
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-complete suppression of surface losses and total internal quantum efficiency in BiVO4 photoanodes†
Bartek J.
Trześniewski
a,
Ibadillah A.
Digdaya
a,
Tetsuro
Nagaki
a,
Sandheep
Ravishankar
b,
Isaac
Herraiz-Cardona
b,
David A.
Vermaas
a,
Alessandro
Longo
cd,
Sixto
Gimenez
b and
Wilson A.
Smith
 *a
*a
aDelft University of Technology, Faculty of Applied Sciences, Department of Chemical Engineering, Materials for Energy Conversion and Storage (MECS), Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: W.Smith@tudelft.nl
bUniversitat Jaume I, Institute of Advanced Materials (INAM), Av. de Vicent Sos Baynat, s/n, 12071 Castelló de la Plana, Spain
cNetherlands Organization for Scientific Research (NWO), The European Synchrotron Radiation Facility (ESRF), CS40220, 38043 Grenoble Cedex 9, France
dISMN-CNR, UOS Palermo, Via Ugo La Malfa, 153, 90146 Palermo, Italy
First published on 26th May 2017
Abstract
Bismuth vanadate (BiVO4) is one of the most efficient light absorbing metal oxides for solar water splitting. BiVO4 photoanodes immersed in an electrolyte in an open circuit configuration and exposed to simulated solar illumination for prolonged time achieve superior photoelectrochemical (PEC) activity. This photocharging (PC) effect is capable of almost completely overcoming the surface and bulk limitations of BiVO4. Herein we show that alkaline conditions favor the PC effect; specifically BiVO4 photoanodes subjected to PC treatment at pH 10 achieve a record high photocurrent for undoped and uncatalyzed BiVO4 of 4.3 mA cm−2 @ 1.23 VRHE, an outstandingly low onset potential of 0.25 VRHE, and a very steep photocurrent onset. Alkaline conditions also facilitate excellent external and internal quantum efficiencies of 75 and 95% respectively (average in the 440 nm > λ > 330 nm range). Moreover, impedance spectroscopy and in situ XAS study suggest that electronic, structural and chemical properties of the bulk of these films remain unchanged during the PC treatment. However, appreciable changes in the surface-related properties take place. Ultimately, our results indicate that the improved activity of PC-BiVO4 is enhanced by surface reaction pathways of the semiconductor–liquid junction, which is directly correlated with the electrochemical environment in which it is modified.
Broader contextPhotoelectrochemical (PEC) water splitting is an attractive route to capture, convert and store the energy from the sun in the form of chemical bonds. This approach allows for sustainable production of solar fuels (typically H2), and thereby addresses the challenges of new energy technologies needed in the pathway towards a carbon-free economy. Solar water splitting relies on PEC cells, consisting of semiconducting photoelectrodes, which directly utilise photo-generated charge carriers to drive the water oxidation and reduction half-reactions on the corresponding solid/liquid interfaces. Metal oxides have been heavily studied as a class of photoelectrode materials for PEC devices due to their low cost and stable operation. Specifically, bismuth vanadate (BiVO4) has proven to demonstrate significant activity as a photoanode material. However, due to its inherent bulk- and surface-related limitations, complex strategies such as doping or co-catalysis are typically necessary for BiVO4 to reach high yields for O2 production. Herein, we investigate photocharging (PC), a simple and novel treatment that can dramatically improve the water splitting capabilities of BiVO4. We demonstrate that illumination combined with alkaline conditions alters the chemistry at the semiconductor–electrolyte interface to form a BiVO4 surface with superior activity for the oxygen evolution reaction. |
Introduction
The storage of intrinsically intermittent solar energy in the form of chemical fuels (e.g. H2) has the potential to simultaneously combat global environmental and energy problems. Photoelectrochemical (PEC) water splitting is one of the most promising approaches for such renewable and sustainable production of hydrogen.1–4 Metal oxides have been extensively used5 as a promising class of photo-electrode materials for PEC devices, due to their high (photo)chemical stability, abundance and low cost, as well as suitable optical and electronic properties. However, out of the wide selection of metal oxides, so far only a few materials, Fe2O3,6 WO3,7 BiVO48 and Cu2O,9 have demonstrated reasonable performance under simulated solar illumination (i.e. photocurrent over 2 mA cm−2 and a large photovoltage).Among the metal oxides, bismuth vanadate (BiVO4) is a material with the highest reported solar to hydrogen (STH) conversion efficiency to date (5.2%).10,11 BiVO4 is an n-type photoanode material, with a bandgap energy of 2.4 eV,12 and a theoretical efficiency of ∼9% STH,13 is made of cheap, relatively earth abundant, non-toxic elements and is reasonably stable under neutral conditions. However, it suffers from surface and bulk recombination losses that limit its performance to well below its theoretical maximum.13 The deposition of a co-catalyst (Co–Pi,13–16 Co–Bi,17 Ni–Bi,18 FeOOH/NiOOH,19 Fe2O3/ZrO220) has been reported to successfully mitigate the surface-related limitations of BiVO4 and has enabled the ability to achieve close to 100% catalytic efficiencies for the oxygen evolution reaction (OER). Complementarily, the low electronic conductivity of BiVO4 has been partially overcome via doping with high-valence cations such as W13,14,21 or Mo21 which substitute for vanadium. Moreover, fabricating BiVO4 films in a nanostructured morphology has been demonstrated19,22,23 to overcome the transport limitations caused by the intrinsically short (70 nm)24 diffusion length of photogenerated charge carriers. Finally, surface passivation of BiVO4 films with thin layers of TiO2 has also been shown25,26 to significantly improve its stability. While these techniques are mostly successful in improving the overall PEC activity of BiVO4, they add extra steps to the material processing and introduce significant complexity in optimizing a practical PEC water splitting device.
In our recent work27 we have reported for the first time on a photoelectrochemical procedure called photocharging (PC), which enables the ability to successfully improve both bulk and surface limitations of BiVO4 photoanodes. We have demonstrated that prolonged exposure of BiVO4 photoanodes in an aqueous solution under open circuit (OC) conditions to simulated solar irradiation can greatly increase its solar water oxidation efficiency via a reduced onset potential and an increased maximum photocurrent density. A similar change in various properties of light active materials exposed to illumination has been widely recognized in the photovoltaic (PV) field as a so-called light soaking effect.28–31 However, to date, no analogous effect has been identified in the PEC field. Moreover, it seems that the mechanism of the photocharging effect observed for BiVO4 may be quite different from PV light soaking. For instance, the light soaking effect observed in PV devices is a purely solid-state phenomenon, i.e. it concerns only the interaction between the solid-state materials and the incident electromagnetic radiation. Accordingly, the light soaking effect in the PV cells has been usually ascribed28–31 to the alteration of the defect structure, change in DOS of trap states or interdiffusion within the light active materials. In contrast, according to our results, the presence of a liquid electrolyte is essential to the PC effect, and no PC can take place without it, i.e. the PC effect does not occur when the BiVO4 films are illuminated in air. These findings indicate that the PC effect should be studied in the context of the semiconductor–liquid junction (SLJ), the key interface in a PEC system.32–36 The significance of the SLJ stands out by the number of important processes in which it is involved: mediation of the transport of charge carriers, formation and passivation of surface states, and catalysis of the oxygen evolution reaction (OER) via the formation of reaction intermediates.
Herein we provide new insights into the possible mechanisms of photocharging in BiVO4 photoanodes, and how it directly leads to improved PEC performance. We investigate changes in the PEC, optical, structural, chemical and electronic properties of photocharged BiVO4 in the context of the SLJ, to provide further insights into the chemical and physical mechanisms behind the PC phenomenon. Firstly, we investigate the effect of the electrolyte pH on the PC efficiency as a function of time to understand whether the PC effect is related to the formation of oxy/hydroxyl-based water oxidation intermediates (HO*, O*, HOO*). Since protons/hydroxyl groups are directly involved in all the steps that constitute the OER,37 we expect that if the PC treatment promotes the formation of any of the water oxidation intermediates, then a different pH could heavily affect the PC efficiency. We show that alkaline conditions favor the PC effect, and specifically that BiVO4 photoanodes subjected to the PC treatment at pH 10 achieve a record high photocurrent for undoped and uncatalyzed BiVO4 of 4.3 mA cm−2 @ 1.23 VRHE, an outstandingly low onset potential of 0.25 VRHE, and excellent external and internal quantum efficiencies of 75 and 95% respectively (average in the 440 nm > λ > 330 nm range). Secondly, we perform an in situ UV-vis spectroscopic study to examine if PC leads to changes in the optoelectronic properties of BiVO4, and hence possibly to different electronic transport properties. We observe that PC leads to increased absorption outside the bandgap energy region of BiVO4 (λ > 520 nm), especially under the PC-facilitating alkaline conditions. However, this increased absorption does not seem to be directly responsible for the photocurrent enhancement, since the external quantum efficiency (IPCE) of PC-BiVO4 for λ > 520 nm remains negligible. In addition, we use impedance spectroscopy to investigate the effect of PC on the electronic properties of BiVO4 and observe only negligible changes in the bulk electronic properties of BiVO4 films before and after the treatment. However, we observe a significant change in surface-related properties; an increase in capacitance and a decrease in resistance. Lastly, we use in situ electrochemical X-ray absorption spectroscopy (XAS) to study the chemical and structural properties of BiVO4 under PEC conditions. Our findings suggest that neither applying a bias potential nor the photocharging treatment alters the structure/chemistry of the bulk of our BiVO4 films; specifically, the oxidation state of vanadium does not seem to change. Overall, our results suggest that PC is related to a light-driven alteration of the surface of BiVO4, and is highly preferential under alkaline conditions. Those changes are most likely related to the formation of highly active water oxidation intermediates and/or changes in the chemistry of the surface. We propose a model in which the surface of BiVO4 becomes hydrogenated under alkaline conditions and under illumination, coupled with creation of oxygen vacancies and reduction of vanadium species, and possibly accompanied by creation of oxo-water intermediates. Consequently, electrocatalytic properties become more beneficial for solar driven water splitting applications. Furthermore, any bulk-related effects that we identify are mediated by phenomena occurring at the surface, which highlights the importance of the SLJ for the efficiency of PEC devices.
Results and discussion
Influence of pH on PC BiVO4 under illumination
To understand the importance of the SLJ during photocharging treatments, the effect of the electrolyte pH on the PC efficiency was studied first. Fig. 1 presents J–V curves of 200 nm thick BiVO4 photoanodes under AM1.5 illumination in PBA buffer at pH 4, 7 and 10 collected before (dashed lines) and after (solid lines) PC for 20 hours. For untreated BiVO4, J–V curves recorded at pH 4, 7 and 10 show little difference. Given the sample-to-sample variation, they can be considered similar, though pH 4 gives consistently the lowest activity in terms of photocurrent density. It is apparent that pH has little influence on the photocurrent generation of untreated BiVO4. Consequently, we can conclude that different concentrations of H+ or OH− do not significantly influence the light driven oxidation of water on untreated BiVO4 photoanodes. | ||
| Fig. 1 J–V curves of BiVO4 in PBA buffer, pH 4, 7 and 10; a stable 10th anodic scan is shown in each case. Dashed curves – untreated films, solid curves – photocharged films. Each PC treatment was performed in the same pH as the corresponding CV experiment. Inset: E–t OC trends collected during the PC treatment. Stars in both plots denote the final photocharging OC potential. Experimental conditions: back-side AM1.5 illumination, films of 200 nm thickness, scan rate of 50 mV s−1. | ||
In contrast, for photocharged BiVO4, J–V curves recorded at pH 4, 7 and 10 show striking differences. There seemingly exists a very strong relationship between the alkalinity of the electrolyte and PEC activity of the photocharged electrodes; in particular, a high pH promotes a high photocurrent and a low onset potential. We observe photocurrent densities of 1.0, 3.3 and 4.3 mA cm−2 @ 1.23 VRHE at pH 4, 7 and 10 respectively. In our initial study27 we have reported 3.3 mA cm−2 @ 1.23 VRHE at pH 7, herein we were able to achieve 4.3 mA cm−2 @ 1.23 VRHE using a pH 10 electrolyte. This is by far the highest reported photocurrent at 1.23 VRHE for undoped and uncatalyzed BiVO4 photoanodes. Moreover, there is also a very clear difference in the slope of the photocurrent in different media. Slightly acidic conditions at pH 4 results in a very sluggish photocurrent onset; initially the J–V curve is almost flat and a significant overpotential is required to achieve reasonable photocurrent densities. On the other hand, alkaline conditions at pH 10 lead to a very steep photocurrent onset from the very beginning, with no observable inflection point in the J–V curve, a trend more typical for measurements where no surface limitations are present,38 for example when a sacrificial hole scavenger such as H2O2 is employed.27 The different slopes suggest that alkaline conditions facilitate major suppression of surface recombination39 in PC BiVO4. Interestingly, the J–V curves at pH 4 recorded before and after the PC treatment overlap, showing that at this pH no PC effect takes place. It seems that there is a threshold pH for the PC to take place and under sufficiently acidic conditions (pH < 7) no PC can occur. This implies that a certain concentration of hydroxyl groups is necessary for the PC treatment to be successful.
Moreover it seems that no specific anion or cation is responsible for the PC effect. As presented in S1 (ESI†), electrolytes with compositions different than K-Pi or PBA buffer can successfully facilitate the PC effect, following a pH trend similar to what is demonstrated in Fig. 1. In addition, a series of experiments were conducted to measure the photocurrent density as a function of thickness (S2 (ESI†) black curves). As expected, due to low absorption of BiVO4, the photocurrent density produced by our photoanodes scales up with thickness of the films, up to ca. 200 nm limit. There’s no significant gain in current for films thicker than 200 nm since 200 nm thick films already absorb most of the available light40 and going beyond 200 nm may be detrimental due to the 70 nm diffusion length of charge carriers in BiVO424 and consequently any gain related to increased absorption could be potentially consumed by bulk transport and recombination losses. S2 (ESI†) also presents the time evolution trends of the PC process (curves red to blue). JV curves collected every 2 h show a steady increase in photocurrent, cathodic shift of the photocurrent onset and improvement in the fill-factor. It seems that the biggest changes occur in the first 6 h of the treatment, and further improvements are more moderate.
The inset in Fig. 1 shows the time evolution of the BiVO4 potential vs. RHE recorded during the PC treatment at pH 4, 7 and 10. The dark open circuit potentials for each pH are shown in ESI† (S3), where the pH does not show a strong influence (EOC,dark). It can be seen that more strongly alkaline pH values favor lower light induced equilibrium potentials (closer to the BiVO4 conduction band-edge). Accordingly, we measured the equilibrium open-circuit (OC) potentials of 0.58, 0.32 and 0.27 VRHE for pH 4, 7 and 10 respectively. Remarkably, the final OC potentials (EOC, marked with stars in both the inset and the main plot) and the photocurrent onset potentials (Eonset) seem to be identical for the corresponding pH values. A correlation between EOC and Eonset is something to be expected, since measurements of EOC can be used to determine the photovoltage (Vph); any changes in Vph are usually reflected in a shift of Eonset35 since Vph can be simply viewed as the amount of bias voltage provided by a photoelectrode itself. However, this exact coincidence of the long-term equilibrium OC potential after photocharging and the photocurrent onset potential of the J–V curves under solar illumination is a unique feature of our photoanodes. It seems that the onset of PEC activity corresponds directly to the photocharged OC potential, and thus the match between these values shows that there has been a significant reduction in the overpotential required to evolve oxygen in photocharged samples. It may imply very fast exchange current densities at the SLJ. Importantly, this proposed correlation seems to hold in the whole pH 4–10 range, and is therefore not a signature of PC-enhanced performance, but rather a unique feature of our material. Notably, measurements collected at different pH values have very different photocurrent slopes at the onset: very sluggish at pH 4 and very steep at pH 10. Therefore, while the exchange current densities may be very fast in the whole pH range, the losses at the interface still vary significantly depending on the pH. To further prove that the OC potential under illumination correlates directly with the OER onset potential, we performed an i(t) experiment at a potential slightly (0.1 V) above the OC potential of a photocharged BiVO4 anode (S4 and S5, ESI†). Our results show that a small anodic bias of 0.1 V vs. the OCV (potential too low to drive OER on untreated BiVO4, see S6 and S7, ESI†) can result in some non-negligible current over 30 hours, suggesting that the observed current is indeed related to the OER.
The potential of a photoelectrode probed by a potentiostat can be considered to be equivalent to its Fermi level position EF or quasi-Fermi level position of the electrons EF,n.36 While the OC potential measured by the potentiostat corresponds effectively to the back contact and not to the surface, it is assumed that any change in one would trigger a similar change in the other. Therefore, we consider the ‘E vs. t’ measurements (Fig. 1, inset) as a technique to monitor in situ the effect of the PC treatment on the EF,n of BiVO4. Our data show that under PC conditions facilitating the highest enhancement in the photocurrent (pH 10), the OC potential strongly shifts towards the water reduction potential (0 VRHE) during the PC time (Fig. 1, inset, black curve). Therefore, we can conclude that a successful PC treatment leads to a shift of the BiVO4EF,n towards the conduction band edge.
The observed shift in the OC potential can be related to pinning of the electron Fermi level by the surface capacitive layer. Pinning of EF has been reported to have a strong effect on the obtainable photovoltage (Vph).35,36,41 Our previous results27 suggest that the photocharging treatment may be related to changes in the chemical compositions of the surface. Consequently the surface of BiVO4 altered during the photocharging seems to give rise to an energetic level at which the Fermi level pins. Our data indicate that Fermi level pinning at the energy level related to this newly formed capacitive layer is more beneficial for water oxidation via an increase of Vph. Furthermore, it has been proposed that under heavy Fermi level pinning conditions, an extra bias voltage may be necessary to start driving the OER.42 This agrees well with our J–V data, which shows that the overpotential to drive OER with untreated BiVO4 is significantly higher than those for photocharged films (supposedly diminished Fermi level pinning). The electronic properties of this proposed surface capacitive layer are further discussed in the impedance spectroscopy section.
One other possible explanation of this OC potential shift could be related to the formation of some oxy/hydroxyl-based water oxidation intermediates at the surface of BiVO4. In general, the chemical nature of the surface adsorbates can be expected to depend strongly on the electrolyte properties. Specifically, in water based electrolytes, the pH will heavily affect the type of aqueous-based adsorbates on the surface of a photoelectrode. Correspondingly, the properties of the SLJ, in particular the surface adsorbates, can affect in some way the chemical reaction mediated by the surface, in our case water oxidation. It seems plausible that under alkaline conditions, high OH− concentration produces adsorbates that can be readily transformed with the charge carriers excited during the PC treatment into more active water oxidation intermediates, simultaneously resulting in an altered EF,n. Importantly, it has been proposed36 that the adsorption of H2O molecules and its derivatives is responsible for the surface states observed typically on MOx photoanodes. Therefore, both presented hypotheses may in fact share a common mechanism.
Furthermore, on the contrary to what has been recently reported,43 our BiVO4 photoanodes do not show any changes in PEC activity when subjected to prolonged illumination in air (S8, ESI†), establishing the necessity of the electrolyte to facilitate the PC effect. Clearly, different deposition techniques used to fabricate BiVO4 films (spin coating in Berlinguette's work43 and spray pyrolysis in our case) inevitably produce films of different compositions and different defect structures, which is further supported by our XPS study presented in Fig. 2. In the work of Berlinguette, a strong peak at ca 532 eV can be observed for untreated samples, which is ascribed to surface defect sites (e.g., dangling oxo or hydroxy groups). However, we observe exactly the opposite; our initial material demonstrates a relatively weak signal at 532 eV. Further on, since the starting material is quite different, we believe that both treatments lead to a different modification and facilitate some different chemical transformation at the surface. In the work of Berlinguette the oxygen peak at 532 eV diminishes over the UV-curing treatment time, which is ascribed to removal of the surface states. On the contrary, we observe that it is only once the oxygen peak at 532 eV becomes more pronounced that an enhanced activity for photoelectrochemical water oxidation by our photoanodes can be achieved. We tentatively ascribe this 532 eV peak to hydroxyl groups.44 Moreover, we observe a significant decrease in the surface vanadium concentration of photoanodes subjected to photocharging treatment. Overall, our XPS study clearly demonstrates that our initial material is different from that of Berlinguette and that we are driving a different chemical transformation with our photocharging treatment. The surface of the photocharged BiVO4 seems to be terminated with some extra hydroxyl groups, to have a decreased concentration of vanadium and to be enriched with oxygen, which happens to make it more active for OER.
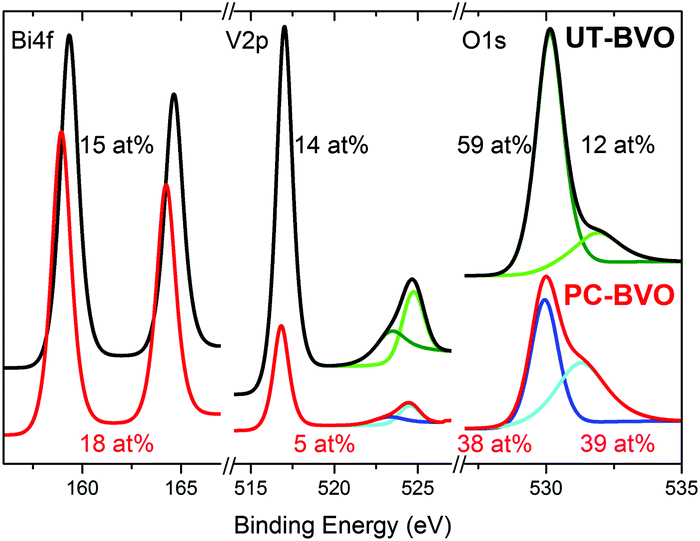 | ||
| Fig. 2 XPS spectra of BiVO4 photoanodes before and after photocharging in 0.1 M PBA buffer, pH 10, under AM1.5G simulated illumination. | ||
Spectroscopic study of PC BiVO4
To further probe the optoelectronic properties of BiVO4 photoanodes during photocharging, we performed in situ UV-vis spectroscopy to measure the change in optical transmittance of the photoanodes during electrochemical testing. Fig. 3 shows the change in transmittance of the films at 550 nm (above the 520 nm bandgap of BiVO4), collected in situ during the PC process. It was observed that for all the 3 pH values studied, the PC treatment leads to a gradual decrease in transmittance at 550 nm, meaning more absorption in the above bandgap region. Moreover, we detect a strong change in transmittance at pH 10 and a very slight one at pH 4. Therefore, regardless of the mechanism for this optical change, the hydroxyl groups seem to be facilitating the darkening effect. | ||
| Fig. 3 Relative change in optical transmittance at 550 nm wavelength as a function of PC time of BiVO4 in PBA buffer, pH 4, 7 and 10. Inset – relative change in optical transmittance in the above-bandgap spectral range as a function of PC time of BiVO4 in PBA buffer, pH 10, shown with a time step of 2 h, the arrow indicates the trend. | ||
Our spectral data suggest that there is a direct correlation between the pH and strength of the observed optical effect, i.e. alkaline conditions favor the darkening of BiVO4 outside the band gap region. In the previous section we identified a similar correlation between the electrolyte pH and the strength of the PC effect, i.e. the higher the pH the more the photocurrent extracted from a photocharged BiVO4 sample. This implies that a ‘darker state’ of BiVO4 can be associated with higher photocurrents. However, there is no clear plateau in the ΔT/dt trend, suggesting that a full transition to a ‘darker state’ is not a necessary condition to fully photocharge BiVO4.
To understand whether the extra above-the-bandgap absorption of photocharged BiVO4 yields an improved photocurrent density, i.e. if it creates charge carriers useful for water oxidation, external quantum efficiency (EQE), or incident photon to current conversion efficiency (IPCE) experiments were performed. Fig. 4(a) shows the results of the EQE measurements of BiVO4 photoanodes before and after PC. All the EQE curves show the onset of the photocurrent at a wavelength of about 520 nm, in good agreement with the bandgap energy of 2.4 eV typically reported for BiVO4.12 All the curves reach a quasi-plateau at a wavelength of about 440 nm. This slow rise in EQE is a direct consequence of optical properties of BiVO4. It is widely believed that a range of weak valence to conduction band transitions in the near bandgap wavelength range45 is responsible for a slow rise in the absorption coefficient of BiVO4, and consequently the observed trend in EQE.
 | ||
| Fig. 4 (a) External and (b) internal quantum efficiency measurements of 200 nm BiVO4 in PBA buffer, pH 7 and 10, before and after PC collected at 1.23 VRHE bias voltage. Green curve – absorption trend of a BiVO4–FTO sample. | ||
Our EQE results clearly show that PC has a remarkable impact on the quantum efficiency of BiVO4 photoanodes. Untreated BiVO4 demonstrates an average IPCE of 20%, while PC BiVO4 shows average IPCE of 75% and 50% for pH 10 and pH 7 respectively (440 nm > λ > 330 nm range). The EQE results follow a trend similar to the voltammetry measurements, i.e. a high pH favors a high quantum efficiency. Fig. 4(b) presents the internal quantum efficiency (IQE), or absorbed photon to current conversion efficiency (APCE) data obtained by integrating the absorption spectra of the films with the EQE/IPCE. For pH 10, we can calculate the average internal quantum efficiency to be around 95% in the 330 to 440 nm wavelength range. This remarkable result clearly shows that the PC treatment allows our BiVO4 photoanodes to convert almost all of the absorbed photons into extractable photocurrent, suggesting that the PC treatment at pH 10 can eliminate both catalytic and bulk limitations almost entirely. This is an unprecedented result for a non-doped, non-catalyst modified photoelectrode, and represents a benchmark for photo-to-chemical conversion for solar water oxidation.
To further strengthen our claim about near complete suppression of surface losses we also present the catalytic efficiency trends of our BiVO4 photoanodes as a function of potential under different conditions (S9, ESI†). We believe that the observed near unity quantum efficiency is also facilitated by the morphology of our films. Although the investigated photoanodes are 200 nm thick, they're not fully compact and flat. As demonstrated in S10 (ESI†), BiVO4 films fabricated via spray pyrolysis are highly porous and intrinsically nanostructured, such that the distance for the charge carriers to travel is significantly less than 200 nm and close to the diffusion length of photogenerated charge carriers reported for BiVO4 (70 nm). Furthermore, it is also possible that the photocharging treatment positively affects the diffusion length of the charge carriers in BiVO4, without affecting the surface morphology at any pH (S10, ESI†).
Interestingly, the quantum efficiency in the above-bandgap wavelength region remains zero after PC. Therefore we can conclude that the additional portion of light absorbed by PC BiVO4 in the above-bandgap region, demonstrated as the previously described darkening effect, does not contribute to the increased photocurrent. Increased absorption in the above-bandgap range and unchanged EQE onset could suggest incorporation of H into the near surface layer of our films, since similar optical effects have been reported for hydrogenated BiVO4.46 Likewise, the darkening effect could possibly be explained by reduction of vanadium species in BiVO4, similarly to what has been reported by Qin et al.,47 who observed enhanced absorption beyond the bandgap in reduced monoclinic BiVO4.
The most likely way for hydrogen to get incorporated into the BiVO4 lattice is via intercalation into interstitial sites,46 leading to the formation of positively charged 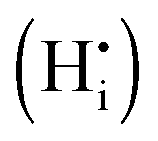 defects. For the sake of conservation of charge, it is possible that the formation of those interstitial hydrogen impurities is accompanied by reduction of vanadium, leading to the formation of negatively charged vanadium species
defects. For the sake of conservation of charge, it is possible that the formation of those interstitial hydrogen impurities is accompanied by reduction of vanadium, leading to the formation of negatively charged vanadium species  . This mechanism is consistent with the XPS study of PC-BiVO4 which we have presented in our initial report,27 and which shows that surface vanadium undergoes reduction from the 5+ to the 4+ oxidation state during the PC treatment. If we now consider self-doping of the V2O5 sublattice with VO2 species, we may expect that oxygen ions will occupy four out of five oxygen sites in the V2O5 sublattice (O×O), leaving the fifth oxygen site empty and leading to the formation of an oxygen vacancy
. This mechanism is consistent with the XPS study of PC-BiVO4 which we have presented in our initial report,27 and which shows that surface vanadium undergoes reduction from the 5+ to the 4+ oxidation state during the PC treatment. If we now consider self-doping of the V2O5 sublattice with VO2 species, we may expect that oxygen ions will occupy four out of five oxygen sites in the V2O5 sublattice (O×O), leaving the fifth oxygen site empty and leading to the formation of an oxygen vacancy 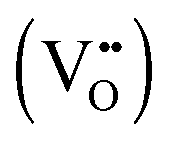 . In this particular case special care needs to be taken in order not to confuse the vanadium and vacancy descriptor (
. In this particular case special care needs to be taken in order not to confuse the vanadium and vacancy descriptor (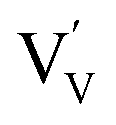 and
and  ). Overall, we can propose the mechanism of light-triggered hydrogenation of the BiVO4 surface layer coupled with the reduction of vanadium surface species; eqn (1) illustrates the possible defect chemistry reaction using the Kröger–Vink notation:
). Overall, we can propose the mechanism of light-triggered hydrogenation of the BiVO4 surface layer coupled with the reduction of vanadium surface species; eqn (1) illustrates the possible defect chemistry reaction using the Kröger–Vink notation:
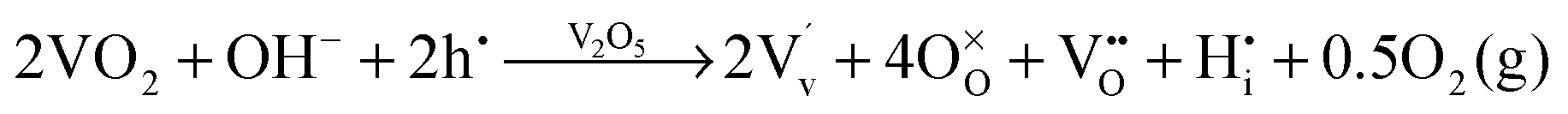 | (1) |
Since our experimental results indicate that both light and alkaline conditions are critical for the PC effect to take place, photogenerated holes (h˙) and hydroxyl groups (OH−) are included as reactants with VO2. The alteration of the surface chemistry via the formation of hydrogen interstitials and oxygen vacancies could lead to increased absorption outside the BiVO4 band gap energy and improve the catalytic properties of our photoanodes, which is in agreement with the results from Wang et al.,46 which show that hydrogen treatment leads to improved photocurrents in BiVO4 photoanodes.
In situ X-ray absorption spectroscopy (XAS)
It is unclear whether the whole bulk of BiVO4 films becomes affected by the photocharging treatment. Therefore, in order to investigate any possible impact of the PC treatment on the chemical and structural properties of bulk BiVO4, specifically to identify any changes in the average bulk oxidation state of vanadium and in the local environment, we performed an in situ XAS study.Fig. 5 presents vanadium K-edge XAS spectra collected in situ as functions of the (a) PC treatment time and (b) applied bias potential. For BiVO4 films subjected to the PC treatment for 8 h, no changes in the XAS spectrum were observed throughout the experiment. Likewise, ex situ conditions and dark OC conditions yield the same spectra (Fig. 5(a)). Similarly, for BiVO4 films poised at potentials of 0.3, 0.7 and 1.1 VRHE there was essentially no shift in the XANES edge energy (Fig. 5(b)). Ultimately, under all the studied experimental conditions, the XAS spectra in the XANES range show a nearly perfect overlap. Therefore, we conclude that neither the photocharging treatment nor applying bias potentials (0.3–1.1 VRHE) alters the structure/chemistry of the bulk of BiVO4 films; specifically, our films do not display any signatures of changes in the average bulk vanadium oxidation state, despite possessing significantly different PEC properties.
 | ||
| Fig. 5 Vanadium K-edge XAS spectra collected (a) ex situ and in situ under OC conditions, in the dark and under illumination; (b) ex situ and under illumination and with applied bias potentials. | ||
Fig. 5 also includes reference spectra of V2O3, VO2 and V2O5. Clearly, the edge position of vanadium oxides shifts to higher energies with increased formal oxidation state (approx. 5470, 5474 and 5476 eV for V3+, V4+ and V5+ respectively). Based on the spectra of vanadium oxides the oxidation state of V in the BiVO4 films was estimated. Notably, the edge position of a XANES spectrum is only an approximate indicator of the oxidation state of metals owing to the several parameters that contribute to the XANES spectra (i.e., charge density, ligand symmetry, and spin density).48,49 However, given significant differences between edge positions of different vanadium oxides and a similar chemical environment of vanadium atoms both in VOx and in BiVO4, the oxidation state of V in our films can be determined quite confidently. The edge position of the BiVO4 films is similar to that of V2O5, indicating a mean oxidation state of +5. Since, as discussed in the previous paragraph, all the BiVO4 XAS spectra are essentially the same, we can conclude that bulk vanadium in our films always preserves the +5 oxidation state.
To verify the local arrangement of vanadium in our films and to monitor any possible changes in the spectrum, XANES calculations have been performed. BiVO4 is known to exist in three polymorphs: orthorhombic pucherite, tetragonal dreyerite, and monoclinic clinobisvanite. However, the only thermodynamically stable phase is clinobisvanite. According to the simulations presented in S11 (ESI†) the main atomic arrangement shown by our films is typical of the crystallographic structure of clinobisvanite. Therefore, we can further confirm that bulk vanadium in our films has a dominant oxidation state of +5.
Impedance spectroscopy (IS)
In an effort to find the correlation between the photocharging treatment and charge transfer behavior, we performed cyclic voltammetry (CV) on the untreated (UT) and photocharged (PC) BiVO4, as shown in Fig. 6. In the presence of trap states (i.e., surface states), one or more reduction peaks are usually present in the cathodic voltammetry scan, which can be attributed to charging or filling of electrons from the conduction band to the surface state as the electron Fermi level shifts upward during the cathodic sweep.50–52 In the dark voltammetry, thus in the absence of photogenerated holes, such trap-related reduction peaks are irreversible. The irreversibility of these capacitive cathodic peaks has been related to the equilibration between the trap states and conduction band.52 In order to unambiguously identify any trap-related peaks, the CV is typically performed in the dark, to avoid masking the possible trap capacitive effects by the photocurrent. The CV scan of the UT sample shown in Fig. 6(a) depicts both anodic and cathodic peaks, which cannot be assigned to deep surface traps (which are irreversible). The reversible peaks, however, can be ascribed to the reduction and re-oxidation of the redox system of V5+ to V4+. It is noticeable that these redox peaks are barely visible in the dark and significantly more pronounced under illumination (S12, ESI†), since the photogenerated charge also contributes to the oxidation and reduction of these redox states. Moreover, slightly higher capacitance can be derived from the hysteresis of the PC cyclic voltammetry curve in Fig. 6(a). | ||
| Fig. 6 Cyclic voltammetry of untreated (UT) and photocharged (PC) BiVO4 (a) in the dark and (b) under illumination. | ||
Consequently, we carried out impedance spectroscopy (IS) measurements to characterize the carrier dynamics in the bulk and on the surface of the untreated (UT) and photocharged (PC) BiVO4 in the dark and under illumination using physical models (i.e., equivalent circuit, EC) that have been used extensively for n-type metal oxide semiconductors when surface states are present (see S13, ESI†).53–55Fig. 7(a) shows the bulk capacitance of UT and PC BiVO4 in the dark and under illumination as a function of applied potentials obtained from the IS measurements. This capacitance shows a peak at ca. 0.7 VRHE which increases both under illumination and after the PC treatment. This behavior is consistent with the cyclic voltammetry study demonstrated in Fig. 6 and Fig. S12 (ESI†), and can be ascribed to the redox process of V4+/V5+. However, we found that the photocharging treatment involving reduction of V5+ to V4+ does not affect the bulk electronic properties, as indicated by the constant bulk resistance for the untreated and the photocharged samples, shown in Fig. 7(b). This likely suggests that the onset potential shift and increase of the photocurrent observed in our photocharged samples are not related to the bulk conductivity of the material.
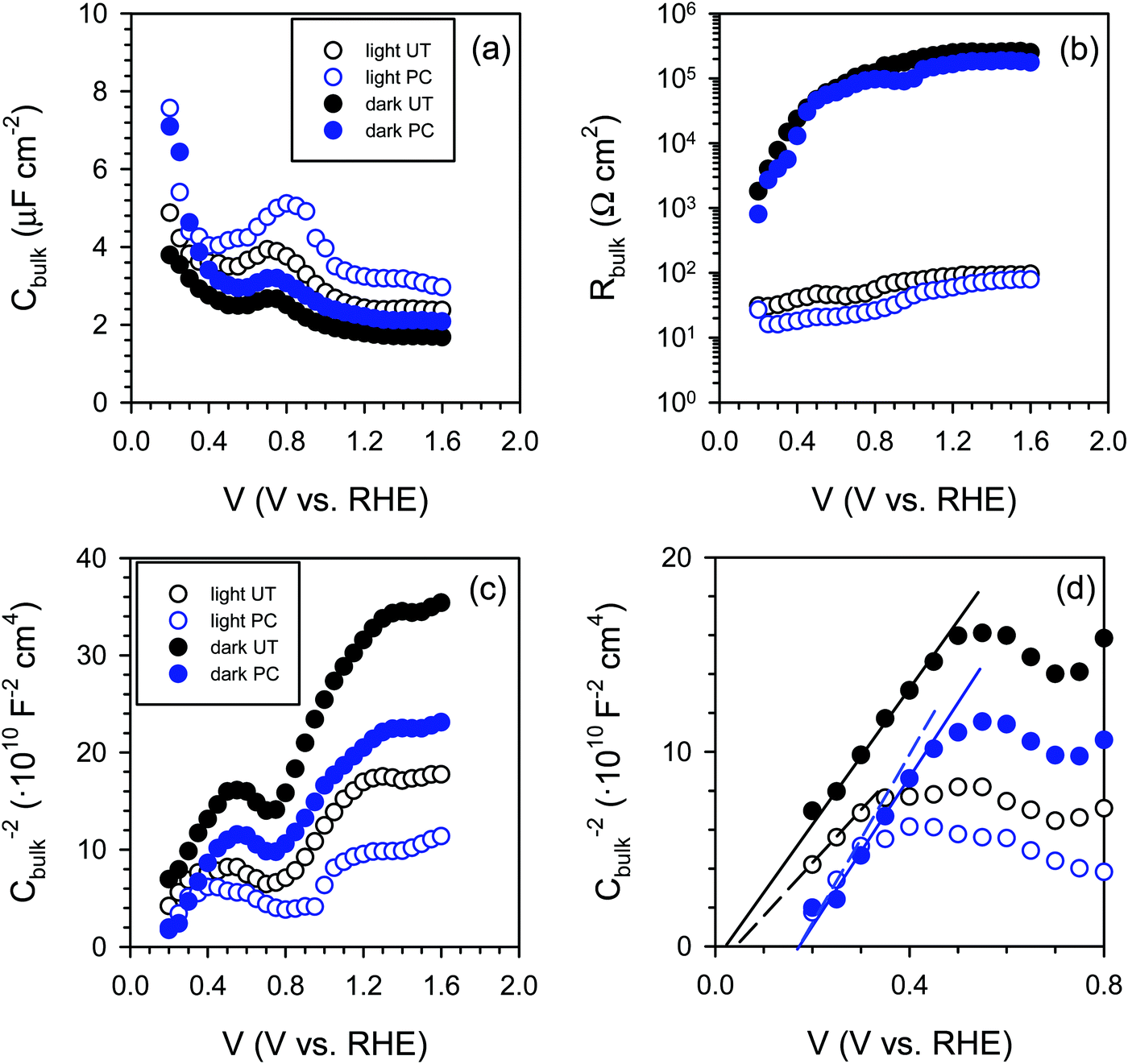 | ||
| Fig. 7 (a) Bulk capacitance (Cbulk) and (b) bulk resistance (Rbulk) as a function of potential, (c and d) Mott–Schottky plots obtained in the dark and under illumination under different PC conditions measured at pH 10. | ||
Further on, we looked for signatures of any changes in the band structure of BiVO4 occurring during the photocharging process. Fig. 7(c) presents the bulk capacitance versus applied voltage, plotted as a Mott–Schottky dependence. The Mott–Schottky plot allows for estimation of the flat band potential (Vfb) of our samples. Due to a significant overlap of the redox capacitance (the peak at 0.7 VRHE) with the space charge capacitance of BiVO4, the potential window available for the linear Mott–Schottky dependence is very narrow. However, the extraction of the space charge capacitance is still possible, as shown in Fig. 7(d). We noticed that the flat band potential shifts anodically upon photocharging treatment, which is consistent with hole accumulation at the capacitive layer. In any case, the positive shift does not contribute to the cathodic shift of the photocurrent onset potential of the photocharged BiVO4. This cathodic shift of the photocurrent is related to reduced surface recombination, improving the performance of the photoanodes.
The IS response of illuminated BiVO4 photoanodes also showed some distinctive feature in the low frequency regime. Such a feature can be usually attributed to a surface signature, such as charging/discharging of the surface states54 and/or catalytic layers.55,56 In order to properly address this phenomenon, the evolution of both surface capacitance (Cs) and charge transfer resistance (Rct) with potential was studied, as depicted in Fig. 8. From Fig. 8(a) we note that the values of the surface capacitance increase on the photocharged sample, suggesting that surface modification indeed took place during the photocharging treatment. The surface capacitance of PC samples does not demonstrate the typical Gaussian distribution that is generally obtained for a system containing localized surface states.54,57 In fact, it remains almost constant in a wide potential window (ca. 1 V), which is similar to that reported in water splitting systems when a catalytic layer is deposited on the surface of a photoanode.55,56 Hence, we attribute this capacitance to the formation of a surface layer formed by the photocharging process due to the adsorption of oxy/hydroxyl species as previously discussed, rather than a localized surface trap. This hypothesis is further supported by the evolution of charge transfer resistance shown in Fig. 8(b). The resistance does not significantly evolve over a large potential window and its minima do not correspond to the maxima of the surface capacitance as expected for a surface trap. The surface capacitive layer pins the electron Fermi level which leads to the anodic shift of the flatband potential as observed from Fig. 7(c) for the photocharged samples.
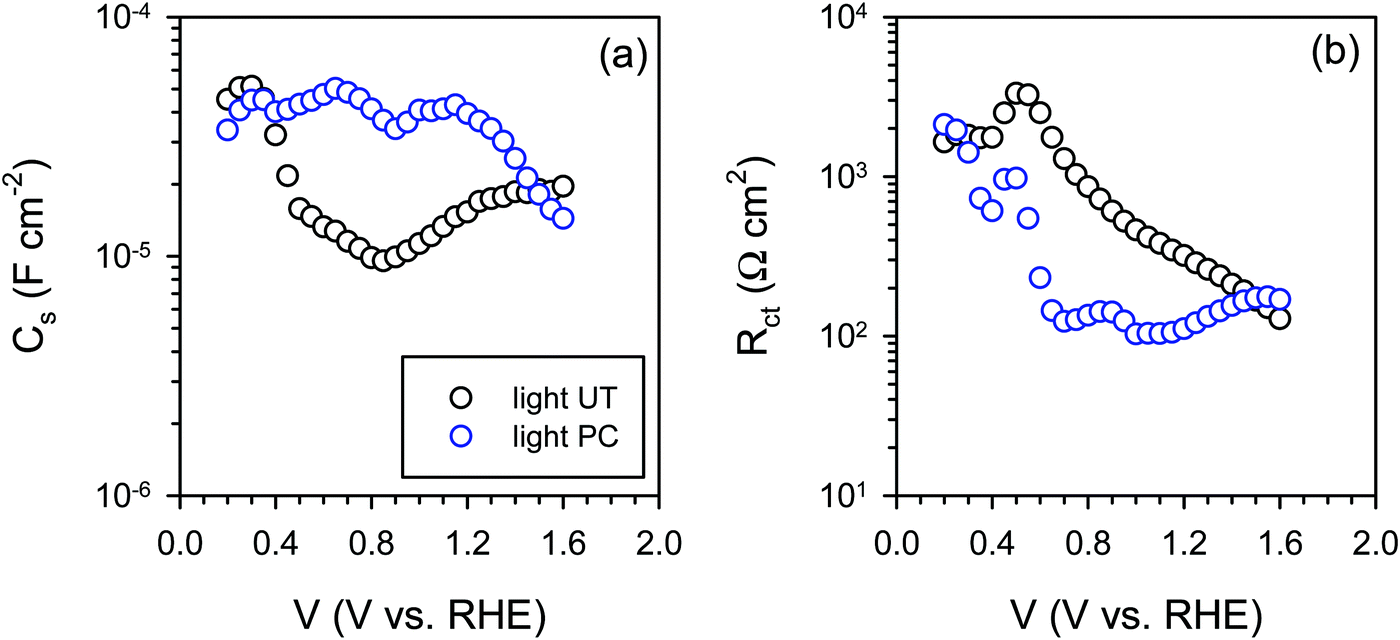 | ||
| Fig. 8 (a) Surface capacitance (Cs) and (b) charge transfer resistance (Rct) of the untreated (UT) and the photocharged (PC) BiVO4 under illumination at pH 10. | ||
This has been illustrated in the cartoon of Fig. 9, highlighting the proposed mechanisms involved in the photocharging process: intercalation of hydrogen and reduction of surface vanadium favoured under alkaline conditions and formation of the capacitive layer. In the mechanistic cartoon of Fig. 9, the reduced band bending in the photocharged material is due to the shift of the Fermi level towards the conduction band of BiVO4, as a result of the photocharging effect. The shift of the flatband potential to positive potentials is consistent with the accumulation of holes at the interface. The diagram also depicts the increase in the photovoltage of PC BiVO4 (Vph2) vs. the photovoltage of the untreated material.
 | ||
| Fig. 9 Band diagrams of BiVO4 in the dark and under illumination before and after the photocharging treatment, including the simplified photocharging mechanism model. The green star indicates the energetic level related to the surface capacitive layer, causing the Fermi level pinning. | ||
Though the flatband potential moves to more anodic potentials, the capacitive layer acts as a ‘hole reservoir’ and minimises recombination, leading to an onset potential of about 0.4 V vs. VRHE, as observed from Fig. 6(b). From this study it can be concluded that the photocharging treatment accelerates the creation of the OER intermediates on the BiVO4 surface which act as a hole reservoir, in the same manner as a catalytic layer. The origin of this early formation of reaction promoters can be connected to the V5+ reduction process under favourable pH electrolyte conditions. The mechanism of the photocatalytic effect by the existence of V4+ has been largely described on V multiple valence materials such as V2O5 doped TiO2, CaV2O6 and BiVO4 nanorods.58–60 Furthermore, it has been demonstrated on BiVO4 nanorods that the presence of V4+ defects induce oxygen vacancies (VO) on the surface of nanoparticles.60 VO can strongly adsorb O2− and OH− species on the surface, which leads to an enhanced photocatalytic effect.
To further elucidate the capacitive behavior of our photoanodes, a chopped light chronoamperometry experiment was performed. A similar study55 has demonstrated that Co–Pi deposited onto α-Fe2O3 can efficiently collect and store photogenerated holes from the hematite electrodes. Co–Pi layers of different thicknesses have different charge storage capacities and therefore demonstrate different decay times in chopped light transients. An analogous study performed on Fh/Ta3N561 describes a “surface charging” process, demonstrated by an increase in the photocurrent. This effect is attributed to accumulation of surface trapped holes, resulting in the enhancement of photocurrent. S14 and S15 (ESI†) present respectively the anodic and cathodic transients of the untreated and photocharged BiVO4 photoanodes. The fast decay observed for the untreated films (S14(a), ESI†) could suggest55 hole accumulation/trapping, charging of the surface states or oxidation of some surface species. On the other hand, photocharged films demonstrate a build-up of photocurrent (S14(b), ESI†), which suggests61 a capacitance-related effect such as surface charging. Cathodic transients show much faster decay in the case of untreated BiVO4 (S15(a), ESI†), when compared with the photocharged material (S15(b), ESI†). It could be indicative of the higher charge storage capacity of PC-BiVO4, in agreement with our EIS results. Overall, photocharged BiVO4 seems to demonstrate some traits typical of catalyzed photoanodes.
A more in depth analysis of the anodic transients is troublesome since within the potential range used in our study the capacitive and faradaic currents are convoluted, therefore it is difficult to extract the integrated capacitive charge. However, the total charge stored in the case of the cathodic transients can be calculated using eqn (2):
 | (2) |
 | (3) |
Conclusions
BiVO4 photoanodes immersed in an electrolyte in the OC configuration and exposed to simulated solar illumination for prolonged time achieve superior PEC activity. This photocharging effect is capable of dramatically improving both bulk and surface functionalities of BiVO4. New insights into the mechanisms of PC in BiVO4 photoanodes and how it leads to improved PEC performance are necessary for its application in real devices.There seems to exist a strong correlation between the pH of the electrolyte and PEC properties of PC BiVO4: alkaline conditions facilitate the PC effect while acidic conditions prevent it from happening. Specifically PC performed under alkaline conditions at pH 10 yields high photocurrents, low onsets and high fill-factors, as well as very high quantum efficiencies (near 100% internal quantum efficiency), suggesting that both bulk and surface limitations of BiVO4 can be overcome with PC alone. The strong pH dependence of the PC effect suggests that the SLJ plays a key role in the PC mechanism.
The IS study reveals that the photocharging effect is connected with the development of a capacitive layer, which has the ability to accumulate holes, and hence leads to reduced surface recombination. In addition, optical properties of BiVO4 subjected to the PC treatment change, namely, PC BiVO4 starts to absorb light beyond the bandgap range. However, this apparent increase in absorption does not lead to the formation of charge carriers useful for water oxidation but it is rather a signature of the transformation of the material into a more PEC active state. This darkening effect could be potentially related to the intercalation of hydrogen in conjunction with the reduction of vanadium in the surface layer of the films. Importantly, in situ XAS study quite convincingly rules out the possibility that the PC treatment leads to reduction of vanadium from the 5+ to the 4+ state within the whole bulk of the films.
Overall, our results suggest that PC is related to light-driven alteration of the surface of BiVO4, preferentially under alkaline conditions. Those changes result in properties becoming more beneficial for solar driven water splitting applications. Furthermore, any bulk-related effects are mediated by phenomena occurring at the surface, which highlights the importance of the SLJ for the efficiency of PEC devices. From a technological perspective, the advantages of the PC effect can be fully exploited under continuous operation of a BiVO4-based photoelectrochemical device, and consequently, redesign of the operation protocols of these devices is imperative to successfully implement this PC effect.
Experimental
Preparation of BiVO4 thin film photoanodes
Thin films of 50, 100, 200 and 300 nm thick BiVO4 were prepared by spray pyrolysis on FTO coated glass substrates. Details of the deposition procedure are described elsewhere13,16 and are also available in the ESI.† Prior to deposition of BiVO4, a SnO2 interfacial layer (∼80 nm) was deposited onto the FTO substrate at 425 °C to prevent recombination at the FTO/BiVO4 interface.62 The substrate temperature during spraying of BiVO4 was maintained at 450 °C. After deposition, the samples were further annealed for 2 h in a tube furnace at 450 °C in air.Photoelectrochemical (PEC) measurements
A 0.1 M phosphate buffer (K-Pi) and a 0.1 M phosphate-borate-acetate (PBA) buffer were used as the electrolyte. K-Pi was prepared by dissolving a mixture of KH2PO4 (Sigma Aldrich, 99.0%) and K2HPO4 (Sigma Aldrich, 99.0%) in Milli-Q water (18.2 MΩ cm), to obtain pH 7.2 and to preserve 0.1 M overall concentration of PO43−. PBA buffer was prepared by dissolving KH2PO4 (Sigma Aldrich, 99.0%), H3BO3 (Sigma Aldrich, 99.5%) and acetic acid (Sigma Aldrich, 99.7%) in Milli-Q water (18.2 MΩ cm). The concentration of each component was set to 0.1 M. The pH was adjusted to a desired value by titrating the PBA solution with KOH (Sigma Aldrich, 99%), and with a pH meter immersed. Throughout this work the PBA buffer, pH 4, 7 and 10, was used.PEC characterization of BiVO4 photoanodes was carried out in an electrochemical cell using a three-electrode configuration. The potential of the working electrode was controlled by a multi-channel potentiostat (Parstat MC, Princeton Applied Research). An Ag/AgCl electrode (XR300, saturated KCl + AgCl solution (KS120), Radiometer Analytical) and a coiled Pt wire were used as the reference and counter electrodes, respectively. Measurements under illumination were performed with a Newport Sol3A Class AAA solar simulator (type 94023A-SR3) producing simulated AM1.5 solar illumination (100 mW cm−2). In all the experiments involving illumination, BiVO4 samples were illuminated from the back-side, i.e. the light came through the substrate side first. Cyclic voltammetry scans were taken at a scan rate of 50 mV s−1 unless stated otherwise.
The photocharging of BiVO4 photoanodes was performed under OC conditions under illumination. BiVO4 samples were placed in the electrochemical cell and exposed to AM1.5 light until the plateau of VWE(t) was reached (10 hours on average).
In situ UV-vis spectroscopy
The spectral data were collected using an OceanOpics USB2000+ spectroradiometer and the SpectraSuite software. The spectroradiometer was placed behind the cell to measure the intensity of light transmitted through a BiVO4 sample.Quantum efficiency
Monochromatic photocurrents were measured with a 300 W Xe lamp (Oriel LSE340 interface, LSH302 lamp housing, LSN252 power supply, LSB530 lightbulb) coupled into a grating monochromator (ActonSpectraPro 150i). An electronic shutter (Uniblitz LS6) was used, and a long-pass colored glass filter (Schott, 3 mm thick) was placed between the monochromator and the sample to remove second-order diffracted light. The shutter was actuated every 10 s, and the photocurrent was taken as the difference between the current when the shutter is opened and closed (3 s integration time and ∼1.5 nm step size). The light intensity was measured with a calibrated photodiode (Ophir PD300-UV) and ranged between 0.3 and 8 mW cm−2. The incident photon to current efficiency (IPCE, sometimes called the external quantum efficiency or EQE) was calculated based on formula (2):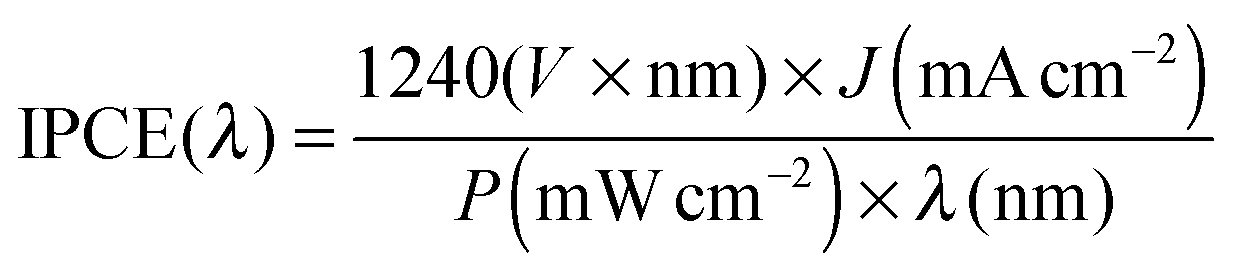 | (4) |
X-ray absorption spectroscopy (XAS): X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS)
V K-edge (5465 eV) XAFS spectra of the thin films of BiVO4 were collected at the Dutch-Belgian Beam Line (DUBBLE) at the European Synchrotron Radiation Facility (ESRF).63 The energy of the X-ray beam was tuned by a double-crystal monochromator operating in fixed-exit mode using a Si(111) crystal pair. All the measurements were performed at ambient temperature and pressure. Spectra of vanadium foil needed for energy calibration and reference spectra of vanadium oxides (V2O3, VO2 and V2O5) were collected in transmission mode using Ar/He-filled ionization chambers. EXAFS spectra of the thin films of BiVO4 were collected in fluorescence mode using a 9-element Ge detector (Ortec Inc.).The XAS investigation of BiVO4 was performed using a cell specially designed for in situ PEC measurements. For the sake of probing the vanadium K-edge, characterized with a relatively low energy of 5465 eV, the electrolyte layer thickness was maintained at ∼10 μm. The cell was equipped with a standard 3-electrode configuration, controlled with a potentiostat and continuously flushed with the electrolyte (0.1 M K-Pi). BiVO4 samples were illuminated from the back-side using a Xe lamp equipped with an optical fiber.
The XANES spectra, three scans per sample, were energy-calibrated, averaged and further analysed using the FDMNES package program using multiple scattering theory based on the muffin-tin approximation on the potential shape.64,65 The muffin-tin radii were tuned to have a 10% overlap between the different spherical potentials. The Hedin–Lundqvist exchange potential was used. The XANES spectrum of a BiVO4 crystal was calculated, considering in the calculation all the atoms surrounding the absorber V within a 7 Å radius sphere. The approximation of non-excited absorbing atoms was used, which better reproduces the experimental data. A similar calculation with the FEFF8 code66 gave similar results. We have not considered the effect of the structural disorder in the XANES simulations. To simulate the XANES spectra, a core-hole broadening of 1.0 eV was used.
Impedance spectroscopy (IS)
IS measurements were carried out in the same set up configuration as the PEC measurements by applying a 20 mV AC signal and scanning in a frequency range between 1 MHz and 50 mHz, at different applied bias. The numerical fitting of the impedance data was carried out using the Zview software (Scribner Associates).Scanning electron microscopy (SEM)
The morphology of thin films was investigated using SEM microscopy (JEOL JSM-6010LA instrument equipped with a tungsten hairpin filament). Samples were investigated in the secondary electron imaging mode (SEI), using an Everhart–Thornley type detector. SEM images were collected using an accelerating voltage of 20 kV, at a working distance of 10 mm.X-ray photoelectron spectroscopy (XPS)
XPS experiment was performed using the Thermo Scientific K-alpha apparatus equipped with an Al K-alpha X-ray Source and a Flood Gun. Parameters used for the measurements were: spot size of 400 mm, pass energy of 50 eV, energy step size of 0.1 eV, dwell time of 50 ms, 10 scans in the vicinity of the orbital binding energy of the elements of interest. XPS spectra were corrected for the C peak position.Acknowledgements
We acknowledge the financial support of the Foundation for Fundamental Research on Matter (FOM TNW 10.327). This work has been done under the agenda of the BioSolarCells Consortium supported by the Dutch Ministry of Economic Affairs, Agriculture and Innovation. We acknowledge The Netherlands Organisation for Scientific Research (NWO) for financial support of the measurements at ESRF. We also thank financial support from the University Jaume I through the project P1·1B2011-50.References
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- Photoelectrochemical Hydrogen Production, ed. R. van de Krol and M. Grätzel, Springer, US, Boston, MA, 2012, vol. 102 Search PubMed.
- R. van de Krol, Y. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311 RSC.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- R. Solarska, A. Królikowska and J. Augustyński, Angew. Chem., Int. Ed., 2010, 49, 7980–7983 CrossRef CAS PubMed.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- L. Han, F. F. Abdi, R. van de Krol, R. Liu, Z. Huang, H.-J. Lewerenz, B. Dam, M. Zeman and A. H. M. Smets, ChemSusChem, 2014, 7, 2832–2838 CrossRef CAS PubMed.
- J. W. Ager III, M. Shaner, K. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 Search PubMed.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- F. F. Abdi, N. Firet and R. van de Krol, ChemCatChem, 2013, 5, 490–496 CrossRef CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- T. H. Jeon, W. Choi and H. Park, Phys. Chem. Chem. Phys., 2011, 13, 21392–21401 RSC.
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CAS.
- C. Ding, J. Shi, D. Wang, Z. Wang, N. Wang, G. Liu, F. Xiong and C. Li, Phys. Chem. Chem. Phys., 2013, 15, 4589–4595 RSC.
- S. K. Choi, W. Choi and H. Park, Phys. Chem. Chem. Phys., 2013, 15, 6499–6507 RSC.
- T. W. Kim and K. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- M. Haro, C. Solis, V. M. Blas-Ferrando, O. Margeat, S. Ben Dhkil, C. Videlot-Ackermann, J. Ackermann, F. Di Fonzo, A. Guerrero and S. Gimenez, ChemSusChem, 2016, 9, 3062–3066 CrossRef CAS PubMed.
- K. P. S. Parmar, H. J. Kang, A. Bist, P. Dua, J. S. Jang and J. S. Lee, ChemSusChem, 2012, 5, 1926–1934 CrossRef CAS PubMed.
- P. M. Rao, L. Cai, C. Liu, I. S. Cho, C. H. Lee, J. M. Weisse, P. Yang and X. Zheng, Nano Lett., 2014, 14, 1099–1105 CrossRef CAS PubMed.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. van de Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS.
- D. Eisenberg, H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 14011–14014 CrossRef CAS PubMed.
- M. T. McDowell, M. F. Lichterman, J. M. Spurgeon, S. Hu, I. D. Sharp, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2014, 118, 19618–19624 CAS.
- B. J. Trześniewski and W. A. Smith, J. Mater. Chem. A, 2016, 4, 2919–2926 Search PubMed.
- M. Gostein and L. Dunn, in 2011 37th IEEE Photovoltaic Specialists Conference, IEEE, 2011, pp. 003126–003131.
- A. Listorti, C. Creager, P. Sommeling, J. Kroon, E. Palomares, A. Fornelli, B. Breen, P. R. F. Barnes, J. R. Durrant, C. Law and B. O'Regan, Energy Environ. Sci., 2011, 4, 3494 CAS.
- F. J. Lim, Y. T. Set, A. Krishnamoorthy, J. Ouyang, J. Luther and G. W. Ho, J. Mater. Chem. A, 2015, 3, 314–322 CAS.
- C. Liu, J. Fan, X. Zhang, Y. Shen, L. Yang and Y. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 9066–9071 CAS.
- F. Lin and S. W. Boettcher, Nat. Mater., 2014, 13, 81–86 CrossRef CAS PubMed.
- T. J. Mills, F. Lin and S. W. Boettcher, Phys. Rev. Lett., 2014, 112, 148304 CrossRef PubMed.
- F. Lin, B. F. Bachman and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 2427–2433 CrossRef CAS PubMed.
- C. Du, X. Yang, M. T. Mayer, H. Hoyt, J. Xie, G. McMahon, G. Bischoping and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 12692–12695 CrossRef CAS PubMed.
- C. Du, M. Zhang, J. Jang, Y. Liu, G. Liu and D. Wang, J. Phys. Chem. C, 2014, 118, 17054–17059 CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- X. Shi, I. Herraiz-Cardona, L. Bertoluzzi, P. Lopez-Varo, J. Bisquert, J. H. Park and S. Gimenez, Phys. Chem. Chem. Phys., 2016, 18, 9255–9261 RSC.
- J. Reichman, Appl. Phys. Lett., 1980, 36, 574–577 CrossRef CAS.
- F. F. Abdi, N. Firet, A. Dabirian and R. van de Krol, MRS Proc., 2012, 1446, mrss12-1446-u02-05 Search PubMed.
- X. Yang, C. Du, R. Liu, J. Xie and D. Wang, J. Catal., 2013, 304, 86–91 CrossRef CAS.
- K. Sivula, J. Phys. Chem. Lett., 2013, 4, 1624–1633 CrossRef CAS PubMed.
- T. Li, J. He, B. Peña and C. P. Berlinguette, Angew. Chem., Int. Ed., 2016, 55, 1769–1772 CrossRef CAS PubMed.
- M. Mullet, V. Khare and C. Ruby, Surf. Interface Anal., 2008, 40, 323–328 CrossRef CAS.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, P.-A. Glans, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, Chem. Mater., 2014, 26, 5365–5373 CrossRef CAS.
- G. Wang, Y. Ling, X. Lu, F. Qian, Y. Tong, J. Z. Zhang, V. Lordi, C. Rocha Leao and Y. Li, J. Phys. Chem. C, 2013, 117, 10957–10964 CAS.
- D.-D. Qin, T. Wang, Y.-M. Song and C.-L. Tao, Dalton Trans., 2014, 43, 7691–7694 RSC.
- A. Bianconi, X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, Wiley, New York, 1988 Search PubMed.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- B. Klahr and T. Hamann, J. Phys. Chem. C, 2014, 118, 10393–10399 CAS.
- O. Zandi and T. W. Hamann, J. Phys. Chem. Lett., 2014, 5, 1522–1526 CrossRef CAS PubMed.
- L. Bertoluzzi, L. Badia-Bou, F. Fabregat-Santiago, S. Gimenez and J. Bisquert, J. Phys. Chem. Lett., 2013, 4, 1334–1339 CrossRef CAS PubMed.
- L. Bertoluzzi and J. Bisquert, J. Phys. Chem. Lett., 2012, 3, 2517–2522 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS PubMed.
- L. Badia-Bou, E. Mas-Marza, P. Rodenas, E. M. Barea, F. Fabregat-Santiago, S. Gimenez, E. Peris and J. Bisquert, J. Phys. Chem. C, 2013, 117, 3826–3833 CAS.
- S. Gimenez, H. K. Dunn, P. Rodenas, F. Fabregat-Santiago, S. G. Miralles, E. M. Barea, R. Trevisan, A. Guerrero and J. Bisquert, J. Electroanal. Chem., 2012, 668, 119–125 CrossRef CAS.
- Z. Zhang, C. Shao, L. Zhang, X. Li and Y. Liu, J. Colloid Interface Sci., 2010, 351, 57–62 CrossRef CAS PubMed.
- R. Yu, N. Xue, S. Huo, J. Li and J. Wang, RSC Adv., 2015, 5, 63502–63512 RSC.
- Y. Zhang, Y. Guo, H. Duan, H. Li, C. Sun and H. Liu, Phys. Chem. Chem. Phys., 2014, 16, 24519–24526 RSC.
- G. Liu, J. Shi, F. Zhang, Z. Chen, J. Han, C. Ding, S. Chen, Z. Wang, H. Han and C. Li, Angew. Chem., Int. Ed., 2014, 53, 7295–7299 CrossRef CAS PubMed.
- Y. Liang, T. Tsubota, L. P. A. Mooij and R. van de Krol, J. Phys. Chem. C, 2011, 115, 17594–17598 CAS.
- S. Nikitenko, A. M. Beale, A. M. J. van der Eerden, S. D. M. Jacques, O. Leynaud, M. G. O’Brien, D. Detollenaere, R. Kaptein, B. M. Weckhuysen and W. Bras, J. Synchrotron Radiat., 2008, 15, 632–640 CrossRef CAS PubMed.
- O. Bunău and Y. Joly, J. Phys.: Condens. Matter, 2009, 21, 345501 CrossRef PubMed.
- Y. Joly, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 125120 CrossRef.
- A. Ankudinov, B. Ravel, J. Rehr and M. Newville, FEFFFIT manual within the FEFF project, University of Washington, Seattle, USA, 1992-1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee03677e |
| This journal is © The Royal Society of Chemistry 2017 |