
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linker-controlled polymeric photocatalyst for highly efficient hydrogen evolution from water†
Yiou
Wang
 a,
Mustafa K.
Bayazit
a,
Savio J. A.
Moniz
a,
Qiushi
Ruan
a,
Chi Ching
Lau
a,
Natalia
Martsinovich
*b and
Junwang
Tang
*a
a,
Mustafa K.
Bayazit
a,
Savio J. A.
Moniz
a,
Qiushi
Ruan
a,
Chi Ching
Lau
a,
Natalia
Martsinovich
*b and
Junwang
Tang
*a
aDepartment of Chemical Engineering, UCL, Torrington Place, London, WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
bDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: n.martsinovich@sheffield.ac.uk
First published on 2nd June 2017
Abstract
Polymeric photocatalysts have been identified as promising materials for H2 production from water due to their comparative low cost and facile modification of the electronic structure. However, the majority only respond to a limited wavelength region (λ < 460 nm) and exhibit fast charge recombination. Our density-functional theory (DFT) calculations have identified an oxygen-doped polymeric carbon nitride structure with heptazine chains linked both by oxygen atoms and by nitrogen species, which results in a reduced band gap and efficient charge separation. A novel synthetic method has then been developed to control both surface hydrophilicity and more importantly, the linker species in a polymer, which highly influences the band gap and charge separation. As such, the synthesized polymer can be excited from UV via visible to even near-IR (λ = 800 nm) wavelengths, resulting in a 25 times higher H2 evolution rate (HER) than the previous benchmark polymeric g-C3N4 (λ > 420 nm), with an apparent quantum yield (AQY) of 10.3% at 420 nm and 2.1% at 500 nm, measured under ambient conditions, which is closer to the real environment (instead of vacuum conditions). The strategy used here thus paves a new avenue to dramatically tune both the light absorption and charge separation to increase the activity of polymeric photocatalysts.
Broader contextSunlight-driven water splitting is a promising solution to current energy and environmental issues by storing the solar energy in hydrogen. To reach the 10% solar to hydrogen efficiency requirement for practical application, ideal photocatalysts should utilize a large portion of photons from sunlight with effective charge separation. Therefore, many strategies have been developed to discover satisfactory photocatalysts, including heteroatom doping and junctions. Controlling the doping position has been proven to significantly influence the materials' activity, e.g. N-doped TiO2 and Cr-doped SrTiO3. Very recently, polymeric photocatalyst materials such as graphitic carbon nitride have attracted substantial attention due to their earth abundance and chemical stability, and the easy modification of their framework and electronic structure. Directed by theoretical calculations, a strategy to control the doping position of oxygen in a polymer, e.g. at the linker position between heptazine units, has been successfully applied. By controlling the linker species, the modified polymer represents a rather narrow band-gap, a hydrophilic surface, and mitigated charge recombination, leading to highly efficient H2 evolution under UV, visible light and even IR (λ = 800 nm) with a high quantum yield. The linker-controlled strategy could be applied to other polymer photocatalysts in an attempt to improve their activity. |
Introduction
Artificial photosynthesis, different from fossil fuel reforming and water electrolysis, is a promising method to generate H2 in a much greener way by utilizing inexhaustible solar energy and water.1 As visible light (λ = 500–750 nm) accounts for ∼50% of the solar energy hitting the Earth's surface, efficient harvesting of visible photons must be achieved to bridge the gap between the current unsatisfactory activity and the required minimum 10% energy conversion efficiency.2,3 In general, the exploration of semiconductor photocatalysts is deemed as the key target, which should meet the band position requirement for water splitting, but also possess the ideal properties of being efficient, robust, and low-cost.4,5 After decades of investigation, tuning the structure of inorganic semiconductor photocatalysts for efficient photocatalysis still remains very challenging due to their low processability.6 For organic/polymeric semiconductors with better tunability, the issue of poor stability remains a primary shortcoming.7Ever since the demonstration of stable visible light driven H2 production from water on polymeric graphitic carbon nitride (g-C3N4) in 2009, polymeric photocatalysts have attracted much attention, and have stimulated recent studies of covalent organic frameworks and porous organic polymers.8–11 Among these polymers, g-C3N4 is arguably the most stable photocatalyst thermodynamically and optically for H2 production and solar energy storage.12 Very recently it has been demonstrated that with proper co-catalysts, g-C3N4 could split water into H2 and O2.13,14 However, the current energy conversion efficiency on g-C3N4 is moderate due to its large band gap and fast charge recombination.15 Furthermore, a Z-scheme water splitting system composed of two photocatalysts is predicted to have a solar to fuel conversion efficiency of over 40%, which is much higher compared with a single photocatalyst system (ca. 30%).16–20 Thus many efforts have been made to improve the performance of g-C3N4 for H2 production, which can be incorporated in a Z-scheme for pure water splitting. Some successful examples of efficient g-C3N4 for H2 production include g-C3N4 nanosheets, crystalline g-C3N4, cyanamide-defected g-C3N4 and our highly polymerized g-C3N4, which all still possess relatively wide band gaps near 2.7 eV, limiting the light absorption to just a small portion of visible photons (λ < 460 nm).21–24 Other approaches such as element doping, reduced crystallinity, copolymerization, introduction of carbon quantum dot sensitizers, and nitrogen defects were reported to shift the overall absorption towards longer wavelengths.25–33 However, the apparent quantum yield (AQY) was still not satisfactory, which might be due to defect-based recombination centers induced by doping. Therefore an effective strategy to improve the utilization of visible light photons combined with excellent charge separation is urgently needed.
In the rational design of materials, theoretical prediction is widely used to facilitate experimental research by providing beneficial guidance. For example, density of states (DOS) calculations indicated that substitutional doping of N into TiO2 instead of interstitial doping is effective for photocatalysis and hence these target materials were successfully synthesized accordingly.36 Calculations of functionalized surfaces also directed a synthetic route for producing high-energy facets, which influenced photocatalytic activities.34,35 Although doping is an efficient approach to manipulate the band structure of a semiconductor, different doping positions always cause different consequences for the final physical and chemical properties. For example, as mentioned earlier, substitutional doping by N in TiO2 generates an efficient photocatalyst driven by visible light, but interstitial doping by N does not have a positive effect.36 Another example is Cr-doped SrTiO3. (Sr0.95Cr0.05)TiO3 exhibited 100 times higher activity than Sr(Ti0.95Cr0.05)O3.37 Extending this approach from crystalline semiconductors to polymeric materials and developing the strategy of controlling doping position for a polymer based on the benchmark photocatalyst g-C3N4 is both novel and challenging. In this study, modeling was first used to predict the effect of doping position in g-C3N4 on light absorption and charge separation. The calculation results indicate that the band gap and band positions of the polymer are determined by the nature and distribution of the linker species between heptazine units. The linking of heptazine units can be controlled during the polymerization process.38,39 Inspired by such knowledge, we have been experimentally successful in controlling the linker species in a polymer via a monomer-directed polymerization strategy (Scheme S1, see ESI†). This oxygen- and nitrogen-linked heptazine-based polymer (ONLH) exhibits an outstanding response to UV up to near-IR irradiation (λ = 800 nm), which clearly distinguishes it from other reported polymers, and leads to a 25 times higher HER than our reference polymer photocatalyst g-C3N4 under visible irradiation (λ > 420 nm) together with a high AQY of 10.3% at 420 nm. More importantly, and to the best of our knowledge, this is the first report of a single polymer photocatalyst which can produce H2 in such a wide operation window without the need of a sensitizer or a complicated junction structure, which results in a record AQY of 2.1% at 500 nm. Importantly, its activity is highly reproducible after storage under ambient conditions for over 1 year.
Experimental
Materials preparation
In a typical ONLH synthesis, 80–120 g of semicarbazide hydrochloride was placed in a lidded high-quality alumina crucible, then placed inside a muffle furnace, and heated at a ramp rate of 5 °C min−1, and finally held at 500, 550, or 600 °C for 4 h. The products were denoted as ONLH-500, ONLH-550, and ONLH-600. For comparison of photocatalytic activity, the widely used dicyandiamide-derived g-C3N4 was synthesized according to the literature by heating dicyandiamide (DCDA) at 550 °C for 4 hours at a ramp rate of 2.3 °C min−1.40 Water, HCl, and NaOH were used to wash the produced powders to remove all unreacted and potentially detrimental surface species.Material characterization
Powder X-Ray Diffraction (PXRD) measurements were taken using a Bruker D4 diffractometer, at 40 kV, 30 mA, using a Cu source with Kα1 = 1.540562 Å and Kα2 = 1.544398 Å, accompanied by a nickel filter. Diffuse reflectance spectra were obtained using a Shimadzu UV-Vis 2550 spectrophotometer fitted with an integrating sphere. Standard barium sulfate powder was used as a reference. Absorption spectra were calculated from the reflection measurements via the Kubelka–Munk transformation. ATR-FTIR spectroscopy was performed using a Perkin-Elmer 1605 FT-IR spectrometer in the wavenumber range of 500–4000 cm−1 with a resolution of 0.5 cm−1. Raman spectroscopic measurements were performed on a Renishaw InVia Raman Microscope, using a 325 nm excitation laser and a wavenumber range of 100–2000 cm−1. Scanning electron microscopy (SEM) images were gained from a JEOL JSM-7401F high resolution Field Emission SEM operating at 2–3 kV. Due to the low conductivity of the semiconductor materials, an Au coating was sputtered onto the samples to improve the image quality. Specific surface area measurements were taken using the BET method (N2 absorption, TriStar 3000, Micromeritics). XPS measurements were obtained using a Thermoscientific XPS K-alpha surface analysis machine using an Al source. Analysis was performed using CasaXPS.Photocatalytic analysis
In a typical H2 evolution reaction, a certain amount of photocatalyst with 5 wt% of Pt (H2PtCl6 as a precursor) was well dispersed in a 50 ml aqueous solution containing 10 vol% TEOA as a sacrificial electron donor, which was combined inside a 130 ml top-irradiated photoreactor. The reactor was sealed, purged with argon gas (99.99%) for 1 hour, and then irradiated using a 300 W xenon light source (Newport 66485-300XF-R1) to photodeposit the Pt co-catalyst. During the photodeposition, periodic measurements were taken to determine if hydrogen was produced at a stable rate to make sure the photodeposition occurred correctly. The reactor was then re-purged with Argon before irradiation under either full arc or visible irradiation with different filters (Comar Optics). For the quantum yield measurement, 100 mg of polymer was used. The final results used the average activities calculated from five runs. The apparent quantum yield (AQY) (Φ) was calculated by using the following formula:| AQY = (2 × number of evolved hydrogen molecules)/(number of incident photons) × 100% |
Computational method
Candidate structures for g-C3N4 and ONLH were modeled as three-dimensional periodic structures, using density-functional theory calculations as implemented in the CRYSTAL09 software.41,42 The hybrid B3LYP exchange–correlation functional was used,43,44 together with all-electron basis sets: 6-21G* for C and N,45 6-31G* for O,46 and 31G* for H.46 Dispersion interactions between the layers were described by means of Grimme's D2 correction.47 A 2 × 2 × 2 k-point grid (8 k-points) was used in all optimization calculations, while a 8 × 8 × 8 k-point grid (reduced by symmetry to 260 k-points) was used in all density of states (DOS) calculations. Full cell optimization was performed for all structures; no symmetry was applied. Vibrational frequencies and infrared (IR) intensities were calculated using CRYSTAL14 software,48,49 for g-C3N4 and for the proposed model of ONLH. IR absorption spectra were simulated from these frequencies and intensities by superposition of Lorentzian peaks. The damping factor (the parameter characterizing peak width) was chosen as 50 cm−1 in the region 500–2500 cm−1, and 200 cm−1 in the region 2500–4000 cm−1. Molecular orbital plots were calculated using CP2K software for structures optimized using CRYSTAL09, using the same B3LYP functional, the auxiliary density matrix method (ADMM) for calculating the Hartree–Fock exchange part of the hybrid functional, with Goedeker–Teter–Hutter pseudopotentials, double-zeta valence polarized (DZVP) scalar-relativistic basis sets, and auxiliary pFIT3 basis sets for the Hartree–Fock exchange calculations.50–53Results and discussion
As outlined in the Introduction, doping can have a dramatic effect on the electronic structure and optical absorption of host materials, and computational modeling can be used to explore hypothetical doped structures before attempting to synthesize them. Here, DFT calculations were performed to predict the effect of oxygen doping in g-C3N4, a layered two-dimensional polymeric heptazine-based material (see ESI† for computational details). Our calculations indicate an oxygen-doped structure containing two types of heptazine chains linked by oxygen linkers (O-chain) and nitrogen linkers (NH-chain). Only the doped structure with both O-chains and NH-chains is stable, nonplanar and has a reduced band gap compared to g-C3N4 (Fig. 1). Molecular orbital plots (Fig. 1a–c) and density of states plots (Fig. 1d) explain the origin of the band gap narrowing: in this O and NH co-linked structure, the top of the valence band (VB) and the bottom of the conduction band (CB) are spatially separated: the VB top is dominated by heptazine N atoms of the NH-chains, while the CB bottom is composed of nitrogen and carbon states of the O-chains and is shifted to more negative energies (lower CB position) compared to the CB of pure g-C3N4. This means that the O-chains overall have an acceptor character, while the NH-chains have a donor character (similar to the recently reported polymeric donor–acceptor photocatalysts).54 Oxygen atoms themselves make negligible contributions to the VB and CB (Fig. 1d), but cause the down-shift of the CB edge. This gap narrowing and charge separation is not observed if O atoms are placed equally in both chains (Fig. 1c and d). Thus, it is the co-existence of the two types of chains (or distinct N-rich and O-rich regions), rather than O atoms themselves, that gives rise to the gap narrowing. Furthermore, molecular orbital plots in Fig. 1a–c visualise this behaviour: in g-C3N4 and in the O-doped structure with O placed equally in both chains, the highest occupied molecular orbital (the HOMO, or the top of the VB) and the lowest unoccupied molecular orbital (the LUMO, or the bottom of the CB) are spread equally over all chains. In contrast, in the ONLH structure with two inequivalent chains, the HOMO is localized on the NH-linked chains, and the LUMO is localized on the O-linked chains. Therefore, in addition to the reduction in the band gap, the spatial separation of photogenerated electrons and holes in ONLH is likely to reduce electron–hole recombination. In other words, ONLH has the potential to solve the key drawbacks existing in the benchmark g-C3N4. In addition, it is statistically unlikely that some chains will contain only oxygen linkers and others only nitrogen linkers; it is on the other hand likely that many O-linked and N-linked regions are formed in a disordered manner, and act as electron-accepting and electron-donating regions. Again, the donor–acceptor nature of ONLH is expected to give rise to separated photoelectron and photohole states, which should lead to a dramatic enhancement in charge separation efficiency and visible light utilization.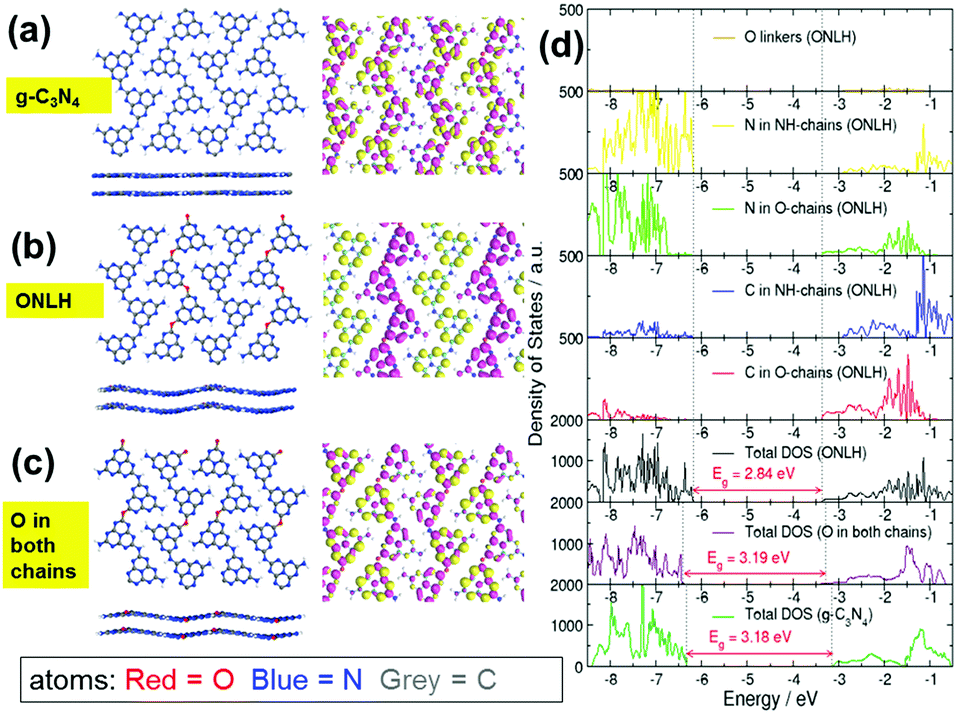 | ||
| Fig. 1 (a–c) g-C3N4, ONLH, and the structure with O linkers in both chains. Left: Proposed structures of g-C3N4 and ONLH according to DFT calculations (front and side views). Middle: Their highest occupied molecular orbitals (yellow) and lowest unoccupied molecular orbitals (pink). (d) Total density of states of g-C3N4 (lowest panel), oxygen-containing material with O atoms placed equally in both chains (second-lowest panel) and ONLH (third-lowest panel) and density of states projected on N, C, and O atoms in different chains in ONLH (upper 5 panels). | ||
Inspired by theoretical predictions, we controlled the polymerization pathway and succeeded in synthesizing the oxygen and nitrogen co-linked heptazine (ONLH) from semicarbazide hydrochloride (NH2CONHNH2HCl), via a one-pot polycondensation route. Samples synthesized at different temperatures (500, 550, and 600 °C) are noted as ONLH-500, ONLH-550, and ONLH-600. For comparison, the widely used dicyandiamide-derived g-C3N4 was synthesized according to the literature.40 To confirm the structure of our new polymer, a systematic process of material characterization was undertaken. From elemental analysis (EA), the bulk atomic stoichiometry of ONLH was found to be C3N4.4O0.3H2.1(ONLH-500), C3N4.3O0.4H1.9(ONLH-550) and C3N4.3O0.5H1.8(ONLH-600), whilst g-C3N4 was noted as C3N4.5O0.1H1.6 (Table 1), showing less nitrogen and much more oxygen in the new polymer. With the polymerization temperature increasing, the amount of O in ONLH goes up, indicating that more O species exist in the polymers' structure at high temperatures.
| Samples | Composition | Surface area/m2 g−1 | Band gap/eV | HER/μmol h−1 | |
|---|---|---|---|---|---|
| >420 nm | Full arc | ||||
| ONLH-600 | C3N4.3O0.5H1.8 | 32.9 | 1.55 | 10.2 | 25.79 |
| ONLH-550 | C3N4.3O0.4H1.9 | 16.7 | 1.65 | 2.44 | 7.5 |
| ONLH-500 | C3N4.4O0.3H2.1 | 14.6 | 1.80 | 1.00 | 3.9 |
| g-C3N4 | C3N4.5O0.1H1.6 | 13.8 | 2.72 | 0.4 | 2.0 |
Powder X-ray diffraction (PXRD) (Fig. 2a) was used to compare the crystal structure of ONLH-500, -550, and -600, and reference g-C3N4. The peaks at 13.0° in the ONLH samples, attributed to the approximate dimension of heptazine units, exist at the same position as in g-C3N4 but are generally much weaker, implying that the conjugated system in the ONLH polymers might not be as well defined as that in g-C3N4.33 The peak at 27.4° for g-C3N4 is assigned to the interlayer stacking distance (0.326 nm).33 As the oxygen concentration in ONLH goes up, this peak shifts left and reaches 27.1° (0.329 nm) in ONLH-600, indicating that the oxygen species account for the enlarged space between the ONLH layers. The widened layer-to-layer distance is consistent with the distorted structure inferred from modeling of g-C3N48 and of ONLH. Through Raman spectroscopy (Fig. 2b), the heptazine structural backbone was confirmed in the ONLH polymers. The main peaks in the Raman spectra (from 1200 to 1700 cm−1) correspond to the dominant C–N stretching vibrations. The peaks at 980 and 690 cm−1 match well with the symmetric N-breathing mode of heptazine units, and the in-plane bending vibrations of the tri-heptazine CNC linkages, respectively.55 The ONLH structure appears a little more disordered because the peaks are not as intense as those of g-C3N4, correlating with the XRD results. Scanning electron microscopy (SEM, Fig. S1, ESI†) indicates a sheet-like structure for both materials, however, ONLH appears more porous (Table 1).
 | ||
| Fig. 2 Characterization of ONLH and g-C3N4: (a) powder XRD pattern, (b) Raman spectra (325 nm excitation), and (c) FT-IR spectra. | ||
Furthermore, the FT-IR spectra (Fig. 2c) support our assignment of the ONLH structure. The peaks of ONLH are generally much broader and less sharp than for g-C3N4 due to it having more C–O bonds in addition to C–N and due to a greater disorder in the structure. This difference between g-C3N4 and ONLH is consistent with the modeling results of IR spectra (Fig. S2, ESI†). The most solid evidence comes from the peaks between 3000 and 3300 cm−1 in g-C3N4, assigned to the stretching modes of the –NHx moieties,24,56 which transform into a very broad low-intensity band assigned to –OH species in the ONLH samples. Moreover, the C–N peaks at 1202 and 1455 cm−1 in g-C3N457 (Fig. 2c) are much less prominent in the FT-IR spectra of the ONLH samples, meaning less C–NHx bonding in ONLH. This is consistent with two intense peaks (1290 and 1540 cm−1) in the calculated IR spectrum of g-C3N4, whose intensity is greatly reduced in ONLH (Fig. S2, ESI†). All these FT-IR features confirm that there are fewer C–NHx and more C–O and C–OH species in the ONLH samples compared with g-C3N4. The FT-IR spectra also rule out some alternatives, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, because no sharp peak is observed in the ∼1700 cm−1 region of the spectrum shown in Fig. 2c.
O groups, because no sharp peak is observed in the ∼1700 cm−1 region of the spectrum shown in Fig. 2c.
X-ray photoelectron spectroscopy (XPS) was used to investigate the chemical states of the elements in these materials (Fig. 3a and Fig. S3, ESI†). Consistent with the EA results, higher O and lower N percentages were found in the ONLH samples compared to g-C3N4 according to the survey spectra of the etched samples (Fig. S3a and b, ESI†). No chlorine content was observed, meaning no residual Cl species from the precursor remain in ONLH. In the O1s XPS spectra of the ion-etched g-C3N4, the only obvious peak at 532.5 eV is assigned to adsorbed H2O.24 In etched ONLH, two new peaks at 531.5 and 533.2 eV are observed, which were attributed to C–OH and C–O–C species (Fig. 3a).58,59 The C1s spectra for both samples (Fig. S3c and d, ESI†) exhibit 3 main peaks at 288.0 eV (NC–N), 286.2 eV (C–O), and 284.6 eV (C–C).24,60 Specifically, the peak at 286.2 eV (attributed to C–O)60 is very weak in g-C3N4, suggesting only slight contamination with oxygen groups, but is much more intense in ONLH, indicating that a large number of carbon atoms are bonded to O atoms, forming C–O bonds. By comparison, CO bonds cannot be responsible for the increased peak at 286.2 eV, because they are expected to give C1s peaks at higher binding energies (287.0–288.0 eV) than C–O (285.5–287.0 eV)61,62 in a similar range to NC–N.60 The N1s spectra illustrate the bonding situation of N in g-C3N4, with peaks at 398.7, 400, 401, and 404 eV which are assigned to heptazine CN–C, N–(C)3, –NHx and π excitation of the C–N conjugated structure, respectively (Fig. S3e and f, ESI†).24 However, in ONLH there are only 3 obvious N1s peaks, while the NHx peak at 401 eV becomes very small. No new N1s peaks appear in ONLH, inferring that no new chemical state of N appears, i.e. N is not directly bonded to O. This decrease of terminal and linking –NHx species, together with increasing C–O, indicates that the oxygen moiety in ONLH (C–OH and C–O–C) might be located at the –NHx positions of g-C3N4. In other words, there are many –OH terminals and C–O–C linkages in the new polymer, resulting in an O/N-linked structure of ONLH (i.e. a mixture of both O and N linkers instead of all N linkers), similar to the computational model. The possibility of O-doping into the heptazine unit, which was proposed/hypothesized before,27 is excluded here as the features of heptazine remain unchanged, both in the Raman, XRD and XPS characterization, and the only remarkable difference is that ONLH exhibits decreased –NHx groups and emerging C–O signals. On the other hand, doping into heptazine might result in issues concerning the stability of the polymer, which we wanted to avoid.
 | ||
| Fig. 3 Characterization of ONLH and g-C3N4: (a) O1s XPS spectra of etched samples, (b) 13C solid state nuclear magnetic resonance (ssNMR) spectra, and (c) UV-Vis spectra. | ||
Carbon-13 solid state nuclear magnetic resonance (13C ssNMR) was used to identify the structural differences (Fig. 3b). The signals at 157 ppm in both samples were assigned to CN3 moieties.63 However, the low field signals related to CN2(NH) and CN2(NH2) at 163 and 165 ppm for g-C3N4 (C bonded to linker N)63 shifted to 164 ppm in ONLH. Moreover, both high and low field signals in ONLH are broadened compared to g-C3N4. These changes are indicative of increased disorder in the system; especially the shift in the high field signal indicates a change in the nature of the linker atoms – this is attributed to fewer NHx species and to the presence of additional O linkages and terminal groups in ONLH, which influences the chemical environment of carbon.63
The UV-Vis spectra of the ONLH samples reveal a dramatically strong red-shift moving into the IR region (Fig. 3c). Compared with typical g-C3N4 with a bandgap of 2.7 eV, much narrower band gaps were determined for ONLH using a Tauc plot (Fig. S4a, ESI,† and Table 1). The band gap narrowing is consistent with our theoretical modeling results, which predict band gap reduction in ONLH due to the NH-linked and O-linked regions, respectively, and a consequent down-shift of the CB (Fig. 1d). In the UV-Vis absorption spectrum of g-C3N4, the band gap absorption around 400 nm is assigned to the characteristic π–π* transitions in heterocyclic aromatics, while the long wavelength absorption is due to the n–π* electronic transitions involving lone pairs of electrons, which is forbidden in a planar structure but allowed in distorted polymeric units.55 Therefore, the ONLH material has a disordered nature as demonstrated by all our characterization results. This disorder is also likely to give rise to separated electron and hole states with energies in the band gap, leading to the Urbach tail in the optical absorption spectrum, which further broadens the absorption. Our calculations, which were performed in a periodic unit cell, cannot fully capture this disorder and the sub-band states, and therefore are unable to reproduce the Urbach tail; therefore, the ONLH absorption is expected to be red-shifted compared to the calculated gap. A higher concentration of oxygen in ONLH results in more distorted structures, and thus narrower band gaps (Table 1), again indicating that the oxygen moieties are responsible for the shift in absorption.
The increased interlayer distance seen in the XRD pattern of ONLH (Fig. 2a) can be ascribed to the O linkers being away from the planar structure, causing structural distortion. The lower intensity of the peaks of ONLH in the XRD and Raman spectra (Fig. 2b) is also due to the disruption of conjugation between neighboring heptazines, caused by oxygen linkers. From FT-IR (Fig. 2c) and XPS spectra on etched samples (Fig. 3a and Fig. S3, ESI†), the decrease of –NHx is confirmed and the oxygen species in bulk ONLH are identified to be C–OH and C–O. The much broader absorption of ONLH polymers in the UV-Vis spectrum (Fig. 3c) is due to more distorted structures caused by the new oxygen linkers. Taking into account the heptazine backbone similarity confirmed by XRD and Raman together with the information from XPS, NMR and FTIR, we can conclude that the structure of the synthesized ONLH material corresponds to the computationally proposed structure shown in Fig. 1b, in which oxygen and nitrogen species link together the heptazine units. This structure has C–O bonds present as ether-type (C–O–C) linkages, replacing some of the –NH– linkers and thus causing a reduction in the number of –NHx groups, while the heptazine rings are preserved intact.
Following the successful preparation of the predicted materials, their photocatalytic hydrogen evolution rates were tested in the presence of a Pt co-catalyst (5%) and TEOA sacrificial electron donor, which is widely used to assess the activity of polymer photocatalysts. No hydrogen was detected in the dark or without photocatalyst, or without TEOA (Fig. S4b, ESI†). It is noted that the H2 evolution rate measured here on g-C3N4 is smaller than that reported in a few papers,22,64 which is due to the conditions used in this study being closer to practical environmental conditions [e.g. 1 bar pressure of inert gas in our reactor instead of vacuum conditions and weak intensity close to one sun (120 mW cm−2)]. The majority of studies reported on H2 evolution by polymer photocatalysts were carried out under circulation-vacuum conditions (∼0.1 bar) which would dramatically improve the energy conversion efficiency by suppressing the back reaction.65 Under our ambient conditions, ONLH-600 exhibits ca. 25 times higher activity under visible light irradiation (λ > 420 nm) than g-C3N440 (Fig. 4a and Table 1). Furthermore, when a 475 nm band pass filter was used (Fig. S4c, ESI†), ONLH-600 showed good photocatalytic activity, however, the benchmark g-C3N4 was not active at all. We also found that ONLH synthesized at different temperatures, and compared to g-C3N4, exhibited a positive correlation between O amount and HER as shown in Fig. 4a and Table 1. Under these ambient and fairly practical reaction conditions, the measured, stable AQY of ONLH-600 calculated after five runs is 10.3% (at 420 nm), corresponding to one of the highest AQYs reported for polymer photocatalysts measured under atmospheric pressure (Tables S1 and S2, ESI†).23,29,66 An unprecedented AQY of 2.1% at 500 nm has also been achieved and the polymer shows activity at long wavelengths extending into the IR region (Fig. 4b).
 | ||
| Fig. 4 (a) Hydrogen evolution rate (HER) and (b) apparent quantum yield (AQY) of ONLH-600 measured at atmospheric pressure under nearly one sun irradiation conditions. | ||
We propose that the pathway of O-terminal/linker formation is related to the acidity of the precursor material NH2CONHNH2HCl. As shown in Scheme S1 (ESI†), during the thermal polymerization process, NH2CONHNH2HCl forms cyanuric acid and liberates ammonia gas.39 Normally, the next step of melamine formation is a dehydration process by replacing –OH species at monomer terminals by NH3.39 However, during the polymerization of NH2CONHNH2HCl, HCl gas is also evolved in the thermal condensation process and can react with NH3, thus competing with the –OH terminals for NH3. Because of the interrupted ideal melamine formation (due to lack of NH3), a certain amount of –OH terminals remain connected to the monomers. Some –OH terminals on heptazine units react with each other to release H2O and form C–O–C linkages, still forming a polymeric system but with a distorted structure instead of a planar one. The hydrochloric acid released in situ is the key to protect the –OH species during the process. This should be an oxygen-maintaining process rather than a replacement process because these O species exist in the precursor. The proposed mechanism leads to the structure of ONLH which agrees well with all of our experimental characterizations and computational modeling.
Thus we controlled the doping position as shown in ONLH in Fig. 1, which is very challenging, resulting in higher photocatalytic activities compared to other polymer photocatalysts reported in the literature (see Tables S1 and S2, ESI†). Any doping into the heptazine ring either makes only small changes to the bandgap or causes concern over stability of the modified polymer, as proven by recent reports from different groups, summarized in Table S2 (ESI†). The importance of the doping position is again emphasized by comparison with another O-containing carbon nitride polymers, obtained by H2O2 treatment of g-C3N4.27 In those reports, oxygen doping was believed to occur in the heptazine units, rather than in the linker positions, as in ONLH. As a result, its electronic properties (band gap ca. 2.49 eV) exhibited only a moderate narrowing compared to pristine g-C3N4 (2.70 eV).27 The prominent differences between the band gaps of H2O2 treated g-C3N4 and ONLH (1.55 eV) strongly suggest that the oxygen linkers in the bulk of the material are essential for achieving a narrow band gap and more efficient visible-light driven photocatalytic activity.
In order to further investigate the reasons for the dramatic increase in H2 evolution by ONLH, photoluminescence (PL) spectroscopy was undertaken using a 325 nm laser probe (Fig. 5a). The PL signal peaks around 450 and 500 nm are assigned to the π–π* transitions and n–π* emission, respectively.55 Strikingly, the PL intensity of ONLH is roughly two orders of magnitude lower than that of g-C3N4 in this region, which indicates that radiative electron–hole recombination is dramatically suppressed in ONLH, consistent with the DFT results. This, to some extent, explains the higher HER of ONLH, caused by the spatially separated electron donor and acceptor regions brought about by the N- and O-linked regions.
 | ||
| Fig. 5 (a) Photoluminescence spectra (325 nm laser excitation) and (b) band alignment of both g-C3N4 and ONLH; (c) the differences in hydrophilicity between ONLH and g-C3N4 using contact angle measurements. | ||
To determine the CB and VB positions for the polymers and their thermodynamic driving force for water splitting, valence band XPS measurements were carried out (Fig. S5, ESI†). The spectra of ONLH and g-C3N4 are similar in shape, however, the edge of ONLH shifted to a lower binding energy compared to g-C3N4. Using evidence from both the valence band XPS and UV-Vis absorption spectra, we can see that the VB of ONLH shifts upward (more negative potential) by 0.25 eV and the CB moves downward (more positive potential) by 0.9 eV compared to pristine g-C3N4 (Fig. 5b). This narrowing of the band gap ensures greater solar absorption but still exhibits sufficient overpotential to drive proton reduction to H2. This is in addition to the improved charge separation illustrated by the PL measurements and theoretical modeling.
We also tested the surface hydrophilicity of the new polymer ONLH-600. Interestingly the active polymer shows a contact angle of 32° compared with 57° measured on g-C3N4, indicating a more hydrophilic surface brought about by OH groups on the new polymer ONLH (see photos in Fig. 5c), which will improve water adsorption and subsequent proton reduction. Regarding the influence of surface area, ONLH-600 only shows twice higher surface area than the others but the HER activity is 4–25 times higher (Table 1). Also, the surface areas of ONLH-500 and ONLH-550 are close to that of g-C3N4, but they are much more active. Therefore, surface area plays a role but is not a dominating factor in the HER activity of the polymers.
Stability of a photocatalyst is a key factor determining its long-term application. We assessed our new polymers’ stability in two ways. One is the widely used multirun test, as shown in Fig. 6a. The activity of hydrogen evolution stably maintains at ca. 10 μmol h−1. The other method is to store the polymer under ambient conditions for a year, then to evaluate its activity. It was found that the activity remains the same as that of the fresh sample (Fig. 6a). Post-testing characterization further proves the stability of the new polymer as shown by Raman and XPS spectroscopies in Fig. 6b and c.
 | ||
| Fig. 6 (a) Stability test of ONLH-600. The first five cycles were typical stability tests and the last run was measured after the sample was stored under ambient conditions for 12 months (>420 nm irradiation). (b) Raman and (c) O1s XPS spectra of ONLH before and after photocatalytic reaction. | ||
Conclusions
In summary, an innovative strategy of linkage controlling polymerization, predicted by DFT calculation, has been developed to synthesize a highly active, robust and narrow band-gap ONLH polymer, which addresses the key challenges facing the benchmark photocatalyst g-C3N4: its small optical operation window and fast charge recombination hampers its efficient photocatalytic H2 evolution but still it must maintain a stable structure. In addition, for the new polymer, the OH terminal groups increase hydrophilicity, overcoming one of the drawbacks of other polymeric photocatalysts. The novel ONLH exhibits ca. 25 times higher HER activity than g-C3N4 under visible light (λ > 420 nm) at atmospheric pressure and moderate light intensity. For the first time, efficient and stable photocatalytic H2 evolution has been observed on a single polymeric photocatalyst using even near-IR excitation, resulting in a record AQY of 10.3% at 420 nm and 2.1% at 500 nm measured in harsh conditions. These excellent properties of the polymer are a result of reduced charge recombination, enhanced hydrophilicity, and narrowed bandgap induced by oxygen linkers and OH terminals which were introduced via a well-designed synthetic pathway. ONLH is potentially applicable in Z-scheme systems for overall water splitting as photoelectrodes.67–70 This polymer can also be used in environmental purification. These findings not only prove the feasibility of polymer modification for wide spectrum photon absorption for efficient H2 fuel synthesis, but should also stimulate fundamental research on the design and tunability of the photophysical properties of polymers through careful control of the polymerization process.Acknowledgements
Y. W. and Q. R. would like to thank the CSC for PhD funding. M. K. B. and J. T. thank the Leverhulme Trust (RPG-2012-582). S. M. and J. T. acknowledge financial support from EPSRC (EP/N009533/1). C. L. thanks the Public Service Department, Malaysia. N. M. acknowledges the use of the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk) accessed via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- G. Hess, Chem. Eng. News, 2005, 83, 12 Search PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- D. J. Martin, P. J. Reardon, S. J. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- S. J. Moniz, S. A. Shevlin, X. An, Z. X. Guo and J. Tang, Chem. – Eur. J., 2014, 20, 15571–15579 CrossRef CAS PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169–180 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- V. W.-h. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS.
- S. Hu, C. Xiang, S. Haussener, A. D. Berger and N. S. Lewis, Energy Environ. Sci., 2013, 6, 2984–2993 CAS.
- H. Doscher, J. F. Geisz, T. G. Deutsch and J. A. Turner, Energy Environ. Sci., 2014, 7, 2951–2956 CAS.
- R. T. Ross and T. L. Hsiao, J. Appl. Phys., 1977, 48, 4783–4785 CrossRef.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- V. W.-h. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Y. Wang, H. Li, J. Yao, X. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819–13826 CAS.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, G. Liu and H.-M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 1605148, DOI:10.1002/adma.201605148.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- A. D. Handoko and J. Tang, Int. J. Hydrogen Energy, 2013, 38, 13017–13022 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- D. Wang, J. Ye, T. Kako and T. Kimura, J. Phys. Chem. B, 2006, 110, 15824–15830 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300–5303 RSC.
- Y. Di, X. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834–838 CrossRef CAS.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr. - Cryst. Mater., 2005, 220, 571–573 CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco and M. Llunell, CRYSTAL09, CRYSTAL09 User's Manual, University of Torino, Torino, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Dovesi, M. Causa, R. Orlando, C. Roetti and V. Saunders, J. Chem. Phys., 1990, 92, 7402–7411 CrossRef CAS.
- C. Gatti, V. Saunders and C. Roetti, J. Chem. Phys., 1994, 101, 10686–10696 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, M. Llunell, M. Causà and Y. Noël, CRYSTAL14, CRYSTAL14 User's Manual, University of Torino, Torino, 2014.
- F. Pascale, C. M. Zicovich-Wilson, F. Lopez Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- M. Guidon, J. Hutter and J. VandeVondele, J. Chem. Theory Comput., 2010, 6, 2348–2364 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- K. Kailasam, M. B. Mesch, L. Möhlmann, M. Baar, S. Blechert, M. Schwarze, M. Schröder, R. Schomäcker, J. Senker and A. Thomas, Energy Technol., 2016, 4, 744–750 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CAS.
- J. Oh, J.-H. Lee, J. C. Koo, H. R. Choi, Y. Lee, T. Kim, N. D. Luong and J.-D. Nam, J. Mater. Chem., 2010, 20, 9200–9204 RSC.
- Z. Zhou, J. Wang, J. Yu, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- C. Clayton and Y. Lu, J. Electrochem. Soc., 1986, 133, 2465–2473 CrossRef CAS.
- D. Briggs and G. Beamson, Anal. Chem., 1993, 65, 1517–1523 CrossRef CAS.
- B. Ozturk, A. de-Luna-Bugallo, E. Panaitescu, A. N. Chiaramonti, F. Liu, A. Vargas, X. Jiang, N. Kharche, O. Yavuzcetin, M. Alnaji, M. J. Ford, J. Lok, Y. Zhao, N. King, N. K. Dhar, M. Dubey, S. K. Nayak, S. Sridhar and S. Kar, Sci. Adv., 2015, 1, e1500094 Search PubMed.
- H.-W. Tien, Y.-L. Huang, S.-Y. Yang, J.-Y. Wang and C.-C. M. Ma, Carbon, 2011, 49, 1550–1560 CrossRef CAS.
- L. Ren, F. Yang, C. Wang, Y. Li, H. Liu, Z. Tu, L. Zhang, Z. Liu, J. Gao and C. Xu, RSC Adv., 2014, 4, 63048–63054 RSC.
- B. V. Lotsch, M. Döblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem. – Eur. J., 2007, 13, 4969–4980 CrossRef CAS PubMed.
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 1700008, DOI:10.1002/adma.201700008.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi and N. Shibata, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- Q. Ruan, W. Luo, J. Xie, Y. Wang, X. Liu, Z. Bai, C. J. Carmalt and J. Tang, Angew. Chem., Int. Ed. DOI:10.1002/anie.201703372.
- J. Xie, C. Guo, P. Yang, X. Wang, D. Liu and C. M. Li, Nano Energy, 2017, 31, 28–36 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- C. Jiang, S. J. A. Moniz, M. Khraisheh and J. Tang, Chem. – Eur. J., 2014, 20, 12954–12961 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee01109a |
| This journal is © The Royal Society of Chemistry 2017 |