
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium trapping in alloy forming electrodes and current collectors for lithium based batteries†
David
Rehnlund
 *a,
Fredrik
Lindgren
a,
Solveig
Böhme
a,
Tim
Nordh
a,
Yiming
Zou
a,
Jean
Pettersson
b,
Ulf
Bexell
c,
Mats
Boman
a,
Kristina
Edström
a and
Leif
Nyholm
*a
*a,
Fredrik
Lindgren
a,
Solveig
Böhme
a,
Tim
Nordh
a,
Yiming
Zou
a,
Jean
Pettersson
b,
Ulf
Bexell
c,
Mats
Boman
a,
Kristina
Edström
a and
Leif
Nyholm
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 538, SE-75121, Uppsala, Sweden. E-mail: David.Rehnlund@kemi.uu.se; Leif.Nyholm@kemi.uu.se
bDepartment of Chemistry – BMC, Uppsala University, Box 599, SE-75124, Uppsala, Sweden
cSchool of Technology and Business Studies, Materials Technology, Dalarna University, Falun, Sweden
First published on 10th May 2017
Abstract
Significant capacity losses are generally seen for batteries containing high-capacity lithium alloy forming anode materials such as silicon, tin and aluminium. These losses are generally ascribed to a combination of volume expansion effects and irreversible electrolyte reduction reactions. Here, it is shown, based on e.g. elemental analyses of cycled electrodes, that the capacity losses for tin nanorod and silicon composite electrodes in fact involve diffusion controlled trapping of lithium in the electrodes. While an analogous effect is also demonstrated for copper, nickel and titanium current collectors, boron-doped diamond is shown to function as an effective lithium diffusion barrier. The present findings indicate that the durability of lithium based batteries can be improved significantly via proper electrode design or regeneration of the used electrodes.
Broader contextNext generation lithium-ion batteries are expected to be based on lithium alloy forming anode materials which can store up to ten times more charge than currently used graphite anodes. This increase in the charge storage capability has motivated significant research towards the commercialisation of anode materials such as Si, Sn and Al. Alloy forming anode materials are, however, known to lose capacity during cycling in a lithium-ion battery. As the origin of these capacity losses for alloy forming anode materials is currently not fully understood it remains a topic of great scientific interest. The leading theory explains the capacity losses by a combination of large volume expansion taking place during reversible lithium alloying and irreversible reactions with the electrolyte involving the formation of a solid electrolyte interphase layer. In the present work it is demonstrated that an alternative failure mechanism effectively can account for the capacity losses seen when cycling alloy forming anode materials versus lithium electrodes. During the cycling small amounts of elemental lithium are trapped within the electrode material due to a two-way diffusion causing lithium to move into the bulk material, which makes the lithium extraction process significantly more time consuming. This mechanism is demonstrated for both alloy forming anode materials (e.g. Si, Sn and Al) as well as for commonly used current collector metals (e.g. Cu, Ni and Ti). |
Introduction
The commercialization of the lithium-ion battery has paved the way for the portable electronics revolution. As the development of electric vehicles requires lithium based batteries with significantly higher energy and power densities there is currently a large interest in silicon,1–4 tin1,3 and lithium anodes.5,6 Lithium is, however, associated with significant safety and cycle life issues due to dendrite formation and short-circuiting of the battery.5,6 Many researchers have therefore chosen to focus on silicon, tin and aluminium electrodes which form lithium alloys with high specific capacities and normally do not exhibit lithium dendrites.1 Silicon is the most interesting material due to its high abundance and high specific gravimetric capacity, i.e. 3579 mA h g−1 for Li3.75Si.1 The formation of lithium alloys, however, gives rise to volume expansion effects (e.g. 280% for Li3.75Si1) which can result in electrode pulverization during cycling as well as continuous solid electrolyte interphase (SEI) layer formation.1,7,8 While significant progress has been made to decrease the volume expansion problem by using e.g. nanoparticles, nanorods and thin films, and/or capacity limitations,1,8,9 capacity losses are still typically seen.10,11 Although these losses generally are ascribed to a combination of volume expansion effects and SEI formation there are also results12–19 indicating that lithium is trapped in the electrodes. The latter hypothesis has, however, received relatively little attention so far and therefore requires further investigation.Results and discussion
Tin nanostructured electrodes
As is seen in Fig. 1, significant capacity loss was seen for a battery composed of a tin nanorod array electrode and a lithium foil electrode. The large reduction peak seen at about 0.25 V vs. Li+/Li on the first cathodic scan can be ascribed to lithium deposition yielding the LixSn alloy (xLi+ + xe− + Sn = LixSn),1,20 while the smaller reduction peaks at about 1.38 and 0.64 V originated from the reduction of the native tin oxide layer.1,20 The anodic current between 0.3 and 1.3 V stem from the oxidation of lithium in the tin alloy. Similar capacity losses are generally seen for alloy forming electrodes and are normally ascribed to a combination of volume expansion effects and SEI formation.1,8,21,22 Since the tin nanorods electrodes were capacity limiting during the cycling versus the Li electrodes (see the discussion below), the present capacity losses can, however, not be explained by SEI formation even though this effect contributed to the difference between the reduction and oxidation charges. Fig. 1b further shows that the capacity increased rather than decreased initially (see Fig. S1 and S2 as well as Section S2 in the ESI†) and that an analogous behaviour also was seen under galvanostatic cycling conditions (see Fig. 1c and d). Note the initial capacity increase for the higher current density and the significantly different shapes of the 1st and 120th cycle chronopotentiograms.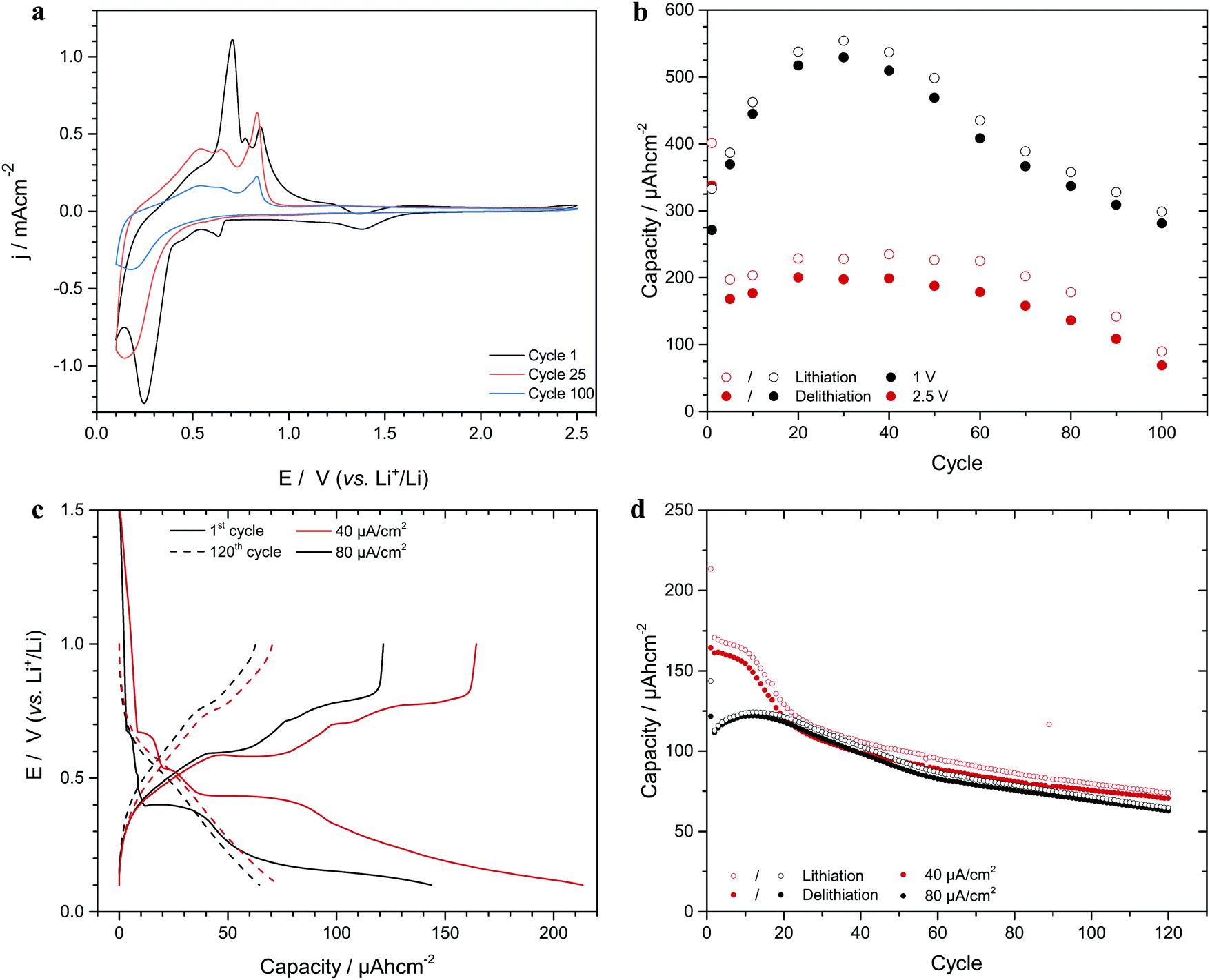 | ||
| Fig. 1 Cyclic voltammograms, chronopotentiograms and areal capacities obtained with tin nanorod electrodes. (a) 1st, 25th and 100th voltammetric cycles for cycling between 0.1 and 2.5 V vs. Li+/Li at a scan rate of 1.0 mV s−1 (the 1st scan was initiated at the open circuit potential, i.e. 2.3 V), (b) footprint areal capacity as a function of the cycle number for scanning between 0.1 and either 1.0 or 2.5 V. (c) 1st and 120th chronopotentiometric curves obtained with current densities of 40 and 80 μA cm−2, respectively between 0.1 and 1.0 V. (d) Footprint areal capacity as function of the cycle number for chronopotentiometric cycling using current densities of 40 and 80 μA cm−2. The electrode footprint area was always 1 cm2. | ||
Based on the results in Fig. 1b and d it is reasonable to assume that the capacities were affected by at least two superimposed phenomena (as is further explained in Section S2 in the ESI†), one giving rise to the initial capacity increase and another yielding a capacity loss. Since it is well-known1,2,8,22,23 that alloy forming electrode materials become amorphous during cycling, we ascribe the initial capacity increase to electrode roughening effects in agreement with recent results for aluminium electrodes.13,24
To study the origin of the capacity loss, the lithium content of the tin electrodes after different numbers of cycles were determined (in their oxidized state) using ICP-AES as described in Section S1 in the ESI.† It was found that the lithium content in the electrodes increased linearly with the cycle number during 120 cycles (see Fig. S3 in the ESI†) indicating that some lithium was trapped on each cycle. Linear plots of the capacity as a function of the square root of the cycling time were also obtained in the region where the capacity decreased (see Fig. S2 in the ESI†). This behaviour, which also can be seen for aluminium nanorod electrode data13 (see Fig. S4 in the ESI†), indicates a diffusion controlled lithium trapping effect. Given an increase in the electrode lithium content of 1.27 μg per cycle (see Fig. S3 in the ESI†), the corresponding lithium trapping charge (i.e. 17.8 mC per cycle) was compared with the difference between the reduction and oxidation charges for each cycle. This calculation (see Section S3 in the ESI†) showed that the trapped lithium could explain approximately 50% of the difference between the reduction and oxidation charges on each cycle. The remaining part was ascribed to the formation of a 3 nm thick SEI layer per cycle, assuming that a 10 nm thick SEI layer was formed on the first cycle. The latter is reasonable as SEI layer thicknesses of up to about 20 nm have been reported.25–27 The calculations further suggest that the continuous SEI growth was due to a partial dissolution of the SEI layer.28–30 The capacity versus cycle number plots seen in Fig. 1 can consequently be explained by superimposed volume expansion and diffusion controlled lithium trapping effects.
Influence of SEI formation on the capacities of Li-half cells
Although the SEI formation clearly affects the Qox/Qred ratio for each cycle, it cannot explain the capacity losses for the half-cells containing lithium electrodes used in this study as the capacity of the lithium electrode was much larger than those of the tin nanorod electrodes as well as those of the silicon composite electrodes discussed below. Since the capacities for such half-cells will be limited by the tin nanorod or silicon electrode capacities (see Section S5 in the ESI†), the SEI formation merely results in a loss of a small part of the lithium foil electrode. In other words, the capacity of the tin nanorod or silicon composite electrode should remain constant in the absence of any lithium trapping and surface area enhancements. It can further be concluded that the SEI formation process is unlikely to result in a decrease in the lithium concentration in the electrolyte as the amount of lithium included in the SEI would be generated at the lithium electrode. There could, in fact, be an increase in the lithium concentration in the electrolyte in the presence of a partial dissolution of the SEI which also may increase the viscosity of the electrolyte. The SEI formation will, nevertheless, still be a problem in full cells in which the capacities of the two electrodes are approximately equal. To decrease the capacity losses for such cells both the lithium trapping and the SEI problems will hence need to be appropriately addressed. It is, however, important to realize that stable capacities for alloy forming electrode materials are very unlikely to be obtained merely by solving the SEI problem.It should also be mentioned that linear plots of the capacity as a function of the square root of the time (such as those seen in the present study), are unlikely to be obtained if the capacity loss is due to SEI formation as can be shown by the following example. If the positive electrode is assumed to be capacity limiting, its full oxidation capacity would be used to compensate for the reductive charge associated with the SEI formation and the reduction of the negative electrode material during the first charge. This means that the negative electrode cannot be fully reduced. On the subsequent discharge step, the oxidative capacity of the negative electrode therefore becomes limiting which means that the positive electrode cannot be fully reduced. After the first cycle, the capacity of the cell has therefore decreased by an amount corresponding to the SEI charge. This procedure may then be repeated on the subsequent cycles and if the charge due to the SEI formation is approximately constant upon the remaining cycles (which is reasonable to assume based on a partial dissolution of the SEI layer), a linear drop in the capacity with the cycle number should be seen. Note that the positive electrode in this case will contain a higher and higher concentration of the oxidized form as it cannot be fully reduced on the cycles. This gradual inactivation of the positive electrode is hence the reason why the cell capacity decreases continuously upon the cycling.
If instead the negative electrode is capacity limiting, the capacity of the cell will remain constant until the capacity of the positive electrode becomes limiting. In this case, the capacity of the positive electrode thus decreases by an amount corresponding to the SEI charge on each cycle. This does, however, not affect the cell capacity until the positive electrode becomes capacity limiting. In a cell containing a lithium foil electrode, the cell capacity should therefore remain constant until the capacity of the lithium electrode becomes limiting.
Silicon nanoparticle composite electrodes
Since lithium alloy formation and large capacity losses also are seen for silicon electrodes, experiments were likewise carried out with silicon nanoparticle composite electrodes. An average particle size of 50 nm was chosen to minimize the influence of the volume expansion effects.1,8,31 In Fig. 2a it is seen that the capacity decrease was about 2000 mA h g−1 during the first 100 cycles (the chronopotentiograms are shown in Fig. S5 in the ESI†). The magnitude of this loss is in good agreement with other results.1,2,23,32,33 While the electrode exhibited an initial capacity of about 3100 mA h g−1 (i.e. 87% of the theoretical capacity for Li15Si434), the average capacity loss was thus about 20 mA h g−1 per cycle. Given that this charge would have corresponded to about 5 μg of trapped lithium per cycle (see Section S5 in the ESI†) it is clear that the lithium would be difficult to detect unless many cycles or very sensitive analysis techniques were employed. Based on the similarities between the silicon and tin nanorod electrode results it is reasonable to assume that the silicon capacity losses were caused by elemental lithium trapping. As seen in Fig. S6 in the ESI,† plots of the capacity versus the square root of the elapsed time were indeed found to be linear after about 25 to 30 cycles in analogy with the tin nanorod electrode results. For the silicon electrode, a small initial capacity increase was also seen, most likely due to the formation of an increased electroactive surface area. Since the capacity retention was about 36% after 100 cycles, the average charge recovery efficiency was about 99% (as 0.99100 ≈ 0.36) suggesting that about 1% of the reduction charge was lost on each cycle. | ||
| Fig. 2 Electrochemical cycling of Si composite electrodes. (a) Specific gravimetric reduction (i.e. lithiation) and oxidation (i.e. delithiation) capacities and Qox/Qred ratios versus cycle number for C/10 cycling. (b) Specific capacities versus cycle number for constant current cycling with a fixed reduction charge of 1200 mA h g−1 followed by a constant current (cc) oxidation step including a constant voltage oxidation step (cccv) after every 10th cycle. (c) Specific capacities versus cycle number for C/10 cycling with constant voltage oxidation steps after 100, 200 and 300 cycles. The first reduction step, as well as the reduction and oxidation steps after the 200th cycle, were carried out at a rate of C/100 and the durations of the 100th, 200th and 300th constant voltage steps were 210, 52 and 223 hours, respectively. (d) Plot of the accumulated difference between the reduction and oxidation charges in (c) versus cycle number. The positions of the constant voltage oxidation steps are marked with asterisks. The voltage cut-off limits were 0.01 and 0.9 V vs. Li+/Li and all constant voltage steps were made to 2.0 V. | ||
Experiments were also carried out to determine the amounts of lithium in silicon composite electrodes after different lithium deposition times. These experiments (see Section S7 as well as Fig. S7 in the ESI†) were carried out using an initial constant current reduction step followed by potentiostatic deposition at 0.01 V vs. Li+/Li for up to 200 hours. The amounts of lithium in the electrodes were determined and compared with the amounts expected based on the deposition charges. The amount of lithium found in the samples was found to correspond to about 92% of the deposition charge. This is important as it suggests that 8% of the charge was consumed by the SEI formation process, and the data also indicates that the SEI dissolution rate was about 1 nm per hour (see Section S7 in the ESI†). The calculated Li/Si mole ratio after 200 hours was about 3.8 in good agreement with the formation of Li15Si4, indicating that the electrode was almost saturated with respect to lithium. Since the oxidation current at the Li electrode must balance the reduction current at the Si composite electrode, it is also clear that the deposited lithium was compensated for by the Li electrode. The plot of the deposition charge versus the square root of the deposition time (see Fig. S7 in the ESI†) was found to be linear and from this plot a lithium diffusion coefficient of 1.7 × 10−11 cm2 s−1 was obtained. Based on this value, a full lithiation of the about 15 μm thick silicon composite electrodes would not be expected at a cycling rate of C/10. Since a deposition time of more than 100 hours was required to reach saturation, it is clear that lithium concentration gradients always should be present in the electrodes during cycling at a C/10 rate or higher.
To further study the capacity losses for the silicon composite electrodes, controlled reduction charge experiments were carried out using a constant charge of 1200 mA h g−1. As is shown in Fig. 2b, the oxidation charge was always smaller than the reduction charge (the average Qox/Qred ratio was 98%) and the accumulated difference between the reduction and oxidation charges therefore increased on each cycle. It was, however, found (see Fig. 2b) that a significant fraction of the difference accumulated over the previous ten cycles could be recovered during a ten hour oxidation step to 1.0 V vs. Li+/Li. Every tenth oxidation step was thus performed with a constant current step followed by a constant voltage pulse. Since the recovery during the constant voltage step was 30% (of the accumulated charge difference) after 10 cycles but 73% after 70 cycles, a larger fraction of the accumulated charge difference could be recovered during the later cycles. Another interesting feature in Fig. 2b is the fact that the oxidation charge on the cycle immediately after each constant voltage pulse was decreased by a value corresponding to the constant voltage pulse charge. The recovery of the deposited lithium was thus significantly decreased after the application of the constant voltage pulse, most likely as the latter increased the lithium trapping ability of the electrode by decreasing the lithium concentration in the surface layer of the electrode. It is therefore reasonable to assume that the lithium trapping effect was linked to the lithium concentration profile in the electrode and that the influence of the trapping effect was smaller for a higher lithium concentration in the electrode.
Additional evidence supporting the diffusion controlled lithium trapping hypothesis is shown in Fig. 2c depicting a plot of the capacity of a silicon electrode as a function of the cycle number for constant current cycling at a rate of C/10 in the presence of constant voltage oxidation steps to 2.0 V vs. Li+/Li on the 100th, 200th and 300th cycles. While it is immediately clear that the capacity decreased significantly during the cycling, a reduction capacity of about 2400 mA h g−1 was, still, obtained after the constant voltage step (to 2.0 V vs. Li+/Li) on the 200th cycle when using a rate of C/100. This capacity should be compared to that of about 110 mA h g−1 obtained at a rate of C/10 immediately prior to the pulse. The capacity increase of almost 2300 mA h g−1 cannot be explained by the oxidation charge associated with the constant voltage oxidation pulse as this corresponded to merely 330 mA h g−1 (see Fig. 2d). The large increase in the reduction capacity must therefore have been due to the longer deposition time associated with the C/100 cycling rate, most likely as this enabled lithium diffusion further into the electrode. This is supported by the fact that the corresponding increase in the C/10 reduction charge after 100 cycles was much smaller despite the longer duration of the pulse. In analogy with the results in Fig. 2b, the oxidation charge on the cycle immediately after the constant voltage step was found to be significantly smaller.
Fig. 2d shows a plot of the accumulated difference between the reduction and oxidation charges in Fig. 2c. The initial rapid increase in the accumulated difference is followed by a region in which the value increases more slowly suggesting a saturation effect. It is also evident that the accumulated difference reached values up to about 5800 mA h g−1 clearly exceeding the theoretical capacity for a silicon electrode (i.e. 3579 mA h g−1 for Li3.75Si34). This means (see Section S3 in the ESI†) that the accumulated charge difference must have contained contributions both from lithium trapping and SEI formation. In Fig. 2d, the negative shifts associated with the constant voltage steps after 100 and 200 cycles stem from the oxidation charge associated with these steps whereas the C/100 deposition step on the 201th cycle resulted in an increased reduction charge and hence an increased accumulated charge difference. The decrease in the accumulated charge difference after 201 cycles is particularly interesting as it shows that the oxidation charge actually exceeded the reduction charge after the C/100 reduction step. This suggests that that lithium deposited during the C/100 step became available for oxidation during the subsequent C/10 oxidation steps, i.e. that lithium diffused from the interior parts of the electrode towards the electrode surface!
The two-way diffusion trapping model
The results presented above as well as previous results obtained with aluminium electrodes12,13,35 indicate that significant capacity losses can be seen due to diffusion controlled lithium trapping. This effect can be explained based on the lithium concentration profiles in the electrodes employing the schematic two-way diffusion model depicted in Fig. 3 (which clearly does not take into account the fact that the real diffusion profiles often have more complex shapes due to e.g. grain boundary diffusion).19,36 This straightforward model can, nevertheless, be used to explain the fundamental aspects of the trapping phenomenon. During the first cycle, lithium diffuses into the electrode during the deposition step whereas the lithium concentration at the electrode surface decreases during the subsequent oxidation step. This gives rise to an intermediate region in which the lithium concentration is higher than both at the electrode surface and in the interior parts of the electrode. The lithium can then hence diffuse both towards the electrode surface and further into the electrode, i.e. two-ways. This implies that the lithium furthest into the electrode can diffuse too far into the electrode to be recovered within the time domain of the subsequent oxidation step and a small part of the deposited lithium therefore becomes trapped in the electrode on each cycle. As the experimental data indicate that less than 1% of the deposited amount of lithium is trapped on each cycle, this effect is initially difficult to detect. There are, nevertheless, several reports19,37,38 indicating the presence of lithium in oxidized electrodes. As is shown in Fig. 2, the trapped lithium is, however, not irreversibly lost and it should hence be possible to regenerate an electrode by merely keeping the electrode in its oxidized state for a sufficiently long time. The actual capacity loss, however, stems from the gradual increase in the lithium concentration in the electrode as slightly less lithium can be deposited on each subsequent cycle. As the electrode becomes saturated with lithium within the electroactive region of the electrode, the capacity approaches zero. It can hence be concluded that the extent of lithium trapping should depend on the time domain of the experiments and the thickness of the electrode since a sufficiently long lithiation step or a sufficiently thin electrode layer should facilitate the attainment of a fully lithiated material. As long as there is lithium deposition (or oxidation of lithium) at the electrode surface, the surface concentration of lithium will, nevertheless, be different from that in the interior parts of the electrode. The electrodes should consequently be designed so that the full capacity of the material always is exploited. This finding could explain why an improved cycling performance generally is seen for electrodes with thin layers of nanoparticles.1,4,8,22 Based on the lithium concentration profiles it can also be concluded that the delithiation (although incomplete) should be inherently faster than the lithiation step in accordance with recent experimental findings.2,39 It is also reasonable to assume that the two-way diffusion model may be applicable to other electrode materials used in lithium based batteries as well. | ||
| Fig. 3 Diffusion controlled lithium trapping. (a) On the first reduction (i.e. lithiation) cycle, the deposited lithium diffuses towards the centre of the particle (steps 1–4). During the subsequent oxidation (i.e. delithiation) the deposited lithium diffuses towards the electrode surface as well as towards the centre of the particle (steps 5–8). Two-way diffusion gives rise to the lithium concentration profile illustrated in step 6 and some lithium remains in the particle even after the oxidation step (step 8). (b) During the second cycle lithiation the deposited lithium diffuses inwards while the lithium in the centre of the particle diffuses outwards (step 1b). As more and more lithium is trapped in the particle, less and less lithium can be deposited on the subsequent cycle. | ||
Lithium diffusion in common current collector materials
Since it is well known that elements forming alloys with lithium are unsuitable as current collectors, materials such as copper, nickel, stainless steel and titanium are generally used together with the negative electrode. Although the amounts of lithium that can be housed in the latter three materials should be about two orders of magnitude lower40 than for copper, these materials can still take up lithium even though this cannot be seen in the phase diagrams.41–43 Significant amounts of lithium were hence found in nickel, copper and titanium pieces (see Fig. 4a) kept in contact with elemental lithium for a week at 50 °C in sealed plastic pouches. Boron-doped diamond (BDD) was also investigated and found to be considerably less permeable to lithium diffusion as seen in Fig. 4. Further evaluation of Li exposed BDD electrodes by HAXPES and TOF-SIMS showed no signs of Li trapping (see Fig. 4 and Fig. S8–S13 in the ESI†). This indicates that the lithium diffusion rate in BDD is much lower than in conventional metal current collectors which is why BDD should be better suited for use in current collectors for negative electrodes based on lithium alloys or lithium films. | ||
| Fig. 4 Li trapping in current collectors. (a) Lithium amounts found in nickel, copper, titanium and boron-doped diamond samples exposed to lithium foils for one week at 50 °C. (b) Lithium amounts found in boron-doped diamond samples exposed to lithium foil for different durations at 50 °C. (c) 6015 eV HAXPES C1s spectra for a cycled and a pristine boron-doped diamond electrode, respectively. (d) TOF-SIMS sputter profile detailing the elemental composition in the boron-doped diamond film. The inset show an SEM image of the surface after sputtering where the scale bars represent 10 μm. | ||
Conclusions
The present results clearly show that the capacity losses generally seen for nanostructured alloy forming materials are more likely explained by diffusion controlled trapping of lithium than volume expansion or SEI formation effects. It can also be concluded that elemental lithium can be trapped both in lithium alloy forming electrode materials (e.g. Si, Sn and Al) and in metals (e.g. Cu and Ni) commonly used as current collectors. The trapping is caused by a two-way diffusion process where some of the deposited lithium diffuses into the electrode bulk and thereby becomes inaccessible during the subsequent oxidation step. The trapping effect depends on the time scale of the experiments and the dimensions of the electrode (i.e. the thickness of the active layer and the size of the particles) and its influence should be decreased by making sure that a complete lithiation and delithiation of the electrode is possible during the cycling.Although the present study only deals with the diffusion of elemental lithium in lithium alloy forming materials and metals it is reasonable to assume that the two-way diffusion model also could be applicable to the lithium ion diffusion in intercalation materials. This suggests that the two-way diffusion effect described in this work may constitute a fundamental problem with respect to many electrode materials currently employed in lithium based batteries.
Acknowledgements
We thank Professor Karin Larsson for her help with the development of the boron-doped diamond substrates and Dr Julia Maibach for her assistance with the HAXPES analyses. Financial support from The Swedish Research Council (VR 2012–4681 and 2015-04421), The Swedish Energy Agency (STEM), Ångström Advanced Battery Center (ÅABC) and StandUp for Energy is gratefully acknowledged. D. R., L. N. and S. B. are the inventors on a patent application PCT/IB2016/055176 entitled “Substrate with doped diamond layer for lithium-based systems”. The authors declare no other financial interest in this work.Notes and references
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- J. Li, N. J. Dudney, X. Xiao, Y.-T. Cheng, C. Liang and M. W. Verbrugge, Adv. Energy Mater., 2015, 5, 1401627 CrossRef.
- V. Aravindan, Y.-S. Lee and S. Madhavi, Adv. Energy Mater., 2015, 5, 1402225 CrossRef.
- M. Gu, Y. He, J. Zheng and C. Wang, Nano Energy, 2015, 17, 366 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- K. Zhang, G.-H. Lee, M. Park, W. Li and Y.-M. Kang, Adv. Energy Mater., 2016, 6, 1600811 CrossRef.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 CAS.
- L. Mai, X. Xu, C. Han, Y. Luo, X. Tian, X. Xu, L. Chang and L. Xu, Chem. Rev., 2014, 114, 11828–11862 CrossRef CAS PubMed.
- G. Zheng, S. W. Lee, Z. Liang, H.-W. Lee, K. Yan, H. Yao, H. Wang, W. Li, S. Chu and Y. Cui, Nat. Nanotechnol., 2014, 9, 618–623 CrossRef CAS PubMed.
- K. Yan, H.-W. Lee, T. Gao, G. Zheng, H. Yao, H. Wang, Z. Lu, Y. Zhou, Z. Liang, Z. Liu, S. Chu and Y. Cui, Nano Lett., 2014, 14, 6016–6022 CrossRef CAS PubMed.
- J. R. Owen, W. C. Maskell, B. C. H. Steele and T. S. Nielsen, Solid State Ionics, 1984, 13, 329–334 CrossRef CAS.
- G. Oltean, C.-W. Tai, K. Edström and L. Nyholm, J. Power Sources, 2014, 269, 266–273 CrossRef CAS.
- K. Ui, K. Yamamoto, K. Ishikawa, T. Minami, K. Takeuchi, M. Itagaki, K. Watanabe and N. Koura, J. Power Sources, 2008, 183, 347–350 CrossRef CAS.
- T. Yoon, C. C. Nguyen, D. M. Seo and B. L. Lucht, J. Electrochem. Soc., 2015, 162, A2325–A2330 CrossRef CAS.
- Y. Eker, K. Kierzek, E. Raymundo-Pinero, J. Machnikowski and F. Béguin, Electrochim. Acta, 2010, 55, 729–736 CrossRef CAS.
- N. Delpuech, N. Dupre, P. Moreau, J.-S. Bridel, J. Gaubicher, B. Lestriez and D. Guyomard, ChemSusChem, 2016, 9, 841–848 CrossRef CAS PubMed.
- A. L. Michan, G. Divitini, A. J. Pell, M. Leskes, C. Ducati and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 7918–7931 CrossRef CAS PubMed.
- A. Bordes, E. De Vito, C. Haon, A. Boulineau, A. Montani and P. Marcus, Chem. Mater., 2016, 28, 1566–1573 CrossRef CAS.
- S. Böhme, K. Edström and L. Nyholm, Electrochim. Acta, 2015, 179, 482–494 CrossRef.
- M. Winter and J. O. Besenhard, Electrochim. Acta, 1999, 45, 31–50 CrossRef CAS.
- X. H. Liu, Y. Liu, A. Kushima, S. Zhang, T. Zhu, J. Li and J. Y. Huang, Adv. Energy Mater., 2012, 2, 722–741 CrossRef CAS.
- G. Kim, S. Jeong, J.-H. Shin, J. Cho and H. Lee, ACS Nano, 2014, 8, 1907–1912 CrossRef CAS PubMed.
- K. Nishio, T. Yanagishita, M. Yoshida, T. Hayakawa and H. Masuda, Electrochem. Commun., 2015, 59, 13–15 CrossRef CAS.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, Chem. Mater., 2013, 25, 394–404 CrossRef CAS.
- K. C. Högström, S. Malmgren, M. Hahlin, M. Gorgoi, L. Nyholm, H. Rensmo and K. Edström, Electrochim. Acta, 2014, 138, 430–436 CrossRef.
- I. Yoon, D. P. Abraham, B. L. Lucht, A. F. Bower and P. R. Guduru, Adv. Energy Mater., 2016, 6, 1600099 CrossRef.
- K. Tasaki, A. Goldberg, J.-L. Lian, M. Walker, A. Timmons and S. J. Harris, J. Electrochem. Soc., 2009, 156, A1019–A1027 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- D. Aurbach, M. Moshkovich, Y. Cohen and A. Schecter, Langmuir, 1999, 15, 2947–2960 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- E. Peled, F. Patolsky, D. Golodnitsky, K. Freedman, G. Davidi and D. Schneier, Nano Lett., 2015, 15, 3907–3916 CrossRef CAS PubMed.
- Z. Wang, C. Xu, P. Tammela, J. Huo, M. Strømme, K. Edström, T. Gustafsson and L. Nyholm, J. Mater. Chem. A, 2015, 3, 14109–14115 CAS.
- T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838–A842 CrossRef CAS.
- W. C. Maskell and J. R. Owen, J. Electrochem. Soc., 1985, 132, 1602–1607 CrossRef CAS.
- A. J. Leenheer, K. L. Jungjohahn, K. R. Zavadil and C. T. Harris, ACS Nano, 2016, 10, 5670–5678 CrossRef CAS PubMed.
- D. X. Liu, L. R. Cao and A. C. Co, Chem. Mater., 2016, 28, 556–563 CrossRef CAS.
- B.-K. Seidlhofer, B. Jerliu, M. Trapp, E. Hüger, S. Risse, R. Cubitt, H. Schmidt, R. Steitz and M. Ballauff, ACS Nano, 2016, 10, 7458–7466 CrossRef CAS PubMed.
- W. Wei, F. Björefors and L. Nyholm, Electrochim. Acta, 2015, 176, 1393–1402 CrossRef CAS.
- A. N. Dey, J. Electrochem. Soc., 1971, 118, 1547–1549 CrossRef CAS.
- C. W. Bale, J. Phase Equilib., 1989, 10, 135–138 CAS.
- W. Klemm and B. Volavsek, Z. Anorg. Allg. Chem., 1958, 296, 184–187 CrossRef CAS.
- K. J. Lee, S. Y. Lee and P. Nash, in Binary Alloy Phase Diagrams, ed. T. B. Massalski, ASM International, 2nd edn, 1990, vol. 3, pp. 2450–2453 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee00244k |
| This journal is © The Royal Society of Chemistry 2017 |