
Highly selective gas separation membrane using in situ amorphised metal–organic frameworks†
Aylin
Kertik
a,
Lik H.
Wee
 *a,
Martin
Pfannmöller
b,
Sara
Bals
b,
Johan A.
Martens
a and
Ivo F. J.
Vankelecom
*a
*a,
Martin
Pfannmöller
b,
Sara
Bals
b,
Johan A.
Martens
a and
Ivo F. J.
Vankelecom
*a
aCentre for Surface Chemistry and Catalysis, University of Leuven, Celestijnenlaan 200f, B3001, Heverlee, Leuven, Belgium. E-mail: likhong.wee@kuleuven.be; ivo.vankelecom@kuleuven.be
bElectron Microscopy for Materials Science, University of Antwerp, Groenenborgerlaan 171, B2020, Antwerp, Belgium
First published on 9th August 2017
Abstract
Conventional carbon dioxide (CO2) separation in the petrochemical industry via cryogenic distillation is energy intensive and environmentally unfriendly. Alternatively, polymer membrane-based separations are of significant interest owing to low production cost, low-energy consumption and ease of upscaling. However, the implementation of commercial polymeric membranes is limited by their permeability and selectivity trade-off and the insufficient thermal and chemical stability. Herein, a novel type of amorphous mixed matrix membrane (MMM) able to separate CO2/CH4 mixtures with the highest selectivities ever reported for MOF based MMMs is presented. The MMM consists of an amorphised metal–organic framework (MOF) dispersed in an oxidatively cross-linked matrix achieved by fine tuning of the thermal treatment temperature in air up to 350 °C which drastically boosts the separation properties of the MMM. Thanks to the protection of the surrounding polymer, full oxidation of this MOF (i.e. ZIF-8) is prevented, and amorphisation of the MOF is realized instead, thus in situ creating a molecular sieve network. In addition, the treatment also improves the filler-polymer adhesion and induces an oxidative cross-linking of the polyimide matrix, resulting in MMMs with increased stability or plasticization resistance at high pressure up to 40 bar, marking a new milestone as new molecular sieve MOF MMMs for challenging natural gas purification applications. A new field for the use of amorphised MOFs and a variety of separation opportunities for such MMMs are thus opened.
Broader contextRemoval of CO2 from natural and biogas is of growing concern with respect to harmful effects of greenhouse gas emissions and technological interests in CO2 as alternative building block for renewable fuels and chemicals. Even though polymer membranes are attractive for molecular-level separations in these industrial-scale processes, commercially available membranes still need to be improved for effective CO2 separations mainly due to their intrinsic permeability/selectivity trade-off, limited thermal stability and high susceptibility to plasticization. Mixed matrix membranes (MMMs), consisting of well-dispersed MOF fillers in a polymeric matrix, could potentially solve these issues. Present work demonstrates that thermally induced oxidative cross-linking of the polymer matrix and improved interactions between the polymer and the amorphised zeolitic imidazolate framework form plasticization-resistant MMMs for highly-selective separations of CO2/CH4 mixed-gas feeds. Obtained membranes could also be of use as molecular sieves for other separation processes, such as hydrocarbons (e.g. olefin/paraffin isomers) separation, or liquid phase separations via pervaporation or solvent resistant nanofiltration. |
Introduction
Nanoporous materials with well-defined and size-selective channels are essential elements for gas sorption, storage, separation and catalysis.1 Metal–organic frameworks (MOFs) with their large surface area, tunable pore size and chemical tailorability, emerged as excellent alternatives to conventional porous materials.2 MOFs are formed by the supramolecular coordination of metal ions/clusters and organic bridging ligands.2,3 These repeating building units exhibit three-dimensional frameworks with long-range ordering and variable pore architectures. Combined with their chemical versatility, MOFs endow via selective adsorption and molecular sieving for numerous potential applications ranging from separation to catalysis, sensing, magnetism, photoluminescence and drug delivery.1,2,4 Self-supporting membranes prepared from such nanostructured porous materials have been prepared for gas separation,5 but making defect-free and mechanically robust membranes has proven to be very challenging. In contrast, polymers are easily processable into large membrane modules, and already widely used in industrial gas separations for e.g. CO2 removal. However, they often suffer from a permeability/selectivity trade-off, as depicted by the Robeson upper-bound.6,7 Commercial polymers can rarely surpass this upper-bound. Despite various attempts to improve the performance of commercial polymers, a reliable approach has not been accomplished to date.8 Designing special macromolecular structures lacks up-scaling potential, and membranes tend to suffer from long-term instability.An alternative approach is mixed matrix membranes (MMMs) consisting of fillers dispersed in a polymer. While the polymer offers mechanical stability and processibility,2 a filler with well-defined sorption properties provide more selective permeation.9 As filler, zeolitic imidazolate frameworks (ZIFs) as a sub-family of MOFs analogous to inorganic zeolites, are particularly interesting.10 ZIF-8 is an archetypal ZIF, consisting of zinc ions and 2-methylimidazole linkers coordinated in a sodalite topology having interconnected pore cavities and a pore aperture of 11.6 Å and 3.4 Å, respectively, capable of separating various gas mixtures.11–14 Poor compatibility between filler and polymer is a notorious problem in preparing MMMs.15,16 Methods such as above-Tg annealing,17 filler surface modification,15 melt-processing,18 priming9 and using low-molecular-weight additives,19 or plasticizers20 have been applied to overcome this problem.
Some MOFs, and specifically ZIFs tend to exhibit a solid–solid phase transformation known as amorphisation. Amorphous MOFs (aMOFs) are networks that retain their basic building blocks but exhibit no long-range ordering.4 aMOFs are obtained by introducing disorder into their parent crystalline frameworks through heating, pressure or mechanical ball-milling.4 Amorphisation is a well-known concept for inorganic materials, but the application of aMOFs is largely unexplored.4,21,22 The current potential application is limited to drug-release23,24 and entrapping harmful substances.25,26 In this work, we show that amorphised MOFs can be created in situ in a ZIF-containing polyimide membrane by a controlled thermal treatment in air. Combined with remarkable changes in polymer properties, this MOF transformation generates MMMs with the highest CO2/CH4 mixed-gas selectivities reported so far for membranes based on commercial polymers, marking a new milestone in the latest Robeson's upper-bound plot, and offering new opportunities to highly selective separations in a variety of related applications.
Results and discussion
Thermal oxidative cross-linking of MMMs
Choice of the right material pair, coupled with the optimisation of the membrane fabrication process is the key to designing a high-performance MMM. For this work, a commercially available polyimide (PI), Matrimid®, was chosen as the matrix polymer. ZIF-8 is a promising candidate for CO2 capture (Fig. S1, ESI†).27 ZIF-8, which has also been reported to undergo an amorphisation process, was chosen as the filler.4 MMMs with different ZIF-8 loadings (20–40 wt%) were prepared and thermally treated at 100, 160, 250 or 350 °C in air for periods up to 24 h.After a prolonged treatment at 350 °C for 24 h, a significant change in membrane color was noted as shown in Fig. 1a–d. The membranes changed from light yellow to dark brown when the temperature was increased. Interestingly, the onset of darkening for the MMMs was 250 °C, whereas it was 350 °C for the unfilled polymer, suggesting that the embedded ZIF-8 has significant influence on the thermal behaviour of the MMMs. According to the TG analysis (Fig. 1e and f), the Matrimid® membrane annealed at 100 °C shows a significant weight loss (10.3 wt%) from 170–470 °C, whereas the unfilled Matrimid® membrane treated at 350 °C only shows a negligible weight loss (1.2 wt%), pointing to an improved thermal stability. A similar trend is observed for the thermally treated MMM (18.2 wt% for 100 °C and 4.2 wt% for 350 °C). More importantly, the embedded ZIF-8 is not degraded upon heating to 340 °C, in contrast to the bulk ZIF-8 powder under similar conditions. The significant enhancement of the thermal stabilities and the change in the visual properties of the unfilled membranes suggests thermo-oxidative cross-linking of the polymer. To confirm this hypothesis, solubility tests were performed and the results are presented in Fig. S2 (ESI†) for the unfilled Matrimid®, and in Fig. 1g–i for the MMMs. Obviously, all membranes treated above 160 °C became largely insoluble in a good solvent for the polymer such as chloroform, with a gel content above 95% after 2 days (Fig. 1h and i).
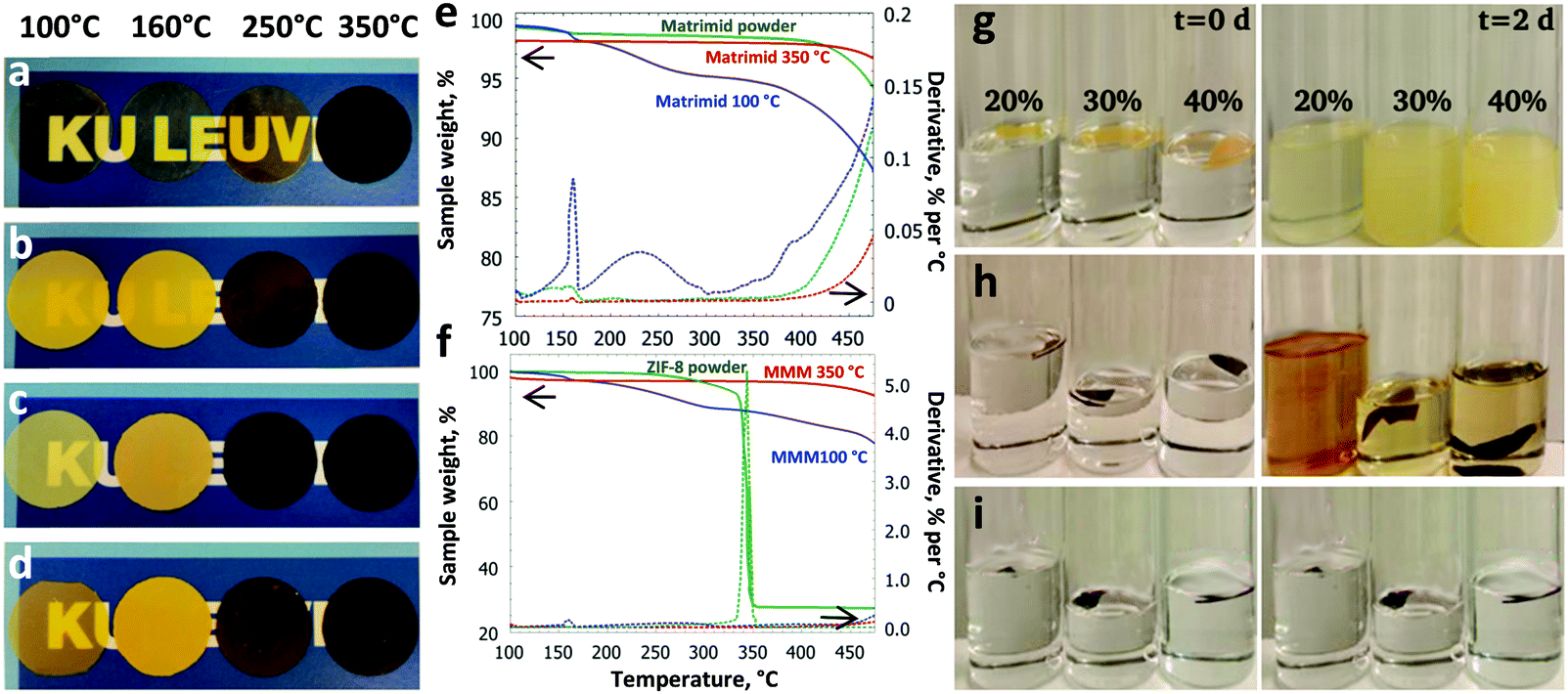 | ||
| Fig. 1 Visual, thermal and solubility tests of the thermally treated membranes. The annealing influences the visual properties of the (a) Matrimid® and (b–d) MMMs with 20–30–40% ZIF-8 loading. (e and f) The TGA curves and the derivative weight loss curves for Matrimid® and the MMM with 40 wt% ZIF-8 loading. (g–i) The solubility tests for MMMs thermally treated at 160, 250 and 350 °C. | ||
In order to elucidate the underlying chemistry of the thermo-oxidative cross-linking of Matrimid®, the membranes were characterized by ATR-FTIR (Fig. 2a). The adsorption bands at 2960, 2927, and 2864 cm−1 assigned to the aliphatic C–H stretching vibration decreased in intensity with increasing temperature in line with the observed darker color of Matrimid®. It has been previously reported that oxidative cross-linking occurs at the methylene bridges of the polymer, which is associated with the ketone and aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations at 1600 and 930 cm−1, respectively.28 A decrease at 823 cm−1 and an increase at 1604 cm−1 are now observed, which could be assigned to methyl and CO oxidation, respectively, with increasing thermal treatment temperature. In general, thermal oxidation of polymers involves free-radical chain reactions. In the present study, a prolonged thermal treatment at 350 °C in the presence of oxygen induces oxidation and cross-linking. The methyl groups in the polyimide are oxidised to provide readily available cross-linking sites via the formation of –CH2 radicals. These free radicals react with O2 to form peroxy radicals (ROO˙), which can further abstract a hydrogen from adjacent polymer chains to generate hydroperoxide (ROOH) moieties. These hydroperoxides then undergo sequential termination steps to induce inter-chain cross-linking.29 The concise mechanism for thermo-oxidative cross-linking is shown in Fig. 2c. A complete chain reaction mechanism including the initiation, propagation and termination steps is illustrated in Fig. S3 and S4 (ESI†). The excess water content found in the cross-linked MMMs (2.6 wt%) versus 0.7 wt% in the MMMs annealed at 100 °C according to TGA (Fig. 1e, f and Fig. S5, S6, ESI†) is mainly due to the water formation as a result of the chain termination reaction. Glass transition temperatures (Tg) increased as the thermal oxidative cross-linking of methylene bridging groups became more prominent.30,31 While the Tg of the unfilled polymer increased to 340 °C, it reached 350 °C for the MMM treated at the same temperature. The tensile strength of the membranes evolved in line with this cross-linking, as shown in Table S1 and Fig. S7 (ESI†). Due to the MOF loading, the tensile strength of the MMMs was overall below that of Matrimid®. However, it still exhibited an exponential increase with thermal treatment temperature, i.e. with increasing cross-linking.
O stretching vibrations at 1600 and 930 cm−1, respectively.28 A decrease at 823 cm−1 and an increase at 1604 cm−1 are now observed, which could be assigned to methyl and CO oxidation, respectively, with increasing thermal treatment temperature. In general, thermal oxidation of polymers involves free-radical chain reactions. In the present study, a prolonged thermal treatment at 350 °C in the presence of oxygen induces oxidation and cross-linking. The methyl groups in the polyimide are oxidised to provide readily available cross-linking sites via the formation of –CH2 radicals. These free radicals react with O2 to form peroxy radicals (ROO˙), which can further abstract a hydrogen from adjacent polymer chains to generate hydroperoxide (ROOH) moieties. These hydroperoxides then undergo sequential termination steps to induce inter-chain cross-linking.29 The concise mechanism for thermo-oxidative cross-linking is shown in Fig. 2c. A complete chain reaction mechanism including the initiation, propagation and termination steps is illustrated in Fig. S3 and S4 (ESI†). The excess water content found in the cross-linked MMMs (2.6 wt%) versus 0.7 wt% in the MMMs annealed at 100 °C according to TGA (Fig. 1e, f and Fig. S5, S6, ESI†) is mainly due to the water formation as a result of the chain termination reaction. Glass transition temperatures (Tg) increased as the thermal oxidative cross-linking of methylene bridging groups became more prominent.30,31 While the Tg of the unfilled polymer increased to 340 °C, it reached 350 °C for the MMM treated at the same temperature. The tensile strength of the membranes evolved in line with this cross-linking, as shown in Table S1 and Fig. S7 (ESI†). Due to the MOF loading, the tensile strength of the MMMs was overall below that of Matrimid®. However, it still exhibited an exponential increase with thermal treatment temperature, i.e. with increasing cross-linking.
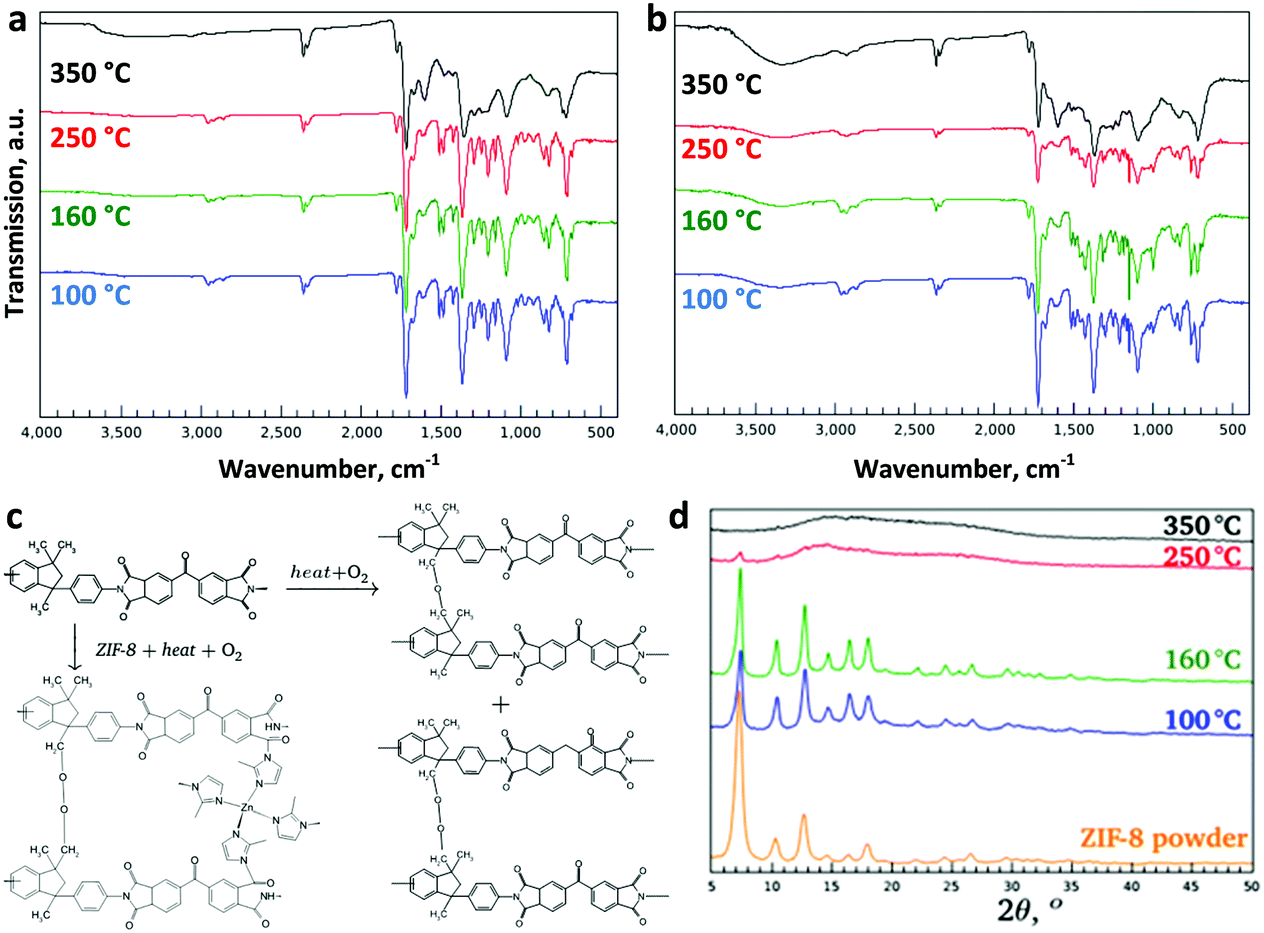 | ||
| Fig. 2 Thermo-oxidative cross-linking of the membranes. The ATR-FTIR patterns of the (a) Matrimid® and (b) MMMs with 40 wt% ZIF-8 loading as a function of temperature. (c) The suggested cross-linking mechanism for the PI and the MMM based on the FTIR results. (d) The characteristic XRD peaks of ZIF-8-containing MMMs. Enlarged versions of ATR-FTIR spectra of (a) Matrimid® and (b) the MMMs with assigned peaks are provided in Fig. S8 (ESI†). | ||
The Matrimid® membranes thermally treated at 250 °C were completely dissolved after 2 days whereas the MMMs treated at the same temperature were more resistant to solvation, especially at high ZIF-8 loadings. The observation suggests that the embedded ZIF-8 actually acts as an additional chemical cross-linker upon heating. Chemical cross-linking is known to realize the reduction of the plasticization tendency of Matrimid® membranes,32 which is critical for their application in CO2 separations. There have also been reports of plasticization-resistant MMMs exhibiting direct cross-linking between MOF and polyimide.33,34 According to ATR-FTIR spectra (Fig. 2b), apart from the previously observed enhanced absorption bands for CO and the decline in C–H stretching vibration, an additional band largely masked by Matrimid® at 1538 cm−1 was noted. This band can be tentatively assigned to the aromatic N–H stretching band of the amide group, confirming the cross-linking of Matrimid® with the imidazolate in ZIF-8. The cross-linking of imidazole from aZIF-8 with Matrimid®, as based on the ATR-FTIR studies, is tentatively shown in Fig. 2c.
XPS analysis clearly illustrated the presence of the peaks assigned to Zn2+ Zn2p3 and Zn2p1 (Fig. S9, ESI†). As ZIF-8 is amorphised, the Zn–N bonds are the first to brake and create unsaturated Zn2+ sites, as reflected in the increased intensity of the XPS peaks for the MMM treated at 350 °C. Recently, cross-linking of the organic ligand in MOF with polymer gel has been reported via acidification with concentrated HCl for breaking the coordinative bonding between the Zn(II) ion and the carboxylate anion.35 However, for our system, the cross-linking of Matrimid® with the molecular building units of ZIF-8 hints of a phase transition upon thermal treatment. Despite the structural transformation of ZIF-8 has been extensively studied by Cheetham21 and Friščić,36,37 amorphisation of ZIF-8 by thermal treatment has not been realised to date mainly due to direct oxidation of ZIF-8 to ZnO in air. In order to evaluate the crystallinity of the thermally treated membranes, XRD was performed (Fig. 2d and Fig. S10a–c, ESI†). For the MMMs thermally treated at 100 °C and 160 °C, the crystalline structures of the ZIF-8 nanoparticles remained intact, but when treated at higher temperatures (250 and 350 °C), the typical XRD diffractions were no longer observed (Fig. 2d). The disappearance of the crystalline peaks suggest the amorphisation of the embedded ZIF-8. The amorphous state of the ZIF-8 embedded in the Matrimid® matrix was further confirmed by TEM electron diffraction (Fig. 3a). No diffraction spots at high camera length that could indicate the presence of ZIF-8 crystals were observed. Electron diffraction only reveals rings representative of amorphous phases at low camera length. Nevertheless, the absorption band at 750 cm−1 assigned to the out-of-plane bending of the imide 5-membered ring is preserved, indicating that the imidazole is thus not oxidized during this amorphisation process. The combined XRD, ATR-FTIR and electron diffraction results confirm that the basic building blocks and the connectivity of the amorphised ZIF-8 counterparts is retained, but the long-range periodic ordering is lacking. From previous studies by Lee et al.,38 direct thermal treatment of ZIF-8 at 350 °C induces ZnO formation at high temperatures. In our MMMs, no ZnO was formed as evidenced from the XRD patterns (Fig. 2d) by the absence of the characteristic diffraction peaks in the region between 30–40° (2θ). In addition, the ZnO band at 500 cm−1 is not observed from the FTIR spectra, which further excludes its formation at high temperature.38 Although ZIF-8 has been reported to undergo amorphisation via ball-milling or compression,4 amorphisation through thermal treatment has not been documented to date yet, since bulk ZIF-8 powder is more likely to undergo a direct phase transition to ZnO upon thermal treatment (Fig. S11, ESI†).38 We assume that the Matrimid® might provide thermochemical protection for the embedded ZIF-8 under a relatively slow heating rate and a prolonged 24 h thermal treatment which is in full agreement with the TGA results.
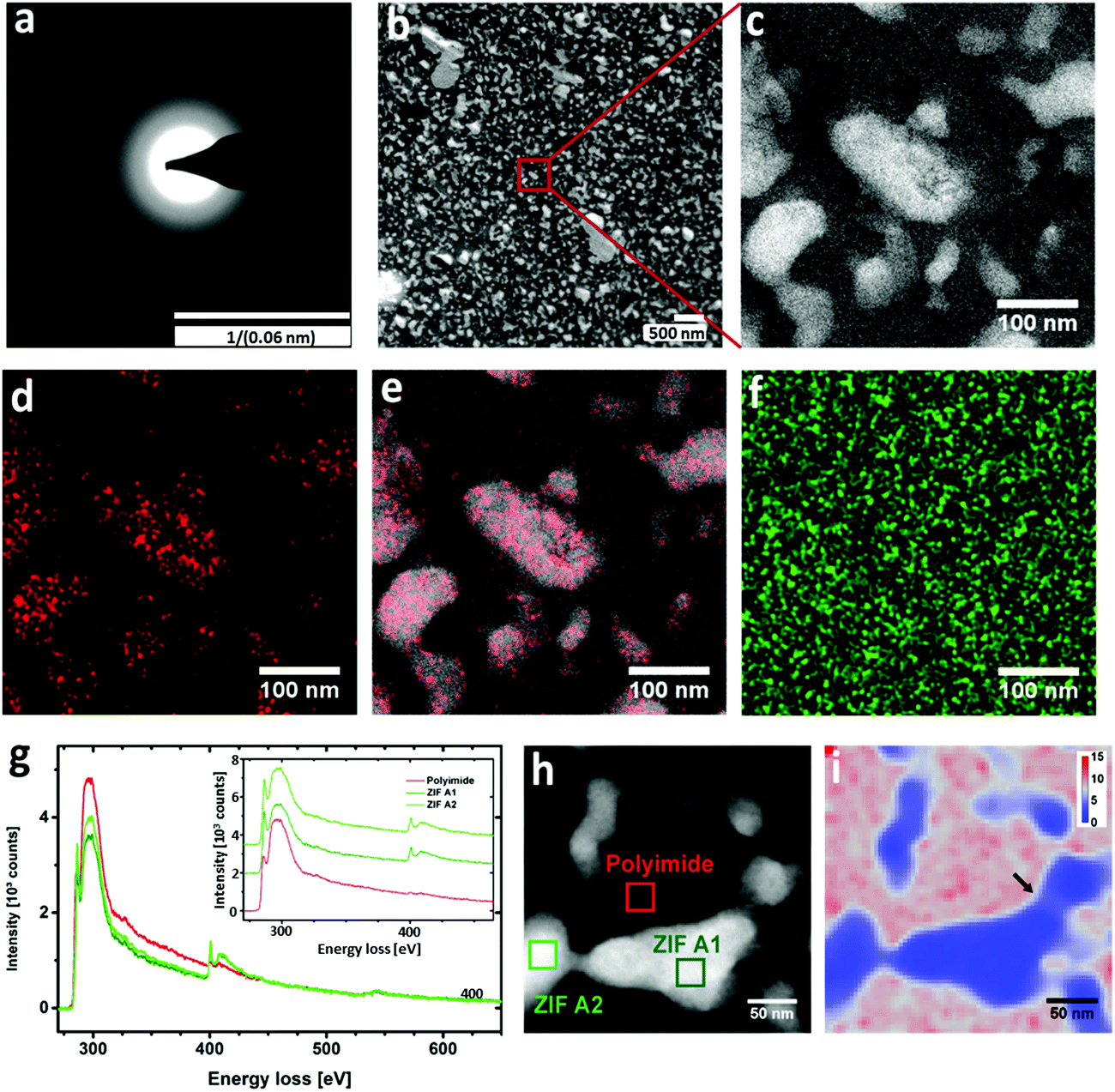 | ||
| Fig. 3 TEM imaging of MMMs with 40 wt% loading. (a) Electron diffraction. (b) HAADF-STEM image of MMMs with 40% ZIF-8. (c) High magnification view with HAADF-STEM. (d) Zn map from EDX measurement corresponding to the area shown in (c). (e) Overlay between HAADF image and Zn map. (f) EDX map showing the O distribution. (g) Average core energy-loss spectra for a polymer region and ZIF-8 regions. (h) HAADF-STEM overview image for STEM-SI map. Boxes indicate regions of averaging for spectra in (g). (i) Map depicting the ratio between C and N contents calculated from spatially resolved core energy-loss spectra of the region outlined by HAADF-STEM in h. | ||
The microstructure of the MMMs
The influence of thermal treatment on the structural morphology of the membranes was studied by SEM (Fig. 4), revealing that the ZIF-8 particles are well-dispersed with excellent filler-polymer adhesion even at high ZIF-8 loadings. No large clusters nor aggregates of ZIF-8 particles were observed. Intriguingly, by increasing the annealing temperature, high quality membranes with a smooth cross-section of homogeneous microstructure were successfully fabricated. In order to gain insights into the internal structural morphology and grain distribution within MMMs, and amorphous ZIF-8-polyimide interaction, advanced STEM analyses were performed on ultrathin cross-sections as shown in Fig. 3. A cross-section of an MMM with 40 wt% ZIF-8 loading was prepared by ultramicrotomy with a thickness of 90 nm for HAADF-STEM and EDX measurements. Overviews at low and high magnification using HAADF-STEM are shown in Fig. 3b and c, respectively. A clear contrast between ZIF-8 (light regions) and polymer (dark regions) allows direct identification of different nanostructure phases. According to the HAADF-STEM images, the ZIF-8 nanoparticles are well distributed. EDX mapping was applied across the area in Fig. 3c to investigate the distribution of certain elements. The resulting map for Zn (Fig. 3d) and its overlay with the HAADF image (Fig. 3e) demonstrates that Zn is only localized in the ZIF-8 areas and not dispersed into the polymer. Mapping of O (Fig. 3f) reveals a homogeneous distribution; no specific enrichment within the particles is observed, which confirms the observation from XRD and ATR-FTIR that the embedded ZIF-8 is not transformed into ZnO upon heating at 350 °C. | ||
| Fig. 4 Effect of MOF loading and annealing temperature on the membrane morphology as observed by SEM. The cross-sectional images of (a–d) Matrimid® and MMMs with (e–h) 20, (i–l) 30, (m–p) 40 wt% ZIF-8 loading, respectively. The annealing temperature is given at the top of each column. | ||
Fig. 3g shows core-loss energy-loss spectra taken for a polyimide area and two ZIF-8 areas as indicated in the HAADF image in Fig. 3h. Spectra are averages from STEM-SI data. A cross-section of 40 nm thickness was used to reduce effects from multiple scattering. To obtain a STEM-SI data set, the focused beam was scanned across the region outlined in Fig. 3h while recording an energy-loss spectrum for each scan position. The energy range is 275–650 eV so that core-loss signals contain information about the C–K edge (ca. 285 eV), N–K edge (ca. 397 eV), and O–K edge (ca. 532 eV). Spectra were corrected for multiple scattering (see Methods section) and the background signal was fitted by a power law function and subtracted. This allows quantitative comparison of the different element edges. It can be clearly seen that ZIF particles show much stronger signals for N and an elevated signal for π* in relation to σ* excitations compared to polyimide. The total integrated peak intensities remain the same. However, sharper peaks for the N-edge and the π* excitation for ZIF-8 nanoparticles indicate a better ordering of amorphous ZIF-8 building units than the amorphous polymer matrix. The O concentration is very low over the entire investigated region. Therefore, the spatially resolved spectra were used to quantify the local C and N compositions yielding a map of C/N ratios as shown in Fig. 3i. The map shows that the amorphous ZIF-8 regions in blue with high N concentrations exhibit a sharp boundary towards the polymer regions of high relative C content (red). Around the boundaries between polymer and ZIF-8 particles there is a small interfacial layer with intermediate C/N ratios (the grey layer around the ZIF-8 regions, Fig. 3i). Since the signals result from integration over the thickness of 40 nm, the interfacial profiles can be induced by the varying C/N ratios along the path of the electrons through the cross-section. However, as indicated by the black arrow in Fig. 3i, such a layer is also visible in a region where only a thin part of ZIF-8 is found. The constant occurrence of the same interfacial C/N ratio provides evidence of a crosslinking between the imidazole in amorphised ZIF-8 and polymer.
The gas separation properties of MMMs
The possibilities of these MMMs exhibiting intrinsic homogeneity achieved via thermally sealed interfacial defects alongside with the incorporated interspersed amorphous ZIF-8 having interesting and unique structural and physicochemical properties for the separation of CO2 from CO2/CH4 (50/50% vol) mixtures were subsequently investigated. The membranes were tested using a custom-built, high-throughput gas separation system (HTGS). Fig. 5 shows the separation performance with respect to temperature and ZIF-8 loading. The Matrimid® membranes (Fig. 5a) showed a significant increase in selectivity from 22 to 40 upon treatment at 250 °C. Astonishingly, the crosslinked pure Matrimid® membranes at 350 °C already achieved a selectivity value of 76, almost 4 times higher than the standard pure Matrimid® membranes (selectivity of CO2/CH4 = 20). It should be mentioned that the Matrimid® membranes thermally treated at 350 °C with plasticization resistance have been previously reported.39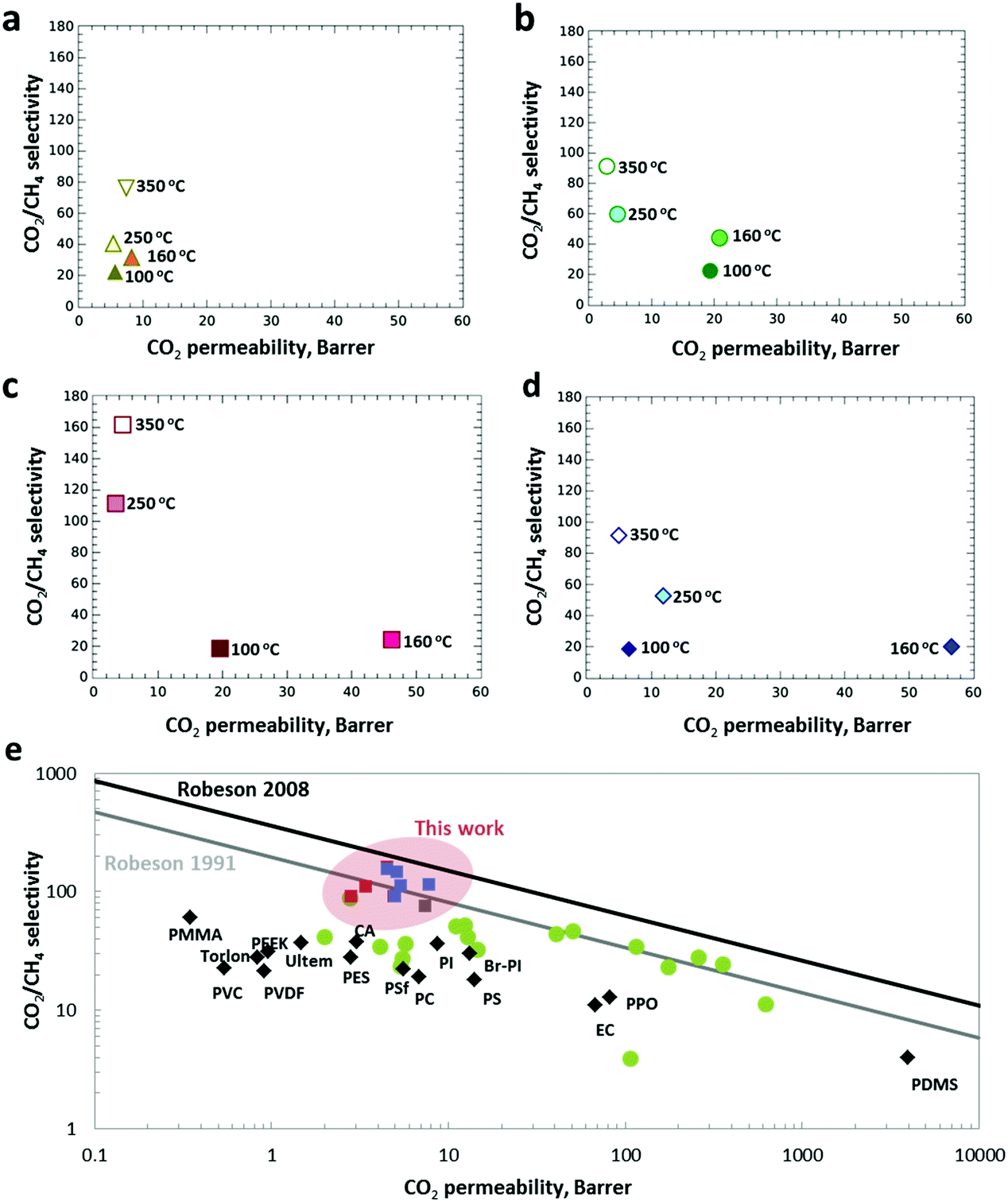 | ||
| Fig. 5 The evolution of the gas selectivity of the thermally treated membranes. (a) Matrimid® and (b–d) MMMs with 20, 30 and 40% ZIF-8 loading with increasing annealing temperature. (e) The gas separation performance of the membranes prepared for this work, primary commercial polymers (black diamonds) and various MOF-based MMMs from literature (green dots) plotted against the Robeson plot of 1991 and 2008. The red and blue squares represent data from MMMs with ZIF-8 and ZIF-7, respectively. A fully detailed comparison of the data in this plot together with the measurements conditions can be found in the Table S3 (ESI†). | ||
However, the lower selectivities of 44 in comparison to our results are probably due to the shorter treatment times (15–30 minutes) applied. In a control experiment, the separation performance of a MMM prepared from pre-amorphised ZIF-8 filler was investigated for CO2/CH4 under similar operating conditions. The amorphised ZIF-8 sample was prepared via ball-milling, as reported by Cheetham and co-workers.40 The characterisation (XRD, SEM and N2 physisorption) of the amorphous ZIF-8 prepared by ball-milling is presented in Fig. S12–S14 and Table S2 (ESI†). The reported method for ZIF-8 amorphisation by ball-milling proved to be highly reproducible. The evidence of polymer–polymer cross-linking is confirmed by ATR-FTIR, as presented in Fig. S15 (ESI†). However, the separation reveals that this MMM with 30 wt% loading failed to achieve high CO2/CH4 selectivity in comparison to the crosslinked MMMs prepared via in situ thermal oxidative treatment (Table S3, ESI†). According to SEM imaging, significant agglomerations and large particles were observed in the pre-amorphised ZIF-8 sample compared to the as-synthesized ZIF-8 (Fig. S13, ESI†). Top view and cross-sectional SEM images show that large particle agglomerations and interfacial voids were clearly visible throughout the MMM (Fig. S16, ESI†). The results explicitly explain the poor separation performance of the MMM prepared from pre-amorphised ZIF-8 filler. The results reveal that thermally induced in situ amorphisation of ZIF-8 in the polymer matrix has distinct advantages over the use of pre-amorphised ZIF-8 for the preparation of defect-free MMMs.
Fig. 5b–d shows the MMM selectivity with regard to ZIF-8 loadings (20–40 wt%) and thermal treatment temperatures (100–350 °C). The thermally treated MMMs (20 wt%) at 100–160 °C exhibit permeabilities up to 20 Barrer which is more than double the permeabilities of the unfilled Matrimid® membranes (8 Barrer). The enhanced permeability is most probably due to the introduction of porosity by the MOF, leading to faster diffusion of the gas molecules by providing easy pathways for the penetrant gases. This phenomenon is more obvious at a higher ZIF-8 loading (30–40 wt%), achieving permeabilities up to 57 Barrer. Similar to the unfilled Matrimid® membranes, MMMs treated at low temperatures (100–160 °C) do not show an improved selectivity, instead an increased in CO2 permeabilities are observed (Fig. 5b), possibly due to the interfacial defects between ZIF-8 nanoparticles and the polymer as well as the residual DMF trapped in ZIF-8. However, with the increase in thermal treatment temperature to 250 °C and 350 °C, a significant increment in selectivities is still achieved. The cross-linked MMMs loaded with 30 wt% of amorphous ZIF-8 achieved the highest selectivity of 162 without sacrificing too much permeability. The superior CO2/CH4 selectivities achieved can be explained by several reasons. (1) Crosslinked polymer networks were achieved via thermal oxidative cross-linking reactions at 350 °C which serve as a molecular sieve network for CO2 separation over CH4 in a mixed-gas condition. (2) The polymer is also cross-linked with the imidazolate linker at the edges of the amorphous ZIF-8 particles, hence eliminating defects at the polymer–filler interface. The cross-linking at the interface of amorphous ZIF-8 with the polymer perfectly sealed the grain boundary at the interface which gave a defect-free MMM, as evidenced by the HAADF-STEM imaging. (3) The improved selectivity may also be attributed by the significant rigidification of the polymer around the filler. The densification of the polymer matrix after cross-linking decreases the CH4 permeability more than the CO2 permeability, which results overall in an improved CO2/CH4 selectivity. Similar phenomena have been reported for cross-linking polymers of intrinsic microporosity (PIMs) with improved selectivity for CO2/CH4 separation.41 Increasing Tg also suggests better interactions between the amorphous ZIF-8 nanoparticles with the polymer chains, restricting motions of the polymer chains, which also adds to the benefit of plasticization resistance at high pressure (40 bar) (vide infra). (4) The abundance of unsaturated Zn2+ and imidazolate linkers, originating from the building blocks of the amorphous ZIF-8 fillers, could also further promote stronger quadrupolar interactions with CO2.42 (5) Last but not least, the embedded crystalline ZIF-8 fillers in the polymer matrix were amorphised as a result of the thermal treatment. According to the EELS imaging, amorphisation of ZIF-8 also leads to a better ordering of the MOF structure in the cross-linked polymer matrix. The amorphous ZIF-8 possessing an interpenetrated, densely packed network structure can serve as an efficient molecular sieve for CH4 molecules.43 An earlier demonstration of the capability of ZIF-8 to trap molecules in the absence of long-range structural periodicity was reported by Chapman et al.,26 with structural evidence for the retention of I2 within the pore network of amorphised ZIF-8. ZIF-7 loaded MMMs show a similar trend in the separation of CO2/CH4 confirming that the demonstrated method is generic (see Table S3 and Fig. S17, S18, ESI†). Based on the separation performance of the MMMs for CO2/CH4 separation, the results suggest that the different chemical structures of ZIF-8 and ZIF-7 have an impact on the separation properties of the MMMs. ZIF-8 is composed of tetrahedral zinc(II) and 2-methylimidazolate, whereas ZIF-7 is composed of tetrahedral zinc(II) and benzimidazolate. The freely available methyl functional groups on the ZIF-8 linkers could enhance polymer-filler crosslinking. As a result, the amorphous MMMs prepared from ZIF-8 filler gave a slightly higher selectivity than ZIF-7 loaded MMMs. Fine-tuning of the surface properties of ZIF for further improvement in CO2/CH4 gas separation performance via e.g. incorporation of accessible amine functionality has been reported.44,45 The incorporation of those mixed-linker ZIFs into the polymer membranes proved to be effective for improving the ideal selectivity for CO2/CH4 separations.45
Over the years, extensive research on improving the performance of commercially available polymer membranes for CO2/CH4 separation via impregnation with MOFs aiming to exceed the Robeson 1991 upper-bound has taken place. Commercial polymers, such as PSF, Ultem®, PPEES or Matrimid® often lead to MMMs with separation properties well below the Robeson upper-bound.46,47 A typical Robeson plot of CO2/CH4 selectivity versus CO2 permeability is presented in Fig. 5e, revealing that the reported cross-linked MMMs containing amorphous ZIF-8 have a separation performance that reached the state-of-the-art upper-bound of 2008 showing the highest selectivities reported so far for MOF based MMMs CO2/CH4 separations. More importantly, gas separation measurements conducted at 40 bar feed pressure confirmed that the implemented post-synthesis treatment induced resistance to plasticization, with selectivities of 90.8 ± 4.2, 134 ± 7.6 and 140 ± 11.5 for the Matrimid®, ZIF-8 and ZIF-7 membranes with 40 wt% loading, respectively (Table S3, ESI†), as a logical consequence of the strong polymer cross-linking.
Conclusions
A high-performance MOF-based MMM was prepared using in situ controlled thermal treatment which remarkably boosts the gas separation performance to the highest CO2/CH4 selectivities ever reported to date for MOF loaded MMMs. The polymer and the MOF become covalently bound, thus decreasing the risk for defects due to grain boundary formation. Advanced TEM imaging of the thin cross-sectional sample prepared via ultramicrotomy successfully provides insights into the crosslinking of the MOF with the membrane which effectively sealed the grain boundary at the interface between the MOF particle and the polymer for the very first time. In addition, the MOF amorphises thanks to the protection of the surrounding polymer, while the polymer phase gets simultaneously cross-linked, resulting to superior separation performances, even under real process conditions for natural gas purification. More importantly, the selectivity and permeability of CO2 could be tuned through simple thermal treatment temperature and time depending on the targeted applications. As demonstrated in this work using a readily available commercial polymer and ZIF-8 filler, the amorphous MMMs has a great potential as stable and highly selective materials for other gas or liquid membrane separations, adsorption, chromatography or catalysis.Methods
Synthesis of MOF particles
ZIF-8 particles were prepared by a modified version of the recipe from Ordoñez et al.,47 using a molar excess of 2-methyl imidazole to Zn(NO3)2·6H2O. Post-synthesis, the solution was centrifuged and washed with DMF, methanol, and CHCl3, respectively. The MOF sludge collected after the final centrifuge step was re-distributed in CHCl3 and stored as such. ZIF-7 was synthesized by dissolving zinc nitrate hexahydrate (0.302 g) and benzimidazole (0.769 g) in DMF (400 mL), and magnetically stirring the mixture for 48 hours at room temperature. The synthesis solution was centrifuged and washed with DMF, methanol, and finally CHCl3. The MOF sludge collected after the final centrifugation step was re-distributed in CHCl3 and stored as such. Amorphous ZIF-8 powder was prepared according to the method reported by Cheetham and co-workers.40 150 mg of ZIF-8 powder was placed inside a 10 mL stainless steel jar alongside a 10 mm stainless steel milling ball at room temperature. The sealed reactor was subjected to 30 Hz milling in a Retsch MM400 grinder mill for 30 min. After milling, the solid product was recovered and characterised by XRD, SEM and N2 physisorption.Preparation of MMMs.
MMMs of 20, 30 and 40 wt% were prepared for this work. An amount of the MOF mixture with known MOF content was weighed, and the Matrimid® (dried overnight at 110 °C) was dissolved in this mixture. The polymer concentration was adjusted around 7 wt% to obtain a reasonably viscous solution that can still be cast and will not suffer from MOF precipitation upon casting. The final mixture was given enough time for the polymer to fully dissolve. As proof of the effectiveness of our membrane fabrication method, additional step of ultrasonication is not needed for obtaining a mixture with a perfect MOF distribution. The MOF loading was calculated as follow:The membrane solution was cast into specially designed Petri dishes that consist of glass rings attached to the flat glass surfaces to ensure uniform membrane thickness was obtained. The cast solution was allowed to vitrify overnight at room temperature in N2 atmosphere to prevent contact with the air humidity. Upon vitrification, the membrane was carefully removed from the petri dish, and placed in an oven for annealing.
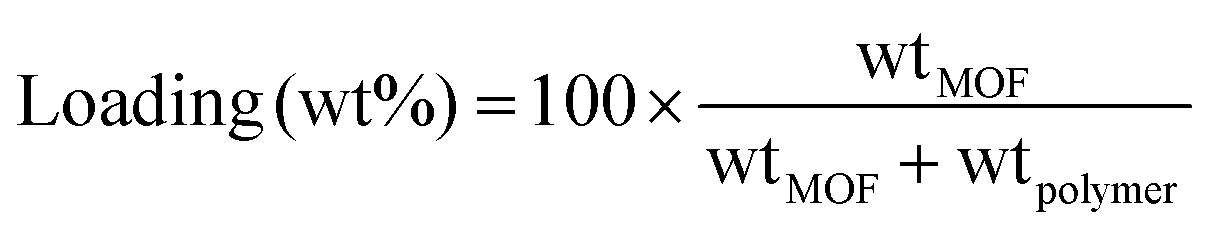
The annealing process
The dried membranes were placed between glass supports to prevent curving and placed in an oven. The oven was heated to 100 (above the boiling point of CHCl3), 160 (slightly above the boiling point of DMF), 250 (above the boiling points of CHCl3 and DMF and below the Tg of Matrimid®) or 350 °C (above the Tg of Matrimid®). The heating process was designed as heating at 1 °C min−1 from room temperature to the final annealing temperature with 50 °C increments. At each increment, the oven was kept isothermally for 2 hours. The membranes remained at the final temperature for 24 hours, and were removed after the oven cooled down to room temperature naturally. Immediate quenching is known to cause the formation of voids between the polymer and the filler due to the difference in the thermal expansion coefficients of the two materials. By allowing the MMMs to cool down naturally, the attachment between the polymer chains and the MOFs was protected.Membrane characterisation
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) was conducted in air using a Varian 620 FT-IR imaging microscope with a Germanium crystal. The samples were analyzed over a wavelength range from 400 to 4000 cm−1 with a spectral resolution of 4 cm−1 and 64 scans. The morphology of the membrane cross-sections was observed with a JEOL JSM-1060LV scanning electron spectroscope (SEM). The membranes that were flexible were freeze-fractured, and the samples that were already too brittle were made to break as evenly as possible in liquid N2. In order to prevent charge build-up due to the non-conductive nature of the polymers, the samples were sputtered with Au/Pd for three cycles of 20 seconds. X-ray diffraction (XRD) patterns of MOF particles and membranes were obtained by using a Stoe-HT X-ray diffractometer, with CuKα radiation, λ = 1.54 Å at room temperature. The thermogravimetric behaviour of the MOFs and membranes was analyzed with TA Instruments TGA-Q500. The samples were heated from room temperature with a heating rate of 5 °C min−1 up to 100 °C, kept isothermally for 10 minutes, then with 2 °C min−1 up to 160 °C, and remained at 160 °C for 10 minutes, and finally with 10 °C min−1 to 500 °C under N2. Differential scanning calorimetry (DSC) measurements were carried out using a TA Instruments DSC Q2000 using Al hermetic closed pans and in N2 atmosphere. The samples were first kept isothermally at 20 °C for 10 minutes, then heated to 370 °C for with a heating rate of 10 °C per minute. After 5 minutes, the samples were cooled down 20 °C at 10 °C per minute, and re-heated to 370 °C using the previously described method. The mechanical strength tests were conducted at room temperature using a Universal Test System (UTS) with 0.0001 mm position resolution and load cells up to 200 N. Scanning transmission electron microscopy (STEM) and spectral imaging (SI) were carried out with an aberration-corrected Titan 60-300 microscope (FEI) operated at 120 kV acceleration voltage, equipped with an Enfinium spectrometer (Gatan) and a high efficiency energy dispersive X-ray (EDX) detector (Super-X). Images of the membrane cross-section were captured with high-angle annular dark-field (HAADF)-STEM. Two samples were prepared with a UC7 ultramicrotome (Leica) with a thickness of 90 nm for imaging and EDX and 40 nm for diffraction and electron energy-loss spectral imaging. For SI convergence and collection angles of 10 mrad and 15 mrad were applied, respectively. TEM diffraction was performed to test for crystallinity using a Philips CM 20 microscope operated at 200 kV. The diffraction pattern was recorded for a previously unexposed area at relatively low magnification comprising several ZIF particles to prevent beam damage. For analysis and quantification of energy-loss spectra HyperSpy was used, an open source Python library.48 Quantification includes removal of plural scattering contributions in the core-loss spectra using low energy-loss spectra. The resulting C/N ratio map was processed by a small Gaussian smoothing filter (size 3 × 3).Analysis of gas separation properties
The gas separation performance of membranes for binary gas mixtures was tested using a custom-built, high-throughput gas separation system (HTGS). The membranes were tested with a 50–50% vol. CO2/CH4 mixture at 35 °C and 10 bar cross-membrane pressure difference. High-pressure measurements were carried out under the same conditions, with the exception of 40 bar cross-membrane pressure difference. The composition of the permeate side was measured by gas chromatography (GC). The membranes were first allowed to reach steady-state overnight. Steady-state was confirmed when consecutive GC measurements gave the same result. Then three more measurements were taken and their average was reported. Each final data point reported is an average of two membranes, and two coupons from each membrane. The permeabilities were measured at steady-state using a constant-volume variable-pressure permeation system. The overall permeability (Po) and the relative permeability of CO2 (PCO2) was calculated by:where α is the rate of pressure increase in the downstream with respect to time, V is the downstream volume, l is the membrane thickness, A is the membrane surface area, R is the ideal gas constant, T is the permeation temperature, and ΔP is the cross-membrane pressure difference
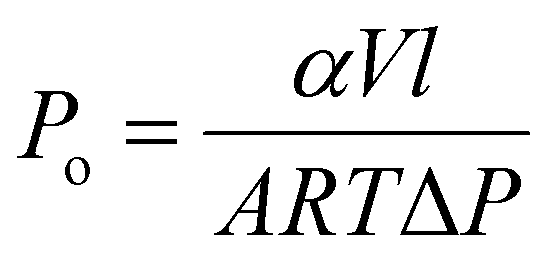
where Po is the overall permeability, yCO2 and xCO2 denote the CO2 content at the permeate and the feed side, respectively.
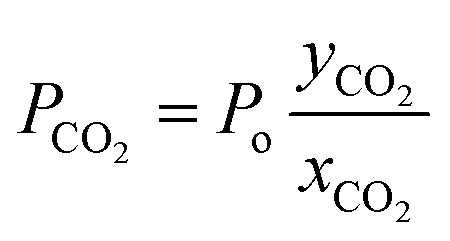
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. acknowledges financial support from the Erasmus-Mundus Doctorate in Membrane Engineering (EUDIME) Programme. L. H. W. thanks the FWO-Vlaanderen for a postdoctoral research fellowship (12M1415N). M. P. acknowledges financial support by the FP7 European project SUNFLOWER (FP7 #287594). S. B. acknowledges financial support from European Research Council (ERC Starting Grant #335078-COLOURATOMS). J. A. M. gratefully acknowledges financial supports from the Flemish Government for long-term Methusalem funding. J. A. M. and I. F. J. V. acknowledge the Belgian Government for IAP-PAI networking. A. K. would also like to thank Frank Mathijs for the mechanical tests, Roy Bernstein for the XPS analysis and Lien Telen and Bart Goderis for the DSC measurements. We thank Verder Scientific Benelux for providing the service of ZIF-8 ball milling.Notes and references
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546 CrossRef CAS PubMed.
- S. Li and F. Huo, Nanoscale, 2015, 7, 7482 RSC.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. Ashraf Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359 CAS.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555 CrossRef CAS PubMed.
- N. Rangnekar, N. Mittal, B. Elyassi, J. Caro and M. Tsapatsis, Chem. Soc. Rev., 2015, 44, 7128 RSC.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390 CrossRef CAS.
- M. Rezakazemi, A. E. Amooghin, M. M. Montazer-Rahmati, A. F. Ismail and T. Matsuura, Prog. Polym. Sci., 2014, 39, 817 CrossRef CAS.
- R. Mahajan and W. J. Koros, Ind. Eng. Chem. Res., 2000, 39, 2692 CrossRef CAS.
- H. Hayashi, A. P. Cote, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS PubMed.
- M. C. McCarthy, V. Varela-Guerrero, G. V. Barnett and H.-K. Jeong, Langmuir, 2010, 26, 14636 CrossRef CAS PubMed.
- S. R. Venna and M. A. Carreon, J. Am. Chem. Soc., 2010, 132, 76 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- H. Bux, A. Feldhoff, J. Cravillon, M. Wiebcke, Y.-S. Li and J. Caro, Chem. Mater., 2011, 23, 2262 CrossRef CAS.
- T. W. Pechar, S. Kim, B. Vaughan, E. Marand, M. Tsapatsis, H. K. Jeong and C. J. Cornelius, J. Membr. Sci., 2006, 277, 195 CrossRef CAS.
- Y. Li, T. Chung, C. Cao and S. Kulprathipanja, J. Membr. Sci., 2005, 260, 45 CrossRef CAS.
- Y. Li, T. Chung, Z. Huang and S. Kulprathipanja, J. Membr. Sci., 2006, 277, 28 CrossRef CAS.
- T. M. Gür, J. Membr. Sci., 1994, 93, 283 CrossRef.
- H. Yong, J. Membr. Sci., 2001, 188, 151 CrossRef CAS.
- R. Mahajan, R. Burns, M. Schaeffer and W. J. Koros, J. Appl. Polym. Sci., 2002, 86, 881 CrossRef CAS.
- T. D. Bennett, A. L. Goodwin, M. T. Dove, D. A. Keen, M. G. Tucker, E. R. Barney, A. K. Soper, E. G. Bithell, J.-C. Tan and A. K. Cheetham, Phys. Rev. Lett., 2010, 104, 115503 CrossRef PubMed.
- J. M. Thomas and L. A. Bursill, Angew. Chem., Int. Ed. Engl., 1980, 19, 745 CrossRef.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, Thomas D. Bennett, N. K. H. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878 RSC.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS PubMed.
- T. D. Bennett, P. J. Saines, D. A. Keen, J.-C. Tan and A. K. Cheetham, Chem. – Eur. J., 2013, 19, 7049 CrossRef CAS PubMed.
- K. W. Chapman, D. F. Sava, G. J. Halder, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 18583 CrossRef CAS PubMed.
- Z. Zhang, S. Xian, Q. Xia, H. Wang, Z. Li and J. Li, AIChE J., 2013, 59, 2195 CrossRef CAS.
- S. Kuroda and I. Mita, Eur. Polym. J., 1989, 25, 611 CrossRef CAS.
- V. Kholodovych and W. J. Welsh, Physical Properties of Polymers Handbook, Springer, New York, 2007 Search PubMed.
- R. A. Jewell and G. F. Sykes, Chemistry & Properties of Crosslinked Polymers, Academic Press, New York, 1977 Search PubMed.
- G. Tillet, B. Boutevin and B. Ameduri, Prog. Polym. Sci., 2011, 36, 191 CrossRef CAS.
- P. Tin, J. Membr. Sci., 2003, 225, 77 CrossRef CAS.
- J. E. Bachman and J. R. Long, Energy Environ. Sci., 2016, 9, 2031 CAS.
- J. E. Bachman, Z. P. Smith, T. Li, T. Xu and J. R. Long, Nat. Mater., 2016, 15, 845 CrossRef CAS PubMed.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427 CrossRef CAS PubMed.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2012, 5, 66 CrossRef PubMed.
- A. D. Katsenis, A. Puškarić, V. Štrukil, C. Mottillo, P. A. Julien, K. Užarević, M.-H. Pham, T.-O. Do, S. A. J. Kimber, P. Lazić, O. Magdysyuk, R. E. Dinnebier, I. Halasz and T. Friščić, Nat. Commun., 2015, 6, 6662 CrossRef CAS PubMed.
- T. Lee, H. Kim, W. Cho, D.-Y. Han, M. Ridwan, C. Won Yoon, J. S. Lee, N. Choi, K.-S. Ha, A. C. K. Yip and J. Choi, J. Phys. Chem. C, 2015, 119, 8226 CAS.
- A. Bos, I. G. M. Pünt, M. Wessling and H. Strathmann, Sep. Purif. Technol., 1998, 14, 27 CrossRef CAS.
- S. Cao, T. D. Bennett, D. A. Keen, A. L. Goodwin and A. K. Cheetham, Chem. Commun., 2012, 48, 7805 RSC.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- Y. Hu, Z. Liu, J. Xu, Y. Huang and Y. Song, J. Am. Chem. Soc., 2013, 135, 9287 CrossRef CAS PubMed.
- A. B. Cairns and A. L. Goodwin, Chem. Soc. Rev., 2013, 42, 4881 RSC.
- J. A. Thompson, N. A. Brunelli, R. P. Lively, J. R. Johnson, C. W. Jones and S. Nair, J. Phys. Chem. C, 2013, 117, 8198 CAS.
- J. A. Thompson, J. T. Vaughn, N. A. Brunelli, W. J. Koros, C. W. Jones and S. Nair, Microporous Mesoporous Mater., 2014, 192, 43 CrossRef CAS.
- R. Abedini, M. Omidkhah and F. Dorosti, RSC Adv., 2014, 4, 36522 RSC.
- M. J. C. Ordoñez, K. J. Balkus Jr., J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28 CrossRef.
- HyperSky, http://https://zenodo.org/record/28025.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee01872j |
| This journal is © The Royal Society of Chemistry 2017 |