
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile generation of iridium PCcarbeneP pincer complexes via water elimination from an alcohol proligand†
Simon
Sung
 and
Rowan D.
Young
*
and
Rowan D.
Young
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: rowan.young@nus.edu.sg
First published on 24th October 2017
Abstract
We report the facile generation of Ir PCcarbeneP pincer systems. These systems are accessed from the reaction between [IrCl(COD)]2 and a bis(diphenyl)phenylene P(OH)P proligand (1) with concomitant dehydration, followed by salt metathesis/ligand exchange in the case of cationic examples. In contrast to previously reported double C–H activation synthetic strategies to access similar complexes, accessing Ir PCcarbeneP complexes through dehydration proceeds rapidly at room temperature and provides the first example of the incorporation of phosphino aryl substituents. The generated complexes are shown to possess the ability to activate inert C–H bonds and partake in ligand cooperativity. Mechanistic evidence suggests that divergent C–H and O–H activation pathways of ligand 1 ultimately lead to the same Ir PCcarbeneP product (2). It is hoped that the stability and synthetic accessibility of these complexes will encourage their increased use in catalyst surveys.
Introduction
Pincer ligands are tridentate meridional ligands that offer unique rigidity, selective activity and complex stability for transition metal centers.1 Such ligands have played an instrumental role in the development of transition metal catalysts capable of performing difficult bond transformations. Among various pincer ligand scaffolds, PCP type pincers, containing a metal–carbon bond, enable the exploits of organometallic chemistry to be invoked for a range of bond transformations and/or catalysis. PCP pincers can be classified based on the hybridization of the metal bound carbon donor, namely as either sp3 or sp2 (sp PCP pincers are yet to be reported).The sp2 PCP pincer ligand class is dominated by aromatic based designs, where the central carbon donor belongs to an aromatic system. However, PCcarbeneP pincers, where the central carbon is an alkylidene donor, have displayed unique reactivity due to their extremely strong trans effect and their ability to partake in ligand–metal cooperativity.2,3
PCcarbeneP pincers have been readily accessible for Ir, Ru and Os centres since early reports by Shaw, and then Gusev demonstrated double C–H activation.4 However, the alkyl backbones of such systems were unstable and conducive to β-hydride elimination. More recently, Ozerov (and later Piers) introduced β-hydride elimination resistant PCcarbeneP ligands.5 Since then, the metals accommodated in PCcarbeneP scaffolds have expanded to include Ni, Pd and Rh.2a,g,6 However, accessing PCcarbeneP pincers through double C–H activation is limited to noble metals well-known for their C–H activation abilities (Ni PCcarbeneP complexes were accessed through HX elimination). Perhaps because of this, much attention has been focused on various designs of iridium PCcarbeneP pincer complexes (Fig. 1).
 | ||
| Fig. 1 (Above) Ir sp2 PCP architectures based on alkylidenyl and aryl carbon attachments. (Below) Examples of selected PCcarbeneP architectures. | ||
Reports have demonstrated the ability of iridium PCcarbeneP pincer complexes to reversibly activate E–H bonds (E = H, O, N, C), perform challenging catalysis, and to partake in difficult redox and catalytic processes.7 The exploration of iridium PCcarbeneP pincer complexes has led to a variety of pincer designs incorporating various carbocycles and heterocycles into the spinal positions, and featuring a range of phosphino alkyl substituents. However, phosphino donors with aryl substituents are yet to be reported, despite their higher stability, affordability and easier synthetic/commercial access as compared to alkyl phosphines. This is likely due to the need for electron rich metal centres to induce double C–H activation. As such, the powerful reactivity presented by iridium PCcarbeneP pincer complexes has only been utilized by synthetic organometallic groups capable of synthesizing and handling alkyl phosphino PCcarbeneP pincer proligands.
We have recently reported the protonolysis of rhodium α-hydroxyalkyl complexes to access PCcarbeneP pincer complexes, avoiding double C–H activation.8 Herein, the formal dehydration of an air-stable bis(diphenylphosphino) alcohol POP pro-ligand to an iridium PCcarbeneP pincer complex is detailed. Competing C–H and O–H activation is suggested with the isolation of a rare α-hydroxylalkyl complex and an iridium alkoxide intermediate. Although other PCcarbeneP iridium complexes featuring diaryl phosphino substituents have yet to be reported in the literature, the activity of the iridium PCcarbenePPh platform is demonstrated with ligand exchange and C–H activation chemistry under mild conditions.
Results and discussion
Addition of compound 1 to [IrCl(COD)]2 results in rapid generation of the PCcarbeneP complex 2 and liberation of H2O and 1,5-cyclooctadiene (COD) (Scheme 1). At room temperature, 31P NMR suggested that conversion to 2 was >50% after 15 minutes. In comparison, double C–H activation approaches to generate Ir PCcarbenePalkyl complexes require prolonged heating (hours) above 100 °C.5b Compound 2 was isolated in high yield (76%) via precipitation with n-hexane. | ||
| Scheme 1 Synthesis of PCcarbeneP iridium complex 2via dehydration of ligand 1. Metathesis of 2 with Na[BArF4] in the presence of PPh3 or PCy3 generates 3 and 4 respectively. | ||
Compound 2 possesses a single 31P NMR resonance at δP 29.0, and a 1H NMR spectrum of 2 displays only aryl proton resonances, which provide little definitive evidence for the identity of 2. However, 13C NMR spectroscopic data for 2 revealed a highly deshielded triplet signal at 207.4 ppm (2JCP = 2.8 Hz), supporting the assigned alkylidene attachment. X-ray quality crystals of 2, grown by vapour diffusion between n-hexane and a concentrated solution of 2 in DCM at room temperature, allowed a diffraction study to be performed. From this, the determined molecular structure of 2 (Fig. 2) confirmed the formation of the PCcarbeneP backbone, with an Ir1–C1 bond length of 1.940(2) Å suggestive of Ir–C double bond character (cf. 1.899(7) Å for the PCcarbenePiPr analogue).5b This Ir![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length lies within the range of previously reported iridium PCcarbeneP complexes, with a minimum observed value of 1.86(1) Å and a maximum value of 2.038(9) Å.6b,7c
C bond length lies within the range of previously reported iridium PCcarbeneP complexes, with a minimum observed value of 1.86(1) Å and a maximum value of 2.038(9) Å.6b,7c
 | ||
| Fig. 2 Molecular structure of 2. Hydrogen atoms omitted, thermal ellipsoids shown at 50%. Selected bond distances (Å) and angles (°): Ir1–P1, 2.291(1); Ir1–C1, 1.940(2); Ir1–Cl1, 2.350(3); C1–Ir1–Cl1, 180.0(1); P1–Ir1–P2, 166.0(1). | ||
The molecular structure of 2 reveals that 2 possesses C2 symmetry, as opposed to C2v symmetry, as often observed in aryl PCP pincer complexes. Consequently, 2 exists as a racemic mixture of R and S conformers.
Cationic iridium PCcarbeneP complexes could be generated via salt metathesis between Na[BArF4] and compound 2 in the presence of a suitable ligand. Such methodology has been previously described by Piers.6b,9 Thus, compounds 3 and 4 were generated by the addition of Na[BArF4] to an equimolar amount of 2 and either PPh3 or PCy3 respectively (Scheme 1). The molecular structures of 3 (Fig. 3) and 4 (Fig. 4) reveal slightly elongated Ir1–C1 distances in the cationic complexes {1.994(8) Å in 3 and 1.953(8) Å in 4}, reflecting the sensitivity of the π-acidic alkylidene linkage to electron density change at the iridium centre due to the π-basic nature of the chloride ligand in 2 and the cationic nature of complexes 3 and 4.
 | ||
| Fig. 3 Molecular structure of 3. Hydrogen atoms and anion omitted, thermal ellipsoids shown at 50%. Selected bond distances (Å) and angles (°): Ir1–P1, 2.298(2); Ir1–C1, 1.994(8); Ir1–P2, 2.282(2); Ir1–P3, 2.394(2); C1–Ir1–P3, 178.3(3); P1–Ir1–P2, 164.1(1). | ||
 | ||
| Fig. 4 Molecular structure of 4. Hydrogen atoms and anion omitted, thermal ellipsoids shown at 50%. Selected bond distances (Å) and angles (°): Ir1–P1, 2.269(2); Ir1–C1, 1.953(8); Ir1–P2, 2.317(2); Ir1–P3, 2.423(2); C1–Ir1–P3, 169.2(2); P1–Ir1–P2, 158.0(1). | ||
The 13C NMR resonances arising from the carbenic carbon positions in compounds 2, 3 and 4 follow the trend that 2 (δc 207.4) < 4 (δc 231.9) < 3 (δc 241.6). This trend roughly correlates with IrC bond lengths, and reflects an increase in ‘free’ carbene character from 2 to 4 to 3.10
Metathesis of 2 in the presence of two equivalents of PPh3 led to the formation of metallacycle 5 (Fig. 5). Compound 5 could also be generated by adding an equivalent of PPh3 to isolated 3 (Scheme 2). Thus, 5 is likely generated via coordination of PPh3 to the electrophilic carbene position in 3 and subsequent cyclometallation at the iridium centre. Cyclometallation of PPh3 is well-documented on iridium,11 but iridium-alkylidene cooperative cyclometallation is less reported.12 Such a ligand directed substrate activation mirrors cooperative PPh3 C–H activation and CO2 activation on related ruthenium vinylidene and carbodiphosphorane complexes.13
 | ||
| Fig. 5 Molecular structure of 5. Hydrogen atoms (except H1) and anion omitted, thermal ellipsoids shown at 50%. H1 was located in a Fourier Difference map. Selected bond distances (Å) and angles (°): Ir1–P1, 2.343(1); Ir1–C1, 2.232(3); Ir1–P2, 2.397(1); Ir1–P4, 2.343(1); Ir1–C43, 2.095(3); C1–P3, 1.840(4); C1–Ir1–P4, 172.5(1); P1–Ir1–C43, 162.53(9). | ||
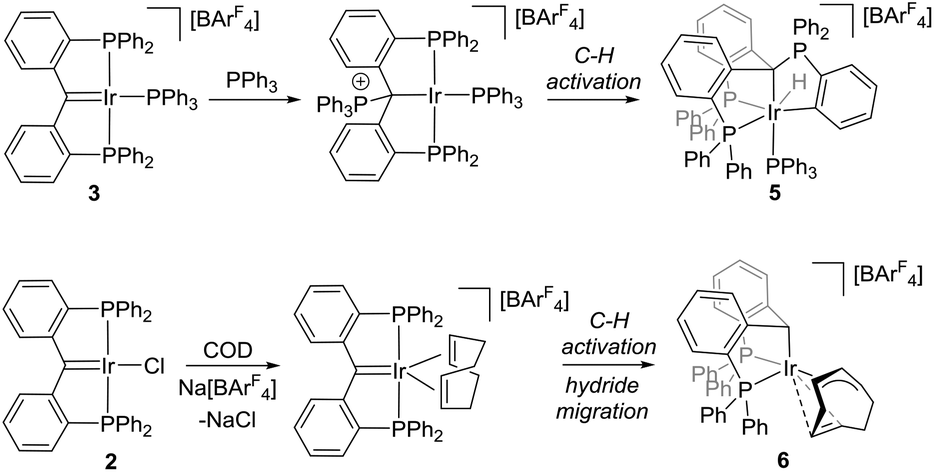 | ||
| Scheme 2 (Above) Reaction of 3 with PPh3 results in ligand cooperative C–H activation of PPh3. (Below) A cationic PCcarbeneP intermediate, generated via metathesis of Na[BArF4] with 2, C–H activates 1,5-cyclooctodiene generating iridium(III) allyl 6. | ||
We recently reported the rhodium analogue of 3, [PCcarbenePPhRh(PPh3)][BArF4].8a Although the rhodium carbene position was found to be electrophilic in this complex, it was stable in the presence of excess PPh3, even upon heating. By comparison, the carbene position in 3 proves to be much more electrophilic than its rhodium analogue. This is somewhat expected, given that iridium stablises the singlet state of the carbene ligand to a greater extend than rhodium.
Metathesis of 2 in the presence of 1,5-cyclooctadiene as the supporting ligand led to product 6 (Scheme 2). A molecular structure of compound 6 (Fig. 6) reveals that the COD ligand had undergone C–H activation, resulting in an allylic coordination. The concomitantly generated hydrido group then transfers from the iridium centre to the carbene ligand transforming the pincer into a facially coordinated PCsp3P ligand, which is also evident by 1H NMR analysis that reveals a resonance at 4.59 ppm correlating to this hydrogen. The resulting Ir–C bond distance in the PCsp3P ligand of 6 is observed at a increased length of 2.158(4) Å (cf. 1.890(4) Å in 2).
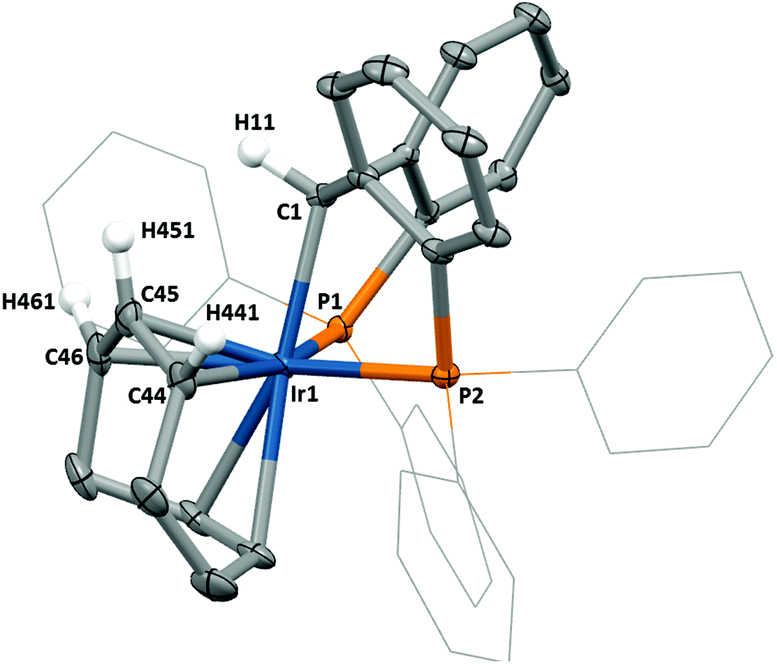 | ||
| Fig. 6 Molecular structure of 6. Hydrogen atoms (except H11, H441, H451 and H461) and anion omitted, thermal ellipsoids shown at 50%. Selected bond distances (Å) and angles (°): Ir1–P1, 2.298(1); Ir1–C1, 2.158(4); Ir1–P2, 2.328(1); C44–C45, 1.422(7); C45–C46, 1.420(7); C1–Ir1–P1, 80.7(1); P1–Ir1–P2, 102.2(1). | ||
The activation of C–H bonds, and also C–C bonds, in iridium dienes is well known in accessing resonance stabilised ligands.14
Compound 6 could also be generated directly by heating compound 1 and [Ir(COD)2][BArF4] at 95 °C for 18 hours. The stability of 6 supports the premise that electron poor iridium centres perform poorly at α-hydrogen elimination. In sharp contrast, cationic iridaepoxide complexes, or intermediates en route to 2 (see below) readily undergo α-hydroxyl elimination, suggesting that this may be a more facile process.9
It was found that the presence of PPh3 arrested the reaction between 1 and [IrCl(COD)]2, and prevented formation of 2. As such, treatment of compound 1 with [IrCl(COD)PPh3] at room temperature followed by heating at 55 °C for 2 days led to a mixture of two products, alkoxide 7a and α-hydroxylalkyl 7b in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio according to the relative integrations by 31P NMR spectroscopy (Scheme 3). Fractional crystallization allowed the isolation and characterization of each compound.
:4 ratio according to the relative integrations by 31P NMR spectroscopy (Scheme 3). Fractional crystallization allowed the isolation and characterization of each compound.
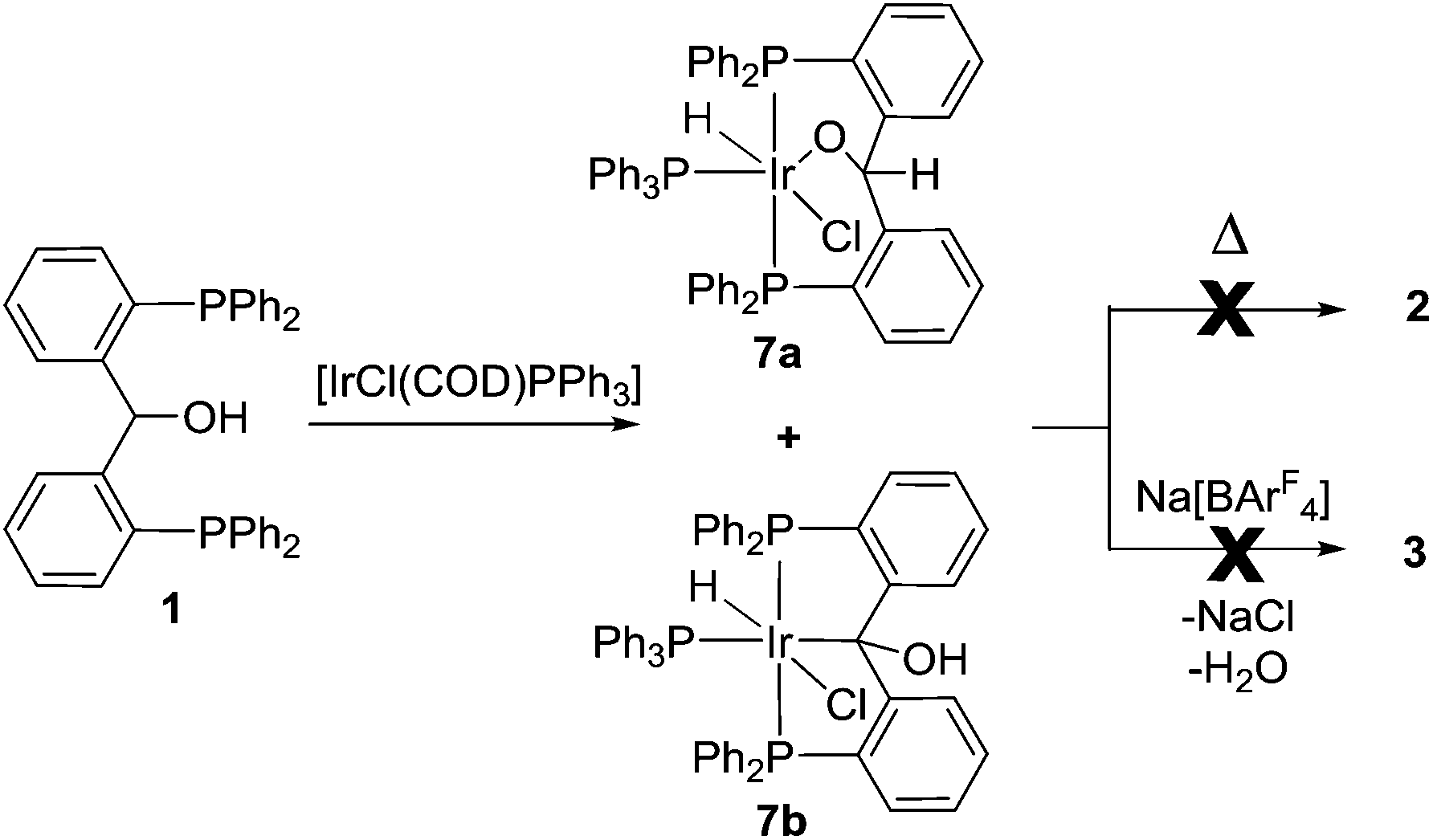 | ||
| Scheme 3 PPh3 arrests the dehydration of 1, with formation of 7a and 7b (1:4 ratio). Heating 7a or 7b failed to generate 2, and reaction with Na[BArF4] failed to generate 3. | ||
The structure of 7a was established via1H, 13C and 31P NMR spectroscopies. Correlation spectroscopy confirmed the formulation of 7a as an alkoxide, with a 13C NMR resonance at δc 78.7 correlating strongly to a methine proton at δH 5.05 in a HSQC experiment (see ESI†). Strong 31P coupling observed for the signal at δH 5.05 suggested a trans PPh3 position. The mer configuration of the pincer was established by 31P NMR, where two signals were observed in a 2:1 ratio at δP −6.1 (2 P, d, 2JPP = 11.4 Hz) and −1.2 (1 P, t, 2JPP = 11.4 Hz).
Compound 7b represents a direct route to access an iridium α-hydroxylalkyl, with previous reported examples either relying on formation of the α-hydroxyalkyl moiety within the metal coordination sphere, or being resonance supported forms better described as protonated β-diketones.15 X-ray diffraction quality crystals of 7b allowed the determination of its molecular structure (Fig. 7). The structure of 7b reveals that it is coordinatively saturated (Oh geometry), preventing potential α-hydroxyl elimination.
 | ||
| Fig. 7 Molecular structure of 7b. Hydrogen atoms (except H1 and H11) omitted, thermal ellipsoids shown at 50%. H1 and H11 were located in a Fourier Difference map. Selected bond distances (Å) and angles (°): Ir1–P1, 2.296(2); Ir1–C1, 2.195(9); Ir1–P2, 2.310(2); Ir1–P3, 2.369(3); Ir1–Cl1, 2.499(2); C1–Ir1–P3, 175.6(3); P1–Ir1–P2, 154.0(1). | ||
In order to investigate which of 7a or 7b represents a more likely model intermediate for the formation of 2, samples of each were heated to promote PPh3 dissociation. However in both cases, our inability to eliminate coordinated PPh3 prevented transformation of 7a or 7b into 2.
Addition of Na[BArF4] to either complexes 7a or 7b readily led to metathesis, but failed to generate a cationic iridium PCcarbeneP complex (i.e.3) even when heated to 80 °C. This is in contrast to previously described rhodium analogues, and may suggest against a proton transfer mechanism for dehydration. In the case of 7a, the known cation fragment [Ir(CO)(PPh3)3][BArF4]16 was generated as the sole product, whereas 7b decomposed into multiple unknown products. Piers has reported the decomposition of [κ3-P′(η2-CO)P′′IrCl] iridaepoxides into related [IrCl(CO)(PR3)2] products, suggesting a plausible decomposition route that proceeds via the β-hydride elimination in 7a.17
Monitoring of the reaction between 1 and [IrCl(COD)]2 at various temperatures between 253 K and 298 K revealed that intermediate complex I forms prior to any bond activation (Scheme 4). The 1H NMR spectrum of complex I at 263 K displays a downfield signal at 11.07 ppm that has been associated with a C–H/metal anagostic interaction in related rhodium intermediates.6a,8a However, 1H–13C NMR correlation experiments, and isotopic labelling experiments suggested the signal was due to the OH motif (see ESI†). Thus, the interaction between the linkage of ligand 1 and the metal centre in complex I can not be defined with any certainty.
 | ||
| Scheme 4 Possible reaction mechanism for the formation of 2. * denotes κ1-COD coordination. N. D. denotes not detected by NMR spectroscopy. | ||
Given that compound 1 has been shown to be susceptible to O–H and C–H activation (i.e. in generation of 7a–b), it is possible that both intermediates II and VI shown in Scheme 4 could be produced upon C–H or O–H activation (respectively) of the chelate ligand in I, representing divergent reaction pathways.
At 253 K, as complex I diminishes in concentration, two independent iridium hydride species are observed, complexes II and VI. Intermediate II was characterized by 1H and 31P NMR spectroscopy, and by reaction with isotopologues 1a and 1b that contained deuterated methine and hydroxyl positions respectively (see ESI†). Intermediate II supports a C–H activation pathway (pathway A, Scheme 4) that proceeds via an α-hydroxylalkyl complex. In contrast to 7b, complex II is characterized by a fac coordination of the α-hydroxyalkyl ligand. The flexibility of PCsp3P ligands, related to 1, to adopt both fac and mer configurations is well-documented.18 As the signal intensities from II diminish, signals for product 2 grow in intensity.
Isomerisation of the tridentate PCP ligand from fac to mer generates III. Concomitant dissociation of COD would allow this process to proceed via a 5-coordinate intermediate, as has been reported for related Rh(III) POP pincer systems.19
As stated above, addition of Na[BArF4] to either complexes 7a or 7b failed to generate a cationic iridium PCcarbeneP complex (i.e.3). This may imply that formation of 2 proceeds via α-hydroxyl elimination in III to give IV, as suggested by Piers,2b,c,9 rather than proton transfer from iridium to the α-hydroxylalkyl position to give V, which was observed for more Brønsted acidic rhodium examples.8a α-Alkyl elimination in closely related iridium PCP complexes has been directly observed by Wendt.20 The product of C–O activation, intermediate IV (Scheme 4), can then undergo H/OH reductive elimination to give 2 and eliminate water.
The hydrido complex VI was observed simultaneously with II, and represents the β-hydrogen elimination product of an initial O–H activation intermediate (V) for pathway B (Scheme 4). Intermediate VI was characterized by 1H, 31P NMR spectroscopy, and reaction with isotopologues 1a and 1b. Although the hydride positions in VI are inequivalent, dynamic exchange between the positions gives rise to a single triplet signal at δH −12.57. The possibility of VI existing as a dihydrogen complex was precluded by the absence of any observable D–H coupling while employing isotopologues 1a–b. Furthermore, VI displays an η2-carbonyl 13C NMR signal at δC 132.1, more indicative of an iridium(III) oxidation state.2c
Intermediate VI can in-principle also be generated from β-hydride elimination from an α-hydroxylalkyl ligand (i.e. from III of pathway A, Scheme 4). Indeed, this likely marks the convergence of pathways A and B. However, using the isotopologue 1a (methine position deuterated), very little of complex II is generated, and much higher concentrations of VI are observed, indicative of a notable kinetic isotope effect for C–H activation. From this reaction solution, X-ray quality crystals of complex VII precipitated. Structural characterization of VII demonstrates it to be an O–H activation product arising from HCl elimination from V (Scheme 4 inset). Indeed, addition of ethereal HCl to crystals of VII generated product 2 and intermediates I, II and VI, which indicates the presence of equilibria between species of pathways A and B.
Further evidence for the identity of VI is garnered from the addition of the dehydrogenated, keto form of 1 (1-H2), with [IrCl(COD)]2 under a H2 atmosphere. At room temperature, intermediates VI and VII are readily identified as the major species after 5–10 minutes, after which time 2 is generated. However, this does not represent a practical synthesis of 2, as it was found that 2 further reacts with H2 to give hydrogenation products.
Conclusions
In conclusion, we have reported a facile method of accessing iridium PCcarbeneP complexes via dehydration of an alcoholic bisphosphino proligand. Mechanistic studies suggest two competing, divergent C–H and O–H activation pathways, ultimately leading to the same iridium PCcarbeneP product (2). The above described reactivity demonstrates the activity of the PCcarbenePPh platform. This system is able to partake in ligand–metal cooperativity to perform difficult bond activations such as C–H activation, as demonstrated in the generation of compounds 5 and 6. Given recent reports of the use of PCcarbeneP iridium complexes in catalysis, the ability to access this ligand platform from stable, commercially available (or simple to synthesise and store) components allows synthetic chemists a potentially new catalytic tool for difficult transformations that have already been demonstrated in-practice using previously reported PCcarbeneP complexes.Experimental
See ESI† for general experimental conditions.Preparation of complex 2
A solution of [IrCl(COD)]2 (134.3 mg, 0.20 mmol) in DCM (3 mL) was treated with a solution of compound 1 (221.0 mg, 0.40 mmol) in DCM (3 mL) at room temperature to give a green coloured solution and then stirred at room temperature for 30 h in a closed vessel. The solution was filtered and then the filtrate was evaporated. The residue was washed with n-hexane (3 × 10 mL) and then dissolved in DCM (3 mL), layered with n-hexane (5 mL) and left to stand at room temperature. The resultant green solid of complex 2 were isolated by filtration and then dried under vacuum (231 mg, 76%).
1H NMR (500 MHz, CD2Cl2, 298 K) δH 6.85 (2 H, t, J = 7.4 Hz, Ar–H), 7.34–7.41 (2 H, m, Ar–H), 7.41–7.57 (12 H, m, PPh2 H′s), 7.77–8.02 (8 H, m, PPh2 H′s), 8.20 (2 H, d, J = 7.6 Hz, Ar–H), 8.26–8.31 (2 H, m, Ar–H). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K) δC 124.6 (t, J = 7.3 Hz), 129.1 (t, J = 5.2 Hz), 129.2 (s), 131.0 (s), 132.1 (t, J = 25.0 Hz), 133.8 (s), 134.6 (t, J = 6.9 Hz), 137.0 (s), 138.5 (t, J = 24.1 Hz), 174.8 (t, J = 19.2 Hz), 207.4 (t, 2JCP = 2.8 Hz, IrC). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K) δP 29.0 (2 P, s, PCP pincer P′s). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C37H29ClIrP2 763.1050; found 763.1021. Elemental analysis: calc. for C37H28ClIrP2: C, 58.3; H, 3.7; found: C, 58.1; H, 3.7%.
Preparation of complex 3
A solution of complex 2 (22.9 mg, 0.030 mmol) in DCM (5 mL) at −78 °C was treated dropwise with a solution of Na[BArF4]·2THF (34.0 mg, 0.033 mmol) in DCM (5 mL) and then a solution of PPh3 (7.9 mg, 0.030 mmol) in DCM (5 mL). The solution was allowed to slowly come to room temperature and was stirred for an additional 2 hours. The reaction mixture was filtered and then the filtrate was evaporated to approx. 1 mL. The solution was layered with n-hexane (10 mL) and then left to stand at room temperature. Dark green crystals of 3 were isolated by filtration and then dried under vacuum (38 mg, 68%).
1H NMR (500 MHz, CD2Cl2, 298 K) δH 6.77 (6 H, ddd, J = 11.2 Hz, J = 8.2 Hz, J = 1.4 Hz, Ar–H), 6.97 (6 H, td, J = 7.9 Hz, J = 2.1 Hz, Ar–H), 7.05 (2 H, t, J = 7.6 Hz, Ar–H), 7.20–7.29 (10 H, m, Ar–H), 7.29–7.49 (15 H, m, Ar–H), 7.56 (4 H, s, [BArF4] Ar–H), 7.73 (8 H, s, [BArF4] Ar–H), 7.96 (2 H, d, J = 7.8 Hz, Ar–H), 8.35 (2 H, td, J = 7.5 Hz, J = 1.3 Hz, Ar–H). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K) δC 117.7–118.1 (m, [BArF4] Ar–C), 121.5–128.3 (m), 128.6–129.0 (m), 129.2 (t, J = 5.2 Hz), 129.3–130.2 (m), 131.0 (d, J = 1.8 Hz), 131.7 (s), 133.3 (s), 133.6–133.8 (m), 134.3 (s), 134.6 (s), 134.6–134.9 (m), 135.2 (s, [BArF4] Ar–C), 135.5 (s), 145.3 (d, J = 7.1 Hz), 162.2 (q, 1JCB = 49.7 Hz, [BArF4] Ar–C), 169.3 (td, J = 18.7 Hz, J = 2.9 Hz), 241.6 (d, 2JCP = 72.7 Hz, IrC). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K) δP 14.0 (1 P, t, 2JPP = 16.4 Hz, PPh3), 30.8 (2 P, d, 2JPP = 16.4 Hz, PCP pincer P's). HRMS (ESI-TOF) m/z: [M]+ calcd for C55H43IrP3 989.2206; found 989.2227. Elemental analysis: calc. for C87H55BF24IrP3: C, 56.4; H, 3.0; found: C, 56.0; H, 3.2%.
Preparation of complex 4
A solution of complex 2 (22.9 mg, 0.030 mmol) in DCM (5 mL) at −78 °C was treated dropwise with a solution of Na[BArF4]·2THF (34.0 mg, 0.033 mmol) in DCM (5 mL) and then a solution of PCy3 (8.4 mg, 0.030 mmol) in DCM (5 mL). The solution was allowed to slowly come to room temperature and was stirred for an additional 2 hours. The reaction mixture was filtered and then the filtrate was evaporated to approx. 1 mL. The solution was layered with n-hexane (10 mL) and then left to stand at room temperature. Dark green crystals of 4 were isolated by filtration and then dried under vacuum (43 mg, 77%).
1H NMR (500 MHz, CD2Cl2, 298 K) δH 0.63–0.74 (5 H, m, Cy–H), 0.87–0.95 (4 H, m, Cy–H), 1.03–1.15 (5 H, m, Cy–H), 1.27–1.52 (16 H, m, Cy–H), 1.74–1.94 (3 H, m, Cy–H), 6.93 (2 H, t, J = 7.4 Hz, 2 H, Ar–H), 7.36 (2 H, dt, J = 7.7, 4.0 Hz, Ar–H), 7.50–7.68 (16 H, m, Ar–H), 7.70–7.82 (10 H, m, Ar–H), 7.99 (8 H, q, J = 5.7 Hz, Ar–H), 8.31–8.43 (2 H, m, Ar–H). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K) δC 26.1 (s, Cy–C), 27.2 (d, 2JCP = 10.2 Hz, Cy–C), 30.7 (s, Cy–C), 38.4 (d, 1JCP = 20.1 Hz), 117.8–118.0 (m, [BArF4] Ar–C), 121.7–128.4 (m), 128.8–129.8 (m), 131.2 (t, J = 25.5 Hz), 132.3 (s), 132.9 (t, J = 3.2 Hz), 134.4 (s), 134.9 (s), 135.3 (s, [BArF4] Ar–C), 135.6 (t, J = 6.0 Hz), 145.7 (td, J = 24.9, 6.3 Hz), 162.2 (q, 1JCB = 49.8 Hz, [BArF4] Ar–C), 170.3 (td, J = 18.5, 2.8 Hz), 231.9 (d, 2JCP = 69.5 Hz, IrC). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K) δP 21.2 (1 P, t, 2JPP = 16.9 Hz, PCy3), 26.7 (2 P, d, 2JPP = 16.9 Hz, PCP pincer P′s). HRMS (ESI-TOF) m/z: [M]+ calcd for C55H61IrP3 1007.3620; found 1007.3620. Elemental analysis: calc. for C87H73BF24IrP3: C, 55.9; H, 3.9; found: C, 55.9; H, 3.4%.
Preparation of complex 5
Method A: A solution of complex 2 (22.9 mg, 0.030 mmol) in DCM (5 mL) at −78 °C was treated dropwise with a solution of Na[BArF4]·2THF (34.0 mg, 0.033 mmol) in DCM (5 mL) and then a solution of PPh3 (19.7 mg, 0.075 mmol) in DCM (10 mL). The solution was allowed to slowly come to room temperature and was stirred for an additional 2 hours. The reaction mixture was filtered and then the filtrate was evaporated fully. n-Hexane was added to the residue, triturated and then decanted. This process was repeated two additional times. The remaining residue was dissolved in DCM (1 mL), layered with n-hexane (10 mL) and then left to stand at room temperature. Light yellow crystals of 5 formed from the solution were isolated by filtration and then dried under vacuum (45 mg, 71%).Method B: PPh3 (1.4 mg, 5.5 μmol) was added to a solution of complex 3 (9.3 mg, 5 μmol) in CD2Cl2 (0.6 mL) at room temperature. The reaction solution was mixed well and then left to stand at room temperature overnight. 1H and 31P NMR analyses confirmed the formation of complex 5 in quantitative yield.
1H NMR (500 MHz, CD2Cl2, 298 K) δH −9.63 (1 H, dddd, JHP = 133.1, 20.6, 17.0, 7.6 Hz, Ir–H), 5.985–6.09 (4 H, m, Ar–H), 6.23–6.33 (1 H, m, Ar–H), 6.46 (1 H, t, J = 8.2 Hz, Ar–H), 6.59–6.72 (3 H, m, Ar–H), 6.79–7.22 (37 H, m, Ar–H), 7.24–7.42 (4 H, m, Ar–H), 7.44–7.65 (8 H, m, Ar–H), 7.66–7.84 (10 H, m, Ar–H), 8.17 (1 H, dd, J = 8.3, 3.6 Hz, Ar–H). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K) δC 55.1 (dd, JCP = 74.8, 27.3 Hz, Ir–C–P), 117.7–118.1 (m, [BArF4] Ar–C), 121.7–128.3 (m), 128.3–130.6 (m), 131.6–131.7 (m), 132.5–134.4 (m), 134.7 (s), 135.1 (s), 135.2 (s), 135.3 (s, [BArF4] Ar–C), 136.2 (s), 137.2 (s), 142.5 (dd, J = 49.6, 8.6 Hz), 144.6–145.3 (m), 146.5–147.7 (m), 151.2 (d, J = 28.3 Hz), 153.6 (dt, J = 22.8, 2.7 Hz), 162.2 (q, 1JCB = 49.8 Hz, [BArF4] Ar–C). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K) δP 3.0 (1 P, apparent dd, JPP = 10.8, JPP = 10.1 Hz, Ir–P), 10.4–11.0 (2 P, m), 36.9 (1 P, apparent dt, 3JPP = 25.9, 3JPP = 8.7 Hz, Ir–C–P). HRMS (ESI-TOF) m/z: [M]+ calcd for C73H58IrP4 1251.3120; found 1251.3144. Elemental analysis: calc. for C105H70BF24IrP4: C, 59.6; H, 3.3; found: C, 59.3; H, 3.4%.
Preparation of complex 6
Method A: A solution of complex 2 (39.5 mg, 0.052 mmol) in DCM (5 mL) at −90 °C was treated dropwise with a solution of Na[BArF4]·2THF (53.4 mg, 0.052 mmol) in DCM (10 mL) and then 1,5-cyclooctadiene (7 μL, 0.057 mmol). The solution was allowed to come to room temperature and was stirred overnight. The reaction mixture was filtered and then the filtrate was evaporated fully. n-Hexane (5 mL) was added to the residue, triturated and then decanted. This process was repeated two additional times. The remaining residue was dissolved in DCM (1 mL), layered with n-hexane (10 mL) and then left to stand at room temperature. Colourless crystals of 6 formed from the solution were isolated by filtration and then dried under vacuum (70 mg, 79%).Method B: A solution of compound 1 (5.5 mg, 0.01 mmol) in 1,2-dichloroethane (0.4 mL) was added dropwise to a solution of [Ir(COD)2][BArF4] (12.7 mg, 0.01 mmol) in 1,2-dichloroethane (0.4 mL) at room temperature. The reaction solution was then heated to 95 °C for 18 h. 31P{1H} NMR spectroscopic analysis confirmed the formation of complex 6 in almost quantitative yield.
1H NMR (500 MHz, CD2Cl2, 298 K) δH 1.15–1.26 (1 H, m, cyclooctadienylium), 1.55–1.68 (1 H, m, cyclooctadienylium), 1.68–1.80 (1 H, m, cyclooctadienylium), 1.80–1.91 (1 H, m, cyclooctadienylium), 2.53 (1 H, dt, J = 14.1, 8.8 Hz, cyclooctadienylium), 2.98–3.11 (1 H, m, cyclooctadienylium), 3.20–3.34 (1 H, m, cyclooctadienylium), 3.69–3.85 (2 H, m, cyclooctadienylium), 3.95 (1 H, t, J = 7.7 Hz, cyclooctadienylium), 4.59 (1 H, s, Ir–C–H), 4.92 (1 H, t, J = 7.9 Hz, cyclooctadienylium), 7.05–7.13 (6 H, m, Ar–H), 7.17–7.59 (26 H, m, Ar–H), 7.73 (8 H, s, [BArF4] Ar–H). 13C{1H} (126 MHz, CD2Cl2, 298 K) δC 20.5 (d, J = 3.8 Hz), 28.4 (d, J = 4.9 Hz), 31.8 (s), 40.5 (s), 49.9 (d, J = 15.5 Hz), 55.4 (d, J = 2.5 Hz), 69.1 (d, J = 32.6 Hz), 100.9 (s), 105.3 (s), 117.7–118.3 (m, [BArF4] Ar–C), 121.7–128.4 (m), 128.5 (s), 128.6–128.9 (m), 128.9–129.1 (m), 129.2–129.3 (m), 129.5 (s), 129.6–129.8 (m), 131.2 (d, J = 9.3 Hz), 131.7 (s), 131.8 (d, J = 11.0 Hz), 131.9 (s), 132.4 (d, J = 9.9 Hz), 134.5 (dd, J = 41.0, 10.6 Hz), 135.1 (s), 135.3 (s, [BArF4] Ar–C), 135.7 (d, J = 39.9 Hz), 137.1 (d, J = 6.7 Hz), 137.6 (d, J = 16.6 Hz), 157.6 (d, J = 29.7 Hz), 159.1 (d, J = 27.0 Hz), 162.3 (q, 1JCB = 49.8 Hz, [BArF4] Ar–C). 31P{1H} (202 MHz, CD2Cl2, 298 K) δP 9.1 (1 P, d, 2JPP 7.7 Hz, PCP pincer P), 17.5 (1 P, d, 2JPP = 7.7 Hz, PCP pincer P). HRMS (ESI-TOF) m/z: [M]+ calcd for C45H40IrP2 835.2232; found 835.2232. calc. for C77H52BF24IrP2: C, 54.5; H, 3.1; found: C, 54.3; H, 3.4%.
Preparation of complexes 7a and 7b
Toluene (20 mL) was added to [IrCl(COD)(PPh3)] (119.6 mg, 0.20 mmol) and compound 1 (110.5 mg, 0.2 mmol) at room temperature and stirred overnight. The solution was then heated at 55 °C for two days. Afterwards, the solution was concentrated to approx. 5 mL and n-hexane (15 mL) was added to precipitate complexes 7a and 7b. The solids were filtered, washed with n-hexane (3 × 10 mL) and then dissolved in toluene (10 mL). The toluene solution was layered with n-hexane (20 mL) and left to stand at room temperature. Initially, colourless crystals of 7b were isolated by fractional crystallization, filtration and then dried under vacuum. The filtrate was evaporated under vacuum, re-dissolved in toluene (5 mL) and then layered with n-hexane (15 mL) and left to stand at room temperature. Crystals of 7a were isolated by filtration and then dried under vacuum (7a: 31 mg, 15%; 7b: 105 mg, 50%).7a: 1H NMR (500 MHz, CD2Cl2, 298 K) δH −18.34 (1 H, td, 2JHP = 19.7, 13.8 Hz, Ir–H), 5.05 (1 H, d, 4JHP = 15.2 Hz, methine C–H), 6.72–6.87 (9 H, m, Ar–H), 6.90 (4 H, td, J = 8.2, 2.3 Hz, Ar–H), 7.00–7.46 (28 H, m, Ar–H), 7.70 (2 H, d, J = 7.7 Hz, Ar–H). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K) δC 78.6–78.8 (m, methine C), 126.0 (t, J = 4.3 Hz), 127.3 (d, J = 10.0 Hz), 127.5 (t, J = 5.2 Hz), 127.8–128.1 (m), 129.4–129.6 (m), 129.7 (d, J = 6.7 Hz), 130.8 (d, J = 2.0 Hz), 134.4 (t, J = 5.4 Hz), 134.7 (s), 134.7–134.9 (m), 135.4 (t, J = 3.0 Hz), 136.1 (d, J = 10.8 Hz), 153.1 (t, J = 3.2 Hz). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K) δP −6.1 (2 P, d, 2JPP = 11.4 Hz, POP pincer P′s), −1.2 (1 P, t, 2JPP = 11.4 Hz, PPh3).
7b: 1H NMR (500 MHz, C6D6, 298 K) δH −20.23 (1 H, td, 2JHP = 16.9, 9.3 Hz, Ir–H), 6.62–7.90 (44 H, m, Ar–H and O–H). 13C{1H} NMR (126 MHz, C6D6, 298 K) δC 84.7 (d, 2JCP = 82.5 Hz, C–OH), 126.5–126.8 (m), 126.8–127.1 (m), 127.4 (d, J = 9.1 Hz), 127.7 (t, J = 4.0 Hz), 128.6 (s), 129.4 (dd, J = 13.3, 8.8 Hz), 132.3 (t, J = 26.1 Hz), 133.5 (s), 133.9 (t, J = 5.3 Hz), 134.6 (t, J = 5.4 Hz), 134.9–135.7 (m), 136.1 (t, J = 27.1 Hz), 143.2 (d, J = 6.8 Hz), 165.5 (t, J = 14.1 Hz). 31P{1H} NMR (202 MHz, C6D6, 298 K) δP −2.1 (1 P, t, 2JPP = 12.7 Hz, PPh3), 13.2 (2 P, d, 2JPP = 12.7 Hz, PCP pincer P′s). HRMS (ESI-TOF) m/z: [M − Cl]+ calcd for C55H45IrOP3 1007.2311; found 1007.2294.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National University of Singapore and the Singapore Ministry of Education for financial support (WBS R-143-000-586-112, R-143-000-697-114 and R-143-000-666-114).Notes and references
- (a) G. van Koten and D. Milstein, Organometallic Pincer Chemistry, Topics in Organometallic Chemistry, Springer, Berlin, 2013, vol. 40 Search PubMed; (b) G. van Koten and R. A. Gossage, The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, Springer, 2015 Search PubMed.
- (a) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776 CrossRef CAS PubMed; (b) L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187 CrossRef CAS PubMed; (c) L. E. Doyle, W. E. Piers, J. Borau-Garcia, M. J. Sgro and D. M. Spasyuk, Chem. Sci., 2016, 7, 921 RSC; (d) R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949 CrossRef CAS; (e) C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048 RSC; (f) C. C. Comanescu and V. M. Iluc, Organometallics, 2015, 34, 4684 CrossRef CAS; (g) C. C. Comanescu and V. M. Iluc, Organometallics, 2014, 33, 6059 CrossRef CAS.
- Ir NHC PCP pincers are known, but their electronic structure differs greatly from PCcarbeneP pincers, as defined herein. See: A. F. Hill and C. M. A. McQueen, Organometallics, 2012, 31, 8051 CrossRef CAS.
- (a) C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020 RSC; (b) H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw and B. Weeks, J. Chem. Soc., Chem. Commun., 1977, 589 RSC; (c) D. G. Gusev and A. J. Lough, Organometallics, 2002, 21, 2601 CrossRef CAS.
- (a) W. Weng, S. Parkin and O. V. Ozerov, Organometallics, 2006, 25, 5345 CrossRef CAS; (b) R. J. Burford, W. E. Piers and M. Parvez, Organometallics, 2012, 31, 2949 CrossRef CAS.
- (a) J. R. Logan, W. E. Piers, J. Borau-Garcia and D. Spasyuk, Organometallics, 2016, 35, 1279 CrossRef CAS; (b) J. D. Smith, J. R. Logan, L. E. Doyle, R. J. Burford, S. Sugawara, C. Ohnita, Y. Yamamoto, W. E. Piers, D. M. Spasyuk and J. Borau-Garcia, Dalton Trans., 2016, 45, 12669 RSC.
- (a) R. J. Burford, W. E. Piers, D. H. Ess and M. Parvez, J. Am. Chem. Soc., 2014, 136, 3256 CrossRef CAS PubMed; (b) A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2015, 6, 2060 RSC; (c) A. V. Polukeev and O. F. Wendt, Organometallics, 2017, 36, 639 CrossRef CAS; (d) A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Organometallics, 2016, 35, 2600 CrossRef CAS; (e) M. S. Balakrishna, Polyhedron, 2017 DOI:10.1016/j.poly.2017.07.026.
- (a) S. Sung, T. Joachim, T. Krämer and R. D. Young, Organometallics, 2017, 36, 3117 CrossRef CAS; (b) S. Sung, J. K. Boon, J. J. C. Lee, N. A. Rajabi, S. A. Macgregor, T. Krämer and R. D. Young, Organometallics, 2017, 36, 1609 CrossRef CAS.
- L. E. Doyle, W. E. Piers and D. W. Bi, Dalton Trans., 2017, 46, 4346 RSC.
- Q. Teng and H. V. Huynh, Dalton Trans., 2017, 46, 614 RSC.
- M. A. Bennett and D. L. Milner, J. Chem. Soc., Chem. Commun., 1967, 581 RSC.
- G. R. Clark, W. R. Roper and A. H. Wright, J. Organomet. Chem., 1984, 273, C17 CrossRef CAS.
- (a) K. Onitsuka, M. Nishii, Y. Matsushima and S. Takahashi, Organometallics, 2004, 23, 5630 CrossRef CAS; (b) L. M. Milner, L. M. Hall, N. E. Pridmore, M. K. Skeats, A. C. Whitwood, J. M. Lynam and J. M. Slattery, Dalton Trans., 2016, 45, 1717 RSC; (c) V. Cadierno, J. Díez, J. García-Álvarez and J. Gimeno, Organometallics, 2005, 24, 2801 CrossRef CAS; (d) R. P. Kamalesh Babu, R. McDonald and R. G. Cavell, Organometallics, 2000, 19, 3462 CrossRef.
- (a) R. H. Crabtree and C. P. Parnell, Organometallics, 1985, 4, 519 CrossRef CAS; (b) R. H. Crabtree, C. P. Parnell and R. J. Uriarte, Organometallics, 1987, 6, 696 CrossRef CAS; (c) R. H. Crabtree, R. P. Dion, D. J. Gibboni, D. V. McCrath and E. M. Holt, J. Am. Chem. Soc., 1986, 108, 7222 CrossRef CAS.
- (a) I. Zumeta, C. Mendicute-Fierro, A. Rodríguez-Diéguez, J. M. Seco and M. A. Garralda, Organometallics, 2015, 34, 348 CrossRef CAS; (b) D. L. Thorn, Organometallics, 1982, 1, 197 CrossRef CAS; (c) D. L. Thorn and J. C. Calabrese, J. Organomet. Chem., 1984, 272, 283 CrossRef CAS.
- J. Peone Jr. and L. Vaska, Angew. Chem., Int. Ed. Engl., 1971, 10, 511 CrossRef.
- J. D. Smith, J. Borau-Garcia, W. E. Piers and D. Spasyuk, Can. J. Chem., 2016, 94, 293 CrossRef CAS.
- J. Arras, H. Speth, H. A. Mayer and L. Wesemann, Organometallics, 2015, 34, 3629 CrossRef CAS.
- R. Dallanegra, A. B. Chaplin and A. S. Weller, Organometallics, 2012, 31, 2720 CrossRef CAS.
- K. J. Jonasson, A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Angew. Chem., Int. Ed., 2015, 54, 9372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1568119–1568125. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03690f |
| This journal is © The Royal Society of Chemistry 2017 |