
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An experimental and theoretical study of an efficient polymer nano-photocatalyst for hydrogen evolution†
Palas Baran
Pati
a,
Giane
Damas
b,
Lei
Tian
a,
Daniel L. A.
Fernandes
a,
Lei
Zhang
a,
Ilknur Bayrak
Pehlivan
c,
Tomas
Edvinsson
 c,
C. Moyses
Araujo
*b and
Haining
Tian
*a
c,
C. Moyses
Araujo
*b and
Haining
Tian
*a
aPhysical Chemistry Division, Department of Chemistry - Ångström Laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: haining.tian@kemi.uu.se
bMaterials Theory Division, Department of Physics and Astronomy, Uppsala University, Box 516, 751 20 Uppsala, Sweden. E-mail: moyses.araujo@physics.uu.se
cDepartment of Engineering Sciences–Solid State Physics, Uppsala University, Box 534, 75121 Uppsala, Sweden
First published on 17th May 2017
Abstract
In this work, we report a highly efficient organic polymer nano-photocatalyst for light driven proton reduction. The system renders an initial rate of hydrogen evolution up to 50 ± 0.5 mmol g−1 h−1, which is the fastest rate among all other reported organic photocatalysts. We also experimentally and theoretically prove that the nitrogen centre of the benzothiadiazole unit plays a crucial role in the photocatalysis and that the Pdots structure holds a close to ideal geometry to enhance the photocatalysis.
Broader contextLight driven water splitting is a promising strategy to convert and store solar energy as a valuable product with high-energy density – hydrogen. The development of efficient photocatalysts for a half reaction of water splitting – proton reduction – is therefore important to improve the overall reaction. In the past few years, organic photocatalysts such as polymers have emerged as promising alternatives to the traditional inorganic and metal-complex materials for the half reaction due to facile structure modification, smaller environmental foot-prints and tunable light absorption. Converting an organic polymer into polymer dots (Pdots) has been proven to be an efficient way to dramatically improve the photocatalytic performance. To rule out the reactive sites and the working mechanism of the Pdots, in this work, we experimentally and theoretically investigated three different Pdot materials in the photocatalysis. Eventually, we concluded that the nitrogen in the benzothiadiazole unit could be the reactive site. Also, an efficient Pdot photocatalyst is found along with this study, which shows an excellent rate of hydrogen evolution. Our result shows that the organic Pdot materials could be a class of efficient organic photocatalysts for light driven proton reduction by a reasonably structural modification. |
By mimicking natural photosynthesis, the abundant solar energy can be stored as H2 for clean and renewable future fuels from an earth abundant source, water.1,2 Since the discovery of the Honda–Fujishima effect in 19723 enormous attempts have been made to develop efficient photocatalysts to generate H2 from light driven water splitting. Developing an efficient photocatalytic system for proton reduction is crucial for future applications of hydrogen generation from water. Molecular catalyst–photosensitizer assemblies4–9 and inorganic material photocatalysts10–14 have been intensively investigated. Organic photocatalysts have also recently attracted intense interest from scientists due to their facile structure modification, smaller environmental foot-prints, tunable light absorption and promising photocatalytic reactivity.15,16 Carbon nitride (g-C3N4) is a successful example of an organic photocatalyst for proton reduction, as reported by Antonietti and coworkers.17 Since then a lot of effort has been devoted to this kind of material through optimization of the preparation method and change of the morphology to improve the proton reduction performance and even use it in the water oxidation reaction.18,19 However, pure g-C3N4 without a co-catalyst has shown an unsatisfactory photocatalytic ability. Normally, inorganic co-catalysts such as Pt and Ni–P have been used to work together with g-C3N4.20 Also, the fixed structure of g-C3N4 limits its light harvesting region as well as the obtainable solar energy-to-hydrogen (STH) conversion efficiency. In order to find an organic photocatalyst with a better light harvesting ability and STH efficiency compared to g-C3N4, scientists have started to consider organic semiconducting polymers as photocatalysts since their optical and physical properties can be easily tuned by changing the different building blocks and thus a sub-nanometer design can be achieved. Actually, before the work on g-C3N4, Yanagida and co-workers used a linear poly(p-phenylene) for hydrogen generation, but it only worked under UV-light irradiation.21 Subsequently, various pure organic materials such as poly(azomethine)s,22 covalent organic frameworks,23 phenyl-triazine oligomers,24 microporous organic nanorods,25 heptazine networks26 and polybenzothiadiazoles27 have been applied in photocatalytic hydrogen production. Recently, Cooper's28,29 and Yu's30 groups further developed the organic polymer photocatalysts and reported a series of microporous polymers (CMPs) by a copolymerization strategy; some of them have shown an improved photocatalytic ability for visible light driven proton reduction in comparison with g-C3N4 without the addition of any co-catalyst. These previous studies reported that organic polymeric materials can be a type of promising photocatalyts for light-driven proton reduction. The particle size of pristine organic polymers regarding the dispersibility issue is a crucial factor influencing the photocatalytic performance. Normally, pristine organic polymeric photocatalysts are not water soluble, suggesting that the photocatalytic experiments have to be carried out in an organic solvent or mixed water/organic solvent in order to increase the dispersibility of organic polymers in the reaction phase.27
We recently proposed that converting an organic polymer into small nanoparticles could eventually solve this problem. A proof-of-concept work of the Pdot photocatalyst prepared by a nano-precipitation method with a light absorbing polymer of poly[(9,9′-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3} thiadiazole)] (PFBT, also named as F8BT) and a co-polymer of polystyrene grafting with carboxyl-group-functionalized ethylene oxide (PS-PEG-COOH) has shown 5-orders of magnitude higher photocatalytic performance of hydrogen evolution than that of the PFBT pristine polymer in the absence of any organic solvent or co-catalyst.31 However, there are still important scientific questions that remain to be answered; where is the reactive site in the Pdots and how does it work? In this work, we approach these questions from both experimental and theoretical viewpoints. Apart from a step forward in the understanding of these systems, a significantly more efficient organic Pdot photocatalyst than PFBT is presented in this study.
Fig. 1 shows the structures of polymers used and compared in this study. The molecular structure of PFBT (1) contains a donor (D)–acceptor (A) backbone, in which fluorene acts as D and benzothiadiazole (BT) acts as A. With the inherently different electron affinities upon light irradiation, the D–A molecule will have an intramolecular charge transfer (ICT) from D to A and the electrons will be preferably accumulated in the BT building blocks, which is further confirmed by density functional theory (DFT) (Table S1, ESI†). In the photocatalytic system, the excited polymers will be subsequently reduced by a sacrificial reductant reagent. The electron concentrated BT building block is important for the proton reduction reaction. More precisely, we hypothesize that heteroatoms such as N and/or S in polymers are the real reactive sites for proton binding and also for the subsequent catalytic reaction. In order to verify this hypothesis experimentally, we compare PFBT with two other polymers, PFODTBT (2)32 and F8T2 (3)33 that separate the effects by varying the building blocks. All three polymers have the same fluorene D, but different A units. Polymer 3 has a thiophene instead of BT in polymer 1. Polymer 2 adopts both thiophene and BT in its backbone to broaden its light absorption in the visible light region in comparison with polymer 1. A critical comparison between these three polymer-based Pdots in photocatalysis would then give us an insight into the effect of the different block units on the photocatalytic performance.
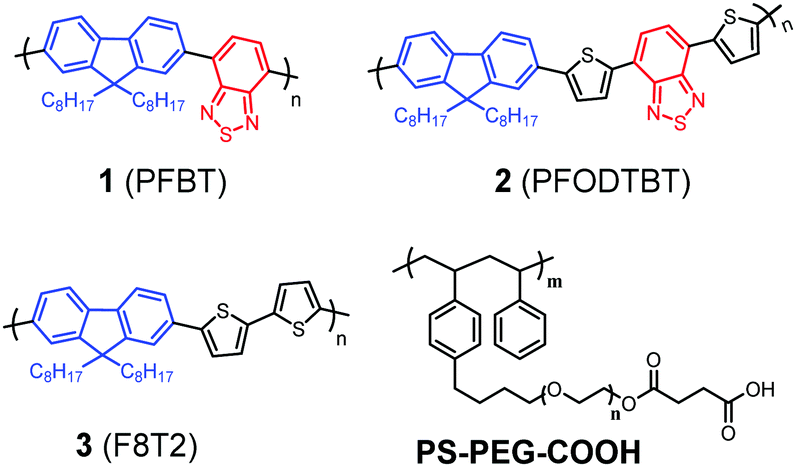 | ||
| Fig. 1 Molecular structures of three organic D–A polymers (1, 2 and 3) and a co-polymer PS-PEG-COOH used for this study. | ||
All polymer-based Pdots were prepared by a nano-precipitation method as described in the previous publication (also shown in Scheme S1, ESI†).31,34 The final amount of polymer in the Pdots aqueous solution was determined from UV-Vis experiments after removing water and re-dissolving in THF. The amounts of the corresponding polymers in Pdots 1, 2 and 3 prepared for this study are 17 μg ml−1, 13 μg ml−1 and 19 μg ml−1, respectively. The absorption and emission spectra of all Pdots in water were measured and shown in Fig. 2a. As compared with the pristine polymers (Fig. S1, ESI†) the formation of nanoparticles broadens the absorption spectra toward the red region, keeping the absorption maxima unchanged which suggests the aggregation of the polymers in the formed Pdots. The particle sizes of the Pdots were measured by dynamic light scattering (DLS), showing the average sizes of all polymer based Pdot particles to be in the interval of 30–40 nm (Fig. 2b) with a tail of both smaller and larger particles. The band gaps of the Pdots 1, 2 and 3 are determined to be 2.38 eV,31 1.98 eV and 2.46 eV, respectively, by the intersections between the absorption and emission spectra. Using two thiophene units instead of a BT unit in polymer 3 does not significantly change the maximum absorption peak of Pdots 3 in comparison with Pdots 1. The Pdots 2 based on polymer 2 includes two extra thiophene moieties and one BT unit to extend the π-conjugated system where the maximum absorption peak shifted up to 550 nm, which is beneficial for photocatalysis from a light harvesting point of view. Minor overlap between the absorption and emission spectra provides us with the possibility of measuring the lifetime of the photoluminescence of all polymers in THF and also polymer-based Pdots in an aqueous solution by time-correlated single photon counting (TCSPC). Interestingly, all Pdots in an aqueous solution showed a shorter photoluminescence lifetime as compared to their pristine polymers in THF probably due to the aggregation of polymers in the Pdot state (Fig. S3, ESI†).
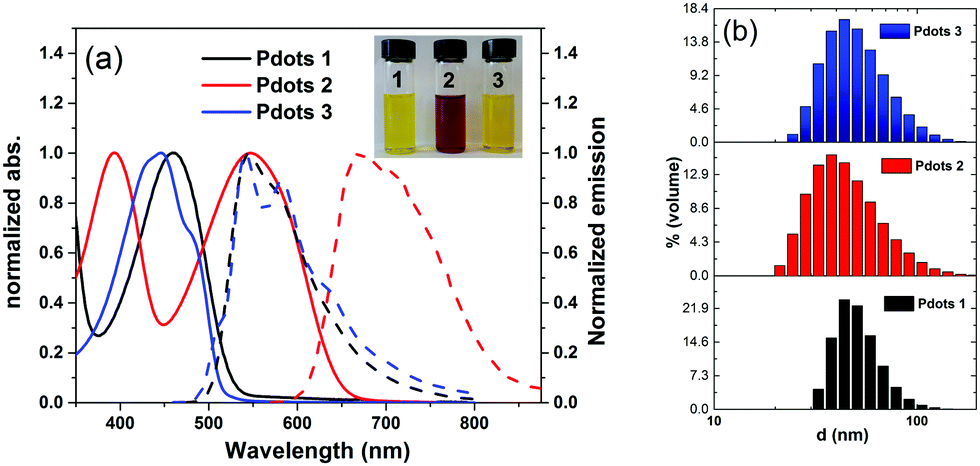 | ||
| Fig. 2 (a) Absorption (solid line) and emission (dotted line) spectra of Pdots 1, 2 and 3 in water (inset: Pdot solution) and (b) distribution of the hydrodynamic diameter of Pdots (1, 2 and 3) measured by DLS. | ||
The energy levels of all Pdots were estimated from cyclic voltammetry (CV) measurements performed in a buffer (pH 4.5) solution and the absorption and emission spectra in an aqueous solution (Fig. S4, ESI†). The corresponding data versus the normal hydrogen electrode (NHE) are shown in Fig. 3a. The reduction potentials (ES/S−) of all Pdots (−1.00 V for 1, −0.90 V for 2 and −0.78 V for 3) are more negative than the thermodynamic proton reduction potential, −0.24 V vs. NHE (pH 4), indicating that there is enough driving force for proton reduction. The energy levels of the excited state (ES*/S−) (1.37 V for 1, 1.08 V for 2 and 1.68 V for 3) are also positive enough to produce reduced Pdots and thus regenerate the system by a sacrificial donor, here ascorbic acid (0.46 V vs. NHE).35
 | ||
| Fig. 3 (a) Energy diagram of three polymer-based Pdots and (b) light-driven hydrogen generation of Pdots 1 (17 μg ml−1), 2 (13 μg ml−1) and 3 (19 μg ml−1) in water at ambient temperature. Conditions: ascorbic acid: 0.2 M; pH 4.0 (adjusted with 1 M NaOH), white LED light (λ > 420 nm, 17 W, 5000 K), inset: H2 bubbles observed during photocatalysis. | ||
To quantify the performance of the Pdots for light driven hydrogen generation, a regular two-component system was employed where ascorbic acid was used as a sacrificial electron donor and Pdots worked as a photocatalyst. The comparative kinetic curves of hydrogen generation are depicted in Fig. 3b. Surprisingly, Pdots 2 rendered a predominant hydrogen generation rate of 50 ± 0.5 mmol g−1 h−1, which is five times faster than that of the previously reported Pdot 1 (8.3 ± 0.2 mmol g−1 h−1). Interestingly, Pdots 3 did not show any considerable reactivity to light driven proton reduction. The stability of Pdots 2 is also dramatically improved by introducing thiophene units, making the photocatalysis work for ca. 4 h. The broad light response and good stability of Pdots 2 made it eventually produce H2 of 63 ± 2 mmol g−1, which is 15 times higher than that from the reference Pdot 1, 4 ± 0.5 mmol g−1. We even observed H2 bubble formation during photocatalysis (see the inset in Fig. 3b). For a scaling up experiment with a 25 ml Pdots 2 solution, we detected 17 μmol of H2 by gas chromatography (GC) after 1 h of light illumination (Fig. S7, ESI†). From the data of the apparent quantum yield (AQY) as a function of wavelength as shown in Fig. 4a, one can see that Pdots 2 shows a much broader light response region compared to that of Pdots 1 due to its broad light harvesting region up to 700 nm. As a consequence, at 550 mm, Pdots 2 rendered a much higher AQY value of ca. 0.6% compared to that of Pdots 1, 0.05%.
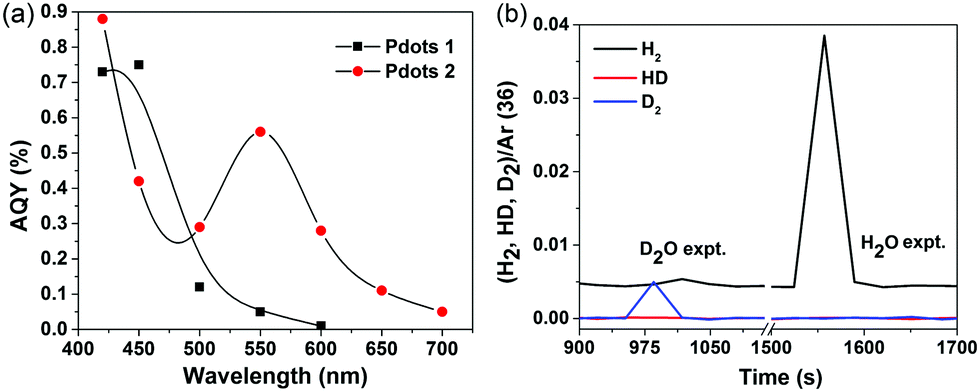 | ||
| Fig. 4 (a) Apparent quantum yield as a function of light wavelength of Pdots 1 and 2, and (b) mass spectra of gases generated from an isotopic labeling experiment. | ||
In order to make sure that the produced H2 is not from the polymer itself, such as degradation or further polymerization, we conducted an isotopic labeling experiment, using D2O and NaOD instead of H2O and NaOH in a photocatalytic experiment of Pdots 2. The mass spectra of the gases generated during photocatalysis were monitored and the corresponding results are shown in Fig. 4b. In the D2O experiment, we mainly observed D2 and slightly DH and H2. As we did not use deuterated ascorbic acid, the H atom in H2 and HD must be from ascorbic acid. As a comparison, the H2O experiment mainly gives H2 instead. This result further confirms the photocatalytic behavior of Pdots for proton reduction.
As a D–A polymer is usually synthesized by a Pd-catalyzing poly-condensation reaction, the effect from any residual Pd in the polymer substrates must also be excluded as Pd is also a very effective hydrogen generation catalyst. To examine the remaining Pd in our samples, we determined the amount of Pd in the polymers by energy-dispersive X-ray spectroscopy (EDX) (Fig. S8–S10 and Table S4, ESI†). Polymer 1 and 2 only contain 0.1% Pd (w/w), which is much less than that detected in the previous publications regarding polymeric photocatalysts for H2 evaluation30 and the co-catalysts added (ca. 3%, w/w) with purpose in other organic photocatalyst systems (listed in Table S2, ESI†). Most importantly, polymer 3 has the most remaining Pd which is 0.5% and its band gap and reduction potential are suitable for visible-light driven proton reduction, but its Pdot did not show any photocatalytic reactivity at all. In our previous work, poly(3-hexylthiophene) (P3HT) Pdots also did not show good activity.31 Therefore, the polymer structure, particularly the heteroatoms, is indeed a crucial factor for influencing the photocatalytic reactivity of the Pdots. Still, in order to figure out if the 0.1% Pd in polymer 1 and 2 is still playing a crucial role as a co-catalyst in the Pdots, we performed Pd poisoning experiments with carbon monoxide (CO) and ethylenediaminetetraacetic acid (EDTA) (Fig. S11, ESI†), which eventually suggests that the Pdots are the real photocatalysts. In addition, we also experimentally proved that the BT unit is playing an important role in photocatalysis where the N or S atom in the BT unit may easily interact with a proton and form a hydrogen bond also from a well-known chemical bonding perspective.
In order to further elucidate the role of N and S atoms in the catalytic activity of the hydrogen evolution reaction (HER), we have firstly employed density functional theory based calculations to assess the hydrogen binding free energy, ΔGH, which has been recognized as a good descriptor for such a reaction.36,37 Here, hydrogen should be adsorbed neither too weakly (to facilitate the protonation process) nor too strongly (to avoid high barriers for H2 formation and release), which is achieved at an optimal value of ΔGH = 0 eV.36,37 As an example, the metallic catalyst Pt exhibits a good value of ΔGH = −0.1 eV, approximately.38 The oligomeric structures (Fig. S13, ESI†) have been used to model the properties of the polymers and ΔGH was computed for hydrogen adsorbed on both S and N sites (see the method section for more computational details). The results are shown in Fig. 5.
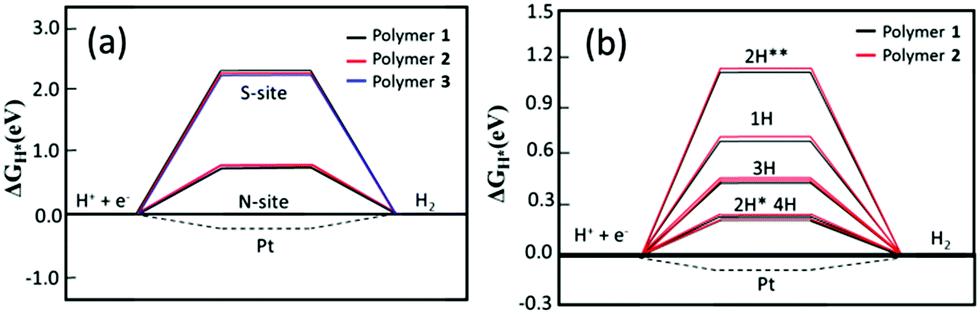 | ||
| Fig. 5 (a) Hydrogen binding free energy, ΔGH, at nitrogen and sulfur sites – the case of one hydrogen atom and (b) hydrogen adsorption free energy ΔGH for different hydrogen concentrations. 2H* refers to two hydrogen atoms added to the same BT block. 2H** refers to two hydrogen atoms added to different BT blocks. | ||
As can be observed in Fig. 5a, there is a distinct difference in ΔGH when different sites are hydrogenated. The hydrogen binding free energy at the sulfur site is about 2.0 eV uphill indicating that there is a high overpotential for the HER; whereas, the hydrogen binding at the nitrogen site decreases ΔGH to about 0.66 and 0.70 eV for polymer 1 and 2, respectively, showing that the addition of BT molecules in the polymeric structure leads to stronger hydrogen adsorption, which in turn favors the energetics toward H2 formation. The solvation effect is also carried out by calculations and the obtained trend (Fig. S15, ESI†) suggests that the calculations performed in the gas phase suffice for our analysis. Such energetics analysis is coherent with the obtained optimized structures where the H–N bond in 1 is 0.39 Å shorter than the H–S bond (1.40 Å), and a similar bond length difference is also found for 2.
We have also investigated the effect of the hydrogen content on the adsorption free energy. The results are displayed in Fig. 5b. A very interesting result is already obtained for the adsorption of two hydrogen atoms. When the adsorption takes place at two different BT units the average ΔGH is found to be about 1.2 eV while this number is significantly reduced to 0.28 eV when the hydrogen atoms are adsorbed on the same BT unit. It still has a very small difference between the configuration of cis and trans but the former is more stable for both the polymers. This suggests that the first protonation in a BT unit favors the second one. Similar results are also obtained for the higher concentration of three and four hydrogen atoms. In the latter case, all N sites are hydrogenated in the model systems.
The HER energy profile along the associative H–H bond formation, in two model systems for the catalytic activity of polymer 2, has also been evaluated within the B3LYP/6-31G* level of theory. A lower barrier height was found when the interaction between the hydrogen atoms takes place from different polymers (1.32 eV), while the barrier is significantly higher (3.12 eV) when the hydrogen comes from the same BT unit (see Fig. 6). Such a result indicates that the formation of molecular hydrogen is kinetically more favorable through the interaction between different BT units in different polymer chains, which corroborates with the experimental outcome where Pdot structures with aggregated polymers make the light driven hydrogen evolution process more efficient.
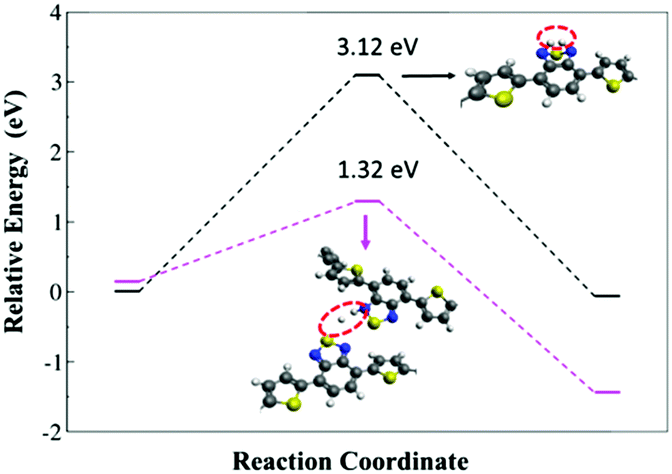 | ||
| Fig. 6 Energy profile of the H2 formation from interacting hydrogen atoms present at the same BT unit and at different polymer units. The values are referenced to the ground-state geometry. The insets indicate the structures of transition states. | ||
In conclusion, we have studied the effect of polymer structures on the performance of a Pdot photocatalyst, from which we have experimentally proved that benzothiadiazole (BT) in Pdots plays an important role in the reduction of photocatalytic protons. By keeping the BT unit and using thiophene units to extend the configuration system, the Pdots 2 based on polymer 2 (PFODTBT) showed a broad light response of up to 700 nm for photocatalysis with an apparent quantum yield of 0.6% at 550 nm and therefore rendered photocatalytic performance with an initial rate of hydrogen evolution up to 50 ± 0.5 mmol g−1 h−1, which is much higher than that reported from the reference Pdot 1, 8.3 ± 0.2 mmol g−1 h−1. The stability of Pdots 2 is also dramatically improved, eventually making Pdots 2 produce an amount of H2, 63 ± 2 mmol g−1, which is 15 times higher than that from the reference Pdot 1, 4 ± 0.5 mmol g−1, under the same conditions. DFT calculations suggest that N atoms in the BT units are actually the catalytic reactive sites in the formation of hydrogen molecules, which corroborates with the experimental outcomes. The first hydrogenation of N-atoms in the BT unit favor the addition of the second hydrogen in the same unit, but the hydrogen formation is favored by the interaction between hydrogen atoms in different polymer chains. The Pdot structure is therefore experimentally and theoretically shown to be beneficial for the photocatalysis. The structural modification of the polymers to further improve and optimize the performance of Pdot based systems for photocatalysts is presently on-going in our lab and, with the knowledge of the importance of the N atom in the active site and its interaction with N on a neighboring polymer, shows promise for further nano-engineering of the active sites. Furthermore, suitable hydrophilic polymers and surfactants have also been considered to increase the concentration of Pdot photocatalysts in an aqueous solution for improving the apparent quantum yield.
Acknowledgements
This work was financially supported by the Swedish Energy Agency, the Knut and Alice Wallenberg Foundation, the Åforsk Foundation (no. 14-452), StandUP for energy and the Olle Engkvist Builder Foundation. L. T. thanks the China Scholarship Council (CSC) for financial support and G. D. thanks the CAPES Foundation for financial support. The computations/simulations were performed on the resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC. We also give our great thanks to Dr Jacinto Sa for his helpful assistance and discussion with DLS and gas mass measurements.Notes and references
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- L. Hammarström and S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1859–1860 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- X. Li, M. Wang, S. Zhang, J. Pan, Y. Na, J. Liu, B. Åkermark and L. Sun, J. Phys. Chem. B, 2008, 112, 8198–8202 CrossRef CAS PubMed.
- T. Kowacs, Q. Pan, P. Lang, L. O'Reilly, S. Rau, W. R. Browne, M. T. Pryce, A. Huijser and J. G. Vos, Faraday Discuss., 2015, 185, 143–170 RSC.
- F. Wang, W.-G. Wang, X.-J. Wang, H.-Y. Wang, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193–3197 CrossRef CAS PubMed.
- T. A. White, S. L. H. Higgins, S. M. Arachchige and K. J. Brewer, Angew. Chem., Int. Ed., 2011, 50, 12209–12213 CrossRef CAS PubMed.
- H. Ozawa, M.-A. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926–4927 CrossRef CAS PubMed.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- I. Tsuji, H. Kato, H. Kobayashi and A. Kudo, J. Am. Chem. Soc., 2004, 126, 13406–13413 CrossRef CAS PubMed.
- J. Huang, K. L. Mulfort, P. Du and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 16472–16475 CrossRef CAS PubMed.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321–1324 CrossRef CAS PubMed.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- V. S. Vyas, V. W.-H. Lau and B. V. Lotsch, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- S. Ye, R. Wang, M.-Z. Wu and Y.-P. Yuan, Appl. Surf. Sci., 2015, 358(Part A), 15–27 CrossRef CAS.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475, 10.1039/C39850000474.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Mullen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- K. Schwinghammer, S. Hug, M. B. Mesch, J. Senker and B. V. Lotsch, Energy Environ. Sci., 2015, 8, 3345–3353 CAS.
- J. H. Park, K. C. Ko, N. Park, H.-W. Shin, E. Kim, N. Kang, J. Hong Ko, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, J. Mater. Chem. A, 2014, 2, 7656–7661 CAS.
- K. Kailasam, J. Schmidt, H. Bildirir, G. Zhang, S. Blechert, X. Wang and A. Thomas, Macromol. Rapid Commun., 2013, 34, 1008–1013 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- S. Admassie, O. Inganäs, W. Mammo, E. Perzon and M. R. Andersson, Synth. Met., 2006, 156, 614–623 CrossRef CAS.
- H. Sirringhaus, R. J. Wilson, R. H. Friend, M. Inbasekaran, W. Wu, E. P. Woo, M. Grell and D. D. C. Bradley, Appl. Phys. Lett., 2000, 77, 406–408 CrossRef CAS.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, TCSPC, Raman, SEM/EDX, GC, Pd poisoning, etc. See DOI: 10.1039/c7ee00751e |
| This journal is © The Royal Society of Chemistry 2017 |