
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unexpected formation of a μ-carbido diruthenium(IV) complex during the metalation of phthalocyanine with Ru3(CO)12 and its catalytic activity in carbene transfer reactions†
Andrey P.
Kroitor
a,
Lucie P.
Cailler
b,
Alexander G.
Martynov
 *c,
Yulia G.
Gorbunova
*cd,
Aslan Yu.
Tsivadze
cd and
Alexander B.
Sorokin
*b
*c,
Yulia G.
Gorbunova
*cd,
Aslan Yu.
Tsivadze
cd and
Alexander B.
Sorokin
*b
aChemical Department, M.V. Lomonosov Moscow State University, Leninskie gory, 1, bldg. 3, 119991, GSP-1, Moscow, Russia
bInstitut de Recherches sur la Catalyse et l'Environnement de Lyon IRCELYON, UMR 5256, CNRS - Université Lyon 1, 2 avenue A. Einstein, 69626 Villeurbanne cedex, France. E-mail: alexander.sorokin@ircelyon.univ-lyon1.fr
cA.N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninskii pr., 31, bldg. 4, 119071, Moscow, Russia. E-mail: martynov.alexandre@gmail.com
dN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninskii pr., 31, 11991, Moscow, Russia. E-mail: yulia@igic.ras.ru
First published on 16th October 2017
Abstract
A μ-carbido diruthenium(IV) phthalocyanine complex was prepared for the first time from the free-base octabutoxyphthalocyanine by direct metalation with Ru3(CO)12. The first examples of the catalytic activity of Ru(IV) binuclear phthalocyanines were demonstrated by the cyclopropanation of aromatic olefins and carbene insertion into the N–H bonds of aromatic or aliphatic amines with turnover numbers of 680–1000 and 580–1000, respectively.
Ruthenium complexes with tetrapyrrolic ligands can be considered as close analogues of iron porphyrins which constitute active sites of hemoproteins playing crucial roles in the transfer of small molecules and oxidation of organic substrates in Nature. To date, ruthenium porphyrins have already found wide catalytic applications in various synthetic procedures,1 including oxidation,2 nitrene and carbene transfer reactions,3 amination4 and other reactions.5 Myoglobin modified through iron cofactor substitution with ruthenium porphyrin showed moderate activity for carbene insertion reactions.6 By contrast, catalytic applications of related ruthenium(II) phthalocyanine analogues are limited by few examples of oxidation7 and cyclopropanation reactions.8 At the same time, due to robust chemical properties of phthalocyanine macrocycles, their ruthenium complexes might also give rise to novel catalytic systems.
Herein we report on the unexpected synthesis and catalytic activity of the μ-carbido diruthenium(IV) phthalocyanine complex in the cyclopropanation of styrenes and in the carbene insertion into the N–H bonds of anilines using ethyl diazoacetate (EDA) as a carbene donor.
Ruthenium phthalocyaninates are typically prepared by the template condensation of phthalonitriles in the presence of ruthenium salts or complexes.9 Direct insertion of ruthenium into the preformed macrocycle is, however, less studied and it is used mainly for the metalation of alkyl-substituted phthalocyanines with Ru3(CO)12 in refluxing benzonitrile, affording complexes with two axially coordinated PhCN molecules.10 Our attempt to apply this method to octa-n-butoxyphthalocyanine H2[(BuO)8Pc] failed.
Nevertheless, the reaction of H2[(BuO)8Pc] with Ru3(CO)12 (Scheme 1) smoothly proceeds in refluxing o-dichlorobenzene (o-DClB) similarly to the previously published preparation of ruthenium porphyrinates.11 The separation of the reaction products by size-exclusion chromatography on Bio-Beads S-X1 revealed that two different complexes with essentially different retention times were obtained as a result of the metalation reaction. The structures of complexes were evidenced by UV-Vis, FTIR, MALDI TOF MS and NMR techniques (Fig. 1 and ESI†). One of them with the longer retention time, obtained with a 62% yield, corresponded to the monomeric Ru(II) complex with an axially coordinated CO molecule – [(BuO)8Pc]Ru(CO), 1. The complex with the shorter retention time, formed with a 22% yield, had an unusual UV-Vis spectrum with the broad low-intensity Q-band centered at 607 nm (Fig. 1a). A set of spectroscopic data allowed us to assign this complex to the dimeric phthalocyaninato-Ru(IV) complex with a μ-carbido bridge [(BuO)8PcRu]2(μ-C), 2. The HR ESI mass spectrum showed a prominent molecular cluster of a doubly-charged molecular ion centered at m/z = 1196.512 (Fig. S6†). Their position and isotopic pattern corresponded to those in the simulated spectrum of C129H160N18O16Ru2 (calcd m/z = 1196.516). In its FTIR spectrum the band of the Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CRu stretching at 1013 cm−1 was observed whereas the band of the CO stretching (1939 cm−1 in the spectrum of 1) was absent (Fig. 1b).12,13 The sandwich structure of this complex was further confirmed by the characteristic splitting of the O-CH2 resonance signal in the 1H NMR spectrum of 2.
CRu stretching at 1013 cm−1 was observed whereas the band of the CO stretching (1939 cm−1 in the spectrum of 1) was absent (Fig. 1b).12,13 The sandwich structure of this complex was further confirmed by the characteristic splitting of the O-CH2 resonance signal in the 1H NMR spectrum of 2.
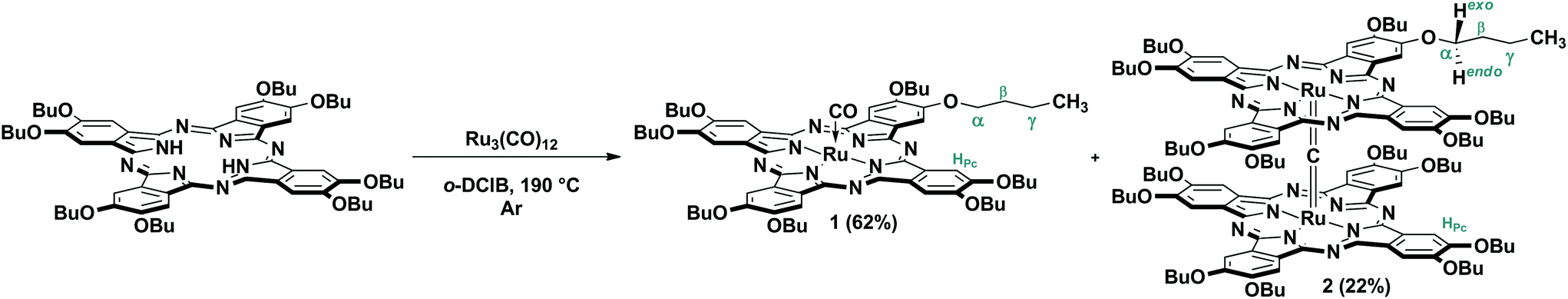 | ||
| Scheme 1 Synthesis of monomeric octabutoxyphthalocyaninatoruthenium(II) carbonyl complex 1 and dimeric μ-carbido-bis[octabutoxyphthalocyaninatoruthenium(IV)] complex 2 with numbering of protons used for the assignment of 1H NMR spectra. | ||
 | ||
| Fig. 1 (a) UV-Vis spectra of complexes 1 and 2 in CHCl3; (b) FTIR (neat) spectra of complexes 1 and 2; (c) 1H NMR spectra of complexes 1 and 2 in CDCl3 + 1 vol% Py-d5, “x” and “#” mark proton resonance signals of chloroform and water. | ||
This splitting is due to the non-equivalence of exo- and endo-protons in this group with respect to the space between the phthalocyanine ligands (Fig. 1c), which was also demonstrated by 1H–13C HSQC NMR spectroscopy (Fig. S11†).14
It is known that thermolysis of Ru3(CO)12 can lead to carbide clusters; among them the most well-studied one is the octahedral cluster Ru6C(CO)17, which is formed upon heating of ruthenium carbonyl in high boiling solvents.15 Therefore, we supposed that complex 2 can be formed either by thermolysis of the carbonyl-containing complex 1, or via the reaction of H2[(BuO)8Pc] with Ru6C(CO)17. However, neither refluxing of the latter reagents in o-DClB nor heating of 1 in the same solvent resulted in the formation of 2. Therefore, we can propose that complex 2 was formed simultaneously with 1via the reaction of H2[(BuO)8Pc] with some intermediate products formed upon the thermolysis of Ru3(CO)12. This tentative mechanism might be corroborated by the results of DFT calculations which suggest that among all the possible products of thermolysis of Ru3(CO)12, Ru6C(CO)17 has the lowest reactivity, while the other complexes Ru6C(CO)23−n should be less stable towards further chemical transformations.16
Although carbido complexes have been well-known in ruthenium chemistry for a long time,17 the herein reported μ-carbido-dimer 2 is exceptionally rare in the chemistry of ruthenium complexes with tetrapyrrolic ligands. It is limited to the single example of the unsubstituted (PcRu)2(μ-C), reported by Homborg et al. in 1997.13,18 This complex was synthesized by the reaction of PcRuCl with chloroform as a source of a carbon atom in the presence of KOH in i-PrOH and it is poorly soluble in organic solvents, which limits its possible applications. In contrast, the μ-carbido complex 2 bearing a substituted ligand formed in an unprecedented one-step procedure starting directly from the phthalocyanine ligand is well-soluble in common solvents which facilitates the investigation of its properties.
Until recently, single-atom bridged bimetallic complexes with porphyrin and phthalocyanine ligands have been regarded as inactive forms in catalysis. However, μ-oxo diiron phthalocyanines were shown to be effective catalysts for the oxidation of aromatic compounds to quinones.19 In turn, μ-nitrido diiron phthalocyanine and porphyrin complexes were found to exhibit a rare catalytic activity20 including oxidation of methane21 and aromatic compounds,22 oxidative dechlorination23 and defluorination24 as well as the formation of C–C bonds.25 However, related μ-carbido diiron macrocyclic complexes have never been reported as catalysts for any reaction. In this context, it is greatly of interest to explore the possible catalytic applications of the novel μ-carbido ruthenium dimer. Thus, the catalytic activity of 2 in the cyclopropanation of styrenes and in the carbene insertion into the N–H bond of anilines using ethyl diazoacetate (EDA) as a carbene donor was investigated (Scheme 2). These reactions provide an access to cyclopropanes or unnatural amino acids and N-heterocyclic compounds, respectively, representing important building blocks used for the synthesis of numerous bioactive molecules.26
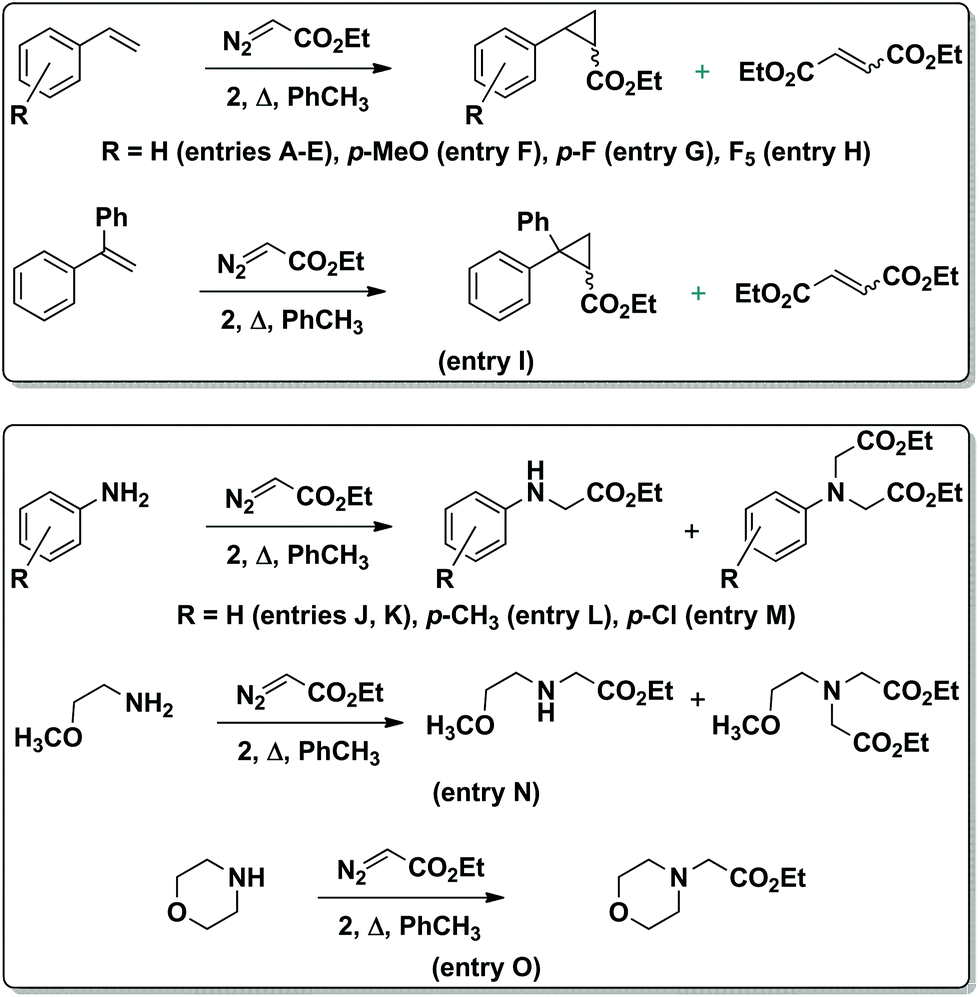 | ||
| Scheme 2 Carbene transfer from EDA to aromatic olefins (top) and amines (bottom), catalyzed by complex 2. | ||
The catalytic activity of 2 (0.1 mol% loading) was initially studied using 1 M styrene solution in toluene and 1.2 equiv. EDA (Table 1). To limit the formation of diethyl maleate and diethyl fumarate byproducts formed from the dimerization of EDA in a ∼90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio, the carbene precursor was slowly added to the reaction mixture. This allowed increasing the yield of ethyl 2-phenylcyclopropane-1-carboxylate from 66% to 84% at 70 °C (Table 1, entries B–D).
:10 ratio, the carbene precursor was slowly added to the reaction mixture. This allowed increasing the yield of ethyl 2-phenylcyclopropane-1-carboxylate from 66% to 84% at 70 °C (Table 1, entries B–D).
a
| Entry | Substrate | T, °C | Addition of EDA, h | Cyclopropanationb | EDA dimerisationc | Cyclopropanes/dimers molar ratio | ||
|---|---|---|---|---|---|---|---|---|
| Yield, % | trans/cis | Yield, % | cis/trans | |||||
| a Reactions were carried out under Ar in toluene using 1 equiv. of olefin (1 M), 1.2 equiv. of EDA (1.2 M) added by using a syringe pump over the desired time and 0.1 mol% catalyst (1 mM). b Yield of cyclopropanation products is based on the amount of the substrate. c Total yield of diethyl maleate and diethyl fumarate is based on the EDA amount. | ||||||||
| A | Styrene | 25 | 2 | 3 | 74:26 |
25 | 88:12 |
0.2:1 |
| B | Styrene | 70 | 2 | 66 | 71:29 |
45 | 88:12 |
2.4:1 |
| C | Styrene | 70 | 4 | 77 | 68:32 |
40 | 88:12 |
3.2:1 |
| D | Styrene | 70 | 6 | 84 | 70:30 |
30 | 89:11 |
4.7:1 |
| E | Styrene | 90 | 6 | 92 | 69:31 |
13 | 89:11 |
11.5:1 |
| F | p-Methoxystyrene | 90 | 6 | 100 | 75:25 |
7 | 90:10 |
25:1 |
| G | p-Fluorostyrene | 90 | 6 | 95 | 70:30 |
9 | 89:11 |
19:1 |
| H | Pentafluorostyrene | 90 | 6 | 68 | 74:26 |
21 | 95:5 |
5.2:1 |
| I | 1,1-Diphenylethene | 90 | 6 | 100 | — | 3 | 92:8 |
50:1 |
The yield of the cyclopropanation product was further improved to 92% at 90 °C (Table 1, entry E). In the absence of catalyst, 4% and 12% styrene conversions were obtained at 70 °C and 90 °C, respectively, thus indicating the involvement of 2 in product formation. The UV-Vis spectrum of the reaction mixture during the cyclopropanation of styrene in toluene exhibited the same maximum at 618 nm as the spectrum of the initial complex 2 indicating that 2 retained the dimeric structure. Aromatic olefins bearing donor and acceptor substituents were cyclopropanated with 95–100% yields. Even in the case of very electron-deficient pentafluorostyrene, a 68% yield of C6F5-containing cyclopropane (entry H) has been achieved. The trans/cis ratio of the cyclopropanation products was insensitive to the olefin nature: ∼70:30 selectivity was observed in all cases. The yield of the cyclopropane product obtained from styrene and EDA in the presence of 2 (92%) was higher than those published for mononuclear Ru(III)PcCl (59%) and Ru(II)PcF16 (80%) complexes.8
The μ-carbido diruthenium complex also catalyzes carbene insertion from EDA into the N–H bonds of aromatic and aliphatic amines (Table 2). Importantly, even with low catalyst loading and very high aniline concentration the μ-carbido ruthenium complex 2 transfers the carbene group to the amine N–H bonds. It should be noted that many catalytic complexes coordinate with the amine substrates and thus become inactivated to reaction with the carbene precursor.3,27 To overcome this limitation, a high concentration of catalyst and low amine concentration should be used, which lead to low turnover numbers and low efficiency. Complex 2 doesn't suffer from this and can be used in low concentration to provide a turnover number up to 1000. The insertion of carbenes into the N–H bonds occurs more cleanly than cyclopropanation. Practically no EDA dimerization products were formed. The reaction mediated by 2 was the most efficient at 90 °C. In the absence of the catalyst the conversion of aniline was limited to 5%. The reaction is tolerant of both electron-donating and electron-withdrawing groups on the aromatic moiety. Substituted anilines converted to mono- and di-insertion products with ∼80:20 ratios. However, 2 is slightly less efficient in the reaction with aliphatic amines (entries N and O) providing a 58% yield of a double insertion product with 2-methoxyethylamine and a 90% yield with morpholine.
a
| Entry | Substrate | [2], mM | Reaction time, h | Yield,b % | RNHCH2COOEt/RN(CH2COOEt)2 ratio |
|---|---|---|---|---|---|
| a Reactions were carried out under Ar in toluene at 90 °C using 1 equiv. of amine (1 M), 1.2 equiv. of EDA (1.2 M) added by using a syringe pump over 2 h. b Yield is based on the amine amount. c Reaction temperature: 70 °C. | |||||
| J | Aniline | 0.2 | 2.5 | 43 | 100:0 |
| K | Aniline | 1 | 2.5 | 100 | 83:17 |
| L | p-Methylaniline | 1 | 17 | 100 | 79:21 |
| M | p-Chloroaniline | 1 | 7 | 100 | 83:17 |
| Nc | 2-Methoxyethylamine | 1 | 30 | 58 | 0:100 |
| O | Morpholine | 1 | 6 | 90 | — |
Previously, dinuclear Ru(I) and Ru(II) complexes with carboxylate, hydrido, silyl, phosphane, arene, cyclopentadienyl and multidentate nitrogen ligands have been used as catalysts for carbene transfer reactions.28 To the best of our knowledge, carbene transfer reactions catalyzed by Ru in the high oxidation state are rare. Simonneaux and coworkers have used Ru(VI) dioxo porphyrin complexes for the cyclopropanation of styrenes, but active ruthenium carbene species was formed after the reduction of Ru(VI) to Ru(II) complexes with EDA.29 Similarly, Gross and co-workers proposed that μ-oxo diiron(IV) and mononuclear Fe(IV) corrole complexes should be initially reduced to the Fe(III) state to react with EDA.30 In the present case, Ru(IV)CRu(IV) phthalocyaninate appears to retain a high oxidation state. For this reason, the activation of EDA occurs only at elevated temperatures. Our attempts to detect a putative active carbene complex formed by 2 were unsuccessful.
In conclusion, although several types of ruthenium complexes have emerged as suitable catalysts for carbene transfer reactions,3,28 μ-carbido diruthenium macrocyclic complexes have not been previously considered as catalysts. The original interest in the μ-carbido complex 2 was fuelled by the observation of the particular catalytic properties of the related μ-nitrido and μ-oxo complexes.19–25 This study represents the first example of the catalytic application of dimeric μ-carbido Ru(IV) phthalocyaninate exemplified by the carbene insertion reactions into olefinic bonds and N–H bonds of amines. Although the catalytic activity is still modest, the results obtained suggest that μ-carbido diruthenium phthalocyanine can be added to the family of catalytically active single-atom bridged bimetallic macrocyclic complexes and might find interesting applications in catalysis. Recent results from Che's group on the remarkable catalytic properties of Ru(II) porphyrin complexes bearing N-heterocyclic carbene ligands in the carbene and nitrene transfer reactions31 support the validity of our approach. Apart from the catalytic applications, the reported approach to μ-carbido ruthenium(IV) complexes might also attract attention from the viewpoint of elaboration of novel optoelectronic materials.32
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Foundation of President of Russian Federation for support of young scientists and leading scientific schools (grant MK-141.2017.3), the Program of RAS (I.14) and Agence Nationale de Recherches, France (grant ANR-16-CE29-0018-01). AGM is grateful to the French Embassy in Moscow for the Mechnikov Fellowship. LPC is indepted to Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche, France, for the PhD fellowship.Notes and references
- (a) D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 2012, 569–580 CrossRef CAS; (b) P. Fackler, S. M. Huber and T. Bach, J. Am. Chem. Soc., 2012, 134, 12869–12878 CrossRef CAS PubMed; (c) K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed.
- (a) Z. Gross and S. Ini, Inorg. Chem., 1999, 38, 1446–1449 CrossRef CAS; (b) C.-J. Liu, W.-Y. Yu, C.-M. Che and C.-H. Yeung, J. Org. Chem., 1999, 64, 7365–7374 CrossRef CAS; (c) C. Wang, K. V. Shalyaev, M. Bonchio, T. Carofiglio and J. T. Groves, Inorg. Chem., 2006, 45, 4769–4782 CrossRef CAS PubMed.
- D. Intrieri, D. M. Carminati and E. Gallo, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2016, pp. 1–99 Search PubMed.
- P. Zardi, D. Intrieri, D. M. Carminati, F. Ferretti, P. Macchi and E. Gallo, J. Porphyrins Phthalocyanines, 2016, 20, 1156–1165 CrossRef CAS.
- (a) I. Nicolas, P. Le Maux and G. Simonneaux, Tetrahedron Lett., 2008, 49, 5793–5795 CrossRef CAS; (b) S. Fantauzzi, A. Caselli and E. Gallo, Dalton Trans., 2009, 5434–5443 RSC; (c) W. Xiao, J. Wei, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2013, 49, 4619–4621 RSC.
- M. W. Wolf, D. A. Vargas and N. Lehnert, Inorg. Chem., 2017, 56, 5623–5635 CrossRef CAS PubMed.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- H.-H. Liu, Y. Wang, Y.-J. Shu, X.-G. Zhou, J. Wu and S.-Y. Yan, J. Mol. Catal. A: Chem., 2006, 246, 49–52 CrossRef CAS.
- (a) T. Rawling and A. M. McDonagh, Coord. Chem. Rev., 2007, 251, 1128–1157 CrossRef CAS; (b) Y. G. Gorbunova, Y. Y. Enakieva, S. G. Sakharov and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2003, 7, 795–800 CrossRef CAS.
- (a) M. S. Rodríguez-Morgade, M. Planells, T. Torres, P. Ballester and E. Palomares, J. Mater. Chem., 2008, 18, 176–181 RSC; (b) A. N. Cammidge, G. Berber, I. Chambrier, P. W. Hough and M. J. Cook, Tetrahedron, 2005, 61, 4067–4074 CrossRef CAS.
- Y. Y. Enakieva, Y. G. Gorbunova, A. Y. Tsivadze, C. Stern and R. Guilard, J. Porphyrins Phthalocyanines, 2007, 11, 883–890 CrossRef CAS.
- S. Omiya, M. Tsutsui, E. F. Meyer, I. Bernal and D. L. Cullen, Inorg. Chem., 1980, 19, 134–142 CrossRef CAS.
- A. Kienast, L. Galich, K. S. Murray, B. Moubaraki, G. Lazarev, J. D. Cashion and H. Homborg, J. Porphyrins Phthalocyanines, 1997, 1, 141–157 CrossRef CAS.
- (a) D. O. Oluwole, A. V. Yagodin, N. C. Mkhize, K. E. Sekhosana, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and T. Nyokong, Chem. – Eur. J., 2017, 23, 2820–2830 CrossRef CAS PubMed; (b) A. G. Martynov, J. Mack, B. P. Ngoy, T. Nyokong, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2017, 140, 469–479 CrossRef CAS.
- D. Braga, F. Grepioni, P. J. Dyson, B. F. G. Johnson, P. Frediani, M. Bianchi and F. Piacenti, J. Chem. Soc., Dalton Trans., 1992, 2565–2571 RSC.
- C. Li, J. Xu, J. Zhao, D. Tian and R. B. King, Dalton Trans., 2010, 39, 10697–10701 RSC.
- (a) L. Ma, G. K. Williams and J. R. Shapley, Coord. Chem. Rev., 1993, 128, 261–284 CrossRef CAS; (b) S. Takemoto and H. Matsuzaka, Coord. Chem. Rev., 2012, 256, 574–588 CrossRef CAS.
- A. Kienast, C. Bruhn and H. Homborg, Z. Anorg. Allg. Chem., 1997, 623, 967–972 CrossRef CAS.
- (a) H. M. Neu, V. V. Zhdankin and V. N. Nemykin, Tetrahedron Lett., 2010, 51, 6545–6548 CrossRef CAS; (b) O. V. Zalomaeva, I. D. Ivanchikova, O. A. Kholdeeva and A. B. Sorokin, New J. Chem., 2009, 33, 1031–1037 RSC; (c) C. Pergrale and A. B. Sorokin, C. R. Chimie, 2000, 3, 803–810 CAS.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- (a) Ü. İşci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063–5075 RSC; (b) A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- Ü. İşci, P. Afanasiev, J.-M. M. Millet, E. V. Kudrik, V. Ahsen and A. B. Sorokin, Dalton Trans., 2009, 7410–7420 RSC.
- P. Afanasiev, D. Bouchu, E. V. Kudrik, J.-M. M. Millet and A. B. Sorokin, Dalton Trans., 2009, 9828–9836 RSC.
- C. Colomban, E. V. Kudrik, P. Afanasiev and A. B. Sorokin, J. Am. Chem. Soc., 2014, 136, 11321–11330 CrossRef CAS PubMed.
- L. X. Alvarez, E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2011, 17, 9298–9301 CrossRef CAS PubMed.
- (a) P. Tang and Y. Qin, Synthesis, 2012, 44, 2969–2984 CrossRef CAS; (b) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- B. J. Anding and L. K. Woo, Organometallics, 2013, 32, 2599–2607 CrossRef CAS.
- G. Maas, Chem. Soc. Rev., 2004, 33, 183–190 RSC.
- E. Galardon, P. Le Maux and G. Simonneaux, Chem. Commun., 1997, 927–928 RSC.
- L. Simkhovich, A. Mahammed, I. Goldberg and Z. Gross, Chem. – Eur. J., 2001, 7, 1041–1055 CrossRef CAS.
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed.
- Y. G. Gorbunova, A. D. Grishina, A. G. Martynov, T. V. Krivenko, A. A. Isakova, V. V. Savel'ev, S. E. Nefedov, E. V. Abkhalimov, A. V. Vannikov and A. Y. Tsivadze, J. Mater. Chem. C, 2015, 3, 6692–6700 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt03703a |
| This journal is © The Royal Society of Chemistry 2017 |