstrated an excellent cycling stability of 97.1% capacitance retention after 20000 charge/discharge cycles. Based on the collective results, such an indole-based conjugated macromolecular structure is hence shown to effectively lower the electronic transfer barrier and thus improve the power density.
2. Experimental section
2.1 Materials and methods
2.2 Characterization
Scanning electron microscopy (SEM) images were recorded on a ZEISS SEM Supra 40 (5 kV). SEM samples were prepared by dripping the sample solutions onto a silicon substrate. Transmission electron microscopy (TEM) was performed on a JEOL-3010 (300 kV acceleration voltage). TEM samples were prepared by dripping the sample solutions onto a copper grid. Raman spectrometry was conducted on a Horiba MicroRaman HR Evolution System using an argon laser beam with an excitation wavelength of 514.5 nm. Surface composition was analyzed by X-ray photoelectron spectroscopy (XPS) using a Kratos Analytical Axis UltraDLD UHV spectrometer with a monochromatized Al Kα X-ray source (1486.6 eV) scanning a spot size of 700 μm by 300 μm. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded on a high resolution FTIR spectrometer (IFS 125HR). The size distribution of the molecules in the BRE0.025M electrolyte was measured using a Malvern Zetasizer Nano ZS. The Brunauer–Emmett–Teller surface area was measured using a Micromerities ASAP 2020 Surface Area and Porosity Analyzer. UV-visible spectra were measured on a UV-1800 Shimadzu UV-VIS spectrophotometer with an optical filter that was calibrated at a bandwidth of 1 nm using a quartz cuvette with a 10 mm light path. Gel permeation chromatography was carried out using an Alliance Waters 2690 system with HPLC grade THF as the eluent and calibrated against poly(methyl methacrylate) (PMMA).2.3 Electrochemical measurements
All electrochemical tests were performed at room temperature. Cyclic voltammetry, galvanostatic charge/discharge measurement, and electrical impedance spectroscopy were conducted using an electrochemical system (Bio-logic VMP 3). To test the electrode materials, porous honeycomb-like carbon (activated carbon or another porous carbon) was mixed with polyvinylidene fluoride (PVDF) and carbon black in a ratio of 8:1:1 with N-methyl-2-pyrrolidone (NMP). The mixture was hand-ground for at least 10 min to obtain a slurry. The slurry was later coated onto carbon paper which served as a current collector. After heating at 80 °C overnight, the carbon paper was cut to 1 cm × 1 cm. For electrolyte preparation, a certain amount of dopamine chloride was dissolved into 50 mL of 0.5 M H2SO4 to obtain 0.01 mol L−1 (0.01 M), 0.025 M, and 0.05 M dopamine solutions, which were labelled as RE0.01M, RE0.025M and RE0.05M, respectively. The transparent RE0.025M electrolyte transformed into a homogenous brown aqueous solution in the three electrode configuration after CV tests at a scan rate of 1000 mV s−1 for 24 h, which was labelled as BRE0.025M. In the three-electrode system, Ag/AgCl was used as the reference electrode, porous honeycomb-like carbon served as the working electrode and counter electrode, and RE0.025M or BRE0.025M was used as the electrolyte. The supercapacitors were characterized in a standard Swagelok 2-electrode cell configuration, which employed titanium as current collectors which were acid-resistant. After matching their capacitance, the two electrodes (1 cm in diameter) were subsequently assembled together with a separator (normal filter paper) sandwiched in between. For both CV and charge/discharge of the full cell test, the measurement voltage was controlled in the range of 0–1.4 V for the aqueous electrolyte test. Scan rates of 25, 50, 100, 200, 500, 1000, 3000 and 5000 mV s−1 were used for CV, while the current densities of 1, 2, 5, 10, 20, 50, 100, 200, 500 and 1000 A g−1 were used for charge/discharge measurement. The mass of the electrodes for the porous honeycomb-like carbon, activated carbon or another porous carbon are approximately similar at ca. 1 mg. When calculating the current density, the weight of the individual electrode was used for the normalization.
The total gravimetric capacitance of a supercapacitor in Farads per gram was calculated from the galvanostatic discharge process according to the following equation:
| F = IΔt/ΔV | (1) |
Based on the total mass of the active materials from the positive and negative electrodes, the energy density (E) and power density (P) were calculated based on the following formula.
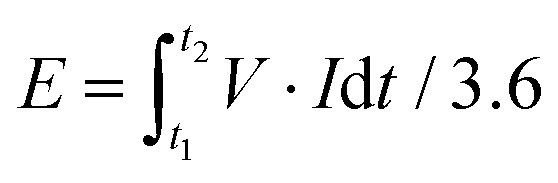 | (2) |
| P = 3600 E/Δt | (3) |
2.4 Calculation
Geometry optimizations was performed using the Vienna ab initio simulation package (VASP) based on density functional theory (DFT).33 Projector augmented wave (PAW) potentials were adopted to simulate electron–ion interactions.34 Generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was employed to approximate exchange and correlation effects for structural relaxation.35,36 In all calculations, the cut-off energy was set at 520 eV for the plane-wave basis restriction. K-Points were sampled under the Monkhorst–Pack scheme for the Brillouin-zone integration.37 All atomic models were fully relaxed. In the atomic structure relaxation, a self-consistency accuracy of 10−5 eV was reached for electronic loops. The free energy change (ΔG) was calculated by the following equation: ΔG = ΔEDFT + ΔEZPE − TΔS. ΔEDFT was calculated by ΔEDFT = E(redox+H) − Eredox − 1/2EH2, where Eredox and E(redox+H) are the total energy of the bare molecular model with the carbonyl group (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the molecular model with the hydroxyl radical (R–OH), respectively, and EH2 is the energy of a single isolated H2 molecule. ΔEZPE and ΔS are the zero-point energy change and the entropy change for the redox reaction, respectively. The redox potential (E0redox) of the redox reaction was calculated using the formula:
O) and the molecular model with the hydroxyl radical (R–OH), respectively, and EH2 is the energy of a single isolated H2 molecule. ΔEZPE and ΔS are the zero-point energy change and the entropy change for the redox reaction, respectively. The redox potential (E0redox) of the redox reaction was calculated using the formula:| E0redox = −ΔG/nF | (4) |
3. Results and discussion
Characterization of the indole-based conjugated redox-mediated electrolyte
Various small redox species have been investigated as redox-additives for redox-active electrolyte supercapacitors.38 In the present work, a new indole-based conjugated redox-mediated electrolyte consisting of 5,6-dihydroxyindole/5,6-quinoneindole motifs was prepared from electrochemical polymerization of dopamine in an acidic supporting electrolyte. The optimum amount of dopamine used to prepare the redox-mediated electrolyte was determined to be 0.025 M (see Fig. S1 (ESI†) for more details). In detail, the transparent dopamine solution (labelled as RE0.025M) was transformed into a homogenous brown aqueous solution (labelled as BRE0.025M) in three electrode configuration by performing CV tests at a scan rate of 1000 mV s−1 for 24 h in the range of −0.45 to 0.5 V (Ag/AgCl as reference electrode, porous honeycomb-like carbon (labelled as PHC) as working electrode and counter electrode). The electrolyte conversion process is recorded in Fig. S2 (ESI†). Note that when PHC was substituted with other non-nitrogen doped carbon materials (such as graphite paper and activated carbon), the electrolyte conversion process rate (indicated by the colour change) was significantly reduced. Such a phenomenon suggested that doping the N heteroatom into the prepared PHC increased the amount of active sites which in turn accelerated the conversion process.After cycling for 24 h, the brown electrolyte, denoted as BRE0.025M, was collected and its composition was investigated under FT-IR spectroscopy and Raman spectroscopy. The FT-IR spectra of RE0.025M and BRE0.025M were acquired as shown in Fig. 1a. RE0.025M displayed characteristic bands in the region of 3000–3600 cm−1, which were assigned to the stretching vibration of O–H and N–H bonds.39,40 The peak at 1191 cm−1 was due to the stretching of C–O bands.40 Besides, the band corresponding to CC in the benzene ring was detected at 1630 cm−1.41 These bands were consistent with the functional groups in dopamine. For BRE0.025M, the bands of O–H, C–O and CC were also detected. However, different from that in RE0.025M, the disappearance of the peaks at 1465 cm−1 (NH2 vibration) and 1297 cm−1 (CH2 bending vibration) verified that an intramolecular cyclization reaction had occurred in dopamine along with the formation of indole derivatives.42 Notably, the wide peak at around 1630 cm−1 might overlap with the peak at 1684 cm−1 which is characteristic of CO bonds. From the Raman spectra (Fig. 1b), two bands at 983 cm−1 and 1055 cm−1 were observed for both the RE0.025M and BRE0.025M electrolytes, which were associated with the O–H out of plane deformation vibration (C–H out of plane deformation vibration) and the C–H in plane deformation vibration, separately.43 A new band was observed at 1390 cm−1 for BRE0.025M, which was assigned to the CC and CN in plane vibration in the pyrrole ring vibration.43 In particular, a wide band appeared at a broad range of 1530 cm−1 to 1700 cm−1, which could be fitted into three bands at 1580 cm−1, 1614 cm−1 and 1648 cm−1. The vibrations at 1580 and 1648 cm−1 indicated the presence of a quinoid carbonyl group (CO),44 and the band at 1614 cm−1 was related to the indole ring vibration.43 Based on the combined results from FT-IR and Raman spectra, BRE0.025M was shown to be composed of 5,6-dihydroxyindole/5,6-quinoneindole motifs. The number weighted molecular weight of BRE0.025M was determined with gel permeation chromatography (GPC) to be ca. 21209 as shown in Fig. 1c, which suggests a polymerization degree of about 143. The polymerization degree of the macromolecule was further verified using a Zetasizer (Fig. S3a, ESI†). The size distribution of the molecules was determined to be in the range of 100–250 nm. In terms of the size of 5,6-dihydroxyindole and 5,6-quinoneindole, the polymerization degree was in the range of 118–260 which is consistent with the GPC result. Hence, such a polymerization may be due to the dopamine first forming the indole skeleton by oxidative ring closure with the subsequent connection of the monomer units to create the indole-based conjugated macromolecule.45 Based on the collective results, dopamine was successfully converted into an indole-based conjugated macromolecule composed of 5,6-dihydroxyindole/5,6-quinoneindole motifs under acidic conditions by a simple electrochemical polymerization method as indicated in Fig. 1d.
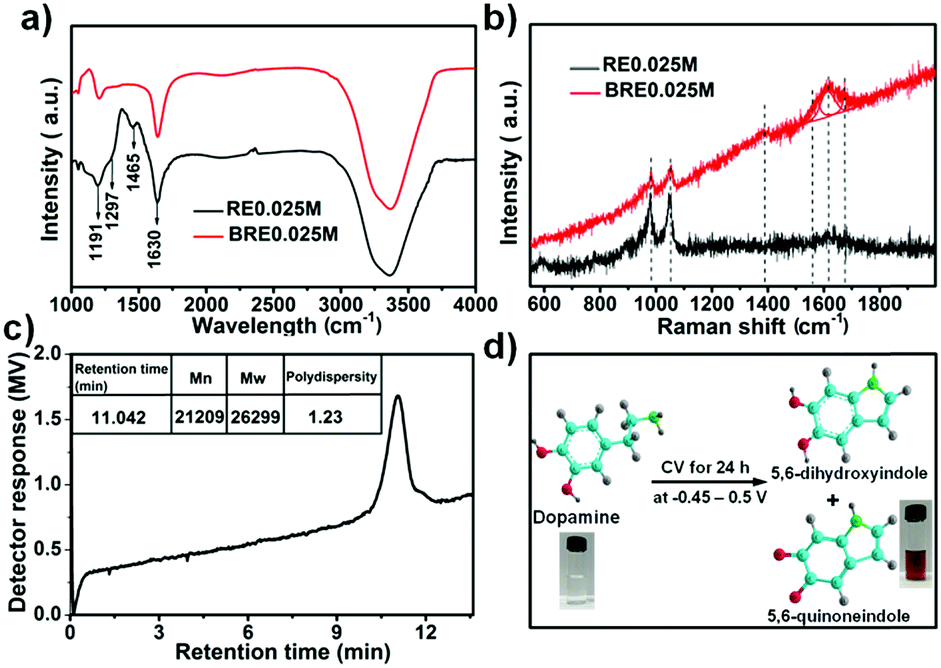 | ||
| Fig. 1 Characteristics of the RE0.025M and BRE0.025M electrolytes. (a) FT-IR spectra; (b) Raman spectra; (c) gel permeation chromatography analysis of the BRE0.025M electrolyte; (d) schematic illustration of the conversion process of dopamine into 5,6-dihydroxyindole and 5,6-quinoneindole motifs. | ||
Characterization of the PHC electrode material
To complement the redox-mediated electrolyte, high performance porous honeycomb-like carbon (labelled as PHC) was prepared. In a typical synthesis, g-C3N4 was firstly prepared by pyrolysis of urea. Subsequently, the raw material wheat flour was mixed with the as-prepared g-C3N4, followed by the addition of KOH. Finally, the dried mixture was annealed at 800 °C for 4 h in N2 flow and subsequently subjected to washing with dilute HCl solution and distilled water. The final product was obtained by drying in an oven at 80 °C. During the process, g-C3N4 served as the nitrogen doping source, while KOH acted as the activating agent.46,47 The three dimensional porous structure and the thin carbon wall were confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) as shown in Fig. 2a and b, respectively. The high-resolution transmission electron microscopy (HRTEM) image (Fig. 2c) at the edge of the PHC suggested a disordered character with a lattice spacing of 0.37 nm. This was further confirmed by the selected-area electron diffraction (SAED) pattern (Fig. 2c, inset) which displayed typical diffusive rings. High surface area and micropores are some essential parameters that the carbon has to possess so as to demonstrate a high capacitive property.48 Hence, in order to investigate the pore structure of the as-prepared PHC, the Brunauer–Emmett–Teller (BET) test was conducted and the results are shown in Fig. 2d. The nitrogen sorption isotherm curve of the as-prepared PHC displayed a typical type I isotherm curve, which indicated that the PHC possessed a microporous structure. Based on the pore size distribution graph (Fig. 2d, inset), the majority of the pores for PHC were in the micropore range (pore width of <2 nm). The presence of micropores could effectively enhance the achievable capacitance, and the presence of an interconnected network could provide favourable pathways for ion penetration and transportation, which greatly increased the appeal of PHC as a supercapacitor electrode material.29–31 Thus as a result, the as-obtained PHC boasted a large surface area of 2341 m2 g−1. The successful doping of N into the PHC was confirmed by the N 1s XPS spectrum (Fig. 2e). Three different peaks centred at 399.1 eV, 400.2 eV and 401.3 eV were observed which corresponded to pyridinic N, pyrrolic N and graphitic N, respectively,46 with a total N content of ca. 2.4 at%. Such an existence of N could contribute positively as enhanced electrolyte wettability and increased capacitance may be achieved in the process.32 The Raman spectra in Fig. 2f displayed signals of the D band (1326 cm−1), G band (1581 cm−1), and a combination of 2D (2651 cm−1) and D + G (2894 cm−1) bands, confirming the graphitic structure. The high ratios of ID/IG (1.07) indicated the presence of structural defects in the prepared PHC due to nitrogen doping.49 This is further confirmed by the Raman spectrum of the non-doped porous carbon (denoted as PC) in Fig. S3b (ESI†). With a high surface area, rich porous structure and N doping, the obtained PHC is expected to be an excellent candidate material for supercapacitors.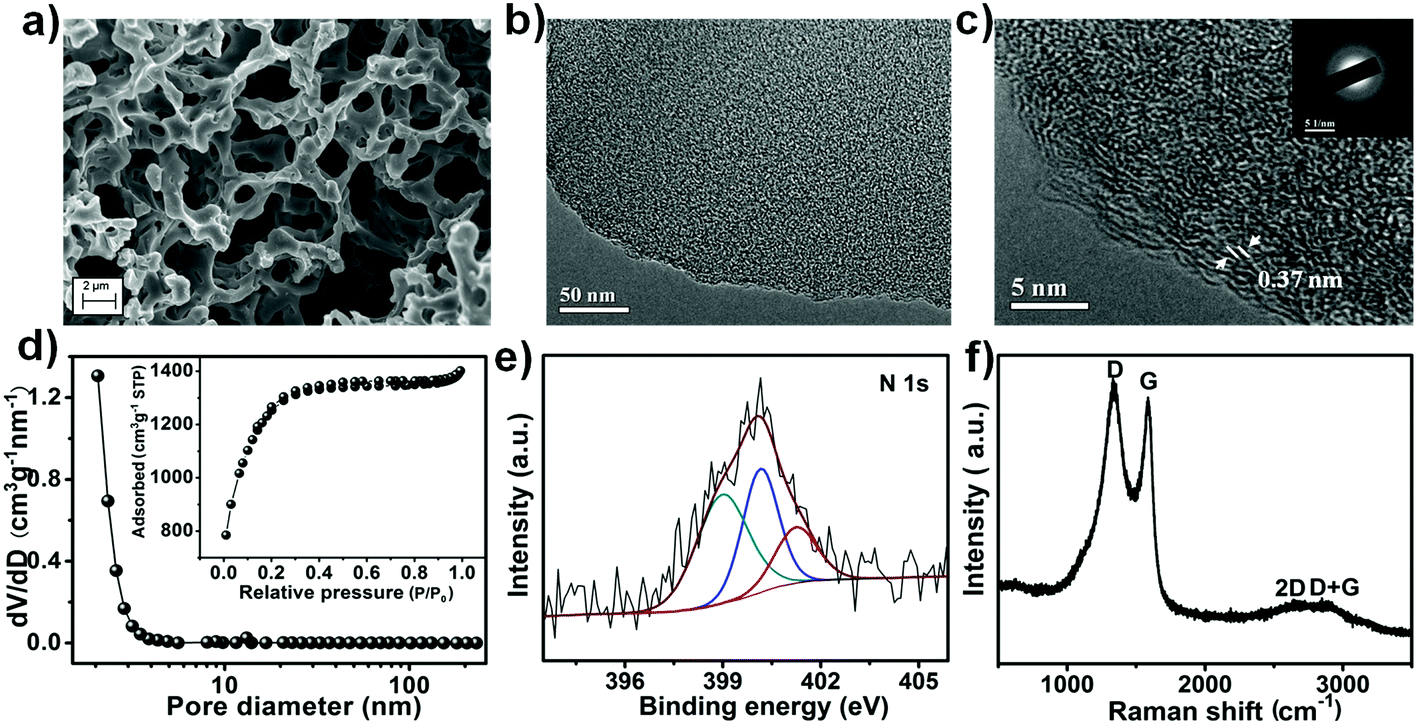 | ||
| Fig. 2 Morphology, porous structure and chemical components of the as-prepared porous honeycomb-like carbon. (a) SEM image; (b and c) TEM image; (d) nitrogen sorption isotherm (inset showing the corresponding pore size distribution); (e) N 1s XPS spectrum; (f) Raman spectrum. | ||
Electrochemical properties of the PHC//PHC BRE0.025M supercapacitor
The stable potential window of the assembled PHC//PHC supercapacitor in the BRE0.025M electrolyte was firstly investigated by conducting cyclic voltammetry (CV) with a step-wise potential increment of 0.2 V from an initial potential window of 0.8 V to a final potential window of 1.8 V (Fig. S4, ESI†). Based on the result, the stable potential window for the PHC//PHC BRE0.025M supercapacitor was ca. 1.4 V. Three electrode electrochemical tests were performed for a full cell potential of 1.4 V (Ag/AgCl as the reference electrode, PHC as the negative and positive electrode) as shown in Fig. S5 (ESI†). The result revealed that the PHC negative electrode operated within a potential window of −0.3 to 0.5 V and the PHC positive electrode operated within a potential window of 0.5–1.1 V. The CV curve (Fig. 3a) showed a quasi-rectangular shape with faradic peaks involved with the phenolic hydroxyl and quinoid carbonyl group at different scan rates. The CV showed an oxidation peak at 0.278 V and a reduction peak at 0.248 V with a redox peak separation of 30 mV. The small redox peak separation suggests an accelerated electron transfer and enhanced reversibility of the redox reaction.50,51 The increase of redox current with scan rate suggested that it had an excellent rate capability with impressive cycling performance relating to the fast ion movement and redox reaction in electrolyte solution. Even under an ultrahigh scan rate of 5000 mV s−1, the CV curve showed a quasi-rectangular shape. The presence of faradic peaks in the CV and the appearance of a nonlinear triangle in the charge–discharge curve agreed well with each other (Fig. 3b).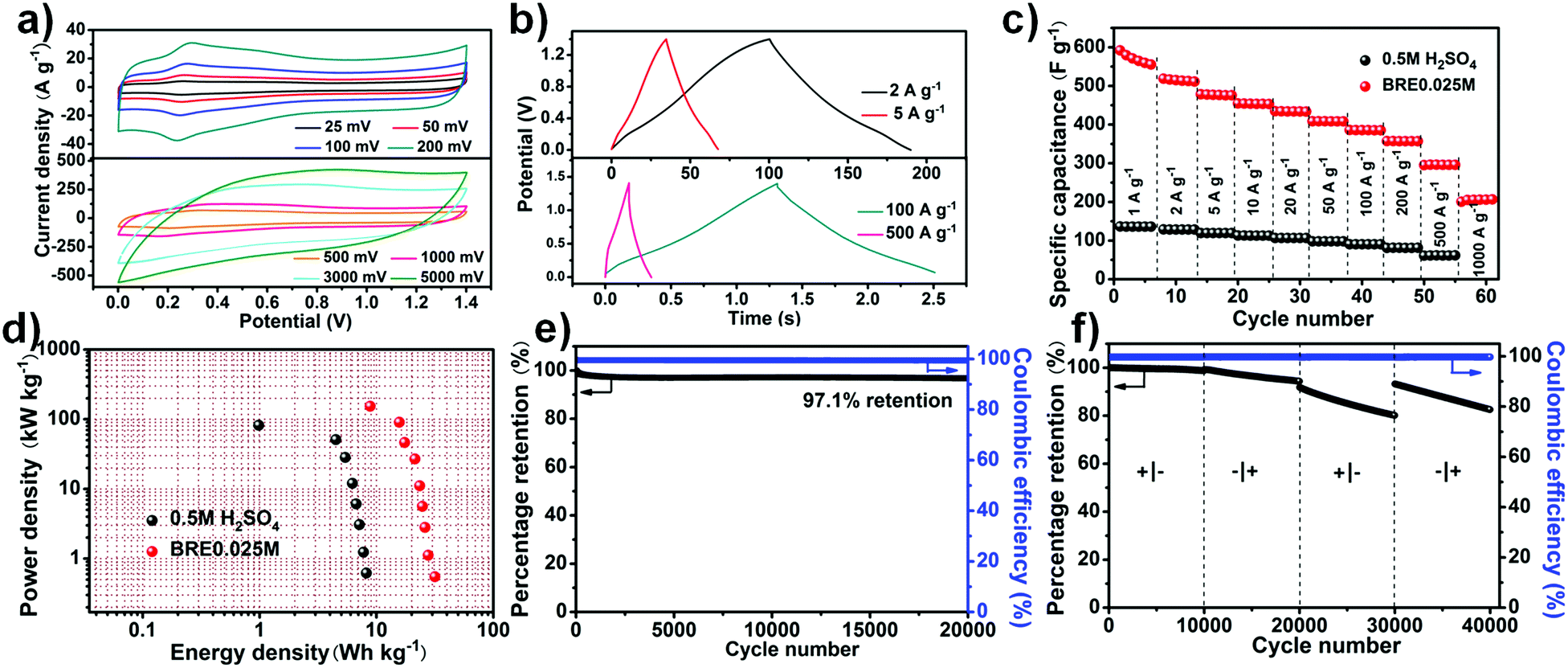 | ||
| Fig. 3 Electrochemical characterization of PHC//PHC using BRE0.025M as redox-mediated electrolyte. (a) CV curves; (b) charge–discharge curves; (c) rate performance at different current densities in 0.5 M H2SO4 and BRE0.025M; (d) Ragone plot for the supercapacitor in 0.5 M H2SO4 and BRE0.025M; (e) cycling stability and coulombic efficiency under a high current density of 100 A g−1 for 20000 cycles; (f) stable cycling with the leads switched every 10000 cycles (at 100 A g−1), repeatedly reversing the polarity. | ||
In contrast, other carbon materials like activated carbon (AC) and non-nitrogen doped porous carbon (PC) were also tested for their performance using the BRE0.025M electrolyte. PC was prepared under the same conditions as PHC, without the addition of g-C3N4. The SEM of PC showed that the prepared carbon also possessed a porous structure like PHC (Fig. S6, ESI†). Similarly, AC and PC presented excellent rectangle-like cyclic voltammetry (CV) curves with a redox peak at low scan rate while presenting rectangle-like CV curves even at a scan rate as high as 5000 mV s−1, indicating ideal capacitive behaviour with a high rate capability (Fig. S7a and b, ESI†). At 1 A g−1, the specific capacitance of PHC reached 550 F g−1, much higher than that in 0.5 M H2SO4 (136 F g−1), and maintained a high specific capacitance of 205 F g−1 at 1000 A g−1, which were much superior to that of the AC (123 F g−1 at 1 A g−1 and 28.2 F g−1 at 500 A g−1) and PC (400 F g−1 at 1 A g−1 and 79.5 F g−1 at 500 A g−1) electrodes (Fig. 3c and Fig. S7c, ESI†). In addition, the Ragone plots further demonstrated the excellent electrochemical performance of PHC. Using 0.5 M H2SO4 as the electrolyte, PHC displayed a maximum gravimetric energy density of 8.2 W h kg−1 and a maximum power density of 81.71 kW kg−1. After introducing conjugated indole-based macromolecule as the redox additive, enhanced power densities and energy densities were obtained, in which the PHC system delivered the highest energy density of 31.6 W h kg−1 at 0.55 kW kg−1. Furthermore, a high energy density of 8.8 W h kg−1 was achieved even at the highest power density of 153.5 kW kg−1 (Fig. 3d). When considering the mass of the redox additive (0.095 mg) transformed into active material, the capacitance is ca. 500 F g−1 at 0.93 A g−1 and it maintained a high specific capacitance of 166 F g−1 at 930 A g−1. Besides, a maximum energy density of about 26.5 W h kg−1 and a maximum power density of 142.5 kW kg−1 were achieved. The excellent power density attained by the PHC//PHC BRE0.025M symmetric supercapacitor was much higher than the previously reported redox-mediated systems that involved small redox-active additives such as hydroquinone, pyrocatechol etc. (see Table S1, ESI†). As shown in Fig. S7d (ESI†), AC and PC showed maximum energy densities of 5.2 and 20.7 W h kg−1 and maximum power densities of 47.0 and 99.5 kW kg−1, respectively, which were inferior to those of PHC. In addition to the ultrahigh power density that was achieved, the cycle stability of the system is far superior to that of the redox-mediated supercapacitor (see Table S1, ESI†). A small amount of capacitance was lost after cycling at 100 A g−1 for 20000 cycles which translates to a high capacitance retention of 97.1% (Fig. 3e). The recorded cycle stability was also shown to be comparable to that of the EDLC supercapacitor (see Table S2, ESI†) but with a high energy density. Interestingly, the cell functioned similarly when operated “backwards” (Fig. 3f). It was nearly undamaged upon polarity reversal due to its symmetric nature (5,6-dihydroxyindole and 5,6-quinoneindole motifs can be converted to one another in acid under electrochemical testing).
4. Discussion
An impressive capacitive performance was observed for the BRE0.025M electrolyte when using PHC as the active material, which indicated that the synergistic design of the electrode material and electrolyte is an essential parameter towards a high performing supercapacitor. The ionic conductivity of BRE0.025M was determined to be ca. 1.60 μS cm−1, which was superior as compared to that of 0.5 M H2SO4 (1.47 μS cm−1) and RE0.025M (1.53 μS cm−1). The high ionic conductivity of BRE0.025M was further supported by electrochemical impedance spectroscopy (EIS) as shown in Fig. 4a. As shown in Fig. 4a, the semicircle in the high frequency range represented the charge-transfer resistance, and the slope of the low-frequency straight line was related to the ion diffusion resistance.52 The charge transfer resistance Rct of BRE0.025M was 0.373 Ω which is 3 times lower as compared to the Rct of RE0.025M (1.21 Ω) and Rct of the 0.5 M H2SO4 supporting electrolyte (1.62 Ω). This indicated that the resistance of PHC in BRE0.025M was much smaller than those in H2SO4 and RE0.025M electrolytes. Such a low ion diffusion resistance might be due to the short ion diffusion path length in the PHC and the high ionic conductivity of BRE0.025M which allowed easy access of the electrolyte to the surface of the electrode.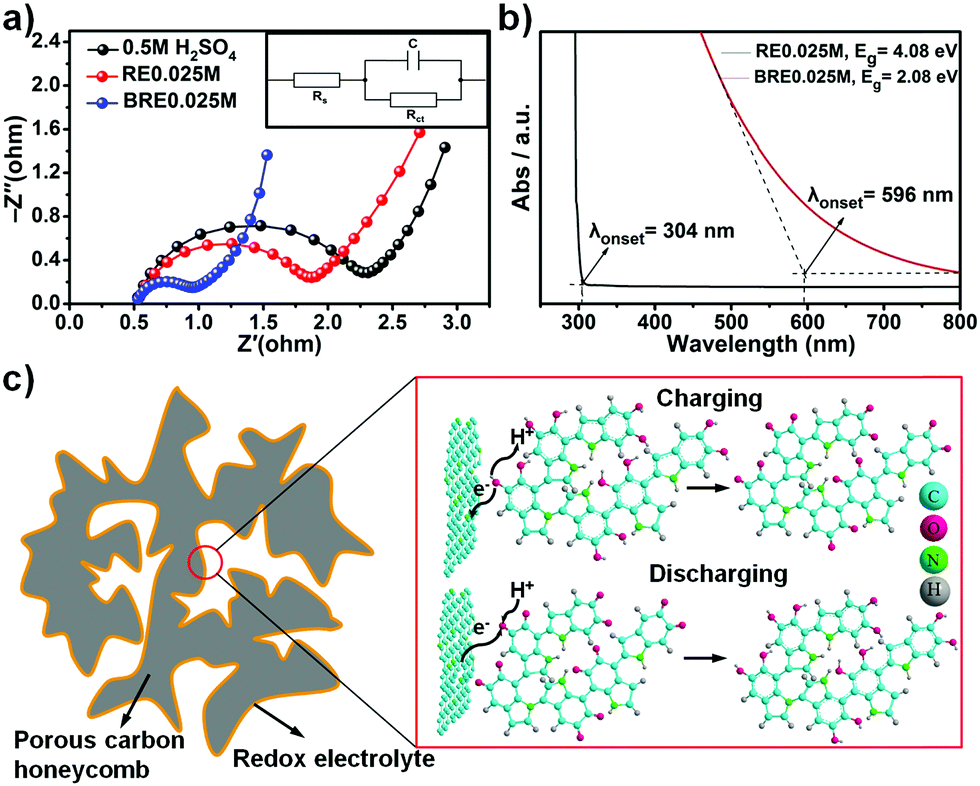 | ||
| Fig. 4 Mechanism study of the enhanced performance of the redox-mediated supercapacitor. (a) Electrochemical impedance spectroscopy of the 0.5 M H2SO4, RE0.025M and BRE0.025M electrolytes; (b) UV-Vis spectra of the RE0.025M and BRE0.025M electrolytes; (c) redox reaction involved with the BRE0.025M electrolyte. | ||
Electronic transfer kinetics can be influenced by the HLG of the molecules. The single 5,6-dihydroxyindole molecule possesses a high HLG which translated to increased difficulty for the delocalized electron to be transferred to an acceptor group.42 In order to facilitate the electron transfer, it is thus prudent to lower the HLG of the molecule. One of the established methods to decrease HLG is through extending the conjugation of the molecule (i.e. polymerization of 5,6-dihydroxyindole monomer).26 Small molecules such as 5,6-dihydroxyindole/5,6-quinoneindole possess a HLG of ca. 5.0 eV. Extending the conjugation towards 3 repeating motifs can lower the HLG to 2.5 eV.42 Hence, by further extension the indole-based conjugated macromolecule containing 5,6-dihydroxyindole/5,6-quinoneindole motifs, based on the trend, should possess even lower HLG. To demonstrate the reduced HLG of the indole-based conjugated macromolecule, UV-VIS spectroscopy was conducted for both RE0.025M and BRE0.025M as shown in Fig. 4b. Based on the onset of the absorbance peak, the HLG of BRE0.025M was calculated to be ca. 2.08 eV which was significantly lower than that of RE0.025M (4.08 eV). Hence this result conclusively suggested that by extending conjugation from one motif, i.e. RE0.025M, to a macromolecule (in this case polymerization degree of ca. 147 for BRE0.025M), HLG can be reduced by half which significantly indicated an improvement in the conductivity. Thus based on the ionic conductivity and EIS results which are further backed by the lower HLG revealed in the UV-VIS spectra, macromolecules with an extensively conjugated structure should possess efficient charge transfer ability and a fast reaction rate as demonstrated in the electrochemical results. The excellent cycling stability shown in Fig. 3e might be accounted for by the extra stability arising due to the high delocalization energy in conjugated systems. In the conjugated system, overlapping of pz orbitals allowed electrons to be delocalized over a larger portion of the macromolecule, which aided in lowering the energy of the molecule in turn making it more stable.27 The redox mechanism was investigated using ΔG (change in Gibbs free energy) calculation, whereby the calculated average redox potential for 5,6-dihydroxyindole to 5,6-quinoneindole was ca. 0.23 V vs. Ag/AgCl, which is close to the experimental value (0.2 V vs. Ag/AgCl). This result indicated that the redox mechanism present in the system involved conversion from 5,6-dihydroxyindole to 5,6-quinoneindole. Hence, the 5,6-dihydroxyindole/5,6-quinoneindole endowed the BRE0.025M electrolyte with redox centres which involved the phenolic hydroxyl group in the 5,6-dihydroxyindole motifs and the quinonoid carbonyl group in the 5,6-quinoneindole motifs (Fig. 4c).
In addition to the high conductivity indole-based conjugated redox-mediated electrolyte, the PHC played a significant role in enhancing the supercapacitor performance. The interaction between the electrolyte and the material is one important aspect that can influence such a performance, and hence the electrolyte contact angle measurement was performed. As shown in Fig. S8 (ESI†), the as-prepared PHC showed better wettability than AC and PC materials in both RE0.025M and BRE0.025M. In particular, the electrolyte wettability of the PHC was better in BRE0.025M (contact angle is ca. 76.9°) as compared to that in RE0.025M (contact angle is ca. 87.9°). Thus, in addition to the large surface area, dense microporous structure, and pseudocapacitive contribution from N heteroatom doping, PHC showed a superior supercapacitive performance as compared to AC and PC. Hence, the synergistic combination of a low HLG and a stable indole-based conjugated macromolecule and a high performance porous honeycomb-like carbon is crucial towards realizing ultra-high power density and a stable supercapacitor.
5. Conclusion
A redox-active indole-based conjugated macromolecule containing 5,6-dihydroxyindole motifs/5,6-quinoneindole motifs was prepared by electrochemical polymerization under acidic conditions. By tapping into the benefit of the conjugation effect, an extensively conjugated macromolecule could improve the structural stability and enhance the electronic transfer kinetics due to the lower HOMO–LUMO gap (HLG). In addition to engineering a highly stable and high electronic conductivity redox-mediated electrolyte, a high performance porous honeycomb-like carbon with N doping was prepared by a facile method, which integrated the features of open porous architecture with interconnected micropores, large specific surface area, high conductivity, high stability, and good electrolyte wettability. The synergism of these characteristics endowed a large surface area for charge storage, additional pseudocapacitance from the redox reaction, and ultrahigh power density due to the low HLG of the extensively conjugated indole-based macromolecule. The assembled supercapacitor exhibited a high specific capacitance of 205 F g−1 at an ultrahigh current density of 1000 A g−1. A maximum power density of 153 kW kg−1 was recorded as one of the highest reported in redox-active electrolyte systems, with a respectable energy density of 8.8 W h kg−1. Combined with an excellent cycling stability of 97.1% capacitance retention after 20000 charge/discharge cycles, this strategy has great potential for the development and large-scale applications of high-performance redox-based supercapacitors.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by Singapore MOE Tier 1 funding R-284-000-162-114 and Singapore NRF CRP funding R284000159281.Notes and references
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publisher, New York, 1999 Search PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- X. Li and B. Wei, Nano Energy, 2013, 2, 159–173 CrossRef CAS.
- J. E. Zuliani, J. N. Caguiat, D. W. Kirk and C. Q. Jia, J. Power Sources, 2015, 290, 136–143 CrossRef CAS.
- A. K. Shukla, A. Banerjee, M. K. Ravikumar and A. Jalajakshi, Electrochim. Acta, 2012, 84, 165–173 CrossRef.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- Q. Wang, J. Yan and Z. Fan, Energy Environ. Sci., 2016, 9, 729–762 CAS.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef.
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef PubMed.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 CAS.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- Q. Meng, K. Cai, Y. Chen and L. Chen, Nano Energy, 2017, 36, 268–285 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 CAS.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386–12394 CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054 CrossRef CAS.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- S. Roldán, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Angew. Chem., Int. Ed., 2011, 50, 1699–1701 CrossRef PubMed.
- J. Wu, H. Yu, L. Fan, G. Luo, J. Lin and M. Huang, J. Mater. Chem., 2012, 22, 19025–19030 RSC.
- Y. Q. Zhu, L. Zhang, X. Y. Chen, Z. H. Xiao and Z. J. Zhang, J. Power Sources, 2015, 299, 629–639 CrossRef CAS.
- S. Roldán, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, Electrochim. Acta, 2012, 83, 241–246 CrossRef.
- S. Roldán, Z. González, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Electrochim. Acta, 2011, 56, 3401–3405 CrossRef.
- J. Roncali, Chem. Rev., 1997, 97, 173–205 CrossRef CAS PubMed.
- M. Arivazhagana and S. Jeyavijayan, Spectrochim. Acta, 2011, 79A, 376–383 CrossRef PubMed.
- J. Zhou, J. Lian, L. Hou, J. Zhang, H. Gou, M. Xia, Y. Zhao, T. A. Strobel, L. Tao and F. Gao, Nat. Commun., 2015, 6, 8503 CrossRef CAS PubMed.
- D. F. Perepichka and M. R. Bryce, Angew. Chem., Int. Ed., 2005, 44, 5370–5373 CrossRef CAS PubMed.
- J. March, Advanced Organic Chemistry Reactions, Mechanisms and Structure, John Wiley & Sons, inc., New York, 3rd edn, 1985, ISBN 0-471-85472-7 Search PubMed.
- C. Peng, G.-H. Ning, J. Su1, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- P. Simon and A. Burke, Electrochem. Soc. Interface, 2008, 17, 38–43 CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- U. N. Maiti, W. J. Lee, J. M. Lee, Y. Oh, J. Y. Kim, J. E. Kim, J. Shim, T. H. Han and S. O. Kim, Adv. Mater., 2014, 26, 40–67 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- T. N. Kumar, N. Chandrasekaran and K. L. Phani, Chem. Commun., 2015, 51, 5052–5055 RSC.
- H. Okuda, A. Nakamura, K. Wakamatsu, S. Ito and T. Sota, Chem. Phys. Lett., 2007, 433, 355–359 CrossRef CAS.
- E. K. Jeon, E. Seo, E. Lee, W. Lee, M.-K. Umb and B.-S. Kim, Chem. Commun., 2013, 49, 3392–3394 RSC.
- F. Yu, S. Chen, Y. Chen, H. Li, L. Yang, Y. Chen and Y. Yin, J. Mol. Struct., 2010, 982, 152–161 CrossRef CAS.
- G. Perna, M. Lasalvia, C. Gallo, G. Quartuccin and V. Capozzi, Open Surf. Sci. J., 2013, 5, 1–8 CrossRef.
- T. Ma, Q. Zhao, J. Wang, Z. Pan and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 6428–6432 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. N. Waterhouse, Y. Zhao, C. Zhou, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- J. Xu, Z. Tan, W. Zeng, G. Chen, S. Wu, Y. Zhao, K. Ni, Z. Tao, M. Ikram, H. Ji and Y. Zhu, Adv. Mater., 2016, 28, 5222–5228 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed structure description and other experimental results. See DOI: 10.1039/c7ee02584j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |