
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExternal oxidant-free cross-coupling: electrochemically induced aromatic C–H phosphonation of azoles with dialkyl-H-phosphonates under silver catalysis†
E. O.
Yurko
 ,
T. V.
Gryaznova
,
K. V.
Kholin
,
V. V.
Khrizanforova
and
Y. H.
Budnikova
*
,
T. V.
Gryaznova
,
K. V.
Kholin
,
V. V.
Khrizanforova
and
Y. H.
Budnikova
*
A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, 8 Arbuzov str., Kazan 420088, Russian Federation. E-mail: yulia@iopc.ru
First published on 21st November 2017
Abstract
A convenient external oxidant-free method of phosphorylation of azole derivatives (benzo-1,3-azoles, 3-methylindole, 4-methyl-2-acetylthiazole) by using dialkyl-H-phosphonates through the catalytic oxidation of their mixture under electrochemical mild conditions (room temperature, normal pressure) in the presence of silver salts or oxide (1%) is proposed. This method allows us to obtain the desired azole dialkylphosphonates with good yield (up to 75%). The transformations of silver and phosphorus precursors and intermediates using cyclic voltammetry, ESR, and NMR spectroscopy were investigated, and a radical process mechanism was proposed. It has been found that AgP(O)(OEt)2 is oxidized earlier than other components of the reaction mixture with the elimination of a radical. The ESR spectrum of this radical's adduct was obtained in the presence of the radical trap PBN. Ag2+ is out of the catalytic cycle.
Introduction
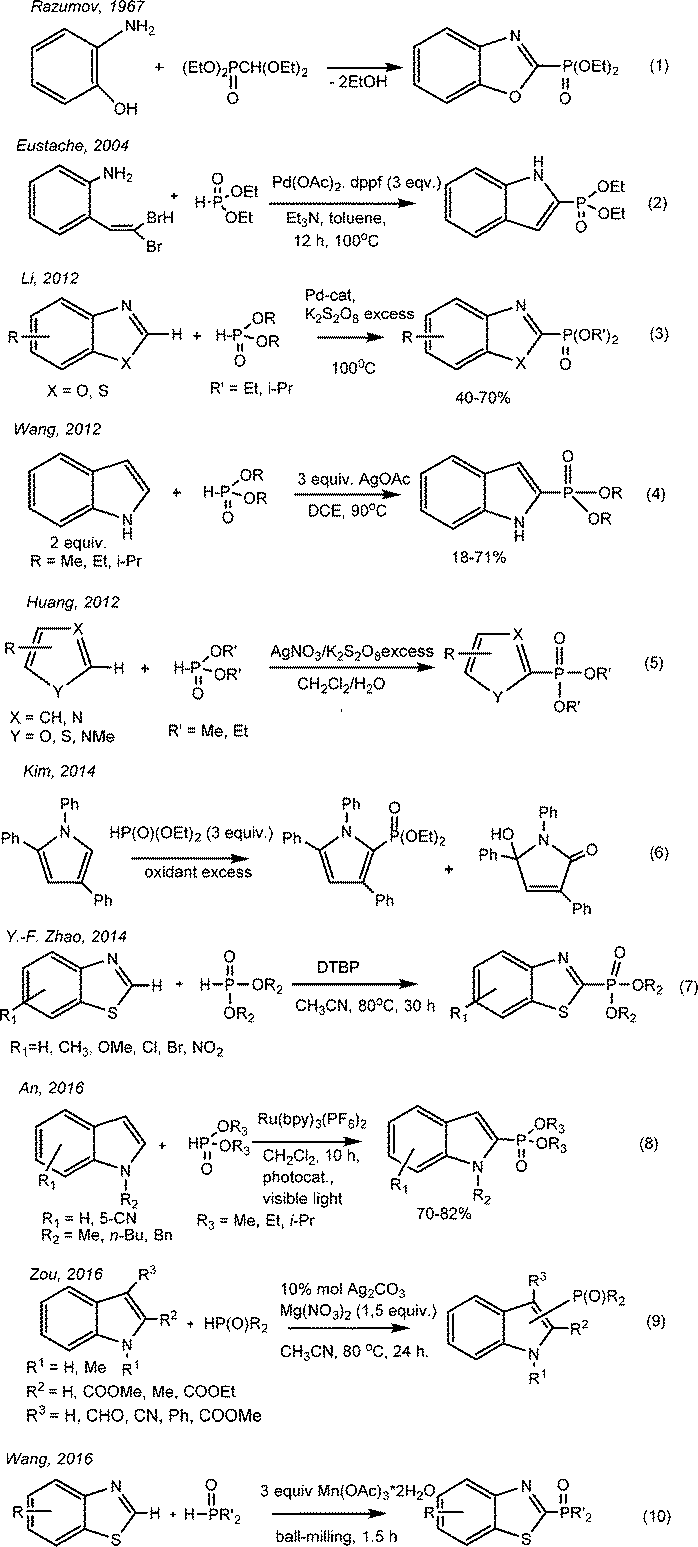
Benzo-1,3-azoles and their derivatives have unique biological properties. Therefore, benzoxazoles possess antitumor activity,1,2 and also are non-nucleoside topoisomerase I inhibitors.3 They can be seen as structural bioisomers of natural nucleotides, such as adenine and guanine, allowing them to easily interact with polymers in living systems.4 Substituted benzoxazoles possess high biological activity. The derivatives of benzoxazole have become the bases for plant protection products (for example, insectoacaricide with contact-intestinal action – “Zolon” – S-2,3-dihydro-(6-chloro-2-oxibenzoxazole-3-ilmetyl)-O,O-diethyltiophosphate5), herbicides,6 antidiabetics, neuroleptics, etc.7 It has been recently found that 3-phosphoindoles are good inhibitors of HIV-1.8 The search for new low-waste ways to obtain the phosphorus-containing derivatives of heterocycles including azoles through the direct phosphorylation of C–H bonds is a relevant topic.The phosphorylated derivatives of benzoxazoles were obtained first in the 60s of the last century by Razumov A. I. through heating ortho-aminophenol and diethyl diethoxymethylphosphonate with alcohol flashing9,10 (Scheme 1, eqn (1)).
 | ||
| Scheme 1 Known approaches towards azole phosphonates. | ||
The method of obtaining 2-diethyl-indol-2-ylphosphonate by the cross-coupling of dibromovinyl-ortho-aminobenzole with diethylphosphite in the presence of palladium salts was described (Scheme 1, eqn (2)).11 Li proposed the method of direct catalytic oxidative phosphorylation of aromatic azoles in the absence of bases and acids with palladium salts as catalysts and an excess of oxidant K2S2O8 (Scheme 1, eqn (3)). The reaction proceeds slowly at 100 °C; the yields of the C-2 phosphorylated derivatives of azoles are 40–70%.12 In 2012, Wang suggested the cross-coupling of 1H-indole and diethylphosphite with C(sp2)–P bond formation with a yield of 18–71%13 (Scheme 1, eqn (4)). Huang realized the phosphorylation of furans, thiophenes, thioazoles, pyrroles and pyridines in the presence of catalytic amounts (0.2 equivalents) of AgNO3 using 3 equivalents of K2S2O8 as an oxidant (Scheme 1, eqn (5)).14 Kim described the dehydrocoupling of substituted pyrrols and dialkylphosphites under similar reaction conditions (Scheme 1, eqn (6)).15 Yu-Fen Zhao has found that depending on the nature of the oxidant – peroxide – one can obtain a variety of products in the reaction of benzothiazoles and H-phosphonates, derivatives of benzothiazoles, 2-acylbenzothiazoles and dialkyl benzothiazole-2-ylphosphonates, respectively, under mild conditions and without the participation of metals16 (Scheme 1, eqn (7)). The regioselective cross-coupling between N-substituted indoles and dialkylphosphites was studied in 2016 (Scheme 1, eqn (8)).17 The reaction was carried out in the presence of the photo-oxidative catalyst Ru(bpy)3(PF6)2 in combination with oxygen as a pure oxidant under exposure to visible light. The products were 2-indolephosphonates with yields of 70–82%.17 Zou suggested the usage of Ag catalysts in the presence of 1.5 equivalents of Mg(NO3)2 as an oxidant for the phosphorylation of indoles substituted at the second and third positions. The yield of 3-indolephosphonate is higher when using 2-substituted indoles rather than 3-substituted indoles (Scheme 1, eqn (9)).18
Wang developed in 2016 a method for constructing C(sp2)–P bonds based on the coupling of benzoazole or azole derivatives with organophosphorus compounds in the presence of a triple excess of Mn(OAc)3 under “ball-milling” (Scheme 1, eqn (10)).19
The disadvantages of the above reactions usually include the need for a large excess of oxidant, elevated temperatures and a long duration of reaction. Of course, the most attractive methods are atom-economical direct C–H/P–H coupling reactions of azoles with dialkyl-H-phosphonates. The development of such environmentally safe and simple catalytic techniques is especially relevant. In this regard, the electrochemical methods that meet the criteria of “green chemistry”, characterized by mild conditions (low temperature, normal pressure), regeneration of catalyst on the electrode, and environmentally safe and low-waste processes are very promising. Electrochemically induced catalytic C–H bond functionalization has become a highly attractive strategic approach to green, clean and efficient transformations in organic synthesis. Thus, new electrochemical reactions of the C(sp2)–H phosphorylation20–27 have been proposed in recent years. The progress of electroorganic synthesis in this field is described in numerous reviews22,23,28–32 and papers concerning some recent advances in C–H functionalization, such as fluoroalkylation,33–38 amination,39–42 aziridination,43 oxygenation,44–46 arylation,47 alkylation,48 amino-oxygenation,49etc. The first electrochemical oxidative phosphorylation of benzoxazoles in the presence of a 3d metal catalyst was reported in 2016.50
The purpose of this work is to create a new method for the phosphorylation of benzo-1,3-azole derivatives with dialkylphosphites through the electrocatalytic activation of C–H bonds in aromatic substrates in the presence of silver salts as catalysts and in the absence of specially added oxidizers at room temperature, as well as to clarify the mechanism of the reaction.
Electrosynthesis
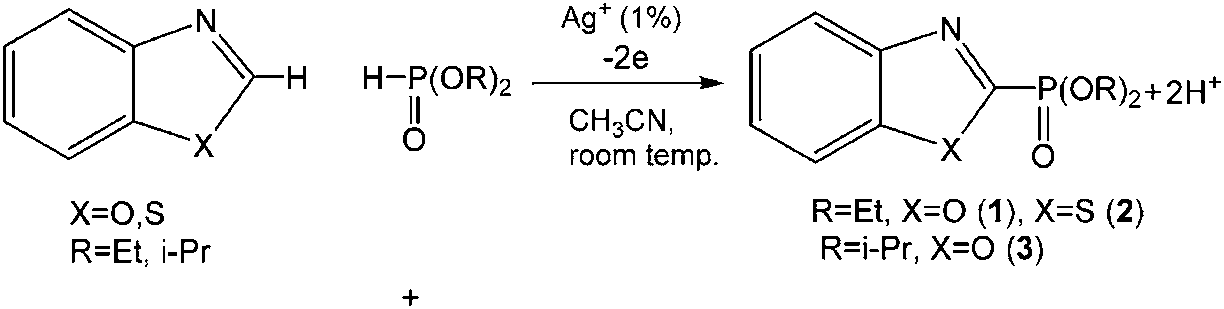
Joint electrolysis of a mixture of benzo(oxa)zoles and dialkyl-H-phosphonates (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in CH3CN under oxidizing conditions in the presence of catalytic amounts of silver salts at room temperature leads to 2-phosphorylated benzo(oxa)zoles (dialkyl-benzothia(oxa)zole-2-yl-phosphonates) (Scheme 2):
:1) in CH3CN under oxidizing conditions in the presence of catalytic amounts of silver salts at room temperature leads to 2-phosphorylated benzo(oxa)zoles (dialkyl-benzothia(oxa)zole-2-yl-phosphonates) (Scheme 2):
Silver salts (nitrate, acetate and carbonate) and silver(I) oxide were tested as catalysts. The electrolysis process was controlled by 31P-NMR. The results of electrolysis are presented in Table 1.
 | ||
| Scheme 2 Electrochemically induced coupling reactions of heteroarenes (benzoxazoles or benzothiazoles) with phosphites under silver catalysis. | ||
| N | Catalyst | Base | Product yield,% |
|---|---|---|---|
| a Results when using diisopropyl-H-phosphonate. | |||
|
|||
| 1 | AgNO3 | — | 41 |
| 2 | AgNO3a |
— | 30 |
| 3 | AgOAc | Processing of t-BuOK after electrolysis | 61 |
| 4 | AgOAc | t-BuOK | 62 |
| 5 | AgOAca | Processing of t-BuOK after electrolysis | 60 |
| 6 | AgOAc | K2CO3 | 63 |
| 7 | AgOAc | Na3PO4 | 75 |
| 8 | AgOAca | Na3PO4 | 74 |
| 9 | AgOAc | NaH2PO3 | 63 |
| 10 | Ag2CO3 | — | 8 |
| 11 | Ag2O | — | 35 |
|
|||
| 12 | AgNO3 | — | 42 |
| 13 | AgNO3a |
— | 45 |
| 14 | AgOAc | Na3PO4 | 51 |
| 15 | AgOAc | NaH2PO3 | 54 |

|
|||
| 16 | AgNO3 | Na3PO4 | 41 |
| 17 | AgOAc | Na3PO4 | 50 |
| 18 | Ag2O | Na3PO4 | 31 |
|
|||
| 20 | AgOAc | Na3PO4 | 74 |
| 21 | AgOAca | Na3PO4 | 71 |
|
|||
| 22 | AgOAc | t-BuOK | Traces |
|
|||
| 23 | AgOAc | t-BuOK | Traces |
The usage of AgNO3 as a catalyst resulted in low yields of phosphorylated benzothia(oxa)azoles. An increase of electricity has no effect on the final product yield; the 31P signal of the source dialkylphosphite has always been present in the reaction mixture. Diisopropyl phosphite turned out to be less reactive under these conditions than diethylphosphite. The catalytic activity of silver acetate AgOAc was higher than that of AgNO3. It can be assumed that OAc− assists in P–H metallation, as was described for OAc− assisted C–H activation51–53 previously. We found that the rate of the key intermediate AgP(O)(OEt)2 formation from AgOAc is higher than that from AgNO3 under other similar conditions. Thus, when AgOAc is mixed with H(O)P(OEt)2 in acetonitrile, a gel-like mass immediately forms after being stirred for an hour, and the conversion is 100%. The yield of Ag(O)P(OEt)2 is quantitative (white powder), a single singlet signal at δ 107 ppm in the 31P NMR spectrum in pyridine is observed, and the signal of the initial H(O)P(OEt)2 is absent. But the interaction of AgNO3 and H(O)P(OEt)2 in acetonitrile is slower at room temperature.
Prior to electrolysis, no products in the reaction mixture including arene, diethylphosphite and silver salt after stirring for 24 hours are observed. The single signal for (EtO)2P(O)H shows constant intensity. During the electrolysis, the transformation of the initial substrates into the products and possible intermediates was monitored. Only after passing more than 2 F of electricity through the reaction mixture does the signal of the source (EtO)2P(O)H disappear. The 31P NMR spectrum contains signals of the products (1–3), phosphorylated arenes in nitrogen-protonated and non-protonated forms. In order to produce a clean reaction product, the mixture was processed with a base. t-BuOK, Na3PO4 and Et3N were tested as bases. Only after processing with t-BuOK could the protonated product form fully convert into the target arene phosphonate. Holding of electrolysis in the presence of a base (K2CO3, Na3PO4 and NaH2PO3 were used) allowed us to obtain the final product at the end of the electrolysis, leaving out the protonated form. Benzothiazole is phosphorylated under these conditions forming 2-substituted derivative 2.
The optimization of synthesis conditions (1–3) through the variation of the nature of the silver catalyst and base showed that the best yields are achieved with silver acetate as a catalyst and Na3PO4 as a base (75%) (Table 1).
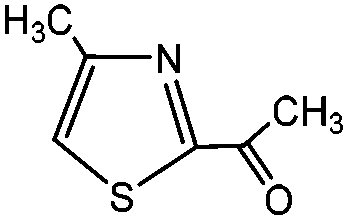
3-Methylindole, 4-methyl-2-acetylthiazole and benzimidazoles were also tested as the substrates in the electrochemical phosphorylation of heterocyclic compounds. It turned out that 2-phosphonated methylindole or 5-phosphonated 4-methyl-2-acetylthiazole is formed under electrocatalytical conditions with yields of 31–50% for the indole and 71–74% for the thiazole (Scheme 3, Table 1). As in the case of benzo-1,3-azoles, the best catalyst is silver acetate (Table 1). Benzimidazoles do not react with (RO)2P(O)H.
 | ||
| Scheme 3 Electrochemically induced coupling reactions of 3-methylindole and 4-methyl-2-acetylthiazole with phosphites under silver catalysis. | ||
Thus, a convenient preparative electrochemical protocol for the synthesis of phosphonated azoles using silver catalysts with low loading (1%) has been developed. Electrolysis takes place in one stage, in the absence of specifically added oxidants at room temperature with good yields.
Cyclic voltammetry and ESR spectroelectrochemistry for mechanistic considerations
In order to investigate the details of the process, we have studied the electrochemical properties of the participants of the aromatic C–H phosphorylation (Table 2).| N | Compound | E oxp (V) |
|---|---|---|
| 1 | AgOAc | 1.89 |
| 2 | AgNO3 | 2.10 |
| 3 | Ag2O | 1.90 |
| 4 | Benzoxazole | 2.26 |
| 5 | Benzothiazole | 2.13 |
| 6 | 3-Methylindole | 2.10 |
| 7 | 4-Methyl-2-acetylthiazole | 1.99 |
| 8 | Diethyl-H-phosphonate | — |
| 9 | Diisopropyl-H-phosphonate | — |
| 10 | AgP(O)(OEt)2 | 1.10 |
It should be noted that there are virtually no literature data about the oxidation potentials of Ag0/Ag+/Ag2+ in organic media, authors often mention the high standard potential of silver(II) in acidic water (E° = 1.98 V vs. normal hydrogen electrode, NHE).54,55 Few reports mentioned the potential of Ag+/Ag0 (it strongly depends on the nature of the solvent, 0.61 V in acetone and 1.08 V in CH2Cl2 (ref. Ag/AgCl)),56 but due to the lack of data on silver salt anions and exact experimental conditions it is difficult to discuss these data. AgNO3 in a solution of HNO3 is oxidized according to ref. 57 at 1.5 V (ref. SSE) or 2.15 V (ref. Ag/AgCl) on boron-doped diamond electrodes, while water is oxidized at almost the same potentials under these conditions, as shown by the authors. At this potential the nitrate ion is decomposed and brown NO gas is formed. However, under the conditions of our electrolysis in the presence of arenes there is no formation of NO.
So, the redox properties of all silver catalysts under electrosynthesis conditions have been studied. The voltammograms of Ag+ (AgOAc or AgNO3) in the oxidative region always have an adsorption peak of oxidation of impurities Ag(0) at relatively low potentials (∼0.3 V), whose height is poorly reproduced and depends on the luminance of samples and the electrolyte mixing time until the registration of the curves (Fig. 1, top). The oxidation peak of Ag+ in Ag2+ is in the region of high positive potentials for AgOAc, AgNO3 and Ag2O (1.89–2.10 V) (Table 2), and voltammograms for all silver catalysts are similar (Fig. 1, top).
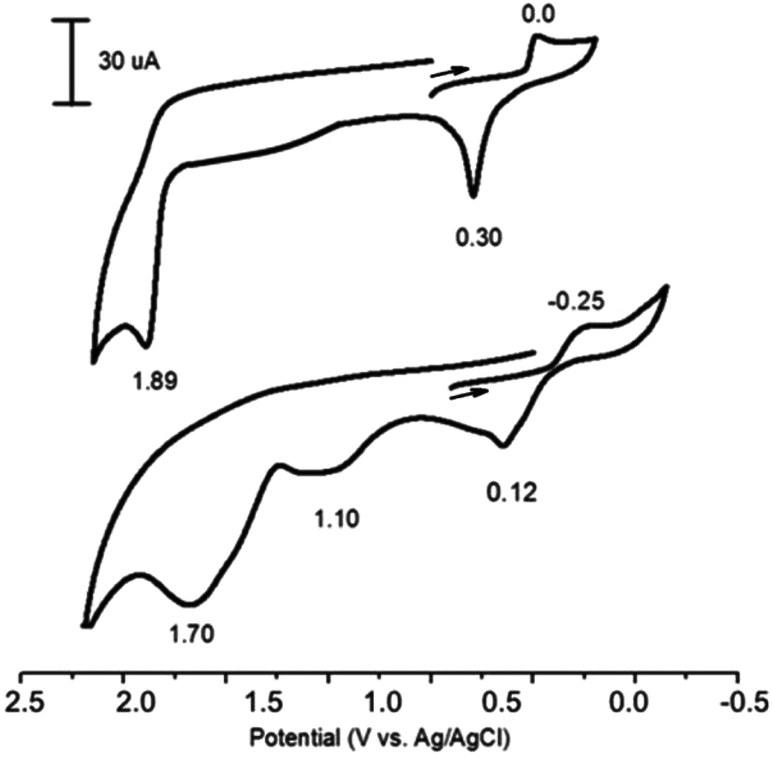 | ||
| Fig. 1 CVs of AgOAc in CH3CN (top) and AgP(O)(OEt)2 in Py + CH3CN (1:10) (bottom). Reaction conditions: 0.1 M Bu4NBF4. Working electrode – GC, reference electrode – Ag/AgCl, scan rate 100 mV s−1. | ||
Aromatic substrates (benzoxazoles, benzothiazoles and others) are oxidized at high anode potentials (1.99–2.26 V) (Table 1). The oxidation of silver acetate, the best catalyst, was also investigated by ESR spectroscopy. A solution of silver acetate (AgOAc) in acetonitrile is ESR silent, but due to its oxidation at a potential of about +2.8 V in an ESR-electrochemistry cell at room temperature an intense ESR spectrum appears. It represents a single homogeneous wide band with the following parameters: g = 2.155 and ΔHp–p = 72 G. This signal appears as a result of the oxidation of the solvated ion Ag(I) to Ag(II). After reaching the maximum intensity the temperature dependence of this spectrum was registered (Fig. 2, left).
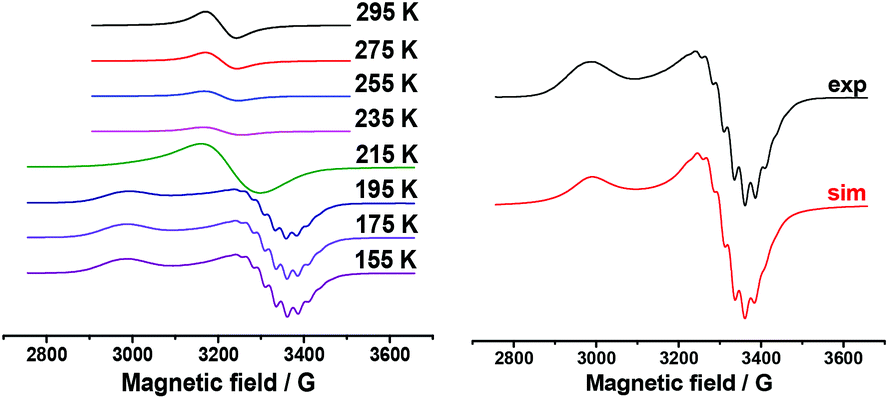 | ||
| Fig. 2 ESR spectrum of AgOAc solution in acetonitrile at +2.8 V. Temperature dependence (left): spectrum of frozen solution (155 K) and its simulation (right). | ||
The spectrum of frozen solution matches the case of the stretched axial symmetry of the environment of the Ag(II) ion (Fig. 2, right). Hyperfine splitting is observed only in perpendicular component of the ESR spectrum. As can be seen from the simulation, this splitting is attributed to hyperfine interaction with four nitrogen nuclei. The spin-Hamiltonian parameters – g∥ = 2.315, g┴ = 2.075, 4:aN = 25 G – close to the literature data for Ag2+ are obtained under other conditions and other surroundings of the metal ion.58–60 However, because the potentials of preparative electrochemical synthesis do not reach the oxidation potentials of Ag+/Ag2+, Ag2+ is not involved in the catalytic cycle under investigation.
Despite the many studies on silver catalysis, the mechanisms of these reactions are investigated very superficially and are usually postulated. It can be assumed that Ag+ reacts at the first stage with (RO)2P(O)H, as suggested in the silver-catalyzed reaction of phosphonation.13 However, nobody has researched the properties of (RO)2P(O)Ag. H. Wang suggested that the reaction of Ag+ with (RO)2P(O)H immediately leads to the formation of the (RO)2P(O)˙ radical,13 and the radical nature of the reaction was based on indirect data – blocking of the reaction in the presence of the radical inhibitor butylated hydroxytoluene.13 Huang,14 Cheng61 and A. Wang62 assumed that Ag+ is oxidized to Ag2+ under the action of the oxidant K2S2O8, which further oxidizes (EtO)2P(O)H to a radical-cation interacting with the arene. When (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), a radical scavenger, was added to the reaction system, the dehydrogenative cross-coupling reaction of arene with diethylphosphite was quenched; so, this result suggests that the reaction may undergo a radical mechanism.14 This plausible mechanism of the dehydrogenative cross-coupling reaction is similar to that proposed by Effenberger et al.63 Zou18 and others14,15 also postulated a radical mechanism of oxidative phosphorylation in the presence of a Ag-catalyst only based on the absence of products in the presence of two equivalents of TEMPO. However, no intermediates were obtained and their reactivity was not proven. So, the failure of the reaction in the presence of TEMPO can be explained by the fact that the latter is easily oxidized by the applied oxidant K2S2O8, for example. Moreover, if TEMPO was taken in a two-fold excess over the oxidant, the entire oxidant was consumed by the side reaction of TEMPO, which is easily oxidized to about 0 V ref Fc+/Fc or +0.42 V ref. Ag/AgCl,64,65 but not by the oxidation of more difficultly oxidized H-phosphonate, arenes or Ag+.
In order to explore the mechanism of the phosphorylation of azoles, we synthesized AgP(O)(OEt)2 and investigated its reactivity. The voltammogram of AgP(O)(OEt)2 has three distinct anodic peaks at 0.12, 1.10 and 1.70 V (Fig. 1, bottom), the first and the third of which can be attributed to the oxidation of Ag0/Ag1+ and Ag1+/Ag2+ (they are observed also for AgOAc at similar potentials), and the second, 1.10 V, can be attributed to the oxidation of the anion (EtO)2P(O)− (similar to the oxidation of (EtO)2P(O)Na66).
In order to establish the character of the catalytic cycle and the character of the intermediate P(O)–Ag bond scission (ionic or radical type), we have carried out a number of ESR experiments under anaerobic conditions. The joint oxidation of the (EtO)2P(O)Ag and spin trap PBN (PBN = N-tert-butyl-α-phenylnitrone) mixture at +0.9 V in an ESR-spectroelectrochemical cell is characterized by appearance and the increase in the intensity of the ESR signal of the PBN bound radical species of the (EtO)2P(O) adduct (Fig. 3 and Scheme 4).
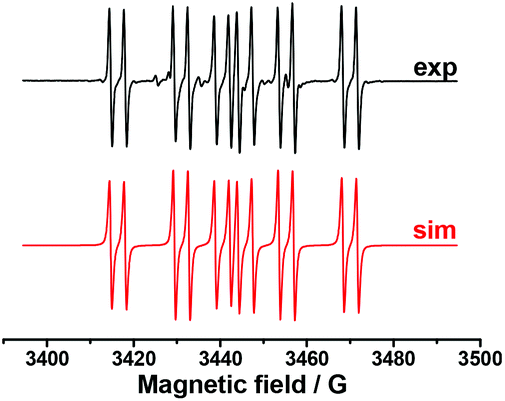 | ||
| Fig. 3 ESR spectra obtained during the anodic joint electrolysis of (EtO)2P(O)Ag and spin trap PBN solution at +0.9 V in CH3CN, recorded at 293 K with simulations. | ||
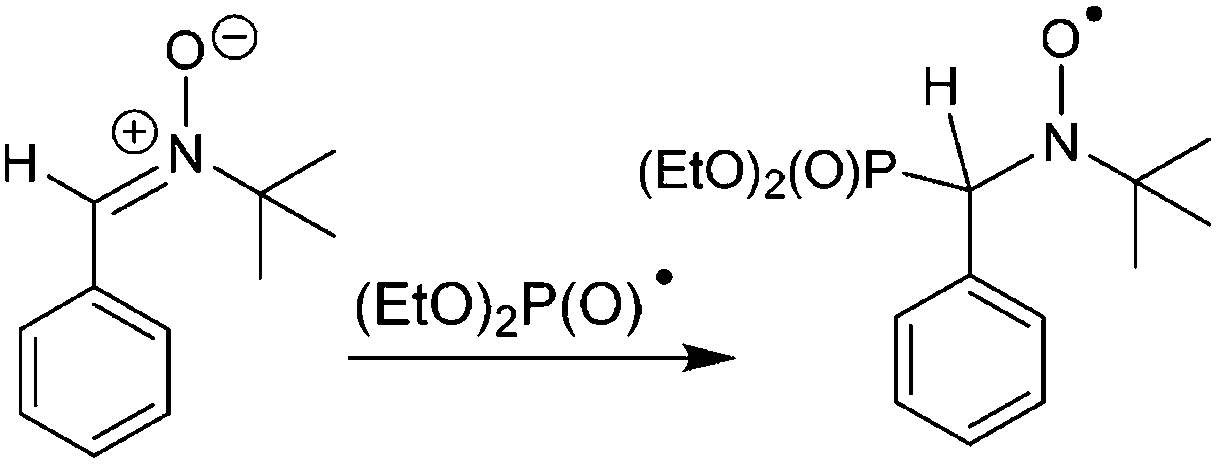 | ||
| Scheme 4 Trapping of the phosphorus-centered radical by PBN. | ||
The spin-adduct spectrum was simulated and the following spin-Hamiltonian parameters were obtained: g = 2.0060, aN = 14.71 G, aP = 24.17 G, and aH = 3.36 G. These parameters almost completely coincide with the parameters of the adduct (EtO)2P(O)-PBN, known from the literature: aN = 14.65 G, aP = 24.33 G, and aH = 3.06 G.67
We can thus conclude that (EtO)2P(O)˙ reacts readily with the spin trap and the spin adduct is quite stable. At a potential of +1.5 V, a new non-basic signal appears and grows, apparently, due to the oxidation of the spin-trap itself.
The oxidation of AgOAc solution and benzoxazole in acetonitrile in an ESR-cell leads to a result similar to the oxidation of a solution of pure AgOAc in acetonitrile – the emergence of a single band in the spectrum at +2.8 V with the same magnetic resonance parameters. Ag2+ participation in electrochemically induced coupling reactions of heteroarenes with phosphites under silver catalysis can be excluded.
We performed a counter electrosynthesis involving Ag(O)P(OEt)2 as a phosphorylation reagent and benzoxazole in a ratio of 1:1. The oxidation of Ag(O)P(OEt)2 during the joint electrolysis leads to the formation of phosphorylated benzoxazole and its protonated form (Scheme 5).
 | ||
| Scheme 5 Joint electrolysis of (EtO)2P(O)Ag and benzoxazole. | ||
The latter, after treatment with a base, passes to the final product. The formation of Ag2+ under the investigated conditions is impossible, since the electrolysis potential is comparatively low. The ESR study of reaction mixtures confirms the absence of Ag2+ at all stages of synthesis.
Since in some studies on the amination of benzoxazoles in the presence of iodide ions (mediators) under oxidizing conditions, including electrochemical conditions, the assumptions about the formation of structures with an open cycle were made previously,68,69 we analyzed the 31P and 1H NMR spectra of the reaction mixtures before and after passing 1 F, 2 F and 2.5 F of electricity (see the ESI†). It was found that no ring opening of the oxazole was observed, at all stages only the initial and final target products were present. Apparently, this is due to the fact that (EtO)2P(O)–H phosphite does not react with protonated benzoxazoles in contrast to dialkylamine (Scheme 6 and ESI†).
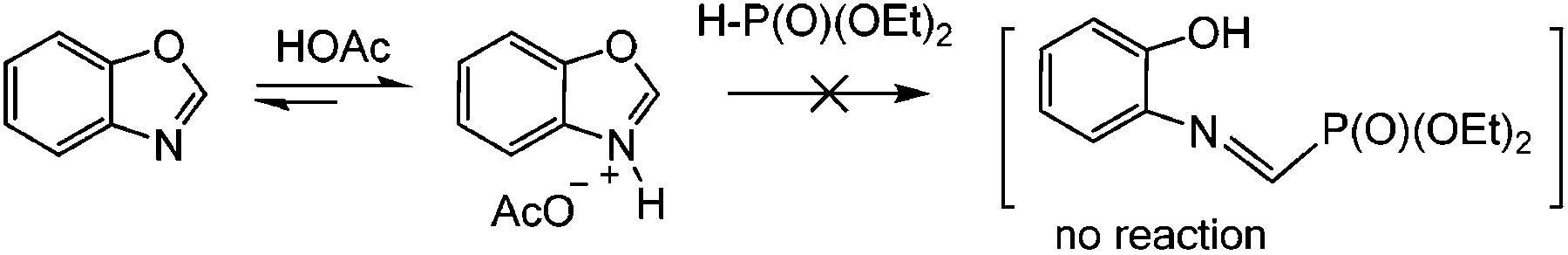 | ||
| Scheme 6 No ring opening reaction under the conditions studied. | ||
The analysis of literature data and conducted research allowed us to propose the following scheme of the electrocatalytic phosphorylation of azoles (Scheme 7), using a benzoxazole reaction as an example. Initially, the silver(I) cation reacts with dialkyl-H-phosphonate, and (EtO)2P(O)Ag is oxidized and yields the (EtO)2P(O)˙ radical. Its addition to heteroarene leads to the radical intermediate, which may lose a hydrogen cation and an electron (at the anode or under Ag+ action), successively giving the desired benzoxazole phosphonate.
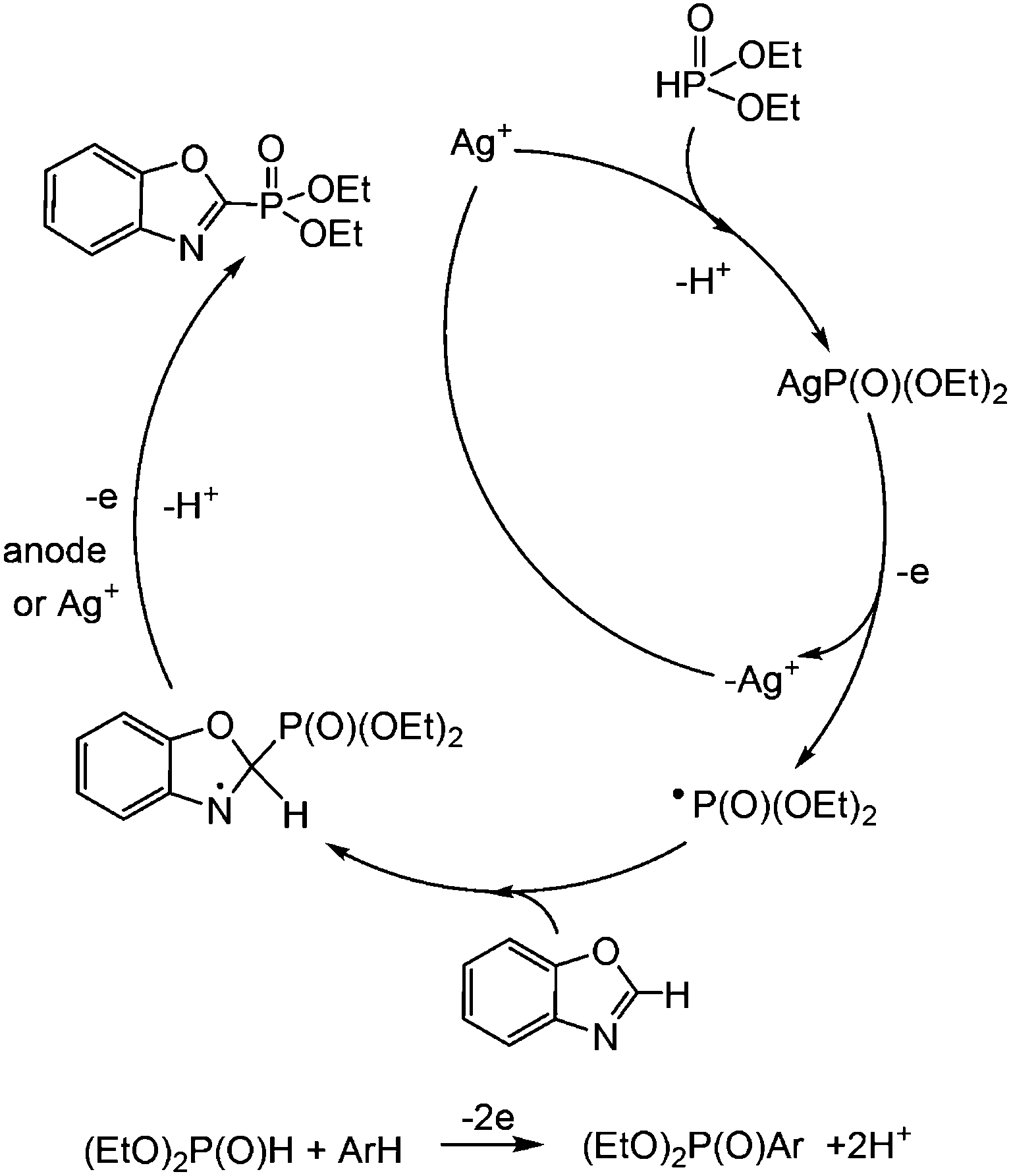 | ||
| Scheme 7 Proposed mechanism of the dehydrogenative cross-coupling reaction (benzoxazole case). | ||
Conclusions
In conclusion, we have developed a silver-catalyzed dehydrogenative cross-coupling reaction of azole derivatives with dialkylphosphites under mild conditions without an excess of external oxidant, affording the corresponding phosphonated products with up to 75% yield and have investigated the catalytic mechanism. The key stage of this electrochemical process is the silver dialkyl phosphonate oxidation yielding the phosphorus-centered radical.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the Russian Science Foundation (grant No. 14-23-00016).Notes and references
- D. Kumar, M. R. Jacob, M. B. Reynolds and S. M. Kerwin, Bioorg. Med. Chem., 2002, 10, 3997 CrossRef CAS PubMed.
- S. M. Rida, F. A. Ashour, S. A. M. El-Hawash, M. M. El-Semary, M. H. Badr and M. A. Shalaby, Eur. J. Med. Chem., 2005, 40, 949 CrossRef CAS PubMed.
- J. S. Kim, Q. Sun, B. Gatto, C. Yu, A. Liu, L. F. Liu and E. J. LaVoie, Bioorg. Med. Chem., 1996, 4, 621 CrossRef CAS PubMed.
- D. W. Dunwell and D. J. Evans, Med. Chem., 1977, 20, 797 CrossRef CAS PubMed.
- N. N. Melnikov and S. R. Belan, Pesticides and plant growth regulators, Reference book, Moscow, 1995 Search PubMed.
- J. Suzuki, K. Takahashi and S. Fukuda, US Pat, 2008/058213, 2008 Search PubMed.
- S. Schunk, M. Rich, M. Engels, T. Germann, J. De Vry, R. Jostock and S. Hers, WO Pat2010/142402, 2010 Search PubMed.
- R. Storer, C. Dousson, F.-R. Alexandre and A. Roland, Patent EP1961757A1, 2008 Search PubMed.
- A. I. Razumov, B. G. Liorber and P. A. Gurevich, Zh. Obshch. Khim., 1967, 7, 2782 Search PubMed.
- A. I. Razumov, P. A. Gurevich, B. G. Liorber and E. D. Borisova, Zh. Obshch. Khim., 1969, 39, 392 CAS.
- S. Thielges, E. Meddah, P. Bisseret and J. Eustache, Tetrahedron Lett., 2004, 45, 907 CrossRef CAS.
- Ch. Hou, Y. Ren, R. Lang, X. Hu, Ch. Xia and F. Li, Chem. Commun., 2012, 48, 5181 RSC.
- H. Wang, X. Li, F. Wu and B. Wan, Synthesis, 2012, 941 Search PubMed.
- C.-B. Xiang, Y.-J. Bian, X.-R. Mao and Z.-Z. Huang, J. Org. Chem., 2012, 77, 7706 CrossRef CAS PubMed.
- S. H. Kim, K. H. Kim, J. W. Lim and J. N. Kim, Tetrahedron Lett., 2014, 531 CrossRef CAS.
- X.-L. Chen, X. Li, L.-B. Qu, Y.-C. Tang, W.-P. Mai, D.-H. Wei, W.-Z. Bi, L.-K. Duan, K. Sun, J.-Y. Chen, D.-D. Ke and Y.-F. Zhao, J. Org. Chem., 2014, 79, 8407 CrossRef CAS PubMed.
- Z. Mina, W. Donga, Z. Penga and D. An, Synth. Commun., 2016, 46, 128 CrossRef.
- W.-B. Sun, J.-F. Xue, G.-Y. Zhang, R.-S. Zeng, L.-T. An, P.-Z. Zhang and J.-P. Zou, Adv. Synth. Catal., 2016, 358, 1753 CrossRef CAS.
- L. Li, J.-J. Wang and G.-W. Wang, J. Org. Chem., 2016, 81, 5433 CrossRef CAS PubMed.
- T. V. Gryaznova, Y. B. Dudkina, D. R. Islamov, O. N. Kataeva, O. G. Sinyashin, D. A. Vicic and Y. H. Budnikova, J. Organomet. Chem., 2015, 785, 68 CrossRef.
- M. N. Khrizanforov, S. O. Strekalova, K. V. Kholin, V. V. Khrizanforova, M. K. Kadirov, T. V. Gryaznova and Y. H. Budnikova, Catal. Today, 2017, 279, 133 CrossRef CAS.
- Y. H. Budnikova, T. V. Gryaznova, V. V. Grinenko, Y. B. Dudkina and M. N. Khrizanforov, Pure Appl. Chem., 2017, 89, 311 CAS.
- Yu. H. Budnikova and O. G. Sinyashin, Russ. Chem. Rev., 2015, 84, 917 CrossRef CAS.
- M. N. Khrizanforov, S. O. Strekalova, K. V. Kholin, V. V. Khrizanforova, V. V. Grinenko, T. V. Gryaznova and Y. H. Budnikova, RSC Adv., 2016, 6, 42701 RSC.
- Y. B. Dudkina, K. V. Kholin, T. V. Gryaznova, D. R. Islamov, O. N. Kataeva, I. Kh. Rizvanov, A. I. Levitskaya, O. D. Fominykh, M. Yu. Balakina, O. G. Sinyashin and Y. H. Budnikova, Dalton Trans., 2017, 46, 165 RSC.
- Yu. B. Dudkina, T. V. Gryaznova, O. G. Sinyashin and Yu. H. Budnikova, Russ. Chem. Bull., 2015, 64, 1713 CrossRef CAS.
- T. Gryaznova, Y. Dudkina, M. Khrizanforov, O. Sinyashin, O. Kataeva and Y. Budnikova, J. Solid State Electrochem., 2015, 19, 2665 CrossRef CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
- Y. B. Dudkina, T. V. Gryaznova, O. G. Sinyashin and Y. H. Budnikova, Russ. Chem. Bull., 2015, 64, 1713 CrossRef CAS.
- Y. H. Budnikova, D. Yakhvarov and O. G. Sinyashin, J. Organomet. Chem., 2005, 690, 2416 CrossRef CAS.
- Y. B. Dudkina, M. N. Khrizanforov, T. V. Gryaznova and Y. H. Budnikova, J. Organomet. Chem., 2014, 751, 301 CrossRef CAS.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwicka and D. C. Smith, Org. Chem. Front., 2015, 2, 823 RSC.
- Y. B. Dudkina, T. V. Gryaznova, Y. N. Osin, V. V. Salnikov, N. A. Davydov, S. V. Fedorenko, A. R. Mustafina, D. A. Vicic, O. G. Sinyashin and Y. H. Budnikova, Dalton Trans., 2015, 44, 8833 RSC.
- M. Khrizanforov, S. Strekalova, V. Khrizanforova, V. Grinenko, K. Kholin, M. Kadirov, T. Burganov, A. Gubaidullin, T. Gryaznova, O. Sinyashin, L. Xu, D. A. Vicic and Y. Budnikova, Dalton Trans., 2015, 44, 19674 RSC.
- M. N. Khrizanforov, S. V. Fedorenko, S. O. Strekalova, K. V. Kholin, A. R. Mustafina, M. Ye. Zhilkin, V. V. Khrizanforova, Y. N. Osin, V. V. Salnikov, T. V. Gryaznova and Y. H. Budnikova, Dalton Trans., 2016, 45, 11976 RSC.
- M. Khrizanforov, V. Khrizanforova, V. Mamedov, N. Zhukova, S. Strekalova, V. Grinenko, T. Gryaznova, O. Sinyashin and Y. Budnikova, J. Organomet. Chem., 2016, 820, 82 CrossRef CAS.
- D. Y. Mikhaylov, Y. H. Budnikova, T. V. Gryaznova, D. V. Krivolapov, I. A. Litvinov, D. A. Vicic and O. G. Sinyashin, J. Organomet. Chem., 2009, 694, 3840 CrossRef CAS.
- D. Mikhaylov, T. Gryaznova, Y. Dudkina, M. Khrizanphorov, Sh. Latypov, O. Kataeva, D. A. Vicic, O. Sinyashin and Y. Budnikova, Dalton Trans., 2012, 41, 165 RSC.
- R. Hayashi, A. Shimizu, Y. Song, Y. Ashikari, T. Nokami and J. Yoshida, Chem. – Eur. J., 2017, 23, 61 CrossRef CAS PubMed.
- P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956 CrossRef CAS PubMed.
- W.-J. Gao, W.-C. Li, C.-C. Zeng, H.-Y. Tian, L.-M. Hu and R. D. Little, J. Org. Chem., 2014, 79, 9613 CrossRef CAS PubMed.
- Y. Jiang, Q.-Q. Wang, S. Liang, L.-M. Hu, R. D. Little and C.-C. Zeng, J. Org. Chem., 2016, 81, 4713 CrossRef CAS PubMed.
- J. Chen, W.-Q. Yan, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, Org. Lett., 2015, 17, 986 CrossRef CAS PubMed.
- Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS PubMed.
- C. Li, C.-C. Zeng, L.-M. Hu, F.-L. Yang, S. J. Yoo and R. D. Little, Electrochim. Acta, 2013, 114, 560 CrossRef CAS.
- Y. Zhu, Z. Chen, J. Zhang, Q. Wu, C. Ma and R. D. Little, Electrochim. Acta, 2016, 207, 308 CrossRef CAS.
- G. Sun, S. Ren, X. Zhu, M. Huang and Y. Wan, Org. Lett., 2016, 18, 544 CrossRef CAS PubMed.
- L.-J. Li, Y.-Y. Jiang, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Org. Chem., 2015, 80, 11021 CrossRef CAS PubMed.
- S. Liang, C.-C. Zeng, X.-G. Luo, F. Ren, H.-Y. Tian, B.-G. Sun and R. D. Little, Green Chem., 2016, 18, 2222 RSC.
- T. V. Gryaznova, M. N. Khrizanforov, S. O. Strekalova, Y. H. Budnikova and O. G. Sinyshin, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1658 CrossRef CAS.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180 CrossRef CAS PubMed.
- Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872 RSC.
- G. Wulfsberg, Inorganic chemistry, University Science Books, Sausalito, 2000 Search PubMed.
- E. Mentasti, C. Baiocchi and J. S. Coe, Coord. Chem. Rev., 1984, 54, 131 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- M. Panizza, I. Duo, P. A. Michaud, G. Cerisola and Ch. Comninellis, Electrochem. Solid-State Lett., 2000, 3, 550 CrossRef CAS.
- Von C. Friebel and D. Reinen, Z. Anorg. Allg. Chem., 1976, 413, 51 CrossRef.
- Z. Mazej, T. Michałowski, E. A. Goreshnik, Z. Jagličić, I. Arčon, J. Szydłowska and W. Grochala, Dalton Trans., 2015, 44, 10957 RSC.
- N. Kanraki and I. Yasumori, J. Phys. Chem., 1978, 82, 2351 CrossRef.
- X. Mao, X. Ma, S. Zhang, H. Hu, C. Zhu and Y. Cheng, Eur. J. Org. Chem., 2013, 4245 CrossRef CAS.
- W. Liu, S. Wang, Z. Li, Y. Huang, S. Li and A. Wang, Synlett, 2015, 2561 CAS.
- F. Effenberger and H. Kottmann, Tetrahedron, 1985, 41, 4171 CrossRef CAS.
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299 RSC.
- S. D. Rychnosky, R. Vaidyanathan, T. Beauchamp, R. Lin and P. J. Farmer, J. Org. Chem., 1999, 64, 6745 CrossRef.
- Yu. M. Kargin and Yu. G. Budnikova, Russ. J. Gen. Chem., 2001, 71, 1393 CrossRef CAS.
- L. D. Haire, P. H. Krygsman, E. G. Janzen and U. M. Oehler, J. Org. Chem., 1988, 53, 4535 CrossRef CAS.
- W.-J. Gao, W.-C. Li, C.-C. Zeng, H.-Y. Tian, L.-M. Hu and R. Daniel Little, J. Org. Chem., 2014, 79, 9613 CrossRef CAS PubMed.
- M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 9613 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General considerations, electrochemical synthesis details, and the spectra of products. See DOI: 10.1039/c7dt03650g |
| This journal is © The Royal Society of Chemistry 2018 |