DOI:
10.1039/C7DT03229C
(Paper)
Dalton Trans., 2017,
46, 17041-17052
Colchicine metallocenyl bioconjugates showing high antiproliferative activities against cancer cell lines†
Received
31st August 2017
, Accepted 20th November 2017
First published on 20th November 2017
Abstract
A series of ferrocenyl and ruthenocenyl conjugates with colchicine bearing a 1,2,3-triazole moiety were synthesized and their anticancer properties were evaluated. We found that the most potent metallocenyl derivatives Rc4 and Rc5 are 6–7 times more cytotoxic toward HepG2 cells, while Fc4 and Fc5 are two times more cytotoxic toward HCT116 cells as colchicine. We also found that compounds Fc4, Fc5, Rc1 and Rc3–Rc5 are able to induce apoptosis, while compound Fc2 arrests mitosis.
Introduction
Colchicine 1 is a naturally occurring tubulin-binding poison isolated from autumn crocus (Colchicum autumnale). 1 is an antimitotic compound, which binds to β-tubulin, destabilizes microtubules and promotes their depolymerisation leading to cell cycle arrest and – consequently – apoptosis and cell death. Microtubules are highly dynamic structures, which play a crucial role in many important intracellular processes including intramolecular transport and cell division. Therefore, a search for new compounds which disrupt the dynamic equilibrium of microtubules/tubulin is an important goal in search for new promising anticancer agents. Several tubulin-binding agents such as paclitaxel 2 or vincristine 3 (Fig. 1) are widely used in cancer chemotherapy while usage of 1 is limited to the treatment of gout1 due to its high systemic toxicity.2 Nonetheless, a large number of new analogues and conjugates of 1 exhibiting promising anticancer activities have been synthesized.3 For example, colchicine-based amines,3h amides,3f 1,2,3-triazoles,3d glycosides,3i and 4-halo derivatives3b demonstrated high cytotoxicity. Colchicine hybrids with α-tubulin binding drugs, e.g. pironetin, were also studied.3c
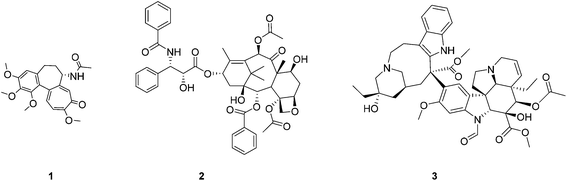 |
| | Fig. 1 Structures of tubulin-binding agents: colchicine 1, paclitaxel 2, and vincristine 3. | |
One of the most efficient methods to prepare new anticancer agents bearing novel and often surprising biological properties is the conjugation of an organic vector with an organometallic moiety.4 This method was successfully applied in the synthesis of organometallic analogues of anticancer (e.g. tamoxifen,5 paclitaxel,6 podophyllotoxin,7 plinabulin8), antiparasitic (e.g. chloroquine,9 praziquantel,10 monepantel11) and antimicrobial agents.12
We demonstrated previously that the introduction of a ferrocenyl moiety into a molecule of a tubulin-binding agent has a positive effect on its biological properties. For example, we found that replacement of the N-benzoyl moiety of paclitaxel with ferrocene not only increased the cytotoxicity of this microtubule-stabilizing agent but also augmented its ability to induce tubulin polymerization.6a We also found that the conjugation of the ferrocenyl moiety with microtubule destabilizers, such as plinabulin and podophyllotoxin, changes their mode of action. For instance, ferrocenyl analogues of plinabulin turned out to be efficient inhibitors of multidrug resistance proteins ABCB1 and ABCG2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 while ferrocenyl conjugates of podophyllotoxin became highly cytotoxic toward multidrug resistant cancer cells.7a These results encouraged us to synthesize metallocenyl conjugates of colchicine and to study their biological properties. Herein we are describing the synthesis and introductory biological assessment of two sets of metallocenyl (ferrocenyl and ruthenocenyl) conjugates of colchicine bearing a 1,2,3-triazolyl linker (Fig. 2). We discuss, in particular, their cytotoxic activity towards cancer cell lines, the impact on the cell cycle and their ability to induce tubulin polymerization.
8 while ferrocenyl conjugates of podophyllotoxin became highly cytotoxic toward multidrug resistant cancer cells.7a These results encouraged us to synthesize metallocenyl conjugates of colchicine and to study their biological properties. Herein we are describing the synthesis and introductory biological assessment of two sets of metallocenyl (ferrocenyl and ruthenocenyl) conjugates of colchicine bearing a 1,2,3-triazolyl linker (Fig. 2). We discuss, in particular, their cytotoxic activity towards cancer cell lines, the impact on the cell cycle and their ability to induce tubulin polymerization.
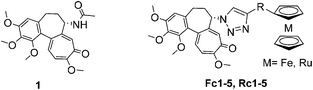 |
| | Fig. 2 Structures of colchicine 1 and its ferrocenyl Fc1–5 and ruthenocenyl Rc1–5 conjugates studied herein. | |
Results and discussion
Synthesis
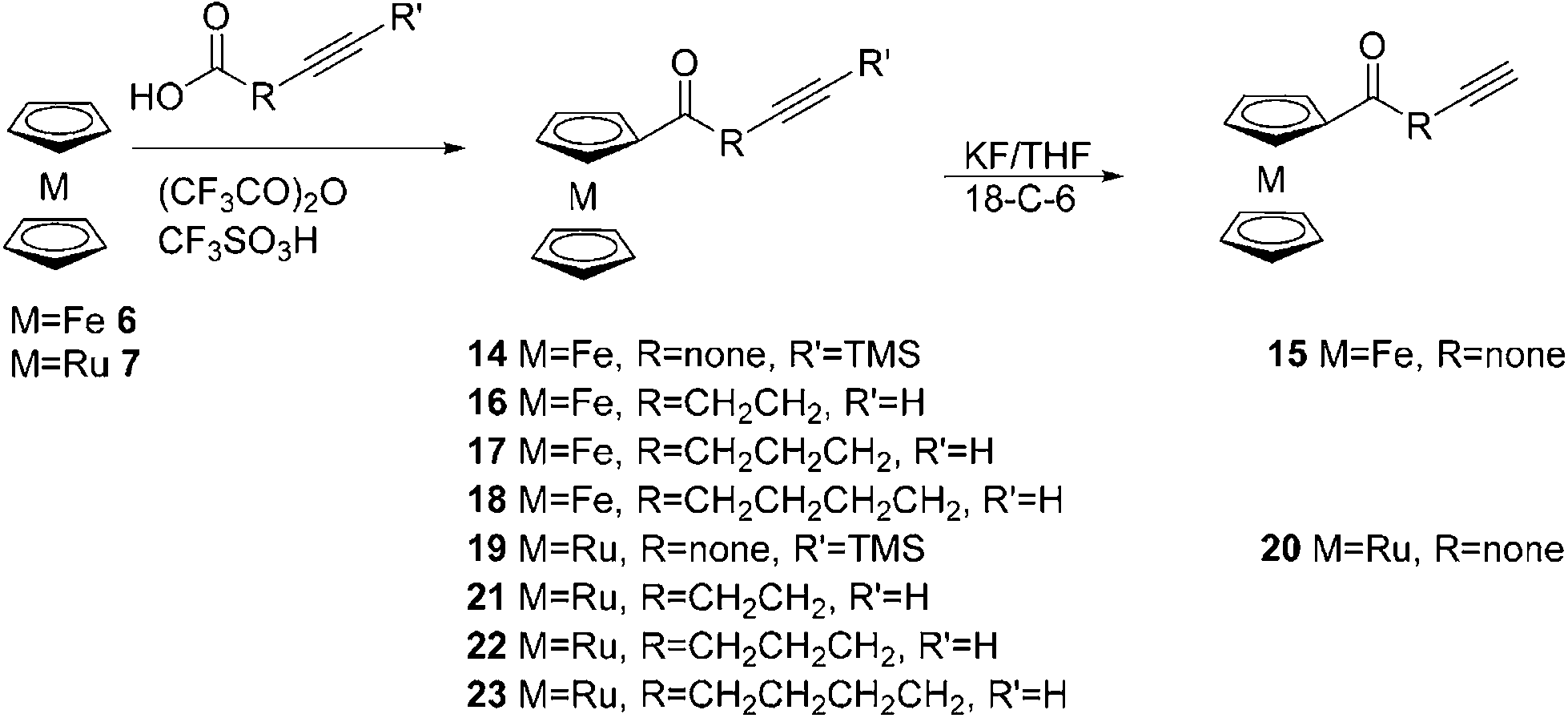
Ferrocenyl, Fc1–5, and ruthenocenyl, Rc1–5, conjugates with colchicine were prepared in a 1,3-cycloaddition reaction of 7-azido-7-deacetamidocolchicine 5 with ethynylmetallocenes 12–13, ϖ-alkynoylferrocenes 15–18 or ϖ-alkynoylruthenocenes 20–23 by following a previously described method.13 Compound 5 was prepared according to a known procedure starting from colchicine (Scheme 1).
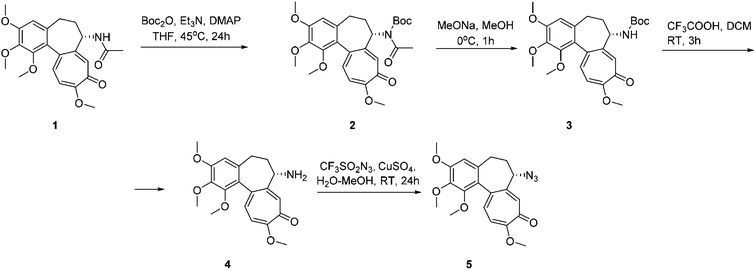 |
| | Scheme 1 Synthesis of 7-azido-7-deacetamidocolchicine 5. | |
Ethynylruthenocene 13 was synthesized by applying the same method as previously described for ethynylferrocene 12 (Scheme 2).14 First, ruthenocene 7 was acetylated in a reaction with acetic acid and trifluoroacetic anhydride catalysed by trifluoromethanesulfonic acid to give acetylruthenocene 9 in 80% yield. Then 9 reacts with phosphorus oxychloride and DMF providing 3-chloro-3-ruthenocenylacroleine 11 in 70%. Further reaction of 11 with potassium hydroxide in 1,4-dioxane gave ethynylruthenocene 13 in 63%.
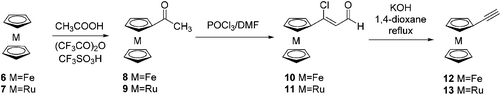 |
| | Scheme 2 Synthesis of ethynylmetallocenes 12–13. | |
The corresponding ϖ-alkynoylmetallocenes were prepared via the Friedel–Crafts acylation of ferrocene and ruthenocene with ϖ-alkynoic acids in the presence of trifluoroacetic anhydride and trifluoromethanosulfonic acid (Scheme 3).
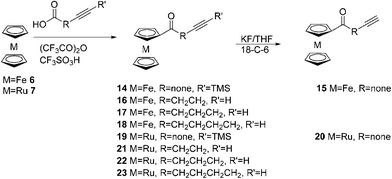 |
| | Scheme 3 Synthesis of ϖ-alkynoylferrocenes and ruthenocenes. | |
Finally, the reaction of 5 with ferrocenyl 12, 15–18 and ruthenocenyl 13, 20–23 acetylenes, catalysed by a freshly prepared copper(II) sulphate and sodium ascorbate copper(I) complex with TTTA, gave the desired products in moderate to good yields (35–81%) (Scheme 4). The structures of the products were confirmed by 1D and 2D NMR spectra.
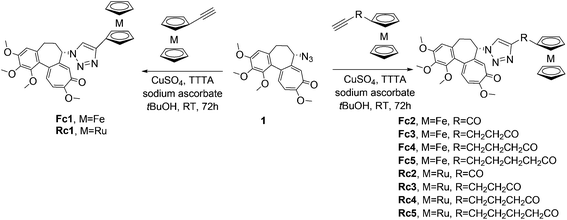 |
| | Scheme 4 Synthesis of ferrocenyl Fc1–5 and ruthenocenyl Rc1–5 conjugates with colchicine. | |
Cytotoxic activity
The cytotoxic potency of the investigated compounds was studied by using the neutral red cell uptake assay in a set of human cell lines derived from different tissues: Colo 205 and HCT116 – colorectal adenocarcinomas; HepG2 – hepatocellular carcinoma; MCF7 – breast adenocarcinoma; and A549 – alveolar basal epithelial cell adenocarcinoma. In general, all of the investigated compounds exhibited biological activities at micromolar or nanomolar concentrations. The cytotoxicity of metallocenyl derivatives was generally comparable to the cytotoxicity of the parental compound 1 and no clear pattern between the linker length, or the kind of the metallocene moiety, and the antiproliferative activity could be demonstrated (see Table 1). The overall toxicity of ferrocenyl compounds (calculated as the sum of ranks for IC50 values for specific compounds across cell lines) could be presented as follows Fc4 > Fc2 > Fc1 ≈ Fc5 > Fc3 while ruthenocene derivatives could be arranged in the following order Rc3 > Rc5 ≥ Rc4 ≥ Rc1 > Rc2. Regarding the cell line sensitivity, MCF7 was the most sensitive line towards ferrocenyl colchicines, followed by HCT116, Colo 205, A549 and HepG2. On the other hand, HCT116 was the most prone towards ruthenocenyl colchicines, followed by MCF7, HepG2, Colo 205 and A549. It should be stressed here that A549 cells were virtually insensitive to Rc4 and Rc5 although the reason for such resistance is unclear and requires further investigation. On the other hand both Rc4 and Rc5 are 6–7 times more cytotoxic toward HepG2, while Fc4 and Fc5 are two times more cytotoxic toward HCT116 than 1, however the explanation of the reason for the high cytotoxicity of these compounds requires further investigation.
Table 1 Cytotoxicity of the ferrocenyl Fc1–5 and ruthenocenyl Rc1–5 colchicine conjugates in comparison with colchicine 1 as determined by using the neutral red uptake assay. 95%-confidence intervals are given below (please note that due to the log-transformation of the data required to perform IC50 calculations, these are asymmetrical). The IC50 values were calculated based on three independent experiments. >100 denotes situations in which the IC50 values were not possible to determine (<50% viability was not achieved in the concentration range used (up to 100 μM))
| Compound |
IC50 |
| A549 |
Colo 205 |
HCT116 |
HepG2 |
MCF7 |
|
Fc1
|
0.42 |
0.16 |
0.14 |
0.89 |
0.059 |
| 0.29–0.59 |
0.08–0.29 |
0.11–0.18 |
0.55–1.44 |
0.051–0.070 |
|
Fc2
|
0.10 |
0.068 |
0.046 |
0.11 |
0.017 |
| 0.07–0.16 |
0.043–0.106 |
0.033–0.065 |
0.06–0.19 |
0.012–0.024 |
|
Fc3
|
1.49 |
0.83 |
0.29 |
2.30 |
0.008 |
| 0.29–4.17 |
0.58–1.17 |
0.20–0.45 |
0.55–4.01 |
0.003–0.016 |
|
Fc4
|
0.37 |
0.11 |
0.006 |
0.045 |
0.002 |
| 0.02–7.44 |
0.01–1.07 |
0.004–0.009 |
0.017–0.121 |
0.001–0.006 |
|
Fc5
|
1.23 |
0.20 |
0.009 |
1.27 |
0.033 |
| 0.15–9.88 |
0.07–0.59 |
0.004–0.019 |
0.48–3.34 |
0.026–0.042 |
| |
|
Rc1
|
0.36 |
0.40 |
0.12 |
0.45 |
0.31 |
| 0.18–0.71 |
0.23–0.68 |
0.07–0.21 |
0.17–2.17 |
0.10–1.01 |
|
Rc2
|
0.43 |
0.64 |
0.39 |
4.65 |
2.96 |
| 0.29–0.62 |
0.44–0.92 |
0.28–0.55 |
1.72–12.6 |
1.71–5.113 |
|
Rc3
|
0.24 |
0.24 |
0.040 |
0.22 |
0.072 |
| 0.15–0.36 |
0.08–0.7 |
0.029–0.054 |
0.12–0.44 |
0.046–0.114 |
|
Rc4
|
>100 |
0.31 |
0.058 |
0.004 |
0.13 |
| 0.05–2.03 |
0.021–0.159 |
0.003–0.007 |
0.03–0.53 |
|
Rc5
|
>100 |
0.090 |
0.048 |
0.003 |
0.71 |
| 0.006–1.47 |
0.007–0.309 |
0.002–0.004 |
0.13–3.78 |
|
1
|
0.061 |
0.017 |
0.016 |
0.023 |
0.005 |
| 0.039–0.097 |
0.013–0.023 |
0.014–0.018 |
0.018–0.030 |
0.004–0.006 |
Effects on the cell cycle
As the principal mechanism of colchicine action is based on tubulin binding, microtubule destabilization and induction of the cell cycle arrest, we wanted to check whether this is also the case for metallocenyl colchicines. We have chosen the Colo 205 cell line as our model for cell cycle analysis as the IC50 value for all the investigated compounds was below 1 μM for these cells. The expected mitotic arrest was observed for 1 and, to a lower extent, for Fc2 (Table 2). However, the subG1 phase (sign of apoptosis) was observed for Fc4, Fc5, Rc1 and Rc3–Rc5. At the same time, the percentage of cells in the G2/M phase was significantly lower for Fc4, Fc5, and Rc3–Rc5 colchicine derivatives. In the case of Rc5 (the most evident case), the results are suggestive for S phase blockage, which may indicate that some vital resources were depleted in cells exposed to this compound. This mechanism however, cannot be excluded for the other compounds mentioned above.
Table 2 Cell cycle phase distribution in Colo 205 cells exposed to 10 nM of ferrocenyl and ruthenocenyl conjugates. Average data ± SEM from 3 independent experiments. Control – cells incubated in a complete medium alone, DMSO – cells incubated in a complete medium with the addition of 0.1% of DMSO (solvent control)
| |
subG1 |
G1 |
S |
G2/M |
|
Control
|
0.0 ± 0.0 |
59.6 ± 3.5 |
30.5 ± 2.9 |
9.5 ± 1.6 |
|
DMSO
|
0.0 ± 0.0 |
60.4 ± 4.4 |
29.2 ± 2.6 |
8.5 ± 1.6 |
|
1
|
3.6 ± 1.3 |
52.2 ± 5.3 |
22.1 ± 3.8 |
20.6 ± 4.4 |
|
Fc1
|
0.0 ± 0.0 |
61.6 ± 4.7 |
30.4 ± 3.8 |
7.1 ± 0.9 |
|
Fc2
|
0.0 ± 0.0 |
50.2 ± 3.0 |
37.1 ± 2.5 |
11.0 ± 1.3 |
|
Fc3
|
0.0 ± 0.0 |
55.3 ± 4.8 |
34.8 ± 3.6 |
8.6 ± 0.9 |
|
Fc4
|
8.0 ± 2.4 |
56.1 ± 4.6 |
30.5 ± 3.4 |
4.6 ± 0.6 |
|
Fc5
|
2.8 ± 0.5 |
64.1 ± 5.6 |
28.9 ± 3.8 |
3.8 ± 0.5 |
| |
|
Rc1
|
6.0 ± 2.3 |
49.8 ± 9.0 |
38.1 ± 5.6 |
5.7 ± 1.4 |
|
Rc2
|
0.0 ± 0.0 |
58.9 ± 6.3 |
32.4 ± 4.9 |
7.9 ± 2.2 |
|
Rc3
|
7.1 ± 1.4 |
59.4 ± 4.7 |
27.3 ± 2.0 |
2.6 ± 1.5 |
|
Rc4
|
7.7 ± 1.6 |
59.3 ± 3.7 |
27.5 ± 2.3 |
2.5 ± 0.4 |
|
Rc5
|
16.3 ± 2.3 |
32.6 ± 4.9 |
44.5 ± 3.4 |
5.4 ± 0.7 |
Influence on tubulin polymerisation
To explain the cell cycle observations in a more detailed way, we analysed the ability of the metallocenyl colchicine conjugates to induce or interfere with tubulin polymerization. Generally, all of the investigated compounds were tubulin polymerization inhibitors although weaker than colchicine. All of the studied compounds were able to completely inhibit tubulin polymerization at a rather high concentration (10 μM, Fig. 3). Fc1 and Fc2 were generally the least potent among the compounds studied although the differences were not overwhelming. Nevertheless, no clear correlation between metallocenyl colchicine cytotoxicity and its ability to disrupt tubulin polymerization could be found. Our previous study has shown that the conjugation of tubulin polymerisation inhibitors such as plinabulin8 and podophyllotoxin7a with ferrocene decreased the ability of the conjugates to inhibit tubulin polymerisation.
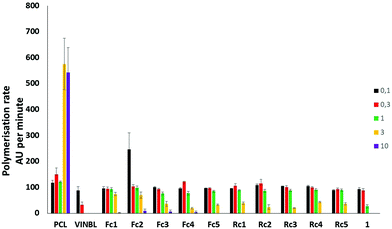 |
| | Fig. 3 Concentration-dependent ability to induce tubulin polymerisation by Fc1–5 and Rc1–5 in comparison with 1, paclitaxel (PCL) and vinblastine (VINBL). Lack of the polymerisation rate bars indicates that the polymerisation rate reached 0 AU per minute (total inhibition). | |
Reactive oxygen species production
It is postulated that the biological effects of metallocenes can be attributed to their redox properties.4f,15 We have previously demonstrated that even though ferrocenyl paclitaxel analogues are able to stimulate intracellular reactive oxygen species (ROS) production, the redox effects are not as important for their biological activity as their ability to bind tubulin.16 We have been intrigued, however, by the differential effects exerted by the investigated colchicine analogues on the cells of different tissue origins (see Table 1). It seems plausible that cells derived from organs that are metabolically active or continuously exposed to reactive oxygen species such as liver17 or lungs18 and thus producing higher amounts of glutathione and other antioxidant barrier elements are less sensitive to redox active compounds. Such effects are readily observed for ferrocenyl colchicines (A549 and HepG2 cells were the most resistant to these compounds) but no such correlation can be inferred for ruthenocene colchicine conjugates. Therefore, we decided to study intracellular ROS production both in less (A549) and in more sensitive (HCT116) cells exposed to metallocenyl colchicines. As a marker of ROS production, we decided to choose dihydrorhodamine 123 (DHR123) – a leuco dye, which is known to be readily oxidized by hydrogen superoxide in a reaction catalysed by peroxidases19 to a highly fluorescent rhodamine 123 further sequestered in the mitochondria. The initial results were quite surprising: ROS production in A549 cells was suppressed by 20 to 40% by metallocene colchicines, irrespective of their chemical structure. Additionally, such an effect was also observed for colchicine itself (Fig. 4A). On the other hand, ROS production was tremendously increased (by 250–475%) in HCT116 cells and again – this effect was observed for all the compounds studied, including parental 1 (Fig. 4C). However, both rhodamine 123 (formed by the oxidation of DHR123) and colchicine are known substrates for ABCB1 – a plasma membrane xenobiotic transporter involved in the multidrug resistance phenomenon.20 Thus, the potential activity of this protein should be excluded, if such results are to be considered biologically relevant. Employing calcein accumulation assay (data not shown) we were able to detect significant ABCB1 activity in HCT116 cells. Therefore, we decided to repeat the experiments but in the presence of 10 μM verapamil – an efficient inhibitor of ABCB1. Such treatment did not significantly alter the results obtained for A549 cells. Both colchicine and its metallocene derivatives (with a notable exception of Rc1 and Rc3 for which the results were statistically insignificant compared to the control) reduced the level of intracellular DHR123 oxidation. The underlying reason for this phenomenon is unknown and requires further investigation. More interesting results were observed in the case of HCT116. The intracellular fluorescence level was much lower than in verapamil-untreated cells, however DHR123 oxidation was statistically more pronounced for Fc4, Rc1, and Rc3–Rc5. It is thus probable that the high antiproliferative activity of Fc4 and Rc3–Rc5 can be at least partially attributed to their redox potential. Another interesting conclusion resulting from the observation of effects of verapamil on rhodamine 123 accumulation in HCT116 cells is that metallocene colchicines do interact with ABCB1. Nevertheless, the ABCB1 activity is not enough to allow the cells escape the fatal effects of metallocene colchicines as HCT116 cells were the second most sensitive cells in terms of antiproliferative potential of the investigated compounds.
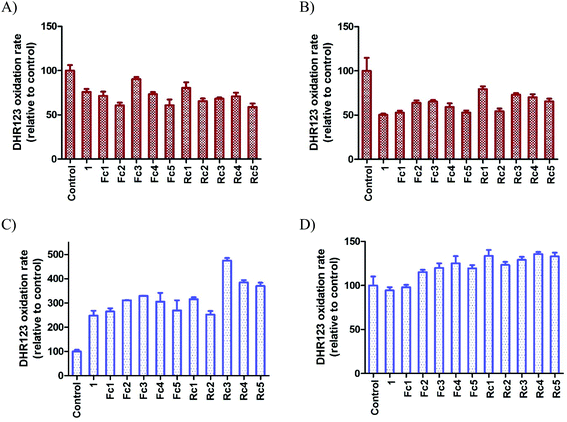 |
| | Fig. 4 Dihydrorhodamine 123 oxidation by A549 (panels A and B) and HCT116 (panels C and D) cells after 4-hour-exposure to 1, Fc1–5 and Rc1–5. Results are expressed as mean ± SEM, n = 3. Fluorescence of control cells (incubated with DHR123 and an appropriate amount of DMSO) was considered 100% for a given experimental set. Panels A and C – cells incubated in the absence of ABCB1 inhibitor, panels B and D – cells incubated in the presence of ABCB1 inhibitor (10 μM verapamil). | |
Partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
Cytotoxicity is generally directly related to the intracellular concentration of the drug. Accumulation of drugs is dependent on their lipophilicity, therefore, increasing the drug lipophilicity may result in an elevation of the intracellular drug concentration. Such an approach was successfully applied in a synthesis of highly lipophilic anthracycline analogs, such as annamycin and idarubicin.21 The octanol–water partition coefficients (logP) of the investigated compounds were determined using the HPLC method.22 All of the studied metallocene conjugates exhibited significantly higher logP values than colchicine (Table 3). Ruthenocene compounds demonstrated slightly higher logP values than their corresponding ferrocenyl counterparts but compound lipophilicity could be arranged as follows for both metallocenes: M5 ≈ M2 > M1 > M4 > M3 (M = Fc, Rc). However, the differences of logP values between compounds were presumably too low to see the effect of drug lipophilicity on the cells.
Table 3 Lipophilicity values of ferrocenyl Fc1–5 and ruthenocenyl Rc1–5 colchicine conjugates in comparison with colchicine 1
| Compound |
logP |
|
Fc1
|
3.203 ± 0.002 |
|
Fc2
|
3.307 ± 0.003 |
|
Fc3
|
2.973 ± 0.001 |
|
Fc4
|
3.037 ± 0.003 |
|
Fc5
|
3.246 ± 0.003 |
| |
|
Rc1
|
3.237 ± 0.004 |
|
Rc2
|
3.393 ± 0.009 |
|
Rc3
|
3.060 ± 0.002 |
|
Rc4
|
3.123 ± 0.002 |
|
Rc5
|
3.317 ± 0.002 |
|
1
|
1.30 (ref. 23) |
Conclusions
We have synthesized two series of metallocenyl, ferrocenyl and ruthenocenyl conjugates with colchicine bearing a 1,2,3-triazolylalkyl linker between the colchicine and metallocenyl moieties. Their cytotoxic properties were studied towards a set of cancer cells of different organ origins. We found that the cytotoxicity of the conjugate was generally similar to that of colchicine. However, the mode of action of metallocene colchicine conjugates differs from the mode of action of the parental compound (interactions of the investigated compounds with tubulin and microtubules as well as their effects on the cell cycle were different from the effects of 1). Additionally, our results clearly demonstrated also that the cellular response to ferrocenyl colchicines differs from the one to ruthenocenyl compounds. Some of the synthesised conjugates exhibit higher cytotoxicity than 1. These effects can be at least partially attributed to metallocene colchicine-induced ROS production. Further studies are required to elucidate these differences as well as to explain the basis of cytotoxicity of some of the investigated metallocenyl colchicines.
Experimental section
General
All reactions were carried out using standard Schlenk techniques. Commercially available solvents (HPLC) and reagents were used as received. Dichloromethane (DCM) was distilled from calcium hydride and stored over activated molecular sieves 4A (8–12 mesh). Column chromatography was performed on FLUKA Silica gel 60 for flash chromatography (0.040–0.063 mm, 230–400 mesh). TLC chromatography was performed on aluminium sheets coated with Merck 5735 Kieselgel 60F254. 1D (1H, 13C{1H}) and 2D (1H–1H COSY, 1H–13C HSQC or HMQC, 1H–13C HMBC) NMR spectra were recorded on a Bruker ARX 600 MHz (spectrometer frequency 600.3 MHz for 1H and 150.9 MHz for 13C). Chemical shifts for the NMR spectra are referenced relative to the residual protons in the deuterated solvent (CDCl3δ = 7.26 ppm for 1H and δ = 77.00 ppm for 13C{1H}). The spectra were recorded at room temperature (301 K), chemical shifts are in ppm and coupling constants in Hz. ESI-MS spectra were recorded in positive mode on a Varian 500-MS LC ion trap spectrometer (samples were dissolved in acetonitrile). HPLC analyses were performed on a Shimadzu Prominence system equipped with a PDA detector and using a Phenomenex Kinetex 5μ XB-C18 100 Å column 150 × 4.6 mm. Detection was recorded at wavelengths 220 nm, 254 nm and 280 nm. The purity was tested at a 1 ml min−1 flow rate using a mixture of A:B 40:60 (v:v), solvents: phase A was 20 mM MOPS, 0.15% 1-decylamine, pH = 7.4; phase B was 0.25% 1-octanol in methanol. Despite numerous attempts, we were unable to obtain correct results of elemental analyses for all colchicine conjugates and the purity of the investigated compound was confirmed by HPLC analysis (purity >95%).
Acetylruthenocene 9.
4.63 g (3.01 ml, 22 mmol) of trifluoroacetic anhydride was added to a solution of 1.31 g (1.25 ml, 21.8 mmol) of acetic acid in 50 ml of anhydrous dichloromethane. Then 4.63 g (20 mmol) of ruthenocene was added and after its complete dissolving 3.0 g (1.76 ml, 20 mmol) of trifluoromethanesulfonic acid was added dropwise. The resulting solution was stirred at RT for 2 h and the reaction was quenched by adding 100 ml of water. The product was extracted with dichloromethane, and the organic solution was washed with water, sodium bicarbonate and brine, dried with sodium sulphate and evaporated. The crude product was purified by column chromatography on silica using dichloromethane as the solvent. A pure product was obtained in 87% yield (4.75 g). This compound was identical with an authentic sample.24
3-Chloro-3-ruthenocenylacroleine 11.
6.28 g (4.0 ml, 41 mmol) of phosphorus(V) oxychloride was added dropwise for 30 min to cool to 0 °C in DMF (25 ml) placed in a round bottom flask. Then a solution of 2.0 g (7.3 mmol) of 9 in a minimal amount of anhydrous DMF was added dropwise and the resulting mixture was stirred at 0 °C for 2 h and poured into 500 ml of water. The resulting mixture was neutralized by adding of solid sodium acetate and the product was extracted with diethyl ether. The organic solution was washed with water, brine and dried over sodium sulphate and evaporated to dryness. Chromatography on silica using dichloromethane–cyclohexane (8–1) as the eluent gave pure 11 in 70% yield (1.64 g). This compound was identical with an authentic sample.25 Mp 97–100 °C; elemental analysis found: C, 49.0; H, 3.8. C13H11ClORu requires C, 48.8; H, 3.5%; IR (KBr) νmax/cm−1: 3306w, 3086m, 2851m, 2742w, 1663vs, 1603vs, 1445m, 1410s, 1384s, 1350m, 1330m, 1253s, 1241s, 1208m, 1133s, 1100m, 1035s, 997s, 817vs, 787s, 691m, 649m, 534w, 456vs; 1H NMR (CDCl3) δ 10.02 (d, J = 7.1 Hz, 1H, CHO), 6.37 (d, J = 7.1 Hz, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 5.08 (t, J = 1.8 Hz, 2H, Cp), 4.81 (t, J = 1.8 Hz, 2H, Cp), 4.62 (s, 5H, Cp), 13C{1H} NMR (CDCl3) δ 191.2 (CO), 153.7 (
), 5.08 (t, J = 1.8 Hz, 2H, Cp), 4.81 (t, J = 1.8 Hz, 2H, Cp), 4.62 (s, 5H, Cp), 13C{1H} NMR (CDCl3) δ 191.2 (CO), 153.7 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CH), 120.2 (CH), 84.8 (Cpipso), 73.3 (Cp), 72.7 (Cp), 70.4 (Cp).
CH), 120.2 (CH), 84.8 (Cpipso), 73.3 (Cp), 72.7 (Cp), 70.4 (Cp).
Ethynylruthenocene 13.
714 mg of powdered potassium hydroxide was added to a solution 1.30 g (4.07 mmol) of 11 in 60 ml of 1,4-dioxane and the resulting mixture was refluxed for 2 h. Then the reaction was quenched by adding 100 ml of water and the resulting solution was neutralized with 5% citric acid. The product was extracted with cyclohexane; the organic solution was washed with brine dried over sodium sulphate and evaporated. A pure product was isolated in 64% yield (660 mg) by column chromatography using cyclohexane as the eluent. This compound was identical with an authentic sample.26 Mp 71–73 °C; elemental analysis found: C, 56.6; H, 4.0. C12H10Ru requires C, 56.5; H, 4.0%; IR (KBr) νmax/cm−1: 3268vs, 3118w, 3102w, 2924w, 2103s, 1442m, 1407m, 1390m, 1260w, 1222s, 1201m, 1098s, 1025s, 904m, 849w, 811vs, 653s, 619s, 556m, 519s, 378m, 428s; 1H NMR (CDCl3) δ 4.86 (br s, 2H, Cp), 4.60 (s, 5H, Cp), 4.55 (br s, 2H, Cp), 2.64 (s, 1H, Hacetylene); 13C{1H} NMR (CDCl3) δ 81.5 (CH), 73.9 (Cp), 73.5 (CH), 71.9 (Cp), 70.6 (Cp).
General procedure A – synthesis of ϖ-alkynoylruthenocenes 19–23
These compounds were prepared by applying the previously described procedure for the synthesis of ϖ-alkynoylferrocenes.27
(3-Trimethylsilyl)propynoylruthenocene 19.
462 mg (305 μl, 2.2 mmol) of trifluoroacetic anhydride was added to a solution of 312 mg (2.2 mmol) of 3-(trimethylsilyl)propynoic acid in 10 ml of anhydrous dichloromethane. To the resulting solution, 462 mg (2.0 mmol) of ruthenocene was added and after its complete dissolution 300 mg (176 μl, 2.0 mmol) of trifluoromethanesulfonic acid was added. The resulting solution was stirred at RT for 2 h and poured into 50 ml of water. The product was extracted with dichloromethane, and the organic solution was washed with water, sodium bicarbonate, brine, dried over sodium sulphate and evaporated. Chromatography on silica using dichloromethane as the eluent gave a pure product in 52% yield (371 mg). Mp 92–94.5 °C; elemental analysis found: C, 54.2; H, 5.2. C16H18ORuSi requires C, 54.1; H, 5.1%; IR (KBr) νmax/cm−1: 3237m, 3110m, 3098m, 2959s, 2900m, 2168m, 2159m, 1629vs, 1590m, 1505w, 1448vs, 1411s, 1392s, 1372vs, 1350m, 1268vs, 1249vs, 1213w, 1102s, 1083vs, 1021s, 1004vs, 894vs, 847vs, 819vs, 761s, 752s, 725s, 705m, 632s, 569m, 482s, 423m; ESI-MS calc. for C16H18ORuSi 356.0 (M+) found 356.9 (M+, 41%); 1H NMR (CDCl3) δ 5.22 (t, J = 1.6 Hz, 2H, Cp), 4.84 (d, J = 1.6 Hz, 2H, Cp), 4.62 (s, 5H, Cp), 0.27 (s, 9H, TMS); 13C{1H} NMR (CDCl3) δ 178.2 (CO), 101.6 (C), 95.6 (C), 85.1 (C), 74.3 (Cp), 72.8 (Cp), 71.8 (Cp), −0.6 (TMS).
(4-Pentyn-1-oyl)ruthenocene 21.
This compound was prepared in 59% yield (369 mg) according to general procedure A starting from 192 mg (2.2 mmol) of 4-pentynoic acid. Mp 79.5–82.0 °C; elemental analysis found: C, 56.6; H, 4.0. C12H10Ru requires C, 56.5; H, 4.0%; IR (KBr) νmax/cm−1: 3306s, 3285m, 3106m, 3090s, 2945m, 2922w, 2905w, 2115m, 1683vs, 1637m, 1456vs, 1426m, 1406s, 1396s, 1477s, 1261vs, 1225m, 1187w, 1098s, 1084vs, 1041w, 1029s, 995s, 980s, 904w, 893w, 863m, 843m, 833m, 815vs, 645vs, 606vs, 544m, 530s, 510m, 450s, 428s; ESI-MS calc. for C15H14ORu 312.0 (M+) found 313.3 (M + H, 100%)+; 1H NMR (CDCl3) δ 5.12 (t, J = 1.7 Hz, 2H, Cp), 4.79 (t, J = 1.8 Hz, 2H, Cp), 4.61 (s, 5H, Cp), 2.87 (t, J = 7.4 Hz, 2H, C2CH2CCH), 2.54 (td, J = 7.3, 2.6 Hz, 2H, CH2C2CCH), 1.97 (t, J = 2.6 Hz, 1H, CH2CH2CC); 13C{1H} NMR (CDCl3) δ 199.7 (CO), 83.8 (CH2CH2CH), 83.3 (Cpipso), 73.6 (Cp), 72.0 (Cp), 70.6 (Cp), 68.6 (CH2CH2CH), 37.6 (H2CH2CCH), 13.4 (CH2H2CCH); elemental analysis calc. for C15H14ORu C-57.9, H-4.5%, found C-57.8, H-4.7%
(5-Hexyn-1-oyl)ruthenocene 22.
This compound was prepared in 75% yield (490 mg) according to general procedure A starting from 246 mg (242 μl, 2.2 mmol) of 5-hexynoic acid. Mp 72–73.4 °C; elemental analysis found: C, 58.9; H, 5.1. C16H16ORu requires C, 59.1; H, 5.0%; IR (KBr) νmax/cm−1: 3253vs, 3111m, 3104m, 3092w, 2957m, 2943m, 2927s, 2906m, 2847m, 2359w, 2343w, 1656vs, 1457s, 1430s, 1402s, 1380s, 1357s, 1316m, 1254s, 1181m, 1166m, 1199s, 1982m, 1052s, 1039m, 1015m, 997m, 987m, 880m, 871m, 841m, 819vs, 796m, 710s, 687s, 648m, 546m, 513s, 455s, 425m; ESI-MS calc. for C16H16ORu 326.0 (M+) found 326.0 (M+, 60%), 327.0 ((M + H)+, 100%); 1H NMR (CDCl3) δ 5.12 (t, J = 1.8 Hz, 2H, Cp), 4.77 (t, J = 1.8 Hz, 2H, Cp), 4.60 (s, 5H, Cp), 2.77 (t, J = 7.1 Hz, C2CH2CH2CCH), 2.74 (td, J = 6.8, 2.6 Hz, 2H, CH2CH2C2CCH), 1.99 (t, J = 2.6 Hz, 1H, CH2CH2CH2CC), 1.90–1.85 (m, 2H, CH2C2CH2CCH); 13C{1H} NMR (CDCl3) δ 201.6, 84.o (2 × C, CH2CH2CH2CH and Cpipso), 73.4 (Cp), 72.0 (Cp), 70.7 (Cp), 68.9 (CH2CH2CH2CH), 37.1 (H2CH2CH2CCH), 23.2 (CH2H2CH2CCH), 17.9 (CH2CH2H2CCH).
(6-Heptyn-1-oyl)ruthenocene 23.
This compound was prepared in 72% yield (488 mg) according to general procedure A starting from 278 mg (2.2 mmol) of 6-heptynoic acid. Mp 87–89.5 °C; elemental analysis found: C, 60.1; H, 5.5. C17H18ORu requires C, 60.2; H, 5.4%; IR (KBr) νmax/cm−1: 3256vs, 3086s, 2948m, 2939m, 2919m, 2904m, 2869m, 2360w, 2343w, 1671vs, 1453s, 1409s, 1393m, 1379s, 1357m, 1303m, 1290w, 1524s, 1213m, 1101s, 1096m, 1050m, 1026m, 1000m, 982m, 892m, 876w, 869w, 821s, 787w, 752w, 684s, 525w, 514m, 460s, 434s, 418m; ESI-MS calc. for C17H18ORu 340.0 (M+) found 341.0 ((M + H)+, 100%); 1H NMR (CDCl3) δ 5.10 (t, J = 1.7 Hz, 2H, Cp), 4.77 (t, J = 1.7 Hz, 2H, Cp), 4.59 (s, 5H, Cp), 2.61 (t, J = 7.4 Hz, 2H, C2CH2CH2CH2CCH), 2.23 (td, J = 7.1, 2.6 Hz, 2H, CH2CH2CH2C2CCH), 1.96 (t, J = 2.6 Hz, 1H, CH2CH2CH2CH2CC), 1.81–1.76 (m, 2H, CH2C2CH2CH2CCH), 1.61–1.56 (m, 2H, CH2CH2C2CH2CCH); 13C{1H} NMR (CDCl3) δ 202.1 (CO), 84.2 (CH or Cpipso), 84.0 (CH or Cpipso), 73.4 (Cp), 71.9 (Cp), 70.7 (Cp), 68.5 (CH2CH2CH2CH2CH), 35.3 (H2CH2CH2CH2CCH), 28.1 (CH2CH2H2CH2CCH), 24.1 (CH2H2CH2CH2CCH), 18.3 (CH2CH2CH2H2CCH).
Propynoylruthenocene 20.
A large excess of powdered potassium fluoride (1.28 g) was added to a solution of 320 mg (0.9 mmol) of 19 and 10 mg of 18-crown-6 in 6 ml of tetrahydrofuran. The resulting solution was stirred at RT for 30 min and poured into 50 ml of water. The product was extracted with dichloromethane, and the organic solution was washed with water, brine, dried over sodium sulfate and evaporated to dryness. Chromatography on silica using dichloromethane as the eluent gave pure 20 in 98% yield (250 mg). This compound was identical with an authentic sample.28 Mp 99–101.2 °C; elemental analysis found: C, 55.1; H, 3.7. C13H10ORu requires C, 55.1; H, 3.6%; IR (KBr) νmax/cm−1: 3212s, 3092w, 2090s, 1625vs, 1449s, 1411w, 1394w, 1375m, 1350w, 1339w, 1324s, 1101m, 1072s, 1034m, 1023w, 1001m, 992m, 882m, 869w, 849w, 841w, 823m, 765m, 725m, 477m, 430m; 1H NMR (CDCl3) δ 5.25 (t, J = 1.8 Hz, 2H, Cp), 4.86 (t, J = 1.8 Hz, 2H, Cp), 4.64 (s, 5H, Cp), 3.11 (s, 1H, Hacetylene); 13C{1H} NMR (CDCl3) δ 177.7 (CO), 84.4 (Cpipso), 81.0 (CH), 76.3 (CH), 74.5 (Cp), 72.8 (Cp), 71.7 (Cp).
General procedure B – synthesis of colchicine-metallocenyl 1,2,3-triazolyl conjugates Fc1–5 and Rc1–5
These compounds were prepared by applying the previously described procedure for the synthesis of 1-aryl-4-ferrocenyl-1,2,3-triazoles.13
7-Deacetamido-7-(4-ferrocenyl-1H-1,2,3-triazol-1-yl)-colchicine Fc1.
75 μl of 0.1 M CuSO4 and 75 μl of 0.1 M sodium ascorbate and 300 μl of 0.01 M of TTTA were placed in a vial. The resulting colourless solution was added to a solution of 30 mg (0.143 mmol) of ethynylferrocene 12 and 55 mg (0.143 mmol) of 7-deacetamido-7-azidocolchicine 5 in 2 ml of tert-butanol. The reaction mixture was stirred at RT for 3 days and the solvents were evaporated. Chromatography on silica using ethyl acetate (or ethyl acetate–cyclohexane 4–1) gave pure Fe1 in 65% yield (55 mg). Mp 143–147 °C; elemental analysis found: C, 63.5; H, 5.6; N, 6.7. C32H31FeN3O5·½H2O requires C, 63.8; H, 5.4; N, 7.0%; IR (KBr) νmax/cm−1: 3102w, 2934m, 2845w, 1618s, 1587vs, 1488s, 1461s, 1431m, 1399s, 1350s, 1323s, 1283m, 1254vs, 1195m, 1181m, 1139s, 1096s, 1046m, 1020m, 1002m, 984m, 818m, 509m; ESI-MS calc. for C32H31FeN3O5 593.2 (M+) found 593.0 (M+, 100%), 594.0 ((M + H)+, 66%); Rf (HPLC) τ = 9.6 min; 1H NMR (CDCl3) δ 7.50 (s, 1H, Htriazolyl), 7.28 (d, J = 10.7 Hz, 1H, H-12), 6.79 (d, J = 10.8 Hz, 1H, H-11), 6.61 (s, 1H, H-4), 6.57 (s, 1H, H-8), 5.41 (dd, J = 11.9, 5.4 Hz, 1H, H-7), 7.72 (m, 1H, Cp), 4.71–4.70 (m, 1H, Cp), 4.29 (br s, 2H, Cp), 4.08 (s, 5H, Cp), 3.97 (s, 3H, 10-OCH3), 3.95 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 2.86–2.81 (m, 1H, H-5), 2.77–2.74 (m, 1H, H-6), 2.67–2.60 (m, 2H, H-5 and H-6); 13C{1H} (CDCl3) δ 178.8 (C-9), 164.3 (C-10), 153.9 (CAry), 151.1 (CAry), 147.6 (CAry), 147.2 (CAry), 141.7 (CAry), 135.2 (C-12), 134.3 (CAry), 133.6 (CAry), 131.9 (C-8), 125.0 (CAry), 119.0 (CHtriazolyl), 111.6 (C-11), 107.5 (C-4), 69.7 (Cp), 68.8 (Cp), 66.8 (Cp), 62.6 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.3 (OCH3), 56.1 (OCH3), 35.3 (C-5), 29.7 (C-6).
7-Deacetamido-7-(4-ferrocenoyl-1H-1,2,3-triazol-1-yl)-colchicine Fc2.
This compound was prepared in 77% yield (118 mg) according to general procedure B starting from 95 mg (0.248 mmol) of 5 and 59 mg (0.248 mmol) of 15. Mp 152–156 °C; elemental analysis found: C, 64.0; H, 5.1; N, 6.6. C33H31FeN3O6 requires C, 63.8; H, 5.0; N, 6.8%; IR (KBr) νmax/cm−1: 3223w, 2927s, 2853m, 1623vs, 1587vs, 1527m, 1488s, 1459s, 1399m, 1377m, 1351m, 1322m, 1254vs, 1195m, 1180m, 1140s, 1097s, 1047m, 1036m, 1022m, 1001m, 984m, 940w, 925w, 841m, 826m, 770m, 500m; ESI-MS calc. for C33H31FeN3O6 621.2 (M+) found 621.0 (M+, 74%), 622.0 ((M + H)+, 100%); Rf (HPLC) τ = 12.3 min; 1H NMR (CDCl3) δ 8.25 (s, 1H, Htriazolyl), 7.32 (d, J = 10.7 Hz, 1H, H-12), 6.81 (d, J = 10.8 Hz, 1H, H-11), 6.61 (s, 1H, H-4), 6.49 (s, 1H, H-8), 5.49–5.46 (m, 3H, Cp and H-7), 4.64 (s, 2H, Cp), 4.17 (s, 5H, Cp), 3.97 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 2.86–2.81 (m, 1H, H-6), 2.79–2.76 (m, 1H, H-5), 2.68–2.60 (m, 2H, H-5 and H-6); 13C{1H} NMR (CDCl3) δ 188.8 (OFc), 178.7 (C-9), 164.4 (C-10), 154.0 (CAry), 151.1 (CAry), 149.1 (CAry), 147.1 (CAry), 141.9 (CAry), 135.5 (C-12), 134.2 (CAry), 133.4 (CAry), 131.8 (C-8), 127.0 (CHtriazolyl), 111.7 (CAry), 107.6 (C-4), 77.7 (Cpipso), 73.0 (Cp), 71.7 (Cp), 71.5 (Cp), 70.3 (Cp), 62.9 (C-7), 61.2 (2 × OCH3), 56.4 (OCH3), 56.1 (OCH3), 35.3 (C-6), 29.7 (C-5).
7-Deacetamido-7-(4-(3-ferrocenyl-3-oxoprop-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Fc3.
This compound was prepared in 80% yield (98 mg) according to general procedure B starting from 72 mg (0.188 mmol) of 5 and 50 mg (0.188 mmol) of 16. Mp 108–112 °C; elemental analysis found: C, 64.7; H, 6.3; N, 5.6. C35H35FeN3O6 requires C, 64.7; H, 5.4; N, 6.5%; IR (KBr) νmax/cm−1: 3095w, 2926s, 2852m, 1664s, 1618s, 1587vs, 1488s, 1457s, 1430m, 1399s, 1379m, 1350s, 1322s, 1254vs, 1195m, 1180m, 1140s, 1096s, 1086s, 1048m, 1020m, 1003m, 983m, 924w, 900w, 824m, 487m; ESI-MS calc. for C35H35FeN3O6 649.2 (M+) found 649.0 (M+, 56%), 650.0 ((M + H)+, 100%); Rf (HPLC) τ = 7.0 min; 1H NMR (CDCl3) δ 7.43 (s, 1H, Htriazolyl), 7.26 (d, J = 10.7 Hz, 1H, H-12), 6.78 (d, J = 10.8 Hz, 1H, H-11), 6.57 (br s, 2H, H-4 and H-8), 5.34 (dd, J = 12.1, 5.9 Hz, 1H, H-7), 4.78 (brs, 2H, Cp), 4.47 (brs, 2H, Cp), 4.13 (s, 5H, Cp), 3.96 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.21–3.19 (m, 2H, CH2C2COFc), 3.14–3.11 (m, 2H, C2CH2COFc), 2.83–2.78 (m, 1H, H-6), 2.70 (dd, J = 14.5, 6.5 Hz, 1H, H-5), 2.62–2.56 (m, 1H, H-5), 2.53–2.47 (m, 1H, H-6); 13C{1H} NMR (CDCl3) δ 203.2 (CH2CH2OFc), 178.9 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 147.7 (CAry), 147.3 (CAry), 141.7 (CAry), 135.2 (C-12), 134.5 (CAry), 133.6 (CAry), 132.0 (C-8), 125.0 (CAry), 122.2 (CHtriazolyl), 111.7 (C-11), 107.4 (C-4), 78.8 (Cpipso), 72.2 (Cp), 69.8 (Cp), 69.3 (Cp), 69.2 (Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 38.7 (CH2H2COFc), 35.4 (C-6), 29.8 (C-5), 20.0 (H2CH2COFc).
7-Deacetamido-7-(4-(4-ferrocenyl-4-oxobut-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Fc4.
This compound was prepared in 75% yield (130 mg) according to general procedure B starting from 100 mg (0.261 mmol) of 5 and 73 mg (0.261 mmol) of 17. Mp 102–105 °C; elemental analysis found: C, 65.4; H, 6.3; N, 5.4. C36H37FeN3O6 requires C, 65.2; H, 5.6; N, 6.3%; IR (KBr) νmax/cm−1: 3096w, 1926s, 2853m, 1663s, 1618s, 1587vs, 1572vs, 1488s, 1457vs, 1379m, 1350s, 1322s, 1283m, 1254vs, 1195m, 1180m, 1140s, 1096s, 1048m, 1020m, 1003m, 984m, 924w, 844m, 824m, 754s, 667w, 533w, 488m; ESI-MS calc. for C36H37FeN3O6 663.2 (M+) found 663.0 (M+, 55%), 664.0 ((M + H)+, 100%), 593.0 (M+, 100%), 594.0 ((M + H)+, 66%); Rf (HPLC) τ = 7.6 min; 1H NMR (CDCl3) δ 7.39 (s, 1H, Htriazolyl), 7.27 (d, J = 11.3 Hz, 1H, H-12), 6.78 (d, J = 10.7 Hz, 1H, H-11), 6.58 (s, 1H, H-4), 6.53 (s, 1H, H-8), 5.37 (dd, J = 12.1, 5.7 Hz, 1H, H-7), 4.79–4.78 (m, 1H, Cp), 4.78–4.77 (m, 1H, Cp), 4.47 (brs, 2H, Cp), 4.17 (s, 5H, Cp), 3.97 (brs, 3H, OCH3), 3.93 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 2.85 (t, J = 7.5 Hz, 2H, C2CH2CH2COFc), 2.81–2.76 (m, 3H, H-6 and CH2C2CH2COFc), 2.72 (dd, J = 13.1, 6.0 Hz, 1H, H-5), 2.60 (dt, J = 13., 6.4 Hz, 1H, H-5), 2.56–2.52 (m, 1H, H-6), 2.12–2.08 (m, 2H, CH2CH2C2COFc); 13C{1H} NMR (CDCl3) δ 204.1 (COFc), 178.9 (C-9), 164.3 (C-10), 153.9 (CAry), 151.1 (CAry), 148.0 (CAry), 147.8 (CAry), 141.8 (CAry), 135.2 (C-12), 134.5 (CAry), 133.6 (CAry), 131.9 (C-8), 125.1 (CAry), 121.4 (CHtriazolyl), 111.7 (C-11), 107.5 (C-4), 79.0 (Cpipso), 72.2 (Cp), 69.8 (Cp), 69.4 (Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 38.7 (CH2H2CH2COFc), 35.4 (C-6), 29.8 (C-5), 25.2 (H2CH2CH2COFc), 23.9 (CH2CH2H2COFc).
7-Deacetamido-7-(4-(5-ferrocenyl-5-oxopent-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Fc5.
This compound was prepared in 57% yield (100 mg) according to general procedure B starting from 100 mg (0.261 mmol) of 5 and 77 mg (0.261 mmol) of 18. Mp 91–96 °C; elemental analysis found: C, 65.4; H, 6.1; N, 6.0. C37H39FeN3O6 requires C, 65.6; H, 5.8; N, 6.2%; IR (KBr) νmax/cm−1: 3092w, 2930s, 2855m, 1735m, 1663s, 1618s, 1586vs, 1488s, 1456s, 1398s, 1376m, 1350s, 1322s, 1282s, 1253vs, 1195m, 1179m, 1139s, 1095s, 1047m, 1019m, 1002m, 983m, 924w, 890w, 823m, 486m; ESI-MS calc. for C37H39FeN3O6 677.2 (M+) found 677.0 (M+, 43%), 678.0 ((M + H)+, 100%); Rf (HPLC) τ = 10.2 min; 1H NMR (CDCl3) δ 7.38 (s, 1H, Htriazolyl), 7.27 (d, J = 9.7 Hz, 1H, H-12), 6.68 (d, J = 10.7 Hz, 1H, H-11), 6.58 (s, 1H, H-4), 6.53 (s, 1H, H-8), 5.36 (dd, J = 11.9, 5.6 Hz, 1H, H-7), 4.76 (br s, 2H, Cp), 4.47 (t, J = 1.6 Hz, 2H, Cp), 4.17 (s, 5H, Cp), 3.97 (br s, 3H, OCH3), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 2.81–2.77 (m, 3H, C2CH2CH2CH2COFc and H-6), 2.75–2.70 (m, 3H, CH2CH2CH2C2COFc and H-5), 2.67–2.52 (m, 2H, H-5 and H-6), 1.78–1.77 (m, 4H, CH2C2C2CH2COFc); 13C{1H} NMR (CDCl3) δ 204.3 (COFc), 171.1 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 148.2 (CAry), 147.8 (CAry), 141.8 (CAry), 135.2 (C-12), 134.5 (CAry), 133.7 (CAry), 131.9 (C-8), 125.1 (CAry), 121.2 (CHtriazolyl), 111.7 (C-11), 107.5 (C-4), 79.1 (Cpipso), 72.1 (Cp), 69.7 (Cp), 69.3 (2 × C, Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 39.3 (CH2CH2CH2H2COFc), 35.4 (C-6), 29.8 (C-5), 28.9 (CH2CH2H2CH2COFc or CH2H2CH2CH2COFc), 25.6 (H2CH2CH2CH2COFc), 23.9 (CH2CH2H2CH2COFc or CH2H2CH2CH2COFc).
7-Deacetamido-7-(4-ruthenocenyl-1H-1,2,3-triazol-1-yl)-colchicine Rc1.
This compound was prepared in 87% yield (145 mg) according to general procedure B starting from 100 mg (0.261 mmol) of 5 and 67 mg (0.261 mmol) of 13. Mp 153–157 °C; elemental analysis found: C, 60.1; H, 5.0; N, 6.3. C34H35N3O5Ru requires C, 60.2; H, 4.9; N, 6.6%; IR (KBr) νmax/cm−1: 3099m, 2930s, 2839m, 1618s, 1586vs, 1488s, 1460s, 1429m, 1398s, 1349s, 1322s, 1282m, 1254vs, 1195m, 1180m, 1138s, 1097vs, 1045s, 1019s, 1001s, 983m, 843m, 810s, 751m, 491m; ESI-MS calc. for C32H31N3O5Ru 638.7 (M+) found 639.0 (M+, 53%), 640.0 ((M + H)+, 100%); Rf (HPLC) τ = 10.1 min; 1H NMR (CDCl3) δ 7.43 (s, 1H, Htriazolyl), 7.26 (d, J = 10.7 Hz, 1H, H-12), 6.78 (d, J = 10.9 Hz, 1H, H-11), 6.59 (s, 1H, H-4), 6.49 (s, 1H, H-8), 5.37 (dd, J = 12.0, 5.6 Hz, 1H, H-7), 5.13 (s, 1H, Cp), 5.11 (s, 1H, Cp), 4.64 (s, 2H, Cp), 4.49 (s, 5H, Cp), 3.96 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 2.80–2.75 (m, 1H, H-6), 2.74–2.71 (m, 1H, H-5), 2.63–2.53 (m, 2H, H-5 and H-6); 13C{1H} NMR δ 178.8 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 147.6 (CAry), 146.4 (CAry), 141.8 (CAry), 135.2 (C-12), 134.3 (CAry), 133.6 (CAry), 131.9 (C-8), 125.1 (CAry), 119.1 (CHtriazolyl), 111.6 (H-11), 107.4 (H-4), 78.8 (Cpipso), 71.5 (Cp), 71.3, 70.6 (Cp), 69.3 (Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.3 (OCH3), 56.1 (OCH3), 35.3 (C-6), 29.7 (C-5).
7-Deacetamido-7-(4-ruthenocenoyl-1H-1,2,3-triazol-1-yl)-colchicine Rc2.
This compound was prepared in 35% yield (61 mg) according to general procedure B starting from 100 mg (0.261 mmol) of 5 and 74 mg (0.261 mmol) of 20. Mp 136–140 °C; elemental analysis found: C, 61.4; H, 5.8; N, 5.7. C33H31N3O6Ru·½C6H14 requires C, 60.9; H, 5.4; N, 5.9%; IR (KBr) νmax/cm−1: 3112w, 2925s, 2852s, 1622s, 1587vs, 1528m, 1488s, 1459s, 1399m, 1376m, 1351m, 1322m, 1255vs, 1195m, 1180m, 1140s, 1088s, 1046m, 1035m, 1022m, 1000m, 984m, 841m, 819m, 768m, 731w, 492w, 468w; ESI-MS calc. for C33H31N3O6Ru 667.1 (M+) found 667.0 (M+, 49%), 668.0 ((M + H)+, 100%); Rf (HPLC) τ = 12.5 min; 1H NMR (CDCl3) δ 8.19 (s, 1H, Htriazolyl), 7.31 (d, J = 10.7 Hz, 1H, H-12), 6.80 (d, J = 10.9 Hz, 1H, H-11), 6.61 (s, 1H, H-4), 6.44 (s, 1H, H-8), 5.78 (t, J = 1.7 Hz, 2H, Cp), 5.44 (dd, J = 12.0, 5.4 Hz, 1H, H-7), 4.90–4.88 (m, 2H, Cp), 4.54 (s, 5H, Cp), 3.98 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 2.82–2.75 (m, 2H, H-5 and H-6), 2.67–2.56 (m, 2H, H-5 and H-6); 13C{1H} NMR δ 186.4 (CORc), 178.7 (C-9), 164.4 (C-10), 154.0, 151., 148.7, 147.1, 141.9, 135.5 (C-12), 134.2, 133.4, 131.8 (C-8), 127.0 (CHtriazolyl), 125.0, 111.7 (C-11), 107.6 (C-4), 82.5 (Cpipso), 74.0 (2 × Cp), 72.9 (Cp), 72.8 (Cp), 72.4 (Cp), 62.8 (C-7), 61.2 (2 × OCH3), 56.4 (OCH3), 56.2 (OCH3), 35.3 (C-6), 29.7 (C-5).
7-Deacetamido-7-(4-(3-ruthenocenyl-3-oxoprop-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Rc3.
This compound was prepared in 45% yield (82 mg) according to general procedure B starting from 100 mg (0.261 mmol) of 5 and 81 mg (0.261 mmol) of 21. Mp 95–99 °C; elemental analysis found: C, 55.8; H, 5.4; N, 5.2. C35H35N3O6Ru·3H2O requires C, 56.1; H, 5.5; N, 5.6%; IR (KBr) νmax/cm−1: 3096w, 2927s, 2853m, 1670s, 1618s, 1578vs, 1488s, 1457s, 1430m, 1399s, 1377m, 1350m, 1322m, 1254vs, 1195m, 1179m, 1139s, 1097s, 1086s, 1048m, 1019m, 1002m, 923m, 844w, 818m, 488w; ESI-MS calc. for C35H35N3O6Ru 695.2 (M+) found 696.0 ((M + H)+, 95%), 718.0 ((M + Na)+ 100%); Rf (HPLC) τ = 7.9 min; 1H NMR (CDCl3) δ 7.38 (brs, 1H, Htriazolyl), 7.26 (d, J = 10.6 Hz, 1H, H-12), 6.78 (d, J = 10.8 Hz, 1H, H-11), 6.57 (s, 1H, H-4), 6.55 (s, 1H, H-8), 5.32 (dd, J = 12.1, 5.9 Hz, 1H, H-7), 5.09 (brs, 2H, Cp), 4.75 (brs, 2H, Cp), 4.51 (s, 5H, Cp), 3.96 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.10–3.04 (m, 4H, C2C2CORc), 2.81 (dt, J = 11.9, 6.5 Hz, 1H, H-6), 2.70 (dd, J = 13.4, 6.5 Hz, 1H, H-5), 2.59 (dt, J = 13.4, 6.5 Hz, 1H, H-5), 2.53–2.49 (m, 1H, H-6); 13C{1H} NMR (CDCl3) δ 201.2 (CORc), 178.9 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 147.7 (CAry), 147.1 (CAry), 141.7 (CAry), 135.2 (C-12), 134.5 (CAry), 133.6 (CAry), 132.0 (C-8), 125.0 (CAry), 122.1 (CHtriazolyl), 111.7 (C-11), 107.4 (C-4), 83.6 (Cpipso), 73.5 (Cp), 72.0 (Cp), 71.9 (Cp), 70.7 (Cp), 70.6 (Cp), 62.5 (C-7), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 37.9 (H2CH2CORc or CH2H2CORc), 35.4 (C-6), 29.7 (C-5), 20.3 (H2CH2CORc or CH2H2CORc).
7-Deacetamido-7-(4-(4-ruthenocenyl-4-oxobut-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Rc4.
This compound was prepared in 86% yield (95 mg) according to general procedure B starting from 60 mg (0.156 mmol) of 5 and 51 mg (0.156 mmol) of 22. Mp 95–100 °C; elemental analysis found: C, 58.7; H, 5.8; N, 5.2. C36H37N3O6Ru·½H2O requires C, 58.8; H, 5.5; N, 5.7%; IR (KBr) νmax/cm−1: 2925s, 2853s, 1668s, 1618s, 1587vs, 1488s, 1457vs, 1398m, 1376m, 1350m, 1322m, 1232m, 1254vs, 1195m, 1179m, 1139s, 1097s, 1086s, 1084m, 1020m, 1001m, 984m, 843m, 818m, 489w; ESI-MS calc. for C36H37N3O6Ru 709.2 (M+) found 709.0 (M+, 72%), 710.0 ((M + H)+, 100%); Rf (HPLC) τ = 8.6 min; 1H NMR (CDCl3) δ = 7.38 (br s, 1H, Htriazolyl), 7.27 (d, J = 11.9 Hz, 1H, H-12), 6.70 (d, J = 10.8 Hz, 1H, H-11), 6.58 (s, 1H, H-4), 6.52 (s, 1H, H-8), 5.36 (dd, J = 12.1, 5.7 Hz, 1H, H-7), 5.10 (brs, 1H, Cp), 5.09 (br s, 1H, Cp), 4.75 (s, 2H, Cp), 4.56 (s, 5H, Cp), 3.96 (OCH3), 3.93 (OCH3), 3.91 (OCH3), 3.75 (OCH3), 2.80–2.77 (m, 3H, H-6 and C2CH2CH2CORc), 2.72 (dd, J = 13.4, 6.5 Hz, 1H, H-5), 2.70–2.67 (m, 2H, CH2CH2C2CORc), 2.63–2.57 (m, 1H, H-5), 2.56–2.51 (m, 1H, H-6), 2.06–2.06 (m, 2H, CH2C2CH2CORc); 13C{1H} NMR (CDCl3) δ 202.2 (CORc), 178.9 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 147.9 (CAry), 147.8 (CAry), 141.7 (CAry), 135.2 (C-12), 134.4 (CAry), 133.6 (CAry), 131.9 (C-8), 125.0 (CHtriazolyl), 121.3 (CAry), 111.7 (C-11), 107.5 (C-4), 83.9 (Cpipso), 73.5 (Cp), 71.9 (Cp), 70.7 (Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 38.1 (CH2CH2H2CORc), 35.4 (C-6), 29.7 (C-5), 25.1 (H2CH2CH2CORc), 24.4 (CH2H2CH2CORc).
7-Deacetamido-7-(4-(5-ruthenocenyl-5-oxopent-1-yl)-1H-1,2,3-triazol-1-yl)-colchicine Rc5.
This compound was prepared in 29% yield (38 mg) according to general procedure B starting from 70 mg (0.183 mmol) of 5 and 62 mg (0.183 mmol) of 23. Mp 87–92 °C; elemental analysis found: C, 61.4; H, 5.7; N, 5.6. C37H39N3O6Ru requires C, 61.5; H, 5.4; N, 5.8%; IR (KBr) νmax/cm−1: 3096w, 2927s, 2850m, 1667s, 1618s, 1587vs, 1488s, 1457s, 1399s, 1376m, 1350s, 1322s, 1254vs, 1195m, 1180m, 1139s, 1096s, 1047m, 1020s, 1002m, 983m, 843m, 817m, 637w, 489w; ESI-MS calc. for C37H39N3O6Ru 723.2 (M+) found 723.0 (M+, 70%), 724.0 ((M + H)+, 100%), 746 ((M + Na)+, 55%); Rf (HPLC) τ = 11.2 min; 1H NMR (CDCl3) δ 7.38 (s, 1H, Htriazolyl), 7.27 (d, J = 11.0 Hz, 1H, H-12), 6.78 (d, J = 10.7 Hz, 1H, H-11), 6.58 (s, 1H, H-4), 6.53 (s, 1H, H-8), 5.36 (dd, J = 12.0, 5.7 Hz, 1H, H-7), 5.07 (brs, 2H, Cp), 4.74 (brs, 2H, Cp). 4.56 (brs, 5H, Cp), 3.96 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 2.81–2.76 (m, 3H, H-6 and CH2CH2C2CH2CORc), 2.72 (dd, J = 13.0, 6.1 Hz, 1H, H-5), 2.64–2.53 (m, 4H, H-5 and H-6 and CH2CH2CH2C2CORc), 1.72–1.71 (m, 4H, C2CH2C2CH2CORc); 13C{1H} NMR (CDCl3) δ 202.4 (CORc), 178.9 (C-9), 164.3 (C-10), 153.9 (CAry), 151.0 (CAry), 148.2 (CAry), 147.8 (CAry), 141.7 (CAry), 135.2 (C-12), 134.5 (CAry), 133.6 (CAry), 131.9 (C-8), 125.1 (CAry), 121.2 (CHtriazolyl), 111.7 (C-11), 107.5 (C-4), 84.0 (Cpipso), 73.4 (Cp), 71.9 (Cp), 70.7 (Cp), 62.5 (C-7), 61.3 (OCH3), 61.2 (OCH3), 56.4 (OCH3), 56.1 (OCH3), 38.5 (CH2CH2CH2C2CORc), 35.4 (C-6), 29.8 (C-5), 28.8 (CH2CH2H2CH2CORc or H2CH2CH2CH2CORc), 25.5 (CH2CH2H2CH2CORc), 24.3 (CH2CH2H2CH2CORc or H2CH2CH2CH2CORc).
Tubulin polymerization
Tubulin polymerization was determined using the commercially available tubulin polymerization assay kit (Cytoskeleton, Inc., Cat. #BK011P). The investigated compounds were dissolved in DMSO and were tested in a range of concentrations (from 0.1 μM to 30 μM). The final concentration of DMSO in all samples was 0.5%. Paclitaxel and vinblastine were used as a positive- and negative-control of tubulin polymerization, respectively. The fluorescence was measured at 37 °C for 120 min at 355/40 nm excitation and 430/8 nm emission wavelengths using an EnVision Multilabel Plate Reader (PerkinElmer).
Viability assay
The toxicity of compounds was determined using the neutral red uptake assay. The cells were seeded at a density of 10000 cells per well in 100 μl of a complete medium in wells of a 96-well plate. After 24 hours the cells were allowed to attach to the surface, and the investigated compounds were added. Stock solutions were prepared in DMSO, and the final concentration of the solvent was kept identical in all samples and did not exceed 0.1% v/v. The tested compounds were studied in a concentration range between 10 nM and 100 μM. Following 70-hour-incubation, neutral red solution was added to the cell to a final concentration of 0.33% w/v. After further 2 hours, the medium was aspirated and the wells were washed once with phosphate-buffered saline for 5 minutes in a gyratory shaker. Then PBS was aspirated and 200 μl of the solubilizer solution (1% v/v acetic acid in 50% v/v ethanol) was added. The plates were kept on a gyratory shaker for another 5 minutes and then the absorbance was measured at 540 nm using the EnVision Multilabel Plate Reader (PerkinElmer). The results were calculated as a percentage of the controls and the IC50 values for each cell line and substance were calculated with GraphPad Prism 5.02 software (GraphPad Inc.) using the four-parameter non-linear logistic regression.
Cell cycle
Exponentially growing Colo 205 cells (100000 cells per well seeded in 6-well plates 24 h before time 0) were treated with 10 nM of the tested compound for 48 h. The cells were then harvested by trypsinisation, washed twice with ice-cold PBS and fixed in 70% ethanol. After storing the cells for at least 8 h at 4 °C, they were stained with a propidium iodide staining solution (75 μM propidium iodide and 50 Kunitz units per ml of RNAse A in PBS) for 30 min at 37 °C. The samples were analysed on an LSRII (Becton Dickinson) instrument (excitation 488 nm, emission 575/26 nm) and the cell cycle phase distribution was determined with FlowJo 7.6.1 software (FlowJo, LLC) using a built-in cell cycle analysis module (Watson pragmatic algorithm).
Reactive oxygen species production
The oxidation rate of dihydrorhodamine 123 (DHR123) was considered a marker of intracellular production of reactive oxygen species. Exponentially growing HCT116 and A549 cells were exposed to 1 μM of the tested compound for 4 h in the presence of 1 μM DHR123 and/or 10 μM verapamil (an inhibitor of ABCB1). The cells were then harvested by trypsinisation and suspended in a complete growth medium. The samples were then collected on an LSRII (Becton Dickinson) instrument and median fluorescence (excitation 488 nm, emission 530/30 nm) was analysed. The median fluorescence of cells incubated with DHR123 and DMSO alone was considered 100% in a given experiment. The statistical significance of the differences was assessed by one-way ANOVA and Bonferroni's post-hoc test, assuming 0.05 as the significance level.
Calcein accumulation assay
The ABCB1 activity in HCT116 cells was measured by performing the calcein accumulation assay as described previously.8 Briefly, the cells were trypsinized and suspended in a complete cell culture medium pre-warmed to 37 °C at a final density of approx. 1 × 106 ml−1. Samples were supplemented with calcein AM at a 70 nM final concentration. A specific inhibitor (10 μM verapamil) was added to the parallel samples. Intracellular fluorescence was measured immediately after dye addition (time 0) and every 3–5 minutes up to approx. 20 minutes using LSRII (Becton Dickinson) instrument set at 488 nm excitation and 530/30 emission. Accumulation curves were plotted and the accumulation rate was assessed with the GraphPad Prism 5.02 software (GraphPad Inc.). The difference between the accumulation rate for dye only and the inhibitor samples was evaluated as a measure of ABCB1 activity.
logP determination
logP values of the investigated compounds were determined by a HPLC method22 on using a Phenomenex Kinetex 5μ XB-C18 100 Å column 150 × 4.6 mm using two mobile phases: phase A was 20 mM MOPS, 0.15% 1-decylamine, pH = 7.4; phase B was 0.25% 1-octanol in methanol. Briefly, samples dissolved in methanol with uracil as an internal reference were injected in the column and eluted with mobile phase B between 55, 60, 65, and 70%. The logP values were calculated as previously described22b using 4-methoxyphenol, naphthalene, acetophenone and paclitaxel as standards. These experiments were repeated three times for each of the compounds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the National Science Centre Poland (NCN) based on decision DEC-2012/05/B/ST5/00300.
References
- P. Richette and T. Bardin, Expert Opin. Pharmacother., 2010, 11, 2933–2938 CrossRef CAS PubMed.
- Y. Finkelstein, S. E. Aks, J. R. Hutson, D. N. Juurlink, P. Nguyen, G. Dubnov-Raz, U. Pollak, G. Koren and Y. Bentur, Clin. Toxicol., 2010, 48, 407–414 CrossRef CAS PubMed.
-
(a) O. N. Zefirova, E. V. Nurieva, D. V. Shishov, I. I. Baskin, F. Fuchs, H. Lemcke, F. Schröder, D. G. Weiss, N. S. Zefirov and S. A. Kuznetsov, Bioorg. Med. Chem., 2011, 19, 5529–5538 CrossRef CAS PubMed;
(b) N. Yasobu, M. Kitajima, N. Kogure, Y. Shishido, T. Matsuzaki, M. Nagaoka and H. Takayama, ACS Med. Chem. Lett., 2011, 2, 348–352 CrossRef CAS PubMed;
(c) C. Vilanova, S. Díaz-Oltra, J. Murga, E. Falomir, M. Carda, M. Redondo-Horcajo, J. F. Díaz, I. Barasoain and J. A. Marco, J. Med. Chem., 2014, 57, 10391–10403 CrossRef CAS PubMed;
(d) P. Thomopoulou, J. Sachs, N. Teusch, A. Mariappan, J. Gopalakrishnan and H.-G. Schmalz, ACS Med. Chem. Lett., 2016, 7, 188–191 CrossRef CAS PubMed;
(e) N. S. Sitnikov and A. Y. Fedorov, Russ. Chem. Rev., 2013, 82, 393 CrossRef;
(f) S. K. Kim, S. M. Cho, H. Kim, H. Seok, S. O. Kim, T. K. Kwon and J. S. Chang, Exp. Mol. Med., 2013, 45(4), e19 CrossRef PubMed;
(g) M. L. Gelmi, S. Mottadelli, D. Pocar, A. Riva, E. Bombardelli, R. De Vincenzo and G. Scambia, J. Med. Chem., 1999, 42, 5272–5276 CrossRef CAS PubMed;
(h) L. Cosentino, M. Redondo-Horcajo, Y. Zhao, A. R. Santos, K. F. Chowdury, V. Vinader, Q. M. A. Abdallah, H. Abdel-Rahman, J. Fournier-Dit-Chabert, S. D. Shnyder, P. M. Loadman, W.-s. Fang, J. F. Díaz, I. Barasoain, P. A. Burns and K. Pors, J. Med. Chem., 2012, 55, 11062–11066 CrossRef CAS PubMed;
(i) A. Ahmed, N. R. Peters, M. K. Fitzgerald, J. A. Watson, F. M. Hoffmann and J. S. Thorson, J. Am. Chem. Soc., 2006, 128, 14224–14225 CrossRef CAS PubMed.
-
(a) A. Vessières, J. Organomet. Chem., 2013, 734, 3–16 CrossRef;
(b) M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715–5727 CrossRef CAS;
(c) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed;
(d) M. F. R. Fouda, M. M. Abd-Eizaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS;
(e) D. Dive and C. Biot, ChemMedChem, 2008, 3, 383–391 CrossRef CAS PubMed;
(f) S. S. Braga and A. M. S. Silva, Organometallics, 2013, 32, 5626–5639 CrossRef CAS.
- S. Top, J. Tang, A. Vessières, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
-
(a) A. Wieczorek, A. Błauż, A. Żal, H. J. Arabshahi, J. Reynisson, C. G. Hartinger, B. Rychlik and D. Plażuk, Chem. – Eur. J., 2016, 22, 11413–11421 CrossRef CAS PubMed;
(b) D. Plazuk, A. Wieczorek, A. Błauz and B. Rychlik, MedChemComm, 2012, 3, 498–501 RSC.
-
(a) A. Wieczorek, A. Blauz, A. Makal, B. Rychlik and D. Plazuk, Dalton Trans., 2017, 46, 10847–10858 RSC;
(b) M. Beaupérin, D. Polat, F. Roudesly, S. Top, A. Vessières, J. Oble, G. Jaouen and G. Poli, J. Organomet. Chem., 2017, 839, 83–90 CrossRef.
- A. Wieczorek, A. Błauż, J. Zakrzewski, B. Rychlik and D. Plażuk, ACS Med. Chem. Lett., 2016, 7, 612–617 CrossRef CAS PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
-
(a) M. Patra, K. Ingram, V. Pierroz, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, J. Med. Chem., 2012, 55, 8790–8798 CrossRef CAS PubMed;
(b) M. Patra, K. Ingram, A. Leonidova, V. Pierroz, S. Ferrari, M. N. Robertson, M. H. Todd, J. Keiser and G. Gasser, J. Med. Chem., 2013, 56, 9192–9198 CrossRef CAS PubMed.
- J. Hess, M. Patra, V. Pierroz, B. Spingler, A. Jabbar, S. Ferrari, R. B. Gasser and G. Gasser, Organometallics, 2016, 35, 3369–3377 CrossRef CAS.
- E. I. Edwards, R. Epton and G. Marr, J. Organomet. Chem., 1975, 85, C23–C25 CrossRef CAS.
- D. Plazuk, B. Rychlik, A. Błauz and S. Domagała, J. Organomet. Chem., 2012, 715, 102–112 CrossRef CAS.
- J. Polin and H. Schottenberger, Org. Synth., 1996, 73, 262–269 CrossRef CAS.
- C. Lu, J.-M. Heldt, M. Guille-Collignon, F. Lemaître, G. Jaouen, A. Vessières and C. Amatore, ChemMedChem, 2014, 9, 1286–1293 CrossRef CAS PubMed.
- D. Plażuk, A. Wieczorek, W. M. Ciszewski, K. Kowalczyk, A. Błauż, S. Pawlędzio, A. Makal, C. Eurtivong, H. J. Arabshahi, J. Reynisson, C. G. Hartinger and B. Rychlik, ChemMedChem, 2017, 12, 1882–1982 CrossRef PubMed.
-
(a) N. Kaplowitz, Yale J. Biol. Med., 1981, 54, 497 CAS;
(b) Z.-Z. Huang, C. Chen, Z. Zeng, H. Yang, J. Oh, L. Chen and S. C. Lu, FASEB J., 2001, 15, 19–21 CAS.
-
(a) A. Meyer, R. Buhl and H. Magnussen, Eur. Respir. J., 1994, 7, 431–436 CrossRef CAS PubMed;
(b) N. S. Gould and B. J. Day, Biochem. Pharmacol., 2011, 81, 187–193 CrossRef CAS PubMed.
- L. M. Henderson and J. B. Chappell, Eur. J. Biochem., 1993, 217, 973–980 CrossRef CAS PubMed.
- S. Kunjachan, B. Rychlik, G. Storm, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2013, 65, 1852–1865 CrossRef CAS PubMed.
- T. Lampidis, D. Kolonias, T. Podona, M. Israel, A. Safa, L. Lothstein, N. Savaraj, H. Tapiero and W. Priebe, Biochemistry, 1997, 36, 2679–2685 CrossRef CAS PubMed.
-
(a) D. J. Minick, J. H. Frenz, M. A. Patrick and D. A. Brent, J. Med. Chem., 1988, 31, 1923–1933 CrossRef CAS PubMed;
(b) K. K.-W. Lo, K. H.-K. Tsang and N. Zhu, Organometallics, 2006, 25, 3220–3227 CrossRef CAS.
-
https://comptox.epa.gov/dashboard/dsstoxdb/results?search=Colchicine
.
- E. Karslyan, A. Konovalov, D. Perekalin, P. Petrovskii and A. Kudinov, Russ. Chem. Bull., 2008, 57, 2032–2033 CrossRef CAS.
- O. Hofer and K. Schlögl, J. Organomet. Chem., 1968, 13, 443–456 CrossRef CAS.
- M. Buchmeiser and H. Schottenberger, J. Organomet. Chem., 1992, 441, 457–464 CrossRef CAS.
- D. Plazuk and J. Zakrzewski, J. Organomet. Chem., 2009, 694, 1802–1806 CrossRef CAS.
- T. Akiyama, T. Hattori, K. Ito, T. Uryu, M. Kajitani and A. Sugimori, Chem. Lett., 1980, 9, 361–364 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt03229c |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Andrzej
Błauż‡
a,
Andrzej
Błauż‡