
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalysis of polysulfide conversion by sulfur-deficient MoS2 nanoflakes for lithium–sulfur batteries†
Haibin
Lin
a,
Liuqing
Yang
a,
Xi
Jiang
a,
Guochun
Li
a,
Tianran
Zhang
a,
Qiaofeng
Yao
a,
Guangyuan Wesley
Zheng
*ab and
Jim Yang
Lee
 *a
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 10 Kent Ridge Crescent, Singapore 119260, Singapore. E-mail: cheleejy@nus.edu.sg
bInstitute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, Innovis, #08-03, Singapore 138634. E-mail: wesley-zheng@imre.a-star.edu.sg
First published on 25th May 2017
Abstract
Lithium–sulfur batteries are promising next-generation energy storage devices due to their high energy density and low material cost. Efficient conversion of lithium polysulfides to lithium sulfide (during discharge) and to sulfur (during recharge) is a performance-determining factor for lithium–sulfur batteries. Here we show that MoS2−x/reduced graphene oxide (MoS2−x/rGO) can be used to catalyze the polysulfide reactions to improve the battery performance. It was confirmed, through microstructural characterization of the materials, that sulfur deficiencies on the surface participated in the polysulfide reactions and significantly enhanced the polysulfide conversion kinetics. The fast conversion of soluble polysulfides decreased their accumulation in the sulfur cathode and their loss from the cathode by diffusion. Hence in the presence of a small amount of MoS2−x/rGO (4 wt% of the cathode mass), high rate (8C) performance of the sulfur cathode was improved from a capacity of 161.1 mA h g−1 to 826.5 mA h g−1. In addition, MoS2−x/rGO also enhanced the cycle stability of the sulfur cathode from a capacity fade rate of 0.373% per cycle (over 150 cycles) to 0.083% per cycle (over 600 cycles) at a typical 0.5C rate. These results provide direct experimental evidence for the catalytic role of MoS2−x/rGO in promoting the polysulfide conversion kinetics in the sulfur cathode.
Broader contextAmong the alternatives proposed to succeed lithium-ion batteries, lithium–sulfur batteries have drawn the most interest because of the very high theoretical capacity of the sulfur cathode (about 1672 mA h g−1). In addition, sulfur also has the benefits of being low cost, naturally abundant and environmentally benign. The development of lithium–sulfur batteries is however met with several technical challenges. The insulating properties of sulfur and its discharge products (Li2S2 and Li2S) resulted in a slow discharge/charge process and a low practical capacity. The intermediate products formed during battery discharge and charge, i.e. lithium polysulfides (Li2Sn, where 3 ≤ n ≤ 8), are electrolyte soluble. The loss of sulfur electrochemically as dissolved lithium polysulfides is the cause of rapid capacity fading during cycling. This article reports the development of an electrocatalyst, MoS2−x/reduced graphene oxide (MoS2−x/rGO), which can accelerate the kinetics of polysulfide conversion reactions to insoluble products. Sulfur deficiencies in the MoS2 nanoflakes were found to be the catalytic centers. The fast conversion of soluble polysulfides can lower their accumulation in the cathode, and hence their effusion from the electrode. Consequently lithium–sulfur batteries using this catalyst in the sulfur cathode could increase the battery rate performance and cycle stability. |
Introduction
Among the next-generation rechargeable batteries proposed to succeed lithium-ion batteries, lithium–sulfur batteries have drawn the most interest because of the high theoretical capacity of the sulfur cathode (1672 mA h g−1, about 10 times of that of a typical lithium-ion battery cathode).1–4 Sulfur also has the advantages of low cost, natural abundance and environmental benignity. The development of lithium–sulfur batteries is however met with several technical challenges. The insulating properties of sulfur and its discharge products (Li2S2 and Li2S) result in a slow discharge/charge process and a low practical capacity. The intermediate products formed during battery discharge and charge, i.e. lithium polysulfides (Li2Sn, where 3 ≤ n ≤ 8), are electrolyte soluble and as such can migrate to the lithium metal anode and deposit there.5,6 The loss of electrochemically active lithium polysulfides leads to a rapid capacity fading during cycling.The strategies developed to date to address these challenges consist mostly of the following: (i) new cathode designs to increase the electrode conductivity and polysulfide retention,7–10 (ii) new electrolyte formulations,11,12 separator structure13–15 and binder chemistry16,17 to minimize polysulfide migration, and (iii) surface engineering of the lithium metal anode to protect against passivation by the lithium polysulfides migrating from the cathode.18–20 Although substantial progress has been made, these strategies are still far from realizing the full potential of the lithium–sulfur batteries.
Some recent research has considered the alternative of improving the kinetics of polysulfide conversion in the sulfur cathode.21–23 During battery discharge and charge, the conversion between sulfur and its end products (Li2S2 and Li2S) has to occur via lithium polysulfides as the intermediate products which are soluble in most lithium–sulfur battery electrolytes used today.24 Since sulfur, Li2S2 and Li2S are insoluble, accelerating the rates of conversion of soluble lithium polysulfides (to S, Li2S2 or Li2S) can reduce the presence of polysulfides in the electrolyte, and hence their impact on the battery performance. This could improve both the sulfur utilization and the battery cycle stability. Although the use of polar compounds such as Magnéli phase Ti4O7 (2 × 103 S cm−1),23 metal-like TiC (104 S cm−1)25 and CoS2 (6.7 × 103 S cm−1)22 as conductive sulfur hosts with good polarity for polysulfide adsorption has been known for some time, the use of platinum, nickel21 and cobalt26 as the “catalysts” for polysulfide conversion is a relatively recent development. As such the catalysis of polysulfide conversion is still in an early phase of research.
In the search for catalysts which can provide good performance at low cost, we discovered MoS2 to be a strong candidate. MoS2 has been shown to be highly effective for the catalysis of several industrially important reactions such as the hydrogen evolution reaction (HER), the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER).27–30 MoS2 with sulfur deficiencies, in particular, has drawn the most research interest because of the high electrochemical activity associated with the presence of sulfur deficiencies.31,32 Indeed, our previous work on using MoS2 as the lithium-ion battery anode has revealed some behavior of the lithium–sulfur batteries, but without the issues of low sulfur conductivity and polysulfide shuttle in discharge and charge.33 Deficiencies such as MoS2 edge sites and terrace surfaces have shown good electrochemical activity for Li2S deposition.34 Herein, rGO decorated with few-layer MoS2 nanoflakes with a controlled amount of sulfur deficiency (MoS2−x/rGO) was used to catalyze the polysulfide conversion in a sulfur cathode. The MoS2 nanoflakes were prepared by the sonication assisted liquid phase exfoliation of commercial MoS2 powder in N-methyl-2-pyrrolidone (NMP). The amount of sulfur deficiencies could be varied by changing the time and temperature in a heat treatment in hydrogen. The experimental results confirmed the involvement of surface sulfur deficiencies in the polysulfide conversion reactions and their catalytic effect on the kinetics of polysulfide conversion. In the presence of a small amount (4 wt% of the cathode mass) of MoS2−x/rGO in the sulfur cathode, the sulfur cathode exhibited both high-rate capability (capacity of 826.5 mA h g−1 at an 8C rate) and good cycle stability (capacity fade rate of 0.083% per cycle for 600 cycles at a 0.5C rate). These performances place MoS2−x/rGO as one of the best (if not the best) polysulfide conversion catalysts reported to date.
Experimental section
Chemicals
N-Methyl-2-pyrrolidone (NMP, 99.5 wt%), polyvinylidene fluoride (PVDF, 99.5 wt%), sulfur (99.5 wt%), lithium sulfide (Li2S, 99.98 wt%), 1,3-dioxolane (DOL, 99.8 wt%), 1,2-dimethoxyethane (DME, 99.5 wt%), molybdenum(VI) oxide (MoO3, 99.5%), lithium bis(trifluoromethanesulfonyl) imide (LiTFSI, 99.95 wt%) and lithium nitrate (LiNO3, 99.99 wt%) from Sigma Aldrich; molybdenum(IV) sulfide (MoS2, 99 wt%) from Alfa Aesar; and Super-P carbon (99.5 wt%) from Timcal were used as received.Preparation of MoS2 nanoflakes and MoS2/GO composite
Few-layer MoS2 nanoflakes were prepared by the sonication-assisted exfoliation of commercial MoS2 powder in NMP.35 In brief, 100 mg MoS2 powder was dispersed in 20 mL NMP, and sonicated for 5 hours under ambient conditions. After centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 minutes, the supernatant containing the MoS2 nanoflakes was diluted with 30 mL water to form the MoS2 stock solution. A graphene oxide (GO) sample prepared using a modified Hummer's method36 was added to this solution and sonically homogenized for 10 minutes. The composite formed as such (MoS2/GO) was recovered by vacuum filtration.
000 rpm for 5 minutes, the supernatant containing the MoS2 nanoflakes was diluted with 30 mL water to form the MoS2 stock solution. A graphene oxide (GO) sample prepared using a modified Hummer's method36 was added to this solution and sonically homogenized for 10 minutes. The composite formed as such (MoS2/GO) was recovered by vacuum filtration.
Preparation of MoS2−x/rGO and MoS2/rGO composites
MoS2 nanoflakes with sulfur deficiencies (sulfur-deficient MoS2 nanoflakes) were formed by heating the MoS2/GO composite prepared above in a 10% H2/Ar mixture. Different combinations of reaction temperature and time were used for the preparation. A MoS2/rGO composite without the sulfur deficiencies was also prepared for performance comparison. Here the rGO was separately prepared by heating a GO sample in a 10% H2/Ar atmosphere at 600 °C for 6 hours. The rGO was dispersed into the MoS2 stock solution to a rGO/MoS2 mass ratio of 8:2 (the same ratio as that of rGO to MoS2−x in MoS2−x/rGO), and sonically homogenized for 5 hours. The MoS2/rGO composite was then recovered by vacuum filtration.
Preparation of rGO/S, MoS2/rGO/S and MoS2−x/rGO/S composites
rGO/S, MoS2/rGO/S and MoS2−x/rGO/S composites (the actual cathode materials for the lithium–sulfur test batteries) were prepared by the conventional melt-diffusion method. In brief sulfur powder and rGO (or MoS2−x/rGO or MoS2/rGO/S) in a 75:25 mass ratio were homogenized by grinding; and then sealed in a vial with Ar. The mixture was then heated at 155 °C for 5 hours to distribute the sulfur uniformly in rGO (or in MoS2/rGO/S or MoS2−x/rGO).
Materials characterization
The morphology of the composites in this study was examined by field emission scanning electron microscopy (FESEM) on a JEOL JSM-6700F SEM, by transmission electron microscopy (TEM) on a JEOL 2100F microscope, and by high-resolution TEM (HRTEM) on a JEOL 2100F system. The composite crystal structures were determined by X-ray diffraction (XRD) on a BRUKER D8 ADVANCE (Germany) instrument using Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) analysis of the samples was performed on a Kratos AXIS Ultra DLD surface analyzer using a monochromatic Al Kα radiation source at 15 kV (1486.71 eV). The XPS peak locations were corrected by referencing the C 1s peak of adventitious carbon to 284.5 eV. Spectral deconvolution was carried out using the XPS Peak 4.1 software. The rGO and sulfur contents of the composites were analyzed by thermogravimetry (TGA) on a Shimadzu DTG-60H analyzer in air (for the measurement of the rGO content) or in N2 (for the measurement of the sulfur content) at a temperature ramp rate of 10 °C min−1.Adsorption properties of lithium polysulfides
Li2S and sulfur in amounts corresponding to the nominal stoichiometry of Li2S6 were added to a 1:1 (v/v) DOL/DME mixture and stirred overnight at 60 °C. The concentration of the Li2S6 solution prepared as such was 3 mmol L−1, and was used as the stock solution for adsorption measurements. 10 mg rGO, MoS2/rGO or MoS2−x/rGO was added to 2 mL each of the lithium polysulfide stock solution. The mixtures were vigorously stirred to facilitate adsorption.
Cell assembly and electrochemical measurements
Symmetric electrochemical cells were assembled by the following procedure: 80 wt% active material (MoS2−x/rGO, MoS2/rGO, or rGO) and 20 wt% PVDF binder were homogenized in NMP to form a consistent slurry, which was then uniformly applied to an Al foil. The foil was cut into 1 cm × 1 cm sheets. The active material loadings on the sheets were about 2–4 mg. CR2025 coin cells were assembled in an Ar-filled M Braun glove box by using two coated Al sheets as the cathode and anode, a Celgard 2400 separator, and 50 μL electrolyte of 1 M LiTFSI and 0.2 M Li2S6 in a 1:1 (v/v) DOL/DME mixture. The counter electrode after the test was disassembled from the cell, rinsed with DOL thrice to remove the lithium salt on the surface; and then evacuated overnight at room temperature for ex situ analysis on the next day. Lithium–sulfur test batteries were assembled by a slightly different procedure: an NMP slurry of 80 wt% active material (MoS2−x/rGO/S, MoS2/rGO/S or rGO/S), 10 wt% Super P and 10 wt% PVDF was applied onto an Al foil to a loading of ∼1.5 mg cm−2. CR2025 coin cells were assembled using the coated Al foil as the cathode, a lithium metal foil anode, a Celgard 2400 separator, and 50 μL 1 M LiTFSI and 2 wt% LiNO3 solution in DOL/DME (1:1 v/v) as the electrolyte. A Neware battery tester was used to regulate the cell discharge and charge. The cathode specific capacities were normalized only by the mass of sulfur, as per the common practice. Cyclic voltammetry (CV) and electrochemical impedance measurements were carried out on an Autolab type III electrochemical workstation.
Results and discussion
Fig. 1A shows the major steps in the preparation of MoS2−x/rGO. The MoS2 nanoflakes and GO (Fig. 1B) were co-dispersed in water; and then the mixed solid phase recovered by filtration was heated in a reducing hydrogen atmosphere at high temperature. The nanocomposite formed as such consisted of MoS2−x nanoflakes on a thin film of rGO. rGO, a common substrate for electrochemical devices,37–39 was used here as a flexible and conductive catalyst support. Fig. 1C is the TEM image of the MoS2 nanoflakes formed by the liquid phase exfoliation of bulk commercial MoS2 particles shown in Fig. S1 (ESI†). MoS2 could be exfoliated into nanoflakes very easily by this procedure and formed a uniform dispersion in the solvent (Fig. S2, ESI†). The HRTEM image in Fig. 1D shows the layer-like structure of the MoS2 nanoflakes, which were about 3–5 nm in thickness and consisted of 6–8 layers. The lattice spacing of 0.62 nm matches well with the (002) diffraction of hexagonal MoS2.40 The small MoS2 nanoflakes were well dispersed on the rGO sheets. The high temperature treatment in hydrogen removed some sulfur atoms in the MoS2 nanosheets to result in the formation of sulfur deficiencies. The geometric compatibility between the two 2D nanomaterials (rGO and MoS2 nanosheets) should improve the quality of the interfacial contact, and the 2D-on-2D construction also allowed a good exposure of the sulfur deficiencies on the catalyst surface for the conversion of adsorbed polysulfides.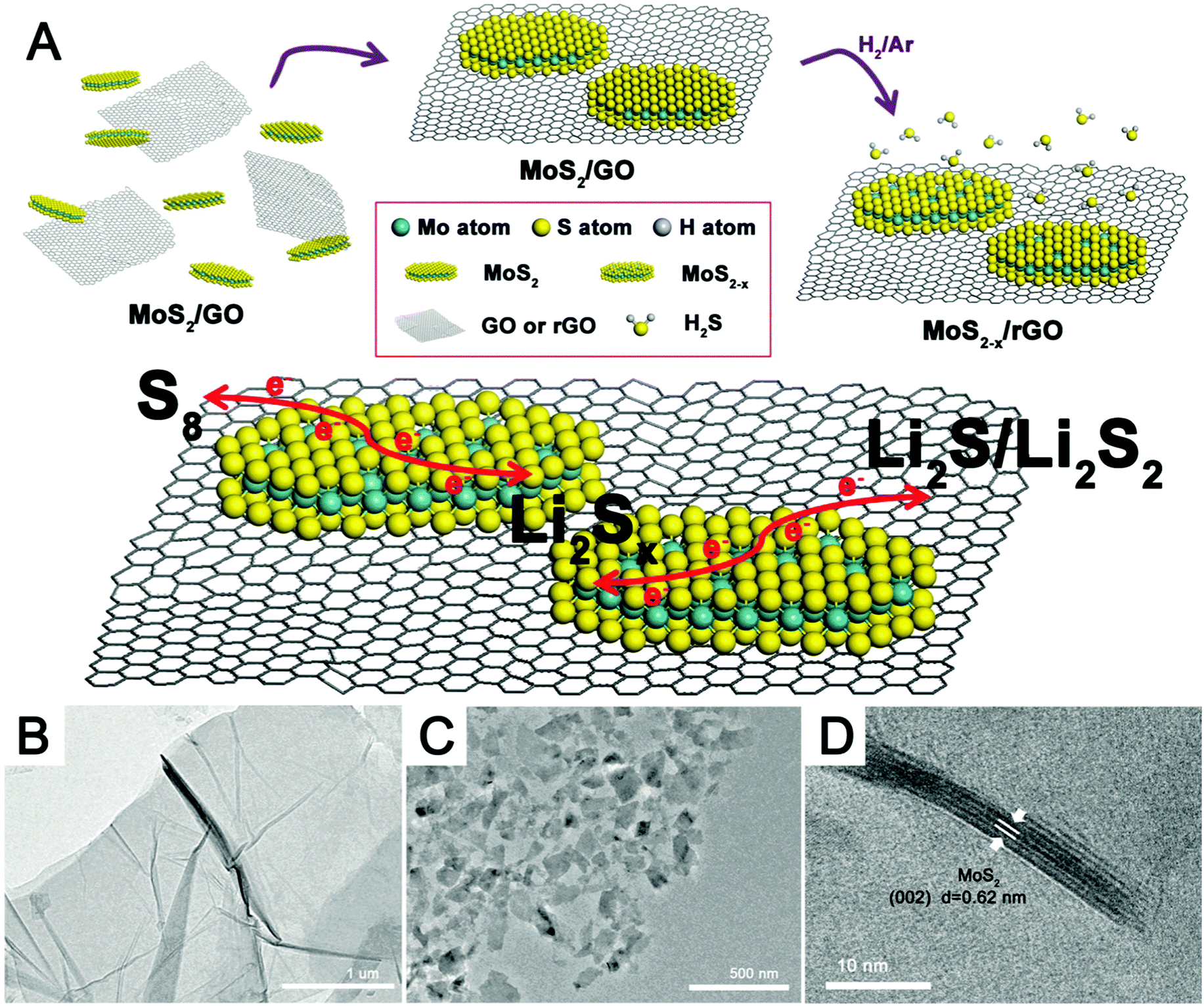 | ||
| Fig. 1 (A) Schematic of the synthesis of the MoS2−x/rGO composite and the conversion of Li2Sx on the MoS2−x/rGO surface. TEM images of (B) a thin GO film and (C) MoS2 nanoflakes. (D) HRTEM image of MoS2 nanoflakes. | ||
The effects of heat treatment temperature and time on the structure of the MoS2−x/rGO composite were analyzed by XRD and XPS. The XRD patterns of samples prepared under different conditions are quite similar (Fig. 2A). The broad diffraction at around 2θ = 20–30° can be attributed to the disorderly stacked rGO sheets. The diffraction peaks of the MoS2 nanoflakes are in good agreement with the 2H phase of MoS2 (PDF#37-1492),41 and hence the phase purity of MoS2 was good. The most intense MoS2 peak was the (002) peak at 2θ ∼ 15°, suggesting [001] as the crystal growth direction. Fig. 2B shows the expanded view of the MoS2 (002) peak. There was a slight shift of this peak to lower 2θ values with the increase in treatment severity (higher temperature or longer heat treatment time). The shift indicates an increase in the lattice parameter42 caused most likely by the removal of sulfur by hydrogen. The resultant reduction of Mo to a lower oxidation state with a larger atomic radius led to the increase in the lattice parameter.
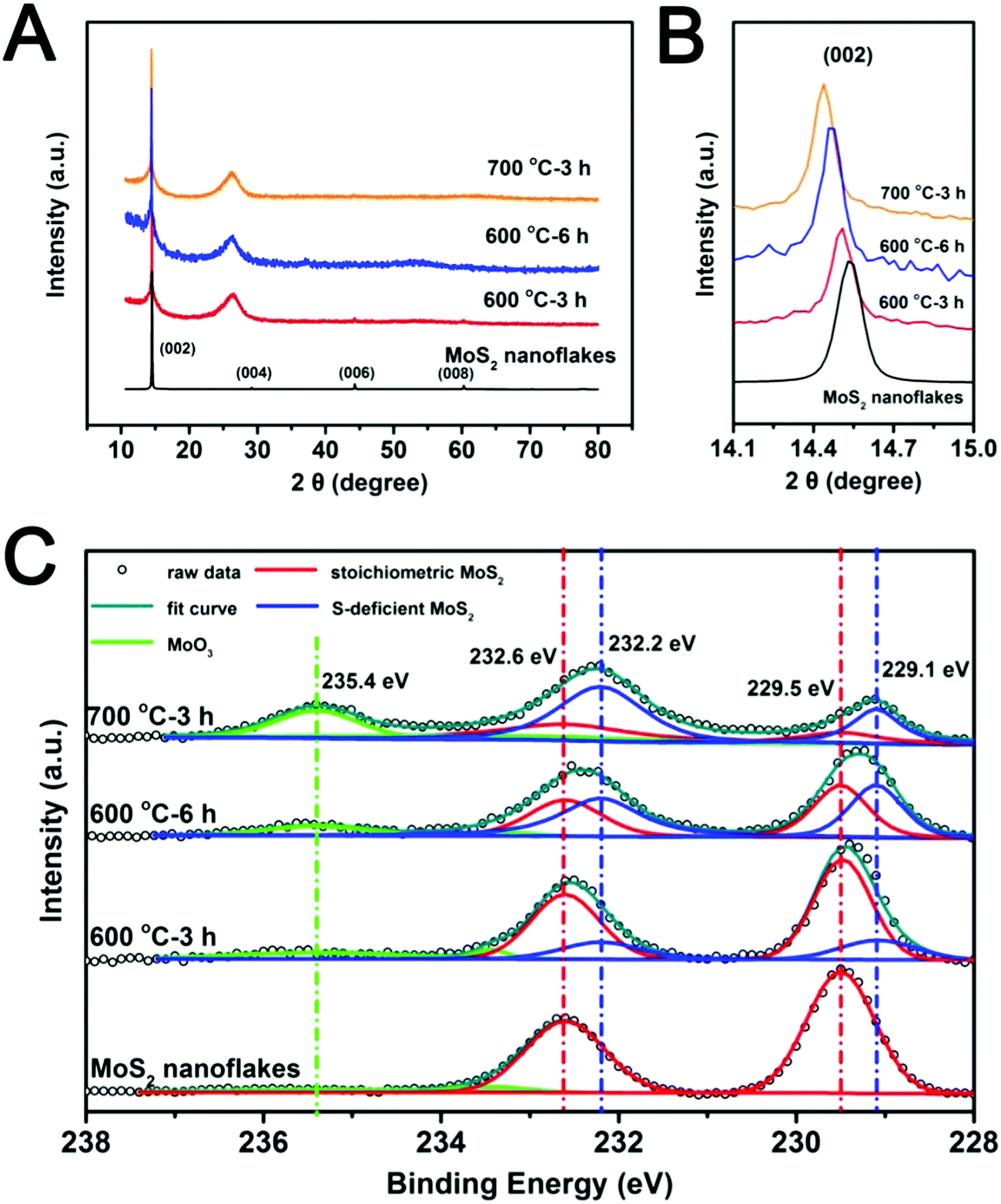 | ||
| Fig. 2 (A and B) XRD patterns and (C) Mo 3d XPS spectra of MoS2 nanoflakes and MoS2−x/rGO composites formed by different combinations of reaction temperature and time in a hydrogen atmosphere. | ||
The surface compositions of MoS2−x/rGO composites prepared under different heat treatment conditions were characterized by XPS. The total molybdenum (Mo) and sulfur (S) contents of the samples as analyzed by XPS are summarized in Table S1 (ESI†). The Mo:S ratio of the as-synthesized MoS2 nanoflakes was 33.2:65.4, close to the 1:2 ratio in stoichiometric MoS2. The Mo:S ratio increased with the increase in reaction temperature and reaction time; indicating the progressive removal of the sulfur element. The Mo 3d spectra were deconvoluted to determine the stoichiometric MoS2 (red), the sulfur-deficient MoS2 (blue) and the MoO3 (green) contents of various samples (Fig. 2C).43,44 Specifically the Mo 3d5/2 and 3d3/2 doublets at ∼229.5 eV and 232.5 eV were deconvoluted into two sets of peaks. The first set of peaks with binding energies of 232.6 eV and 229.5 eV could be attributed to stoichiometric MoS2, while the second set at lower binding energies (232.2 eV and 229.1 eV) could be assigned to sulfur-deficient MoS2. The peak distinctively upstream of the MoS2 peaks may be attributed to MoO3 (green).44 The appearance of MoO3 could be attributed to the oxidation of some low oxidation state Mo atoms in MoS2, as per the previous report.35 The results showed that the increase in temperature and reaction time increased the amount of sulfur deficiency. The presence of MoO3 in the 700 °C sample could be due to the oxidation of Mo metal clusters (which is highly susceptible to atmospheric oxidation). Thermal annealing of MoS2 in a hydrogen environment could lead to the removal of sulfur atoms as H2S gas and hence the formation of sulfur deficiencies. The excess Mo could also form Mo metal clusters.59 The Mo metal clusters were oxidized to MoO3 after the sample was removed from the heating chamber. MoO3 could also be formed directly by the substitution of sulfur in MoS2 with oxygen from GO during the heat treatment. Although the 700 °C sample contained a high sulfur deficiency content, the deep reduction of MoS2 led to an unstable nanoflake structure (Fig. S3, ESI†) caused by the excessive expansion of the lattice parameter. The MoS2−x/rGO composite with the highest sulfur deficiency content which could still preserve the nanoflake structure was prepared at 600 °C for 6 hours (x = 0.42).
The morphology of the stable high sulfur-deficiency MoS2−x/rGO composite (prepared at 600 °C for 6 hours) was examined by both FESEM and TEM. The FESEM image in Fig. 3A shows that the composite mirrored the laminate structure of rGO synthesized under the same conditions (Fig. S4A, ESI†). TEM images (Fig. 3B and C) confirm the presence of MoS2−x nanoflakes on the rGO sheets. The 0.62 nm lattice spacing in the HRTEM image of a MoS2−x nanoflake sample (Fig. 3D) is the same as that of the (002) planes of hexagonal MoS2 in Fig. 1D, indicating that the sulfur deficiencies did not alter the native MoS2 structure. The rGO content in the MoS2−x/rGO composite as calculated by TGA (Fig. S5, ESI†) was about 78 wt%. For comparison, a composite containing MoS2 and rGO (reduced from GO by heating in hydrogen at 600 °C for 6 hours), MoS2/rGO, was also analyzed. The rGO content in the latter was similar, 77 wt%. TGA nonetheless detected higher thermal stability for rGO in MoS2−x/rGO to suggest the stronger interaction between rGO and MoS2 when the latter was sulfur deficient. Since sulfur deficiencies can render the MoS2−x surface more electron rich,45 and rGO formed below 600 °C has general p-type characteristics,46 electron transfer from rGO to MoS2−x may occur to develop a stronger bond between the two at their interface.47 Such electron coupling is expected to contribute positively to the charge transfer at the MoS2−x/rGO interface.
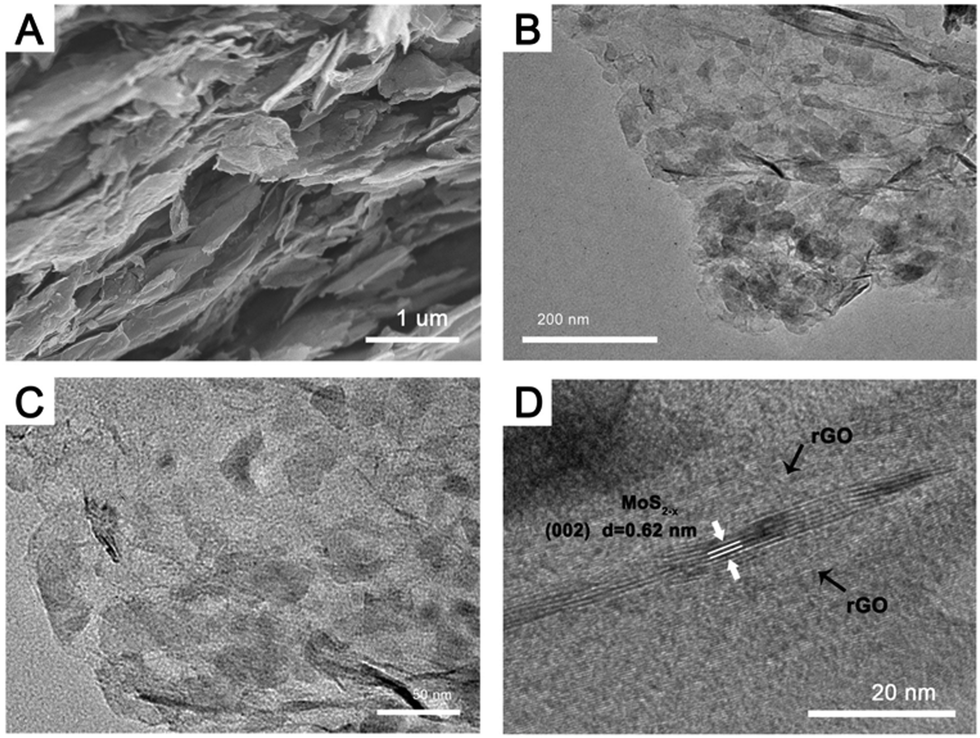 | ||
| Fig. 3 (A) FESEM image, (B and C) TEM images and (D) HRTEM image of the MoS2−x/rGO composite prepared by heating MoS2/GO in a hydrogen atmosphere at 600 °C for 6 hours. | ||
The catalytic effect of MoS2−x on the polysulfide redox reactions was first revealed by CV in symmetric cells with identical working and counter electrodes in a 0.2 M Li2S6 electrolyte (Fig. 4A). MoS2/rGO and rGO prepared under the same conditions were used as the experimental controls (Fig. 4B and C). The CV of a Li2S6-free electrolyte was also measured to correct for capacitive contributions. The voltammogram of the MoS2−x/rGO electrode in the Li2S6 electrolyte exhibited high reversibility with four distinct peaks at −0.047 V, −0.39 V, 0.047 V and 0.39 V respectively (Fig. 4A). The MoS2/rGO electrode exhibited remnants of these peaks as broad redox features at −0.31 V, −0.61 V, 0.31 V and 0.61 V (Fig. 4B). For the rGO electrode, only a very drawn-out reduction peak at −1.22 V and a very drawn-out oxidation peak at 1.22 V were detected (Fig. 4C).
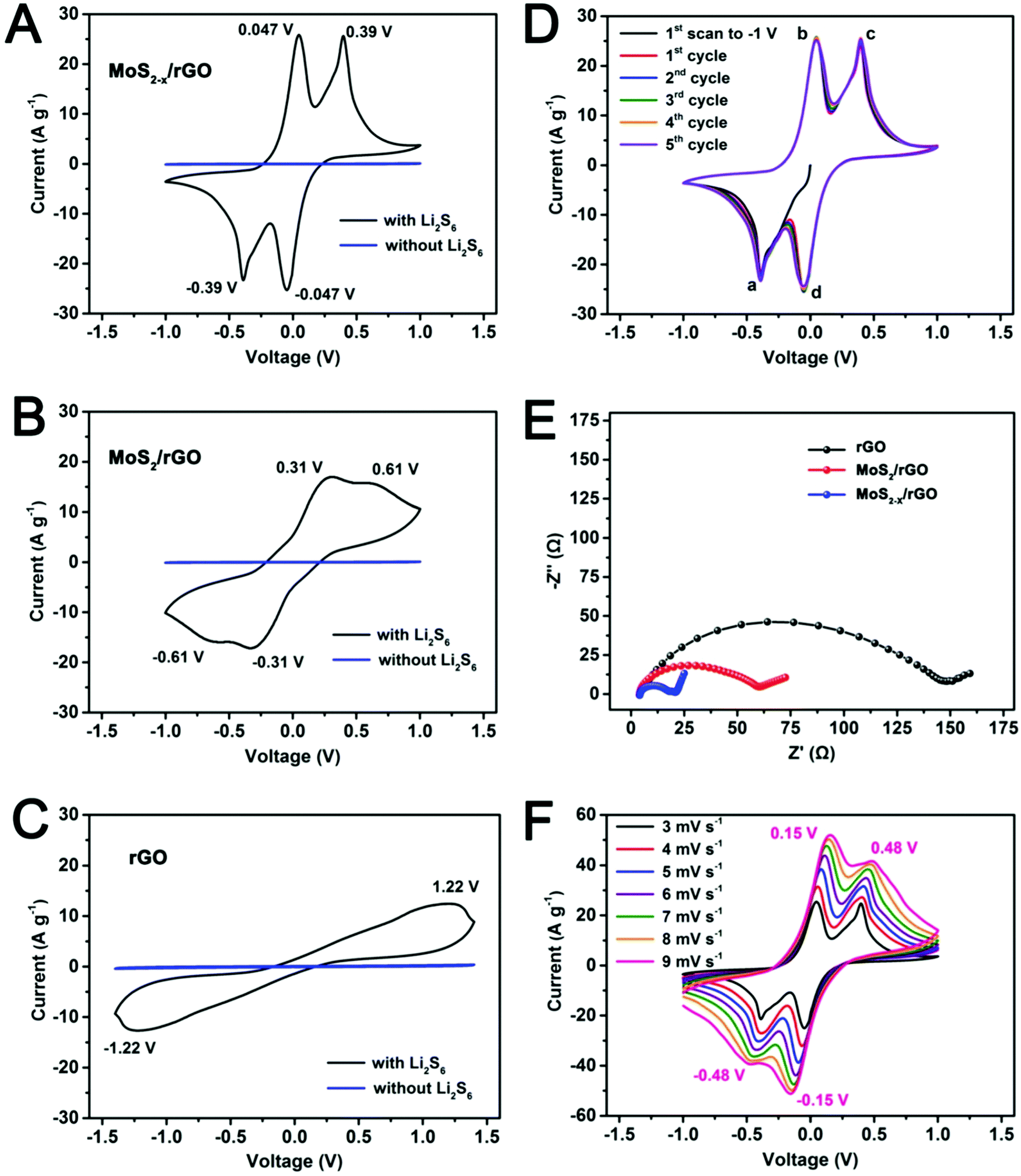 | ||
| Fig. 4 Cyclic voltammograms of symmetric cells with identical electrodes of (A) MoS2−x/rGO, (B) MoS2/rGO and (C) rGO in electrolytes with and without 0.2 M Li2S6 at 3 mV s−1. (D) Multi-cycle voltammograms of the MoS2−x/rGO symmetric cell at 3 mV s−1. (E) Electrochemical impedance spectra of the symmetric cells. (F) Voltammograms of the MoS2−x/rGO symmetric cell at different scan rates. | ||
Fig. 4D shows the first five cycles of the MoS2−x/rGO electrode in CV. The nearly perfect superimposition of the peaks suggests good stability of the sulfur-deficient MoS2−x/rGO electrode. In the first cathodic scan from zero potential between the electrodes, only the peak at −0.39 V (peak a) appeared. The cathodic peak at −0.047 V (peak d) emerged only from the second scan onwards. Since Li2S6 was the only electrochemically active species in the electrolyte, it is reasonable to assume that Li2S6 was reduced to Li2S (or Li2S2) on the working electrode, and oxidized to sulfur on the counter electrode in the cathodic scan. Hence the reduction of Li2S6 on the working electrode which manifested in peak a was complemented by the oxidation of Li2S6 on the counter electrode. Peak b in the following anodic scan was due to the reconstitution of Li2S6 by the oxidation of Li2S (or Li2S2) on the working electrode. Similarly, peaks c and d identical in shape to peaks a and b were due to the oxidation of Li2S6 to sulfur, and the reduction of sulfur to Li2S6 on the working electrode respectively. Therefore, the peaks at −0.39 V/0.047 V and −0.047 V/0.39 V‡ were paired redox features of the symmetric cell. The reactions are summarized in Fig. S6 (ESI†). The sharpness of the peaks and the narrow peak separation in each redox pair indicate good electrochemical reversibility and facile polysulfide conversion. It should be mentioned that the two paired redox peaks related to the Li2S6 conversion reaction were absent in a previous study on CoS2 using the symmetric cell.22 The high scan rate (50 mV s−1) and the narrow voltage range (from −0.7 V to 0.7 V) used in that study could have suppressed the detectability of these redox features. When the electrodes were MoS2/rGO without the surface sulfur deficiencies, the broadened peaks and the increased peak separation are indications of reduced electrochemical reversibility and slower reactions (Fig. 4B).§ Electrochemical reversibility and conversion kinetics were the lowest with the rGO electrodes, resulting in the merging of peaks (Fig. 4C).
The polarity-induced adsorption between the polysulfides and a polar sulfide surface may have contributed to the more facile kinetics of polysulfide conversion on MoS2/rGO and MoS2−x/rGO (the apolar rGO surface is antagonistic to polysulfide adsorption).48 This was demonstrated by a simple visual adsorption test (Fig. S8, ESI†) where the adsorption of Li2S6 on MoS2/rGO and MoS2−x/rGO completely decolorized the polysulfide solution. Electrochemical impedance spectroscopy (Fig. 4E) also registered the smallest charge transfer resistance (the size of the high frequency semicircle in the Nyquist plot) for the MoS2−x/rGO symmetric cell. In the CVs measured at different scan rates (Fig. 4F), there were some slight shifts of the redox peaks with the increase in the scan rate. However, the peak separation in the MoS2−x/rGO cell at a high scan rate of 9 mV s−1 was still significantly narrower than the peak separations in the MoS2/rGO or rGO cells at 3 mV s−1. All the above are evidence for the greatly enhanced kinetics of polysulfide conversion on the MoS2−x/rGO surface.
For additional insights into the reactions of polysulfides on the MoS2−x/rGO surface, the counter electrodes of symmetric cells with MoS2−x/rGO or rGO electrodes after scanning from 0 V to −1.4 V were examined by FESEM.¶Fig. 5 shows the FESEM images of the rGO (A–C) and the MoS2−x/rGO (D–F) counter electrodes before and after scanning to −1.4 V. The larger number of sulfur particles and their more uniform distribution on the MoS2−x/rGO surface could only come from the presence of more electrochemically active sites for polysulfide conversion. XPS was also used to analyze the surfaces of the counter electrodes of symmetric cells after scanning from 0 to −0.14 V, and from −0.14 V to 0 V. Fig. 5G and H show, respectively, the S 2p XPS spectra of the rGO and MoS2−x/rGO counter electrodes. The 163.5 eV and 164.7 eV peaks could be attributed to the sulfur deposited on the counter electrodes, while the very prominent peak at ∼169 eV to the S–O bond in oxidized sulfur species such as –SOx.21 Since sulfur deposition on the counter electrode was mostly completed when the symmetric cells were scanned to −0.39 V (Fig. 4D), the sulfur deposit would be extensively oxidized when the cells were scanned to −1.4 V. The stronger S–O peak from the MoS2−x/rGO cell can then be used as an indirect evidence for more sulfur formation in this cell (Fig. 5G and H). When this symmetric cell was returned to 0 V from −1.4 V, the decrease in the S–O peak intensity was caused by the electrochemical reduction of the oxidized sulfur species on the counter electrode. In contrast, the S–O peak from the rGO symmetric cell underwent very minor intensity changes from −1.4 V to 0 V, an indication of the limited sulfur presence on the rGO surface. The more extensive sulfur formation and reduction reactions in the MoS2−x/rGO cell could only be caused by the existence of catalytically more active sites on the MoS2−x/rGO surface. There was also evidence in the Mo 3d XPS spectra for the interaction between MoS2−x and polysulfides during the polysulfide conversion reactions (Fig. 5I). When the symmetric cells were scanned to −1.4 V, the sulfur-deficient MoS2 component (blue curve) in MoS2−x/rGO was significantly diminished in intensity. It is believed that the deficiencies in MoS2−x rendered the surface of MoS2−x/rGO electron-rich. The XPS results of Fig. 5I indicate a weaker XPS signal from the sulfur deficiencies after sulfur deposition, suggesting the electron transfer from the former to the latter.43,49,50 It has been reported in the oxygen reduction reaction (ORR) research that oxygen adsorption on an oxygen-deficient surface would elongate the O–O bond for an easier reduction.56 The sulfur deficiencies on the MoS2−x/rGO surface may likewise facilitate the reduction of sulfur to polysulfides, probably through the involvement of some metastable Sx˙− species.57,58 When the symmetric cell was scanned back to 0 V, XPS showed that the sulfur deficiencies were restored, and hence the reversibility of the overall process. These changes establish the correspondence between sulfur deficiency and the extent and reversibility of polysulfide conversion, and provide indirect proof for sulfur deficiencies as the origin of enhanced catalytic activity in polysulfide electrochemical reactions.
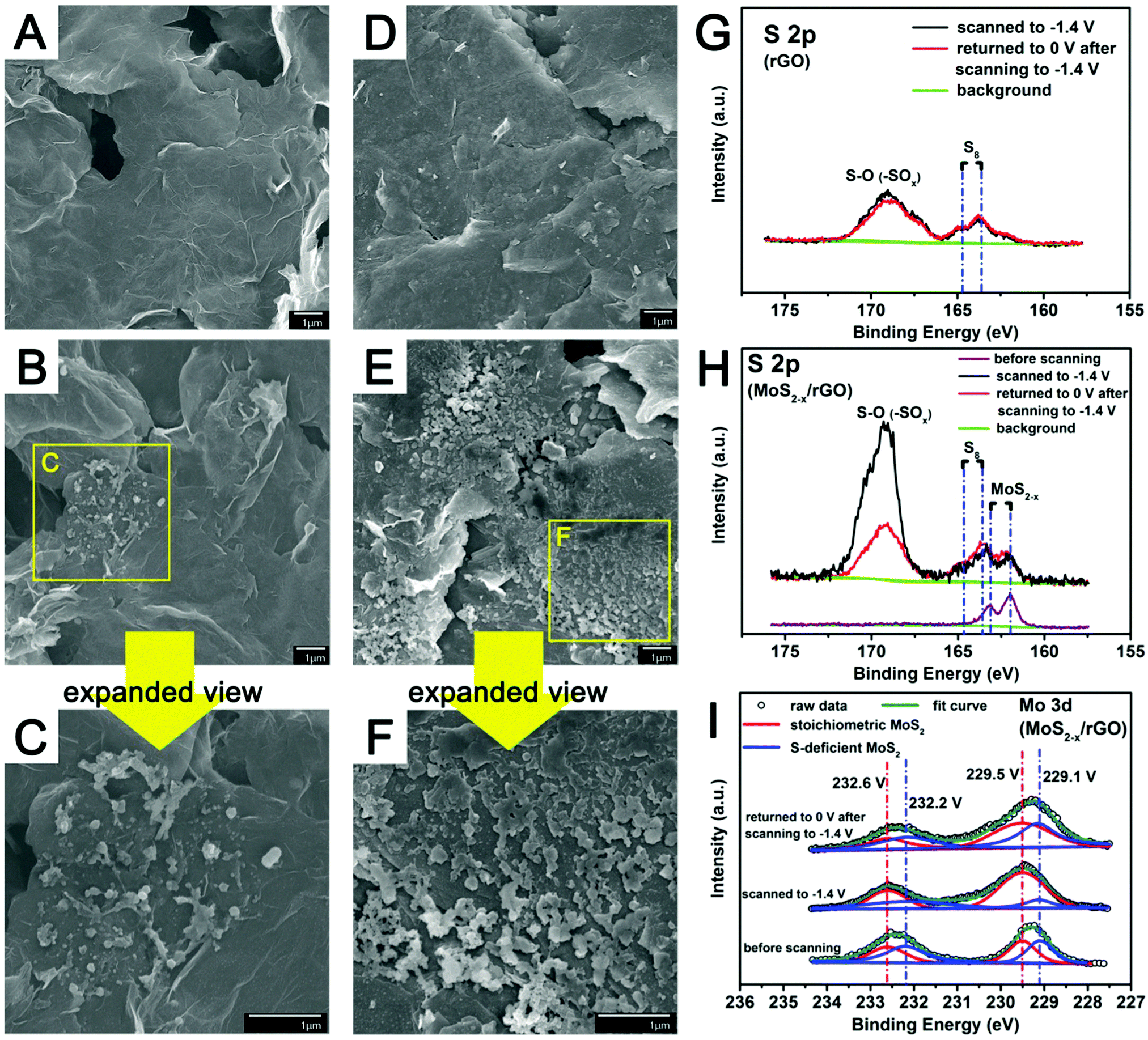 | ||
| Fig. 5 FESEM images of (A) the pristine rGO electrode and (B and C) the rGO counter electrode removed from the symmetric cell after scanning to −1.4 V; (D) the pristine MoS2−x/rGO electrode and (E and F) the MoS2−x/rGO counter electrode after scanning to −1.4 V. XPS spectra of (G) the rGO and (H and I) MoS2−x/rGO counter electrodes of symmetric cells after scanning to −1.4 V, or after scanning to −1.4 V and returning to 0 V. | ||
The actual performance of MoS2−x/rGO as a catalyst in lithium–sulfur batteries was evaluated in coin cells using a MoS2−x/rGO/S composite cathode and a lithium metal anode. Coin cells with a MoS2/rGO/S or rGO/S cathode were also assembled for comparison. The sulfur contents in the composites as assayed by TGA were about 75 wt% (Fig. S9, ESI†). Fig. 6A shows the typical voltammograms and the galvanostatic discharge–charge voltage profile of the MoS2−x/rGO/S cathodes between 1.8 and 2.6 V. Since lithiation of MoS2 occurs below 1.5 V vs. Li/Li+, the MoS2 nanoflakes would not have contributed to any capacity in the 1.8–2.6 V voltage range.51 The two cathodic peaks at about 2.3 V and 2.0 V could be associated with the reduction of sulfur to soluble long-chain lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8), and the subsequent conversion of the latter to insoluble short-chain polysulfides (Li2S2/Li2S) respectively. The two peaks in the reverse anodic scan at about 2.3 V and 2.4 V represent the reverse reactions of the conversion of short-chain polysulfides to sulfur.52,53 The two distinct discharge voltage plateaus (at ∼2.34 V and 2.12 V) at the 0.5C rate could be attributed to the conversion of sulfur to long-chain lithium polysulfides, and the formation of Li2S2/Li2S from the latter. The reverse of these reactions occurred during charge to form two corresponding voltage plateaus at ∼2.23 V and 2.35 V respectively. The galvanostatic discharge and charge curves are therefore in agreement with the voltammograms.54,55 The generally intense peaks on MoS2−x/rGO/S indicate the great extent of the polysulfide conversion due to the fast electrode kinetics.
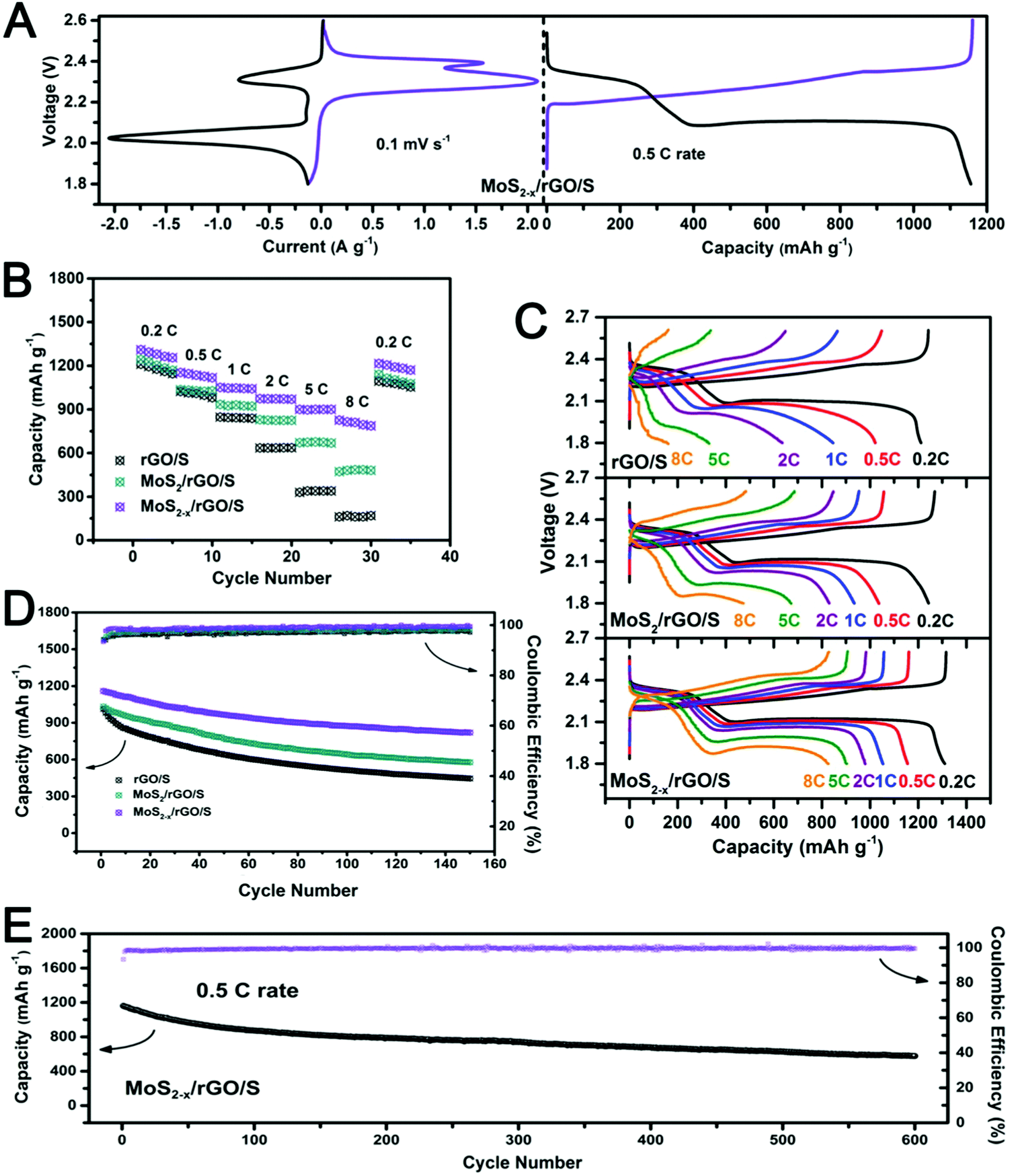 | ||
| Fig. 6 (A) Cyclic voltammograms at 0.1 mV s−1 and the representative galvanostatic discharge–charge voltage profile at 0.5C, (B) comparison of rate performance at different C-rates, (C) galvanostatic discharge–charge curves and (D and E) cycle stability of rGO/S, MoS2/rGO/S and MoS2−x/rGO/S cells in the 1.8–2.6 V voltage range at 0.5C (1C = 1600 mA g−1 based on the mass of sulfur). | ||
Fig. 6B compares the electrochemical performance of the MoS2−x/rGO/S, MoS2/rGO/S and rGO/S cathodes at different C-rates (from 0.2C to 8C; 1C = 1600 mA g−1). The first cycle discharge capacities of rGO/S, MoS2/rGO/S and MoS2−x/rGO/S at the 0.2C rate were 1210.5 mA h g−1, 1243.2 mA h g−1 and 1310.5 mA h g−1 respectively. The capacity difference deviated more at higher rates, and was 826.5 mA h g−1 for MoS2−x/rGO/S (63.1% of its 0.2C capacity), 473.3 mA h g−1 for MoS2/rGO/S (38.1% of its 0.2C capacity) and 161.1 mA h g−1 for rGO/S (13.3% of its 0.2C capacity) at the 8C rate, which was the test limit of this study. The higher affinity of MoS2−x/rGO for polysulfide adsorption and the catalytic effect of sulfur deficiencies in MoS2 for polysulfide conversion are expected to be the contributive factors although their respective contributions are difficult to resolve at this time. Fig. 6C shows the galvanostatic discharge and charge curves of the cells at different C-rates. An increase in the C-rate caused the charge voltage plateaus to shift positively and the discharge voltage plateaus to shift negatively. The voltage plateaus at the 8C rate were clearly visible in the MoS2−x/rGO/S cell, due to the more facile electrode kinetics on MoS2−x/rGO.
The cyclabilities of the MoS2−x/rGO/S, MoS2/rGO/S and rGO/S cathodes at the typical 0.5C rate are compared in Fig. 6D. Not only were the rGO/S and MoS2/rGO/S cathodes lower in initial capacity (1013.3 mA h g−1 and 1033 mA h g−1), they also exhibited more severe capacity fading endings with 445.3 mA h g−1 and 576.4 mA h g−1 after 150 cycles. In contrast, the MoS2−x/rGO/S cathode exhibited both higher discharge capacity and greater cycle stability (initial discharge capacity of 1159.9 mA h g−1 and capacity of 819.9 mA h g−1 after 150 cycles). The long-term cycling performance of MoS2−x/rGO/S at the 0.5C rate was also evaluated (Fig. 6E). After 600 cycles of continuous cycling, a discharge capacity of 628.2 mA h g−1 remained (a capacity fade rate of 0.083% per cycle). The Coulombic efficiency was as high as 99.6%. Cycle stability was thereof another benefit of the catalysis of polysulfide conversion in the sulfur electrode. A higher conversion rate of soluble polysulfides to insoluble sulfur products could decrease their accumulation in the cathode and consequently, their loss from the cathode by diffusion. Greater cycle stability was therefore realized by suppressing a major capacity loss mechanism. Compared to other catalysts in use today for the lithium–sulfur batteries (Table S2, ESI†), MoS2−x/rGO is clearly a well-rounded choice with a strong performance in almost all functional categories.
Conclusions
In this study, we demonstrated the effectiveness of MoS2−x/rGO as a catalyst for polysulfide conversion in a sulfur cathode. It was confirmed that the surface sulfur deficiencies participated in the polysulfide conversion and catalyzed the kinetics of polysulfide redox reactions. When a small amount of MoS2−x/rGO (4 wt%) was added to the sulfur cathode, high-rate performance and good cycle stability of the batteries were obtained. The high rate performance could be attributed to the acceleration of the polysulfide conversion kinetics on the surface sulfur deficiencies. The fast conversion of soluble polysulfides decreased their accumulation in the sulfur cathode and inhibited their subsequent loss from the cathode by diffusion. The suppression of this loss mechanism led to a more sustained cyclability. The study here not only presented a catalyst candidate which is among the best reported to date, but it also provided experimental evidence for and some new insights into the origin of the catalytic effects.Acknowledgements
H. L. acknowledges the National University of Singapore for his research scholarship.References
- W. Zhou, B. Guo, H. Gao and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502059 CrossRef.
- J. Li, L. Yang, S. Yang and J. Y. Lee, Adv. Energy Mater., 2015, 5, 1501808 CrossRef.
- J. Zhang, H. Hu, Z. Li and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 3982–3986 CrossRef CAS PubMed.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- H.-J. Peng, J.-Q. Huang, M.-Q. Zhao, Q. Zhang, X.-B. Cheng, X.-Y. Liu, W.-Z. Qian and F. Wei, Adv. Funct. Mater., 2014, 24, 2772–2781 CrossRef CAS.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- X. Wang, T. Gao, X. Fan, F. Han, Y. Wu, Z. Zhang, J. Li and C. Wang, Adv. Funct. Mater., 2016, 26, 7164–7169 CrossRef CAS.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- J. Chen, K. S. Han, W. A. Henderson, K. C. Lau, M. Vijayakumar, T. Dzwiniel, H. Pan, L. A. Curtiss, J. Xiao, K. T. Mueller, Y. Shao and J. Liu, Adv. Energy Mater., 2016, 6, 1600160 CrossRef.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- S. H. Chung and A. Manthiram, Adv. Mater., 2014, 26, 1360–1365 CrossRef CAS PubMed.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347–353 CAS.
- P. Bhattacharya, M. I. Nandasiri, D. Lv, A. M. Schwarz, J. T. Darsell, W. A. Henderson, D. A. Tomalia, J. Liu, J.-G. Zhang and J. Xiao, Nano Energy, 2016, 19, 176–186 CrossRef CAS.
- T. Nakazawa, A. Ikoma, R. Kido, K. Ueno, K. Dokko and M. Watanabe, J. Power Sources, 2016, 307, 746–752 CrossRef CAS.
- G. Zheng, S. W. Lee, Z. Liang, H. W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 6, 1552 CAS.
- H. Al Salem, G. Babu, C. V. Rao and L. M. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS PubMed.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- Y. Gorlin, M. U. M. Patel, A. Freiberg, Q. He, M. Piana, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A930–A939 CrossRef CAS.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- Y.-J. Li, J.-M. Fan, M.-S. Zheng and Q.-F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 CAS.
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- D. Kiriya, P. Lobaccaro, H. Y. Nyein, P. Taheri, M. Hettick, H. Shiraki, C. M. Sutter-Fella, P. Zhao, W. Gao, R. Maboudian, J. W. Ager and A. Javey, Nano Lett., 2016, 16, 4047–4053 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, C. Liu, P. Phillips, P. Yasaei, A. Behranginia, P. Zapol, R. F. Klie, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2016, 10, 2167–2175 CrossRef CAS PubMed.
- Y. Yin, J. Han, Y. Zhang, X. Zhang, P. Xu, Q. Yuan, L. Samad, X. Wang, Y. Wang, Z. Zhang, P. Zhang, X. Cao, B. Song and S. Jin, J. Am. Chem. Soc., 2016, 138, 7965–7972 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- G. Ji, Y. Yu, Q. Yao, B. Qu, D. Chen, W. Chen, J. Xie and J. Y. Lee, NPG Asia Mater., 2016, 8, e247 CrossRef CAS.
- H. Wang, Q. Zhang, H. Yao, Z. Liang, H.-W. Lee, P.-C. Hsu, G. Zheng and Y. Cui, Nano Lett., 2014, 14, 7138–7144 CrossRef CAS PubMed.
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2016, 28, 337–348 CrossRef CAS.
- W. Tang, B.-M. Goh, M. Y. Hu, C. Wan, B. Tian, X. Deng, C. Peng, M. Lin, J. Z. Hu and K. P. Loh, J. Phys. Chem. C, 2016, 120, 2600–2608 CAS.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854–3859 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X. D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- F. Wu, J. Li, Y. Su, J. Wang, W. Yang, N. Li, L. Chen, S. Chen, R. Chen and L. Bao, Nano Lett., 2016, 16, 5488–5494 CrossRef CAS PubMed.
- M. Thripuranthaka, R. V. Kashid, C. Sekhar Rout and D. J. Late, Appl. Phys. Lett., 2014, 104, 081911 CrossRef.
- X. Fan, P. Xu, Y. C. Li, D. Zhou, Y. Sun, M. A. Nguyen, M. Terrones and T. E. Mallouk, J. Am. Chem. Soc., 2016, 138, 5143–5149 CrossRef CAS PubMed.
- J. Xiao, X. Chen, P. V. Sushko, M. L. Sushko, L. Kovarik, J. Feng, Z. Deng, J. Zheng, G. L. Graff, Z. Nie, D. Choi, J. Liu, J.-G. Zhang and M. S. Whittingham, Adv. Mater., 2012, 24, 2109–2116 CrossRef CAS PubMed.
- D. M. Sim, M. Kim, S. Yim, M.-J. Choi, J. Choi, S. Yoo and Y. S. Jung, ACS Nano, 2015, 9, 12115–12123 CrossRef CAS PubMed.
- I. S. Kim, V. K. Sangwan, D. Jariwala, J. D. Wood, S. Park, K.-S. Chen, F. Shi, F. Ruiz-Zepeda, A. Ponce, M. Jose-Yacaman, V. P. Dravid, T. J. Marks, M. C. Hersam and L. J. Lauhon, ACS Nano, 2014, 8, 10551–10558 CrossRef CAS PubMed.
- L.-p. Feng, J. Su and Z.-t. Liu, RSC Adv., 2015, 5, 20538–20544 RSC.
- N. D. K. Tu, J. Choi, C. R. Park and H. Kim, Chem. Mater., 2015, 27, 7362–7369 CrossRef.
- X. Wang, G. Li, M. H. Seo, F. M. Hassan, M. A. Hoque and Z. Chen, Adv. Energy Mater., 2015, 5, 1501106 CrossRef.
- Q. Pang and L. F. Nazar, ACS Nano, 2016, 10, 4111–4118 CrossRef CAS PubMed.
- H.-J. Peng, Z.-W. Zhang, J.-Q. Huang, G. Zhang, J. Xie, W.-T. Xu, J.-L. Shi, X. Chen, X.-B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551–9558 CrossRef CAS PubMed.
- E. Gracia-Espino, G. Hu, A. Shchukarev and T. Wågberg, J. Am. Chem. Soc., 2014, 136, 6626–6633 CrossRef CAS PubMed.
- J. Ye, W. Chen, Q. Chen, Z. Yu and J. Y. Lee, Electrochim. Acta, 2016, 190, 538–547 CrossRef CAS.
- L. Wang, Y. Wang and Y. Xia, Energy Environ. Sci., 2015, 8, 1551–1558 CAS.
- C. Wang, X. Wang, Y. Yang, A. Kushima, J. Chen, Y. Huang and J. Li, Nano Lett., 2015, 15, 1796–1802 CrossRef CAS PubMed.
- K. Park, J. H. Cho, J.-H. Jang, B.-C. Yu, A. T. De La Hoz, K. M. Miller, C. J. Ellison and J. B. Goodenough, Energy Environ. Sci., 2015, 8, 2389–2395 CAS.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou, Z. Liu, W. Chen, L. Liu, S. Feng and Y. Zhang, Nano Lett., 2015, 15, 5137–5142 CrossRef CAS PubMed.
- F. Cheng, T. Zhang, Y. Zhang, J. Du, X. Han and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 2474–2477 CrossRef CAS PubMed.
- H. L. Wu, L. A. Huff and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2015, 7, 1709–1719 CAS.
- D.-H. Han, B.-S. Kim, S.-J. Choi, Y. Jung, J. Kwak and S.-M. Park, J. Electrochem. Soc., 2004, 151, E283 CrossRef CAS.
- D. Kiriya, P. Lobaccaro, H. Y. Y. Nyein, P. Taheri, M. Hettick, H. Shiraki, C. M. Sutter-Fella, P. Zhao, W. Gao, R. Maboudian, J. W. Ager and A. Javey, Nano Lett., 2016, 16, 4047–4053 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee01047h |
| ‡ These voltage values are potential differences between two redox reactions on two electrodes, and as such cannot be associated with half reactions. |
| § We have also prepared another composite (MoS2−x/rGO-2) by mixing rGO and MoS2−x in water, and used it as the electrodes of a symmetric cell (Fig. S7, ESI†). Its redox performance was slightly inferior to the MoS2−x/rGO electrode due to the poor mixing between MoS2−x and rGO by this preparation route, which increased the likelihood of nanoflake aggregation. Nonetheless, sharper and narrower redox peaks were still obtained relative to the MoS2/rGO electrode. The positive effect of sulfur deficiencies on polysulfide conversion was again demonstrated. |
| ¶ The counter electrode instead of the working electrode was analysed because it was where sulfur was deposited. The Li2S or Li2S2 on the working electrode is prone to decomposition by atmospheric moisture, which could increase the difficulty in result interpretation. |
| This journal is © The Royal Society of Chemistry 2017 |