
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-polymerized dopamine as an organic cathode for Li- and Na-ion batteries†
Tianyuan
Liu‡
a,
Ki Chul
Kim‡
b,
Byeongyong
Lee
a,
Zhongming
Chen
c,
Suguru
Noda
c,
Seung Soon
Jang
*b and
Seung Woo
Lee
 *a
*a
aG. W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332-0405, USA. E-mail: seung.lee@me.gatech.edu
bComputational NanoBio Technology Laboratory, School of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332-0245, USA. E-mail: seungsoon.jang@mse.gatech.edu
cDepartment of Applied Chemistry, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 18th November 2016
Abstract
Self-polymerized dopamine is a versatile coating material that has various oxygen and nitrogen functional groups. Here, we demonstrate the redox-active properties of self-polymerized dopamine on the surface of few-walled carbon nanotubes (FWNTs), which can be used as organic cathode materials for both Li- and Na-ion batteries. We reveal the multiple redox reactions between self-polymerized dopamine and electrolyte ions in the high voltage region from 2.5 to 4.1 V vs. Li using both density functional theory (DFT) calculations and electrochemical measurements. Free-standing and flexible hybrid electrodes are assembled using a vacuum filtration method, which have a 3D porous network structure consisting of polydopamine coated FWNTs. The hybrid electrodes exhibit gravimetric capacities of ∼133 mA h g−1 in Li-cells and ∼109 mA h g−1 in Na-cells utilizing double layer capacitance from FWNTs and multiple redox-reactions from polydopamine. The polydopamine itself within the hybrid film can store high gravimetric capacities of ∼235 mA h g−1 in Li-cells and ∼213 mA h g−1 in Na-cells. In addition, the hybrid electrodes show a high rate-performance and excellent cycling stability, suggesting that self-polymerized dopamine is a promising cathode material for organic rechargeable batteries.
Broader contextLarge-scale electrical energy storage applications demand the development of cost-effective electrode materials. Organic electrode materials have attracted considerable attention owing to their low cost, high theoretical capacity, and chemical diversities. However, the development of organic electrodes has been hindered by their high solubility in nonaqueous electrolytes and their poor electrical conductivity. Here, we study the electrochemical properties of self-polymerized dopamine using both DFT computation and experimental methods, proposing polydopamine as a promising organic cathode material for rechargeable Li- and Na-ion batteries. |
Introduction
Organic electrode materials have been intensively investigated to replace conventional transitional metal based inorganic cathodes for rechargeable batteries.1,2 This is because of their potential advantages, including high theoretical capacity, environmental friendliness, and use of low cost and earth-abundant resources, which are ideal for large-scale electrical energy storage applications, such as grid load-leveling, renewable energy storage, and electric vehicles.2,3 The past few decades have witnessed the development of various organic electrode materials, such as conductive polymers,4,5 organosulfur compounds,6,7 organic free-radical compounds,8,9 and organic carbonyl compounds.10–16 Despite their high theoretical capacities, the development of organic cathodes has been hindered by two factors, which significantly limit their practical applications. First, most of the organic molecules are highly soluble in organic electrolytes, which results in low cycling stability. The second shortcoming is their poor electrical conductivity limiting the rate-performance of the electrodes.A promising strategy to overcome these issues is to polymerize redox-active molecules onto conductive carbon substrates.17–19 In this nanocomposite structure, the linkage of molecules via a covalent bond prevents the dissolution of molecules, while a conductive carbon network supports fast redox reactions in the polymer. Utilizing this strategy, Song et al. synthesized poly(anthraquinonyl sulfide) and polyimide on graphene flakes, demonstrating a significantly enhanced rate performance of the composite electrodes as organic cathodes in Li-cells.17 However, most of the reported polymerization processes of carbonyl compounds are complicated and usually introduce inactive linkers.12,17 In addition, the electrochemical performance of the composite electrodes significantly depends on the morphology of the polymer coatings on the substrate.18 This indicates that there is an optimized interface structure between the polymer coating and the conductive substrate, which can maximize the electrochemical performance of the composite electrodes. Thus, identifying a simple and versatile polymerization process that enables the strong adhesion of redox-active carbonyl compounds on the surface of conductive substrates is highly desirable.
With a similar molecular structure to adhesive proteins in mussel, dopamine can be oxidized and self-polymerized into polydopamine in alkaline aqueous solutions, generating adhesive polymer coatings on various types of substrates.20–24 After a series of cyclisation and oxidation reactions in an alkaline environment, it is proposed that dopamine can be oxidized into 5,6-indolequinone before polymerization (Scheme S1, ESI†).25 We noticed that 5,6-indolequinone has a similar molecular structure to benzoquinone and naphthoquinone, which have two redox-active quinone groups that can store Li- or Na-ions.26–28 Thus, if the polymerized form of 5,6-indolequinone or other dopamine intermediates shows redox-active properties with Li- or Na-ions, this polydopamine can be a promising carbonyl-based cathode for rechargeable batteries (Scheme 1). However, the redox properties of polydopamine with Li- or Na-ions remain unclear. Here, we investigate the electrochemical properties of polydopamine using both computational and experimental methods. Density functional theory (DFT) calculations show the multiple redox reactions of polydopamine with electrolyte ions in the high voltage region of 2.5–4.1 V vs. Li, which is also confirmed by cyclic voltammetry measurements. To maximize its electrochemical performance, we fabricated flexible, free-standing hybrid films via a spontaneous coating process of polydopamine on few-walled carbon nanotubes (FWNTs) and a continuous vacuum-filtration method. Hybrid electrodes consisting of polydopamine-coated FWNTs exhibit enhanced rate-performance and excellent cycling stability owing to the unique combination of the redox-active polydopamine and superior electrical conductive FWNTs.
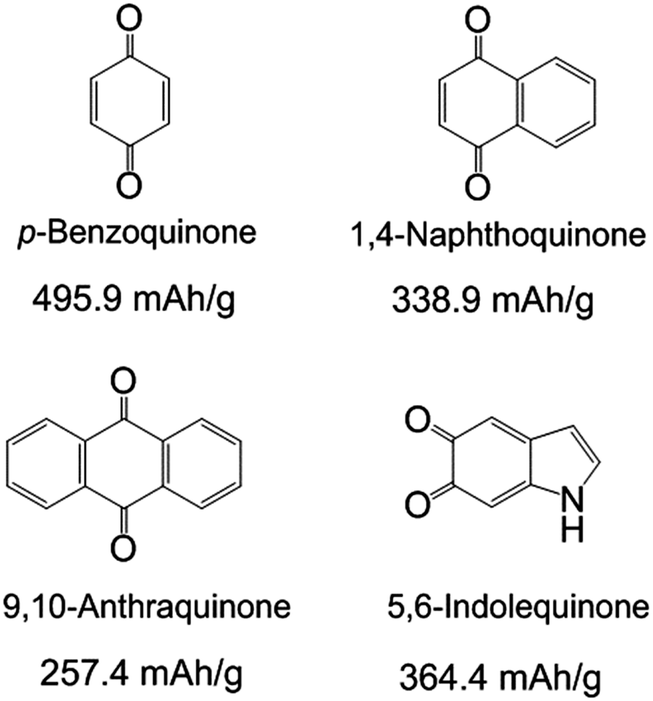 | ||
| Scheme 1 Molecular structures and the corresponding theoretical capacity of p-benzoquinone, 1,4-naphthoquinone, 9,10-anthraquinone and 5,6-indolequinone. The theoretical capacities are calculated based on the redox reactions of carbonyl groups in the molecules. | ||
Results and discussion
In this study, the first principle DFT method was utilized with PBE029,30 and the standard 6-31+G(d,p) basis set31 using Jaguar.32 We investigated the redox-potentials of monomers, and the corresponding oligomers such as dimers and trimers based on the two most reported structural units in polydopamine,33–35 namely, 5,6-indolequinone and 5,6-dihydroxyindole in Scheme S2 (ESI†). The calculated redox potentials are shown in Fig. 1a. 5,6-indolequinone with carbonyl groups showed a high redox potential of 2.83 V vs. Li, while 5,6-dihydroxyindole including hydroxyl groups showed a very low redox potential of −0.26 V vs. Li (Table S1, ESI†). This indicates that only 5,6-indolequinone with a high redox potential can be utilized as the cathode material for rechargeable batteries. The high redox potential of 5,6-indolequinone is consistent with our previous findings of redox-active carbonyl groups on reduced graphene oxides36 and quinone derivatives.26 It is also interesting to note that the redox potentials for both structural units are not significantly dependent on the degree of polymerization: the oligomers containing quinone showed a slightly increased redox-potential to 2.93–2.94 V vs. Li, whereas those of oligomers containing hydroxyl groups increased to 0.11–0.27 V vs. Li. Previous studies utilized the lowest unoccupied molecular orbital (LUMO) energy levels to explain the different redox behaviors of various organic molecules.11,26,36 We find that the redox potentials calculated in this study also show linear correlations not only with the LUMO energy levels but also with the electron affinity (EA) and the gap between the highest occupied molecular orbital (HOMO) and the LUMO (HOMO–LUMO) (Fig. 1b–d). This infers that redox-active molecules with more negative EA and LUMO energy levels would have higher redox potentials due to the preferentially reductive nature of the molecules. In addition, we considered the existence of secondary amine (R–NH–R) in the molecules, which can contribute additional charge capacity to polydopamine through the p-type doping/de-doping process with anions (Scheme S3, ESI†). To confirm this contribution, we computed the redox potentials of the molecules binding with the anion (PF6−). The DFT calculation showed that the doping/de-doping process of the anions (PF6−) occurred at 4.03 and 4.41 V vs. Li for 5,6-dihydroxyindole and 5,6-indolequinone monomers, respectively. These redox potentials decreased to 3.73–3.82 and 3.95–3.97 V vs. Li for the corresponding dimers and trimers, respectively (Table S2, ESI†). Thus, these computational results predict multiple redox reactions of polydopamine with electrolyte ions in the broad voltage range of 2.5–4.1 V vs. Li.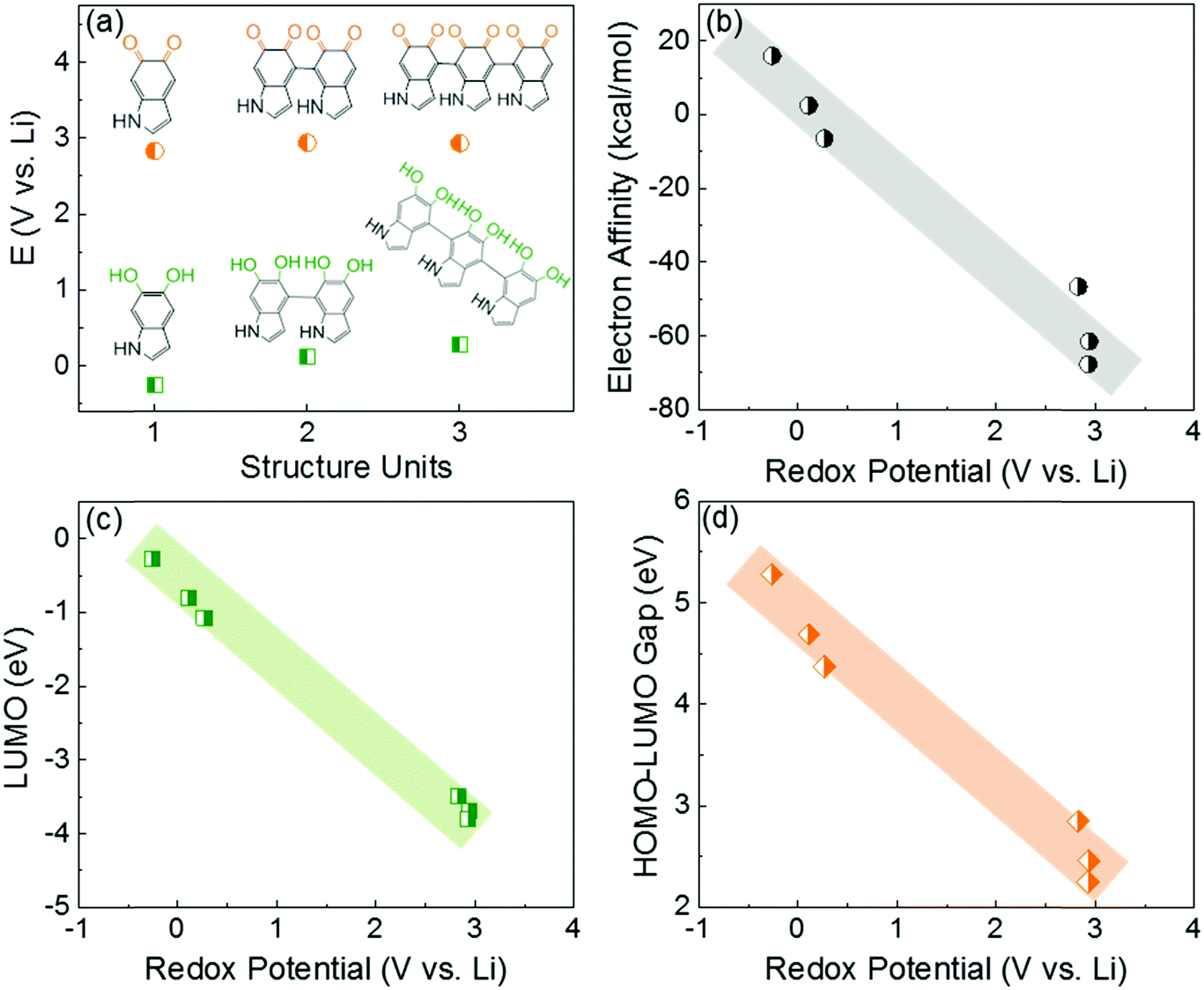 | ||
| Fig. 1 (a) The calculated redox potentials of 5,6-dihydroxyindole and 5,6-indolequinone monomers and their oligomers (dimers and trimers). The correlations of the redox potentials of the molecules in (a) with their electronic properties, namely (b) EA, (c) LUMO, and (d) HOMO–LUMO gap. | ||
Because most organic electrode materials have poor electrical conductivity, they require a mixing process with a large amount of conductive additive to activate the redox reactions. In this study, we utilized a sub-millimeter long FWNT, which was synthesized using a fluidized-bed chemical vapor deposition method, as a conductive substrate owing to its high electrical conductivity and high aspect ratio.37,38 We coated polydopamine on the surface of FWNTs through the self-polymerization process of dopamine in weak alkaline solutions (Scheme 2a). Dopamine and FWNTs were mixed at different concentration ratios (i.e. FWNT/dopamine = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, 1:4, and 1:8) in 2-amino-2-hydroxymethylpropane-1,3-diol (Tris) aqueous solution (1.2 mg mL−1, pH = 8.5). The mixtures were stirred at room temperature for 6–18 h, during which polydopamine was coated on the surface of FWNTs. Flexible and free-standing films were assembled via vacuum-filtering of the aqueous mixture (Scheme 2b). The loading amount of polydopamine within the hybrid films can be controlled by the concentration ratio and stirring time of the aqueous mixture. The hybrid films were labeled with the concentration ratios of FWNTs and dopamine in the aqueous mixtures (i.e. PDA-X indicates the film prepared from the concentration ratio of FWNT/dopamine = 1:X, where X = 1, 2, 4 and 8). We estimated the weight percentage of polydopamine in the hybrid films by measuring the mass difference between the used FWNTs and the hybrid films. As the concentration of dopamine increased, the weight percentage of polydopamine progressively increased (Fig. 2a), whereas their yield percentage from monomer to polymer gradually decreased (Fig. 2b). It is interesting to note that increasing the stirring time can increase both the weight percentage and the yield of polydopamine, indicating that the effective polymerization process takes place under stirring conditions (Fig. 2a and b). After stirring the mixture for 18 h, a maximum loading of 65.6 wt% was achieved for PDA-8, while a maximum yield of 65.5% was obtained for PDA-1. In addition, the normalized electrical conductivity decreased from 209 Scm2 g−1 for the pristine FWNT film to 20 Scm2 g−1 for PDA-8 (65.6 wt%) owing to the insulating nature of the polydopamine coating on the surface of the FWNTs (Fig. 2c).
:1, 1:2, 1:4, and 1:8) in 2-amino-2-hydroxymethylpropane-1,3-diol (Tris) aqueous solution (1.2 mg mL−1, pH = 8.5). The mixtures were stirred at room temperature for 6–18 h, during which polydopamine was coated on the surface of FWNTs. Flexible and free-standing films were assembled via vacuum-filtering of the aqueous mixture (Scheme 2b). The loading amount of polydopamine within the hybrid films can be controlled by the concentration ratio and stirring time of the aqueous mixture. The hybrid films were labeled with the concentration ratios of FWNTs and dopamine in the aqueous mixtures (i.e. PDA-X indicates the film prepared from the concentration ratio of FWNT/dopamine = 1:X, where X = 1, 2, 4 and 8). We estimated the weight percentage of polydopamine in the hybrid films by measuring the mass difference between the used FWNTs and the hybrid films. As the concentration of dopamine increased, the weight percentage of polydopamine progressively increased (Fig. 2a), whereas their yield percentage from monomer to polymer gradually decreased (Fig. 2b). It is interesting to note that increasing the stirring time can increase both the weight percentage and the yield of polydopamine, indicating that the effective polymerization process takes place under stirring conditions (Fig. 2a and b). After stirring the mixture for 18 h, a maximum loading of 65.6 wt% was achieved for PDA-8, while a maximum yield of 65.5% was obtained for PDA-1. In addition, the normalized electrical conductivity decreased from 209 Scm2 g−1 for the pristine FWNT film to 20 Scm2 g−1 for PDA-8 (65.6 wt%) owing to the insulating nature of the polydopamine coating on the surface of the FWNTs (Fig. 2c).
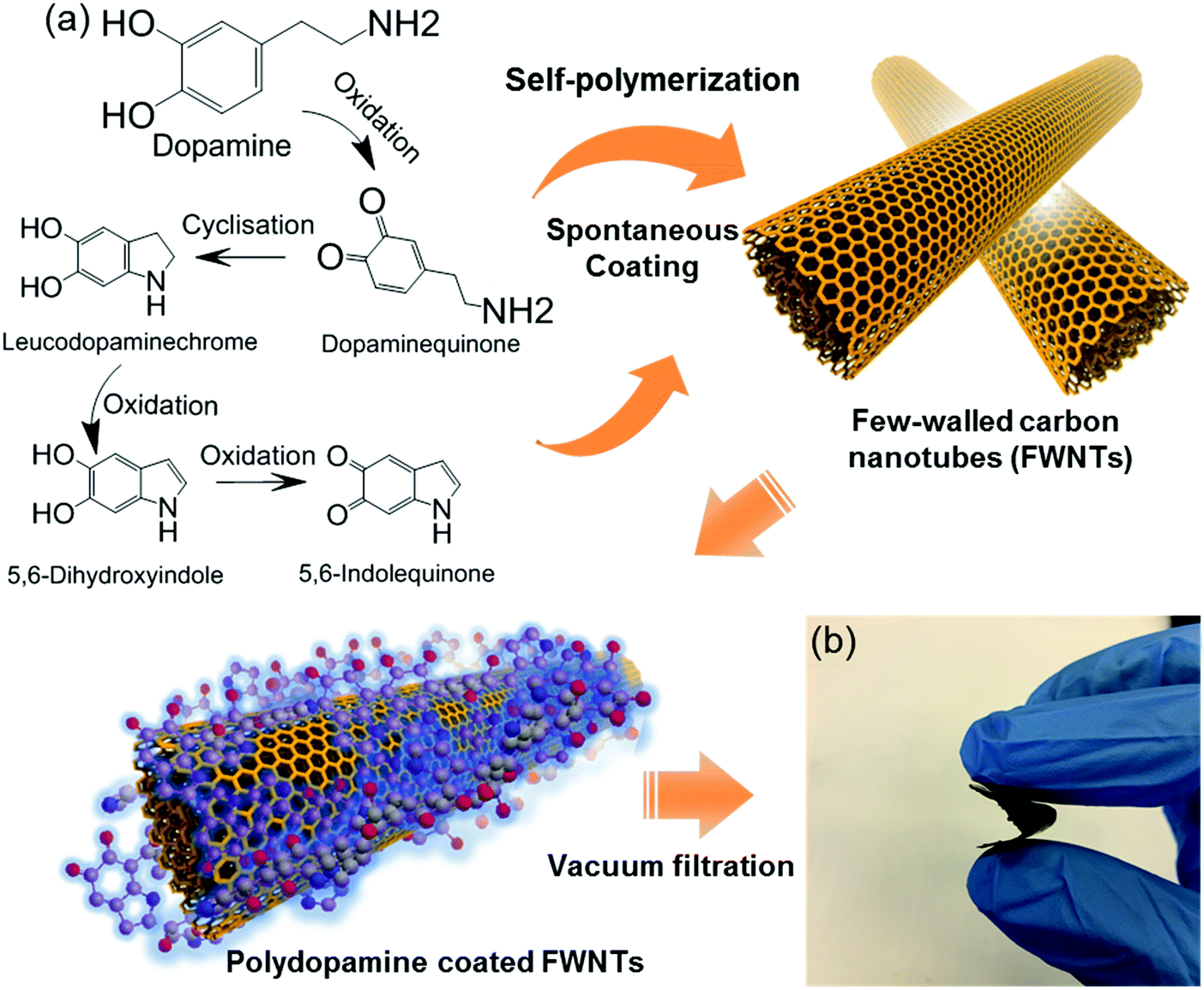 | ||
| Scheme 2 (a) Oxidative self-polymerization reactions of dopamine in weak alkaline solution and a continuous coating process of polydopamine on the surface of few-walled carbon nanotubes (FWNTs). (b) Digital image of a flexible hybrid film consisting of polydopamine coated FWNTs. | ||
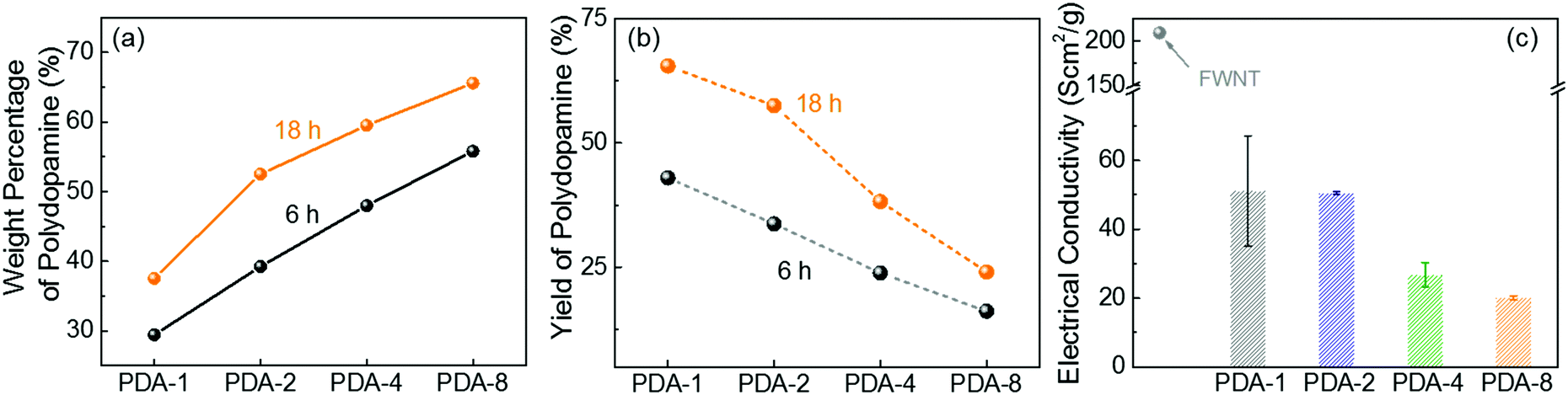 | ||
| Fig. 2 (a) Weight percentage and (b) yield of polydopamine in the hybrid films as a function of the concentration of dopamine and stirring time. (c) Normalized electrical conductivities of hybrid films as a function of the concentration of dopamine (18 h stirring). | ||
The formation of quinone groups in the polydopamine was investigated and the resulting UV-vis spectra are shown in Fig. 3a. Compared with the spectrum of pristine dopamine, the spectrum of the hybrid film (PDA-2, 6 h stirring, 39 wt%) displayed an intensive absorption band at ∼266 cm−1 due to the π-plasmon absorption of carbon nanotubes39 and a shoulder absorption peak at ∼350 nm was ascribed to the formation of the quinone.33 The detailed surface chemistry of the hybrid film (PDA-2, 18 h stirring, 53 wt%) was further analyzed using X-ray photoelectron spectroscopy (XPS) (Fig. 3b–d). A high-resolution C 1s spectrum of pristine FWNTs was dominated by the sp2-hybridzed carbon peak, while the hybrid film displayed a broadened spectrum towards a higher binding energy owing to the introduced polydopamine coating on the FWNTs (Fig. 3b). The C 1s spectrum of the hybrid film can be fitted into four peaks at 284.5 ± 0.1 eV for CHx/C–NH2 and sp2-hybridized carbon, 285.6 ± 0.1 eV for C–O/C–N, 287.7 ± 0.1 eV for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and 290.9 ± 0.3 eV for the π → π* transition satellite for aromatic carbon species.40,41 The O 1s spectrum of the hybrid film was fitted with two main peaks, the peak centered at 533.1 ± 0.2 eV for O–C and 531.2 ± 0.1 eV for OC,40,41 where the percentage of carbonyl species (quinone group) is 26%. In addition, the N 1s spectrum of the hybrid film revealed three peaks at 401.9 ± 0.1 for primary amine (R–NH2), 399.9 ± 0.1 eV for secondary amine (R–NH–R), and 398.5 ± 0.1 eV for tertiary/aromatic (N–R) amine functional groups.40,41 The primary amine peak corresponds to dopamine, the secondary amine peak is associated with polydopamine or oxidized intermediates, and the tertiary/aromatic peak is ascribed to the tautomers of 5,6-dihydroxyindole and 5,6-indolequinone (Scheme 2a).40 A recent study proposed that polydopamine consists of the combination of covalent polymerization of 5,6-indolequinone and a non-covalent self-assembly of dopamine and its intermediates.42 Therefore, the coexistence of different amine groups in the hybrid film is consistent with the proposed structural model of polydopamine.
O and 290.9 ± 0.3 eV for the π → π* transition satellite for aromatic carbon species.40,41 The O 1s spectrum of the hybrid film was fitted with two main peaks, the peak centered at 533.1 ± 0.2 eV for O–C and 531.2 ± 0.1 eV for OC,40,41 where the percentage of carbonyl species (quinone group) is 26%. In addition, the N 1s spectrum of the hybrid film revealed three peaks at 401.9 ± 0.1 for primary amine (R–NH2), 399.9 ± 0.1 eV for secondary amine (R–NH–R), and 398.5 ± 0.1 eV for tertiary/aromatic (N–R) amine functional groups.40,41 The primary amine peak corresponds to dopamine, the secondary amine peak is associated with polydopamine or oxidized intermediates, and the tertiary/aromatic peak is ascribed to the tautomers of 5,6-dihydroxyindole and 5,6-indolequinone (Scheme 2a).40 A recent study proposed that polydopamine consists of the combination of covalent polymerization of 5,6-indolequinone and a non-covalent self-assembly of dopamine and its intermediates.42 Therefore, the coexistence of different amine groups in the hybrid film is consistent with the proposed structural model of polydopamine.
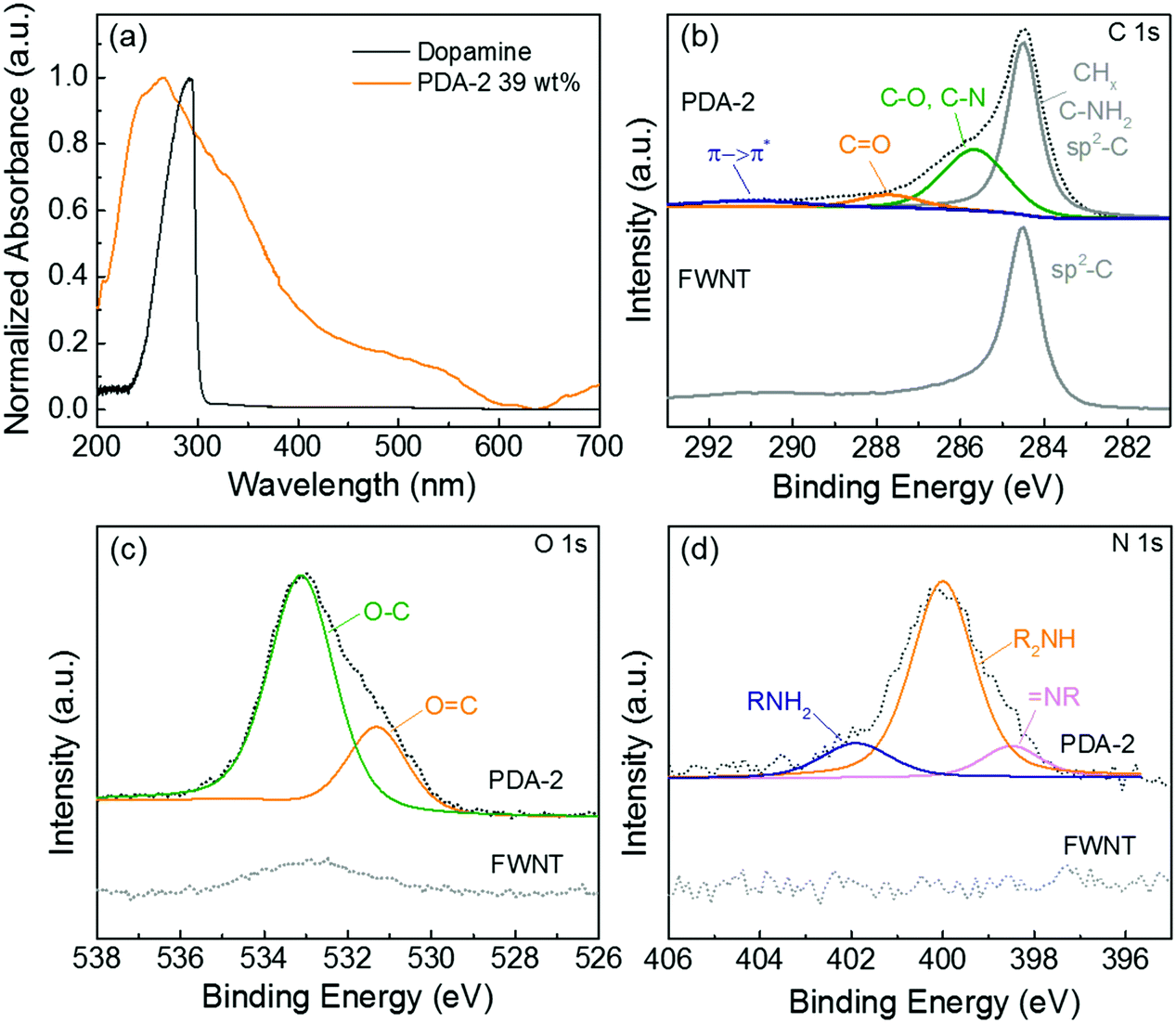 | ||
| Fig. 3 (a) UV-vis spectra of aqueous solutions of pristine dopamine and the hybrid film (PDA-2, 6 h stirring, 39 wt%). High-resolution XPS (b) C 1s, (c) O 1s, and (d) N 1s spectra of the pristine FWNT and the hybrid film (PDA-2, 18 h stirring, 53 wt%). | ||
Both the pristine FWNTs and hybrid films exhibit 3D interpenetrating network structures consisting of FWNTs (Fig. 4a) or polydopamine coated FWNTs in the scanning electron microscopy (SEM) images (Fig. 4b–d). In addition, when the concentration of dopamine is high, randomly distributed polydopamine spheres with diameters of ∼200 nm were also observed within the hybrid films (Fig. 4c and d). These polydopamine spheres were synthesized via self-nucleation reactions in aqueous solution as the concentration of dopamine increased. These polydopamine spheres were also observed from the homogeneous polymerization process of dopamine without FWNTs (Fig. S1, ESI†). We further conducted elemental mapping of oxygen and nitrogen on pristine FWNTs and hybrid films by energy-dispersive X-ray spectroscopy (EDX). Despite the observation of the polydopamine particles within the selected area for EDX mapping, the hybrid film exhibited a homogenous distribution of oxygen and nitrogen with higher density compared to those of the pristine FWNT film, confirming the successful coating of polydopamine on the FWNTs (Fig. S2, ESI†). The conformal coating of amorphous polydopamine on the surface of FWNTs was further confirmed by the high-resolution transmission electron microscopy (TEM) images. Pristine FWNTs exhibited a slightly roughened outer wall owing to the introduced defects during the growth process (Fig. 4e). Synthesized polydopamine formed an irregular coating on the surface of the FWNTs for the hybrid film with a low loading amount of the polymer (39 wt%) (Fig. 4f). This coating became more uniform with a thickness of ∼5 nm as the loading of polydopamine increased to 53 wt% (Fig. 4g).
 | ||
| Fig. 4 Scanning electron microscopy (SEM) images of (a) the pristine FWNT and hybrid films: (b) PDA-2 (53 wt%), (c) PDA-4 (59.5 wt%), and (d) PDA-8 (65.6 wt%). The inset in (a–d) are the SEM images of FWNTs and hybrid films at high magnification. High resolution transmission electron microscopy (TEM) images of (e) pristine FWNT and hybrid films: (f) PDA-2 (39 wt%) and (g) PDA-2 (53 wt%). | ||
The redox-active properties of polydopamine were investigated using cyclic voltammetry (CV) measurements in both Li- and Na-cells. The free-standing hybrid films with mass loadings of 1–3 mg cm−2 were directly used as cathodes against the Li or Na metal anodes. The CV scans of the pristine FWNT films were also measured as references, showing characteristic box-like shapes owing to their electrical double-layer capacitance charge storage mechanism (Fig. S3a and b, ESI†).19,43,44 The first forward scan of the hybrid electrode exhibited a wide oxidation peak with an onset potential of ∼2.5 V vs. Li, which is ascribed to the interactions between the quinone groups in polydopamine and Li-ions. The oxidation peak position of ∼3.3 V vs. Li is similar to that of benzoquinone (∼3.1 V vs. Li)26 (Fig. 5a). The second oxidation peak at ∼3.9 V vs. Li is associated with the p-doping process of anions (PF6−) into the polymer chain or nitrogen atoms, which is also consistent with the DFT calculation results (Table S2, ESI†).45,46 A strong oxidation wave with an onset potential of ∼4 V vs. Li can be attributed to further anodic polymerization of the intermediates within the polydopamine film.47 Upon subsequent CV cycling, the anodic polymerization current progressively disappeared and the broad redox peaks centered at ∼3.1 V vs. Li approached stable current values (Fig. 5a and b), this redox potential is very consistent with those calculated using the DFT method (2.83–2.93 V vs. Li) (Fig. 1a). The pristine polydopamine electrode, which was prepared from polydopamine spheres (Fig. S1, ESI†), exhibited similar redox characteristics in a Li-cell (Fig. S4a, ESI†). Similar to the results in Li-cells, the hybrid electrode displayed steady-state redox peaks centered at ∼2.8 V vs. Na after the anodic polymerization process in Na-cells (Fig. S3c and 5b, ESI†). These high redox potentials suggest that polydopamine is an ideal candidate for organic cathodes for both Li- and Na-ion batteries. The charge storage characteristics of the polydopamine were further analyzed by the potential-dependent CV measurements in Li-cells (Fig. 5c). In a low voltage range of 1.5–2.5 V vs. Li, the CV scan showed a box-like shape, indicative of double layer capacitance from the FWNTs. In contrast, the redox peaks were gradually developed as the voltage increased from 2.5 to 4 V vs. Li, which clearly revealed the redox reactions between the quinone groups and Li ions. In addition, the backward CV scans from 4.5 V to 3.5 V showed a gradual increase in currents, which can be attributed to the p-doping process of anions into the polymer chain or nitrogen atoms.45,46 Moreover, the rate-dependent CV scans in both Li- and Na-cells maintained the redox features at higher scan rates (100 mV s−1 for Li-cells and 25 mV s−1 for Na-cells) owing to the fast surface redox reactions (Fig. 5d and Fig. S3d, ESI†). The linear relationship between the peak current and the scan-rate indicates a surface-controlled redox process of polydopamine within the hybrid films (Fig. 5d inset).
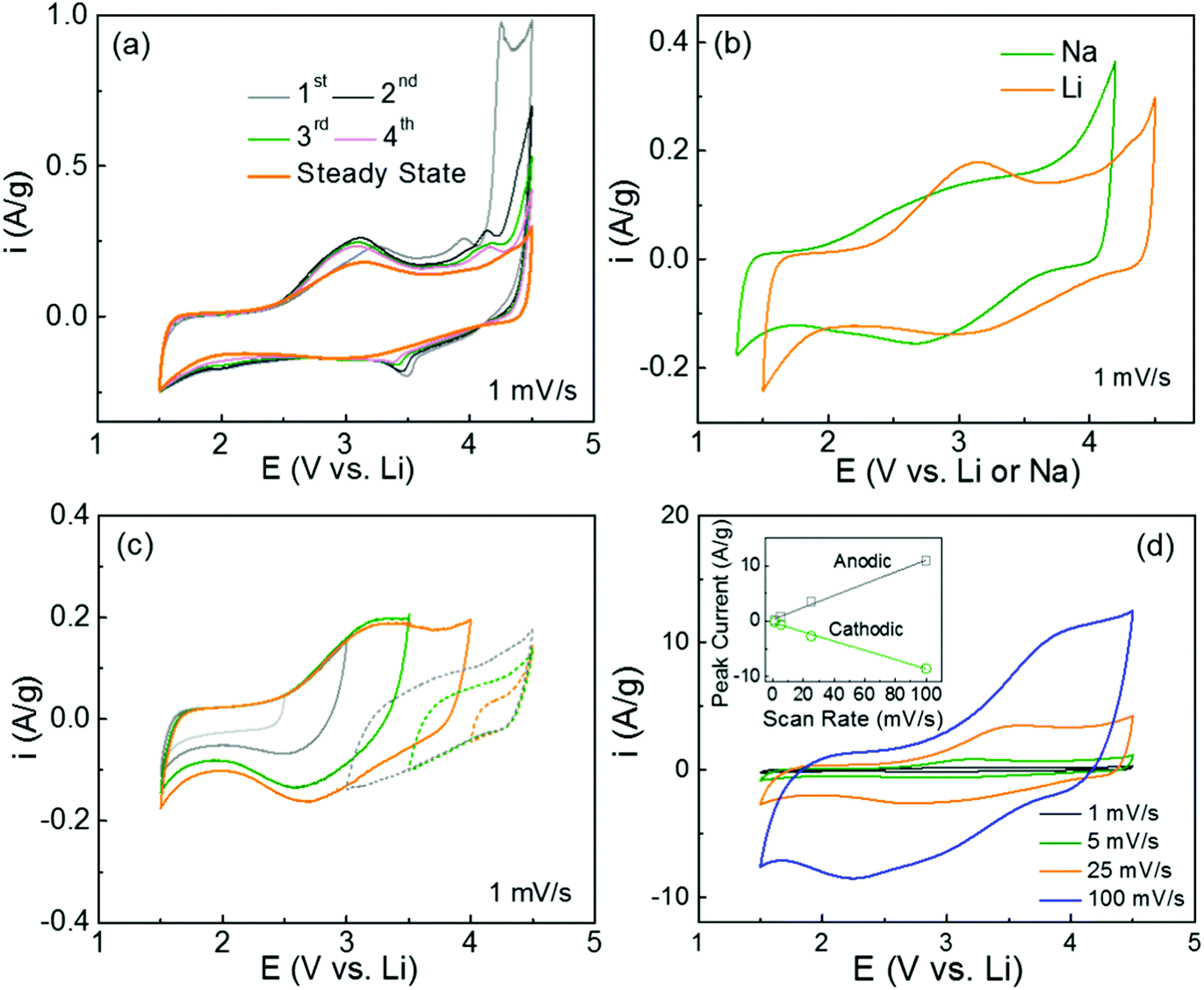 | ||
| Fig. 5 Cyclic voltammetry (CV) measurement of the hybrid electrode (PDA-2, 39 wt%) in Li- and Na-cells. (a) The first four and steady-state CV scans at 1 mV s−1 in a Li-cell. (b) Comparison of steady-state CV scans in Li- and Na-cells. (c) Potential-dependent and (d) rate-dependent CV scans in Li-cells. Peak current of the electrode as a function of scan rate is shown in the inset of (d). The voltage window of the CV scans in the Li-cells was 1.5–4.5 V vs. Li in 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (3:7 volume ratio) and the voltage window in Na-cells was 1.3–4.2 V vs. Na in 1 M NaPF6 in a mixture of EC and DMC (3:7 volume ratio). | ||
To further understand the redox reactions between the quinone groups in polydopamine and Li ions, we conducted ex situ XPS measurements of the hybrid electrode (PDA-2, 53 wt%) after the constant potential holding step and the continuous self-discharge process in Li-cells. The voltage drift of the hybrid electrode was measured at open-circuit for ∼18 h after holding the cell at a target voltage for 60 min (Fig. 6a). The open-circuit potential of the hybrid electrode is ∼3.2 V vs. Li. After the voltage holding process at 4.5 V vs. Li, the open-circuit voltage gradually dropped to ∼3.6 V vs. Li during the rest of the process owing to parasitic leakage current.48 In contrast, after the holding process at 1.5 V vs. Li, the voltage progressively recovered to ∼2.5 V vs. Li with time. The electrode also showed almost constant voltage behavior after holding at 3.5 V vs. Li, which is close to the open-circuit potential of the as-assembled electrode. After the self-discharge test, we washed the electrodes in dimethyl carbonate (DMC) and then measured the XPS Li 1s spectrum to compare the remaining Li ions on the polydopamine (Fig. 6b). The hybrid electrode that was held at 1.5 V vs. Li showed a strong Li 1s peak owing to the adsorbed Li ions on the quinone groups in the polydopamine after the discharge process. The Li 1s peak intensity was found to decrease with increasing hold voltage, indicating the desorption process of the Li ions on the quinone groups during the charge process. This potential-dependent Li adsorption and desorption behavior further supports the reversible redox reactions between quinone groups in the polydopamine and Li ions.
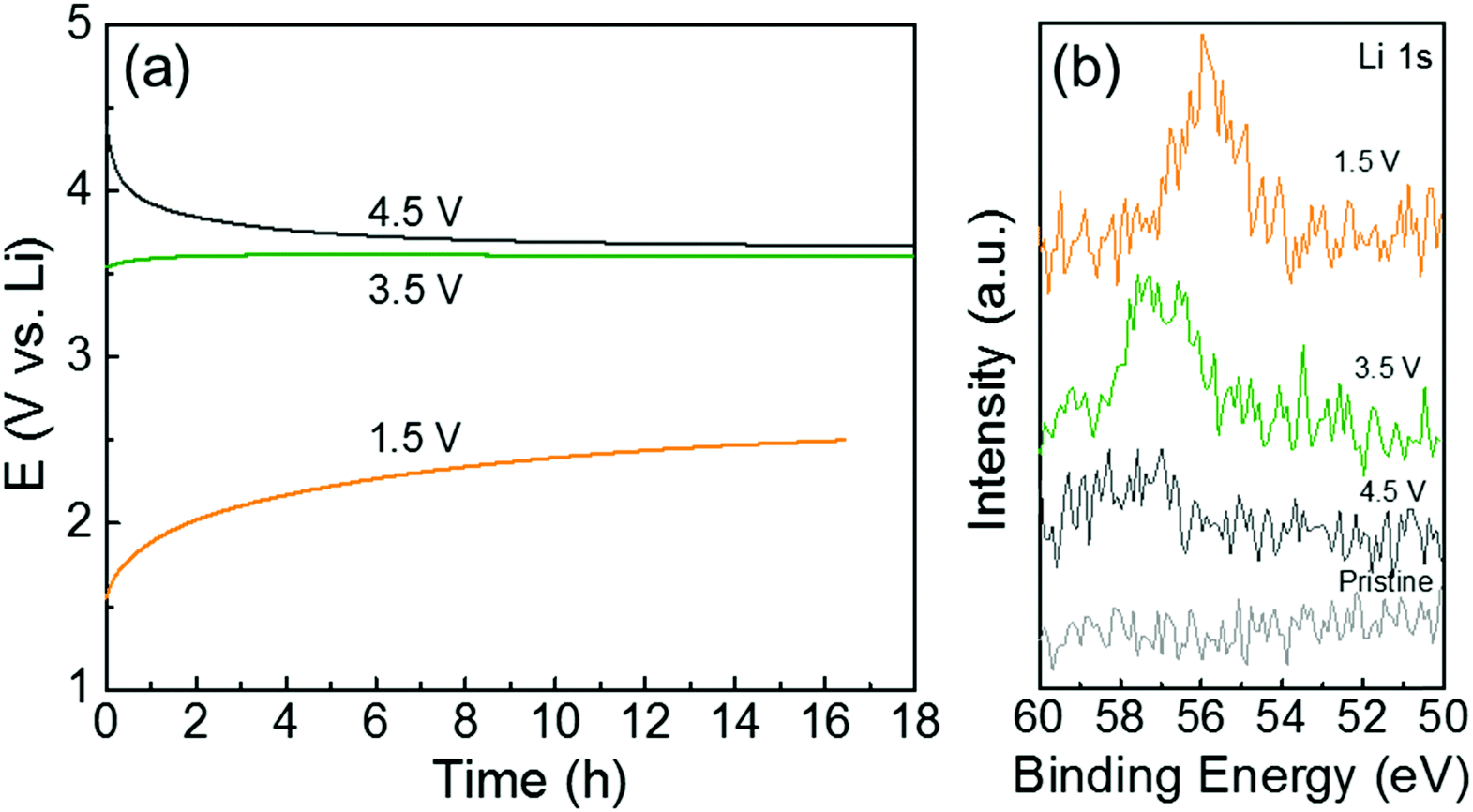 | ||
| Fig. 6 (a) The self-discharge course of the hybrid films after holding at different voltages. (b) XPS Li 1s spectra of the hybrid films (PDA-2, 53 wt%) after holding at different voltages and a continuous self-discharge test. | ||
We further utilized the DFT method to structurally characterize the redox reactions between the quinone groups in polydopamine and Li ions at the molecular level. The changes in the structural and electronic properties due to the Li binding to the monomer constituting the polydopamine are depicted in Fig. S5 (ESI†). The first Li ion positioned between the two carbonyls loses its electrons to the neighboring oxygen atoms, exhibiting a charge of +0.52. The charge transfer between Li and O subsequently affects the bonding characteristics of the carbonyls, partially losing their double bond nature. The second Li ion adsorbed on top of the aromatic ring in the molecule provides additional charge transfer with the neighboring oxygen atoms. This ultimately leads to a further change in the bonding characteristics of the carbonyls, forming in the C–O–Li bonds (Table S3, ESI†). Fig. 7 shows how the redox potentials for the 5,6-indolequinone monomer and dimer consisting of polydopamine are changed with the number of bound Li ions during the discharge process. As expected, the redox potentials are gradually reduced as the number of the bound Li ions increases. The redox potentials of monomer and dimer become negative after binding with two and four Li ions, respectively, losing their cathodic activities.
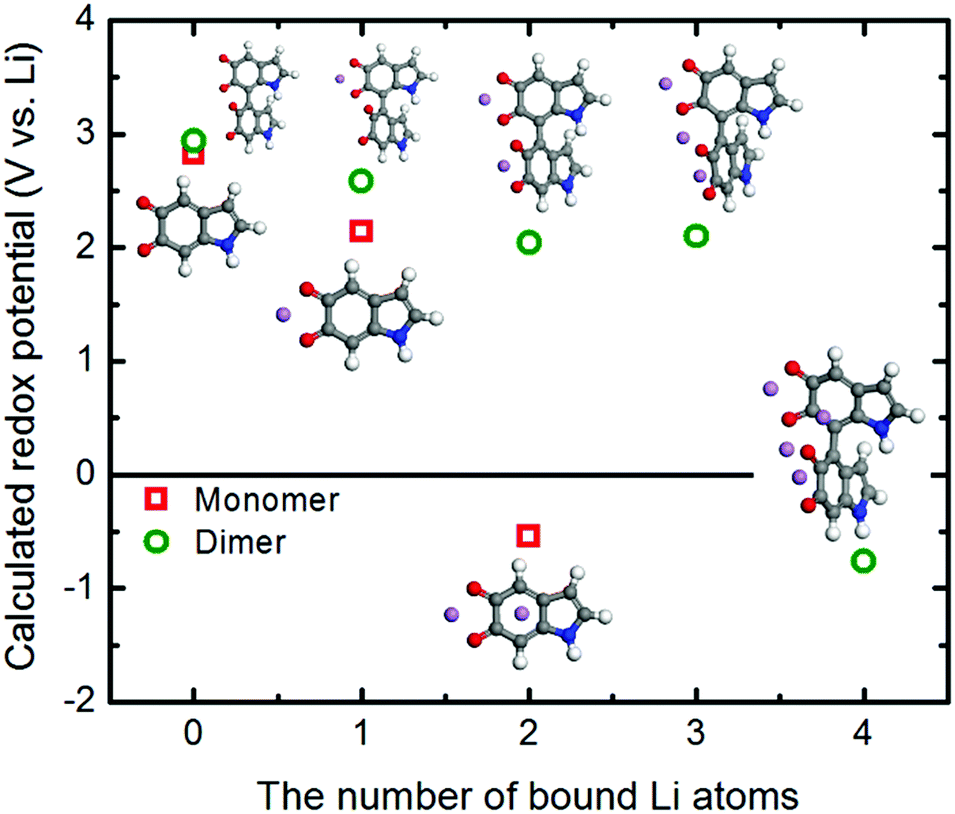 | ||
| Fig. 7 Change in the redox potentials (PBE0 functional) as a function of the Li atoms binding with the 5,6-indolequinone monomer and dimer. | ||
Galvanostatic charge and discharge tests were conducted in both Li- and Na-cells to evaluate the charge storage capabilities of the hybrid electrodes. We compared the discharge profiles of the pristine FWNT and hybrid electrodes in the Li- and Na-cells (Fig. 8a and b). The pristine FWNT electrodes showed small gravimetric capacities of ∼40 mA h g−1 in a Li-cell19,43,44 and ∼32 mA h g−1 in a Na-cell, while the hybrid electrodes (PDA-2, 39 wt%) delivered significantly enhanced capacities of ∼117 mA h g−1 and ∼103 mA h g−1 in Li- and Na-cells, respectively. It is interesting to note that the hybrid electrodes have sloped discharge profiles, which can be attributed to the combination of double-layer capacitance and multiple redox reactions. As the loading of polydopamine increased from 39 to 53 wt%, the gravimetric capacities of the hybrid films slightly increased to ∼133 mA h g−1 in a Li-cell and ∼109 mA h g−1 in a Na-cell. To understand the intrinsic charge storage capability of polydopamine, we calculated the rate-dependent gravimetric capacities of polydopamine in the hybrid electrodes based on the rule of mixtures in the Li- and Na-cells (Fig. 8c, d and Fig. S6, ESI†). At a slow discharge rate of 0.05 A g−1, the polydopamine in the hybrid electrode with a lower loading (PDA-2, 39 wt%) delivered higher capacities of ∼235 mA h g−1 in a Li-cell and ∼213 mA h g−1 in a Na-cell compared to those of the polydopamine with a higher loading (PDA-2, 53 wt%) (∼217 mA h g−1 in a Li-cell and ∼178 mA h g−1 in a Na-cell). In addition, the pristine PDA electrode, which was prepared from polydopamine spheres (Fig. S1, ESI†), delivered a discharge capacity of 158 mA h g−1 and a charge capacity of 102 mA h g−1 at a current density of 0.05 A g−1 (Fig. S4b, ESI†). The lower discharge capacity and the difference between the charge and discharge capacities of the pristine PDA electrode may be attributed to the electrical and ionic resistance through the large polydopamine particles. The capacity of polydopamine in the hybrid electrodes is comparable or even higher than most of the reported carbonyl-based compounds (Table S4, ESI†).12,17,18,49,50 Over higher current densities, the polydopamine with a lower loading (39 wt%) showed an overall enhanced rate-performance compared to that of the polydopamine with a higher loading (53 wt%). At a high current density of ∼10 A g−1, the polydopamine with a lower loading (39 wt%) exhibited high capacities of ∼139 mA h g−1 in a Li-cell and 54 mA h g−1 in a Na-cell, whereas the polydopamine with a higher loading (53 wt%) delivered decreased capacities of ∼87 mA h g−1 and ∼10 mA h g−1 in Li- and Na-cells, respectively. These differences indicate that a portion of the polydopamine is not effectively utilized as the loading amount increases. Such decreased charge storage performance of the polydopamine with a high loading may be attributed to the polydopamine spheres that were separated from the conductive FWNT network (Fig. 4), resulting in poor utilization of their redox activities. A relatively low capacity (121–129 mA h g−1) and poor rate-performance of the hybrid electrodes prepared with a higher concentration of dopamine (PDA-4 and 8) further confirms this trend (Fig. S7c and d). Fig. S8 (ESI†) compares the Nyquist plots for the hybrid electrodes (PDA-1, 2, 4, 8, 18 h stirring). A single semicircle in the high-to-medium frequency region and an inclined line at a low frequency provide information about the charge-transfer resistance Rct and mass-transfer process, respectively. A higher Rct (2051–2370 Ω) of PDA-4 and PDA-8 compared to those of the PDA-1 and PDA-2 electrodes (1336 Ω) further indicates the significantly low charge transfer resistance within the PDA-4 and PDA-8 electrodes including the separated polydopamine spheres. These results suggest that the conformal coating of polydopamine on the conductive substrate is a crucial design factor for maximizing the performance of the hybrid electrodes.
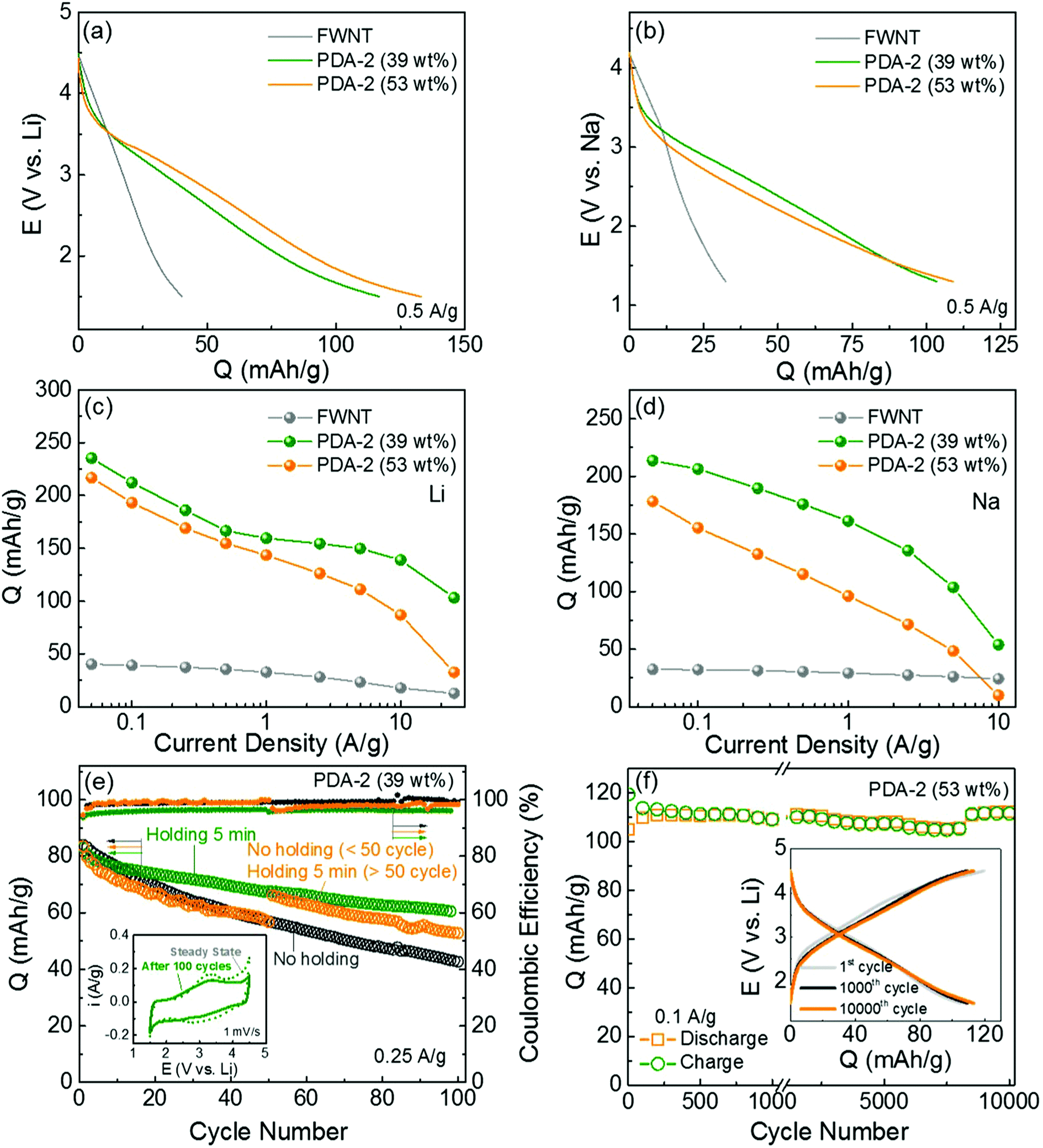 | ||
| Fig. 8 Comparison of discharge profiles of the pristine FWNT (grey) and hybrid electrodes (PDA-2, 39 wt% (olive) and 53 wt% (orange)) at 0.05 A g−1 in (a) Li- and (b) Na-cells. Gravimetric discharge capacities of polydopamine as a function of discharge rate in (c) Li- and (d) Na-cells. Gravimetric capacities of the polydopamine were calculated based on the rule of mixtures. (e) Cycling stabilities of the hybrid electrodes (PDA-2, 39 wt%) and their Coulombic efficiencies. The cells were cycled at 0.25 A g−1 up to 100 cycles using three different methods: a dark grey circle for continuous cycles without any potentiostatic holding process, an olive circle for cycles including a short 5 min potentiostatic holding at 4.5 or 1.5 V vs. Li at the end of each charge and discharge cycle, and an orange circle for the initial 50 cycles without holding and subsequent 50 cycles with 5 min of potentiostatic holding. The inset shows the comparison of CV scans of PDA-2 (39 wt%) at the initial cycle and 100th cycle at 1 mV s−1. (f) Gravimetric charge and discharge capacities of the hybrid electrode (PDA-2, 53 wt%) as a function of cycle number up to 10000 cycles using an accelerating cycling method.43 (f) Inset compares the charge and discharge curves of the 1st and 1000th and 10000th cycles. The capacities were measured based on the total weight of the hybrid electrodes for a, b, e and f, and the weight of polydopamine for c and d. | ||
The cycling stabilities of the hybrid electrodes were evaluated by repeatedly charging and discharging the cells at a constant current density of 0.25 A g−1 (Fig. 8e, Fig. S9 and S10, ESI†). Different cycling test conditions were applied to the same electrode (PDA-2, 39 wt%). The electrode under repeated charge/discharge cycles without any potentiostatic holding process exhibited rapid capacity fading, retaining 50% of its initial capacity after 100 cycles. On the other hand, when a short potentiostatic holding for 5 min at 4.5 or 1.5 V vs. Li was applied at the end of each charge and discharge cycle, the capacity retention was significantly improved, maintaining 78% of its initial capacity. A comparison of the CV scans before and after the cycling test (Fig. 8e inset, PDA-2, 39 wt%) indicates that the loss of capacity was attributed to the decrease of the redox peaks. A control experiment was also conducted in which no potentiostatic holding was applied for the first 50 cycles, and a 5 min potentiostatic holding was applied for the subsequent 50 cycles. The capacity gradually decreased during the first 50 cycles, while the capacity immediately recovered once the potential holding was applied. It is interesting to note that this recovered capacity was similar to that of the cycling test with a continuous 5 min potentiostatic holding test. A similar result was also observed for the electrode with higher polydopamine loading (PDA-2, 53 wt%, Fig. S9, ESI†). This suggests that the gradual capacity loss during cycling due to the kinetic limitation of the redox reactions within the polydopamine film rather than the dissolution of the molecules. We also evaluated the cycling stability of this electrode via an accelerated cycling where the cell was cycled at a high current density of 10 A g−1 and then the capacity was measured at a slow current density of 0.1 A g−1 after a 30 min holding43 (Fig. 8f). The accelerated cycling test revealed a negligible capacity decrease up to 10000 cycles, indicating that most of the capacity of the hybrid electrode is recoverable after a 30 min holding. To further confirm the stability of polydopamine in the organic electrolyte, stability and dissolution tests were conducted following the method published in a previous study.27 A piece of the fresh hybrid electrode including 51.4 wt% of polydopamine was dipped into the electrolyte solvent, a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC). Although the electrode was kept for more than 12 h, no color change of the solvent was observed (Fig. S11a, ESI†). In addition, there was no mass change after completely drying the dipped electrode, indicating that synthesized polydopamine is stable in the electrolyte. Solubility tests of the hybrid electrodes in either fully charged or discharge states also showed no color changes even after keeping the electrodes in the electrolyte for over 12 h (Fig. S11b, ESI†), indicating that polydopamine is also insoluble in the electrolyte in the charged or discharged states. These results further confirm that the self-polymerization process can prevent the dissolution of dopamine molecules, which is a common shortcoming of most organic electrode materials.
Conclusions
In conclusion, we have demonstrated that polydopamine can be utilized as a high-performance organic cathode material for rechargeable Li- and Na-ion batteries. Dopamine was self-polymerized on the surface of FWNTs in alkaline aqueous solutions during which the loading of polydopamine can be simply controlled by the mixing time and concentration of dopamine. The free-standing and flexible hybrid films were assembled using a vacuum filtration method, which were utilized as cathodes in both Li- and Na-cells. Polydopamine within the hybrid films exhibited multiple redox reactions with both electrolyte ions in the high voltage region of 2.5–4.1 V vs. Li, which was consistent with the DFT computation results. The hybrid electrodes exhibited gravimetric capacities of ∼133 mA h g−1 in Li-cells and ∼109 mA h g−1 in Na-cells through double layer capacitance and multiple redox-reactions. We further revealed that the polydopamine itself in the hybrid electrodes can store high capacities of ∼235 mA h g−1 in Li-cells and ∼213 mA h g−1 in Na-cells. Moreover, the hybrid electrodes showed a high rate-performance with excellent cycling stability. Combined with its advantage of a simple polymerization process, the introduced polydopamine based electrodes could be ideal candidates for developing high-performance organic batteries.Acknowledgements
This work was supported by the Samsung Advanced Institute of Technology (SAIT)'s Global Research Outreach (GRO) Program. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542174).Notes and references
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- J.-M. Tarascon, ChemSusChem, 2008, 1, 777–779 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- N. Oyama, T. Tatsuma, T. Sato and T. Sotomura, Nature, 1995, 373, 598–600 CrossRef CAS.
- J. Wang, C. O. Too and G. G. Wallace, J. Power Sources, 2005, 150, 223–228 CrossRef CAS.
- K. Naoi, K. I. Kawase and Y. Inoue, J. Electrochem. Soc., 1997, 144, L170–L172 CrossRef CAS.
- S. J. Visco, C. C. Mailhe, L. C. De Jonghe and M. B. Armand, J. Electrochem. Soc., 1989, 136, 661–664 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- L. Bugnon, C. J. H. Morton, P. Novak, J. Vetter and P. Nesvadba, Chem. Mater., 2007, 19, 2910–2914 CrossRef CAS.
- M. Yao, H. Senoh, S.-I. Yamazaki, Z. Siroma, T. Sakai and K. Yasuda, J. Power Sources, 2010, 195, 8336–8340 CrossRef CAS.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 448–450 RSC.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- H.-G. Wang, S. Yuan, Z. Si and X.-B. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 CAS.
- H.-G. Wang, S. Yuan, D.-L. Ma, X.-L. Huang, F.-L. Meng and X.-B. Zhang, Adv. Energy Mater., 2014, 4, 1301651 CrossRef.
- T. Sun, Z.-J. Li, H.-G. Wang, D. Bao, F.-L. Meng and X.-B. Zhang, Angew. Chem., Int. Ed., 2016, 128, 10820–10824 CrossRef.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS PubMed.
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 7040 CrossRef PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799–6802 CrossRef CAS PubMed.
- F. Bernsmann, V. Ball, F. Addiego, A. Ponche, M. Michel, J. J. D. A. Gracio, V. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819–2825 CrossRef CAS PubMed.
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS PubMed.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- Z. Zhu, H. Li, J. Liang, Z. Tao and J. Chen, Chem. Commun., 2015, 51, 1446–1448 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. Adamo, G. E. Scuseria and V. Barone, J. Chem. Phys., 1999, 111, 2889–2899 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- Jaguar version 7.6 Schrodinger, LLC, New York, NY, 2009 Search PubMed.
- Q. Wei, F. Zhang, J. Li, B. Li and C. Zhao, Polym. Chem., 2010, 1, 1430–1433 RSC.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- M.-H. Ryou, Y. M. Lee, J.-K. Park and J. W. Choi, Adv. Mater., 2011, 23, 3066–3070 CrossRef CAS PubMed.
- T. Liu, K. C. Kim, R. Kavian, S. S. Jang and S. W. Lee, Chem. Mater., 2015, 27, 3291–3298 CrossRef CAS.
- Z. Chen, D. Y. Kim, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2014, 80, 339–350 CrossRef CAS.
- D. Y. Kim, H. Sugime, K. Hasegawa, T. Osawa and S. Noda, Carbon, 2011, 49, 1972–1979 CrossRef CAS.
- V. A. Karachevtsev, A. M. Plokhotnichenko, M. V. Karachevtsev and V. S. Leontiev, Carbon, 2010, 48, 3682–3691 CrossRef CAS.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- M. B. Clark, J. A. Gardella, T. M. Schultz, D. G. Patil and L. Salvati, Anal. Chem., 1990, 62, 949–956 CrossRef CAS.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- T. Liu, R. Kavian, Z. Chen, S. S. Cruz, S. Noda and S. W. Lee, Nanoscale, 2016, 8, 3671–3677 RSC.
- S. W. Lee, B. M. Gallant, Y. Lee, N. Yoshida, D. Y. Kim, Y. Yamada, S. Noda, A. Yamada and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 5437–5444 CAS.
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812–1816 CrossRef CAS PubMed.
- Y. Li, M. Liu, C. Xiang, Q. Xie and S. Yao, Thin Solid Films, 2006, 497, 270–278 CrossRef CAS.
- B. M. Gallant, S. W. Lee, T. Kawaguchi, P. T. Hammond and Y. Shao-Horn, J. Electrochem. Soc., 2014, 161, A1625–A1633 CrossRef CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 CAS.
- W. Walker, S. Grugeon, O. Mentre, S. Laruelle, J.-M. Tarascon and F. Wudl, J. Am. Chem. Soc., 2010, 132, 6517–6523 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee02641a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |