
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-driven azide-induced mononuclear discrete versus one-dimensional polymeric aromatic Möbius cadmium(II) complexes of an N6 tetradentate helical ligand†
Farhad
Akbari Afkhami
ab,
Ghodrat
Mahmoudi
 *c,
Atash V.
Gurbanov
de,
Fedor I.
Zubkov
e,
Fengrui
Qu
b,
Arunava
Gupta
*ab and
Damir A.
Safin
*f
*c,
Atash V.
Gurbanov
de,
Fedor I.
Zubkov
e,
Fengrui
Qu
b,
Arunava
Gupta
*ab and
Damir A.
Safin
*f
aCenter for Material and Information technology, The University of Alabama, Box 870209, 2007 Bevil Building, Tuscaloosa, Alabama 35487, USA. E-mail: agupta@mint.ua.edu
bDepartment of Chemistry, The University of Alabama, Box 870336, 250 Hackberry Lane, Tuscaloosa, Alabama 35487, USA
cDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55181-83111, Maragheh, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk
dOrganic Chemistry Department, RUDN University, Miklukho-Maklaya str. 6, 117198 Moscow, Russian Federation
eDepartment of Chemistry, Baku State University, Z. Xalilov Str. 23, AZ1148, Baku, Azerbaijan
fInstitute of Condensed Matter and Nanosciences, Molecules, Solids and Reactivity (IMCN/MOST), Université catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: damir.a.safin@gmail.com
First published on 2nd October 2017
Abstract
We report the synthesis and structural characterization of a heteroleptic mononuclear discrete complex [Cd(N3)2(L)(MeOH)]·MeOH (1·MeOH) and a one-dimensional coordination polymer of the composition [Cd3(N3)6(L)]n (2), fabricated from Cd(NO3)2·4H2O and the helical organic ligand benzilbis((pyridin-2-yl)methylidenehydrazone) (L) in the presence of two equivalents of NaN3. The formation of different structures is driven by the solvent. The former complex is formed in the presence of MeOH, while the latter complex is formed in EtOH. The CdII centre in 1·MeOH is trapped by the two pyridyl-imine units of the tetradentate ligand L, two azide ligands and one oxygen atom of one methanol ligand with the CdN6O coordination polyhedron yielding a square face monocapped trigonal prism. The asymmetric unit of 2 consists of three symmetrically independent atoms of CdII, six azide anions and one L. The polymeric structure of 2 is realized through chains of the Cd(N3)2 units which are decorated with Cd(N3)2L units. The CdII atoms from the backbone of the coordination polymer have a distorted octahedral coordination, while the remaining CdII atom forms a trigonal prism with two basal planes nearly parallel to each other. In both complexes, the 12π electron chelate ring of the CdL fragment is shown to be aromatic by establishing it as a Möbius object. Hirshfeld surface analysis of 1 in 1·MeOH and L in 2 showed that the structures of both species are highly dominated by H⋯X (X = H, C and N) contacts, of which the latter two are highly favoured, as well as some contribution from highly enriched C⋯C contacts is clearly observed.
Introduction
Nature likes helical molecules.1 Deoxyribonucleic acid, discovered by Watson and Crick in 1953, is the most famous self-organized helicate structure.2 The term helicate for a polymetallic helical double-stranded complex was first suggested by Lehn in 1987.3 However, the first known helical double-stranded dinuclear structure was reported in 1976 for the doubly deprotonated octaethyl formylbiliverdine with ZnII.4 Since then, great efforts have been made towards a detailed study of the self-assembly of helicate structures.5,6 The predesign of coordinating ligands is the most effective strategy to obtain desired helicate metal-containing complexes. However, the use of metal-containing precursors to synthesize helicate systems is much less investigated.4–6Non-helical chelating organic ligands, containing suitable donor sites, may give rise to a helical topology upon binding to metal ions7–10 because in inorganic complexes, the coordination behavior of the metal ions directs the overall topology in a manner that has not been seen in the parent ligand. On the other hand, polydentate Schiff base ligands, comprising two symmetrically related pyridyl coordination sites derived from benzyldihydrazone, in the solid state are monohelical due to the constraint rotation around the C–C bond.11–13 Upon coordination, the rotating freedom of the two N–N bonds is also restrained, and the ligand can be locked in a twist conformation (Scheme 1).
 | ||
| Scheme 1 Synthesis of complexes 1·MeOH and 2. | ||
Based on Heilbronner's rule, a conjugated system, including 4nπ electrons, can be aromatic if it is Möbius.14 A Möbius strip can have N number of half-twists designating its order (Fig. 1). Herges and co-workers have reported the first Möbius molecule with the order N of 1.15 Thereafter, several Möbius molecules of various orders have been reported.16–22 Datta and co-workers have reported a CdII-based mononuclear double helical complex, with fully conjugated ligands, comprising two 13 membered chelate metallocycles with 12π electrons23 and pointed out that the complex is aromatic and eventually Möbius.24
 | ||
| Fig. 1 (Left) A circular strip, (Middle) a Möbius strip with N = 1 and (Right) a Möbius strip with N = 2. The images adopted from ref. 24. | ||
The nature of a selected metal center is a powerful tool to create the targeted structure. Accordingly, using the CdII atom for the formation of hybrid materials is attractive since being a soft metal and exhibiting a variety of coordination numbers it can promote the formation of non-predictable final structures with possible interesting properties. On the other hand, the coordination chemistry of azides had practically not existed prior to the 1960s when the first wide-ranging investigations came out.25–30 An azide ion is a flexible polydentate ligand that can simultaneously bind several metal ions in different coordination modes. As such, the azide anion is a versatile ligand that can bridge metal centers either end-to-end (μ1,3) or end-on (μ1,1) or many other combinations of both possibilities.31
When growing crystals from solution, the solvent of crystallization plays a pivotal role in determining the overall structure, crystal system and lattice arrangement. This role can range from influencing the polymorph that is grown to being built into the crystal structure itself to form a solvate. Whereas, in some solvates, the solvent molecules act as space fillers, and in other solvates, the solvent molecules are essential components of the lattice by interacting with the host molecules by specific intermolecular interactions, such as hydrogen bonds. The former type of solvates are classified as inclusion phases, and the latter type of solvates as cocrystals.32 Certain molecular shapes and intermolecular interactions favor the formation of unsolvated crystals.33 The solvate is stabilized when the incorporated solvent molecule either improves the packing or strengthens the intermolecular interactions in the crystal.34
In this contribution we describe the solvent-driven synthesis and complete structural investigation of mononuclear discrete and one-dimensional polymeric aromatic Möbius Cd(N3)2-based complexes constructed from the N6 tetradentate helical ligand L, derived from benzildihydrazone and 2-acetylpyridine. Notably, it has recently been indicated that an empty s orbital can participate in a Möbius framework of p orbitals.24
Results and discussion
An equimolar one-pot reaction of Cd(NO3)2·4H2O with the organic ligand L in the presence of two equivalents of NaN3 in MeOH at 60 °C in a branched tube apparatus leads to a heteroleptic mononuclear discrete complex [Cd(N3)2(L)(MeOH)]·MeOH (1·MeOH) (Scheme 1). Notably, using the same procedure but in EtOH yielded crystals of the polymeric complex of the composition [Cd3(N3)6(L)]n (2).Both compounds 1·MeOH and 2 were obtained in good yields, and were fully characterized by elemental analysis, FTIR spectroscopy, powder and single-crystal X-ray diffraction, and Hirshfeld surface analysis.
According to the single crystal X-ray diffraction data, 1·MeOH and 2 each crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit of 1·MeOH consists of a single molecule 1 and one lattice methanol (Fig. 2), while the asymmetric unit of 2 consists of three symmetrically independent atoms of CdII, six azide anions and one molecule of L (Fig. 3).
. The asymmetric unit of 1·MeOH consists of a single molecule 1 and one lattice methanol (Fig. 2), while the asymmetric unit of 2 consists of three symmetrically independent atoms of CdII, six azide anions and one molecule of L (Fig. 3).
 | ||
| Fig. 2 (Left) Crystal structure of 1·MeOH. CH-hydrogen atoms are omitted for clarity. (Right) Coordination polyhedron around the CdII atom in the crystal structure of 1·MeOH. Color code: H = black, C = gold, N = blue, O = red, Cd = magenta. | ||
 | ||
| Fig. 3 (Top) Crystal structure of the asymmetric unit of 2. Hydrogen atoms are omitted for clarity. (Bottom) Coordination polyhedra around the CdII atoms in the CdL (left) and Cd2(N3)10 fragments (right) in the crystal structure of 2. Color code: C = gold, N = blue, Cd = magenta. | ||
In the structure of 1·MeOH the CdII centre has a seven coordination geometry bound by the two pyridyl-imine units of the ligand L, two nitrogen atoms of two azide ligands and one oxygen atom of one methanol ligand (Fig. 2). The Cd–N bond distances, corresponding to the four nitrogen atoms of the Schiff-base ligand, range from 2.372(2) Å to 2.533(2) Å, maintaining that Cd–NPy < Cd–Nimine (Table S1 in the ESI†). Notably, the bond lengths of the azide ligands (2.270(2) and 2.327(2) Å) are significantly shorter than the bonds of the nitrogen donor atoms within the L ligand, indicating a certain amount of π-electron donation.35 Both azide ligands in 1·MeOH are quasi-linear as the average of the N–N–N angle is 178.1°. The coordination polyhedron around the CdII center in the structure of 1·MeOH, with a CdN6O chromophore, can be either a distorted pentagonal bipyramid or a face capped octahedron or a square face monocapped trigonal prism. The latter polyhedron seems to be the best description (Fig. 2), which is further supported by the corresponding face N–N–N bond angles close to 90° (see the file 1-MeOH.cif in the ESI†). The two basal planes of N2O are nearly parallel to each other and form a 6.8° dihedral angle and the CdII ion is displaced by 1.55 and 1.62 Å from the two basal planes. The ligand L in the structure of 1·MeOH adopts a twist helical conformation. This conformation gives rise to a torsion angle of 75.03(6)° for the N–C(Ph)–C(Ph)–N moiety and the coordination leads to significantly different torsion angles of 121.7(2) and 137.72(19)° around the bonds C(Ph)–N–N–C. The torsion angle between two phenyl rings from the twist ligand is of 86.46(10)°.
The crystal packing of 1·MeOH is governed primarily by the methanol ligand and the lattice methanol molecule. The molecules of 1·MeOH are bound together to form dimers (Fig. 4) by means of strong hydrogen bonds between the OH proton of the lattice methanol and the coordinated nitrogen atom of the azide ligand as well as the oxygen atom of the same lattice methanol and the OH proton of the methanol ligand of the second molecule 1 and vice versa (Table S2 in the ESI†). These interactions give a synthon of motif R44(12) (Fig. 5). The crystal packing of 1·MeOH is further stabilized by face-to-face π⋯π stacking between the pyridyl rings and C–H⋯π interactions formed by both phenyl and one of the pyridyl fragments (Tables S3 and S4 in the ESI†).
 | ||
| Fig. 4 (Top) Hydrogen bonded dimer in the crystal structure of 1·MeOH. (Bottom) 1D polymeric chain in the crystal structure of 2. CH hydrogen atoms are omitted for clarity. Color code: H = black, C = gold, N = blue, O = red, Cd = magenta. | ||
 | ||
| Fig. 5 Top (left) and side (right) views on a homosynthon of motif R44(12) formed by means of hydrogen bonds between the OH hydrogen atoms of the methanol ligand, lattice methanol and the nitrogen atom of one of the azide ligands and vice versa in the structure of 2. Color code: H = black, C = gold, N = blue, O = red, Cd = magenta. | ||
The polymeric structure of 2 is achieved through the chains of Cd(N3)2 units along the crystallographic a axis which are further decorated with Cd(N3)2L units (Fig. 4). The two symmetrically independent CdII atoms which form the backbone of the coordination polymer each have a distorted octahedral coordination with the Cd–N bond lengths ranging from 2.2702(13) to 2.4428(13) Å (Table S1 in the ESI†). The remaining CdII atom, linked to the ligand L, also has a six coordination geometry bound by the two pyridyl-imine units of the ligand L and two nitrogen atoms of two azide ligands (Fig. 3).
The coordination polyhedron around this CdII center in the structure of 2, with a CdN6 chromophore, is best described as a trigonal prism (Fig. 3). The two basal planes of N3 are nearly parallel to each other and form a 4.6° dihedral angle and the CdII ion is displaced by 1.50 and 1.54 Å from the two basal planes. The Cd–N bond distances corresponding to the four nitrogen atoms of the ligand L are almost identical and range from 2.3512(14) to 2.3894(13) Å, maintaining that Cd–NPy ≈ Cd–Nimine (Table S1 in the ESI†). The bond lengths of the azide ligands within the Cd(N3)2L unit are also shorter, however with a lower degree than in the structure of 1·MeOH, than the bonds of the nitrogen donor atoms within the L ligand (Table S1 in the ESI†).
The torsion angle for the N–C(Ph)–C(Ph)–N moiety is of 76.5(2)° while the same angles for the C(Ph)–N–N–C fragment are of 119.18(15) and 125.21(16)°. The torsion angle between two phenyl rings from the twist ligand L is of 88.44(9)°. The shortest and longest Cd⋯Cd separations within the backbone of the coordination polymer are about 3.54 and 5.40 Å, respectively, while the shortest Cd⋯Cd separations between the symmetrically independent metal atoms arising from the backbone of the coordination polymer and the CdL metal center are about 5.62 and 5.89 Å (Table S1 in the ESI†). The azide ligands exhibit a variety of coordination modes, i.e. μ1,1-Nazide, μ1,3-Nazide, μ1,1,1-Nazide and μ1,1,3-Nazide (Fig. 4).
The crystal packing of 2 is dictated primarily by the face-to-face π⋯π stacking between the pyridyl rings as well as C–H⋯π and N–N⋯π interactions (Table S4 in the ESI†). The latter interactions are formed by the phenyl fragments.
Since the topologies of both complexes, with respect to the organic ligand L, are helical (Fig. 6), enantiomers are expected. Moreover, complexes 1·MeOH and 2 crystallize in the triclinic space group P and therefore each represents a racemic mixture in the solid state (Fig. 6).
 | ||
| Fig. 6 Space-filling models of the left-handed (left) and right-handed (right) helical nature of the CdL fragment in the crystal structures of 1·MeOH and 2. The phenyl and hydrogen atoms are omitted for clarity. Color code: C = gold, N = blue, Cd = magenta. | ||
Bulk samples of the described complexes were each studied by means of X-ray powder diffraction analysis (Fig. 7). The experimental X-ray powder patterns of the complexes are in full agreement with the calculated powder patterns obtained from single crystal X-ray diffraction, showing that the bulk materials are free from phase impurities.
 | ||
| Fig. 7 Calculated (black) and experimental (red) X-ray powder diffraction patterns of 1·MeOH and 2. | ||
The FTIR spectrum of 1·MeOH contains a wide strong band characteristic for the asymmetric stretching vibration, νasym(N3), of the azide ligands at 2032 cm−1 (Fig. 8). Two intense bands at 2062 and 2108 cm−1 in the FTIR spectrum of 2 were assigned to the same stretching modes of the azide anions but of different coordination modes (Fig. 8).36 The FTIR spectrum of 1·MeOH also displays a broad intense band for the coordinated and lattice methanol ranging from about 3100 to 3700 cm−1 (Fig. 8). The symmetric stretching vibrations, νsym(N3), of the azide ligands are observed in the spectrum of 1·MeOH as a single band at 1438 cm−1, while two bands at 1434 and 1443 cm−1, characteristic for the same symmetric stretching vibrations of azides, are shown in the FTIR spectrum of 2 (Fig. 8). The characteristic bands of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching modes appear in the range of 1560–1600 cm−1 in the spectra of both compounds (Fig. 8).
C and CN stretching modes appear in the range of 1560–1600 cm−1 in the spectra of both compounds (Fig. 8).
 | ||
| Fig. 8 FTIR spectra of 1·MeOH (black) and 2 (red). | ||
In order to examine the interactions in the crystal structures of 1 and 2, Hirshfeld surface analysis37 and the corresponding 2D fingerprint plots38 were obtained using CrystalExplorer 3.1.39
Unlike other molecular volumes and surfaces (e.g. van der Waals volumes, solvent-accessible surfaces, solvent-excluded surfaces), Hirshfeld surfaces are not simply a function of the molecular geometry but are only defined within the crystal. Consequently, the Hirshfeld surfaces reflect the interplay between different atomic sizes and intermolecular contacts in the crystal (condensed phase). The Hirshfeld surfaces and volumes are much larger than the conventional ones, generally filling at least 95% of the crystal volume,40 compared with more conventional packing coefficients of between 0.65 and 0.80.41 The Hirshfeld surfaces are obviously packed very tightly in the crystal, at most touching and never overlapping. However, quite unlike any other partitioning or packing scheme, they leave small intermolecular voids, which can be regarded as regions where the crystalline electron density is very low and is not dominated by any single molecule. The use of Hirshfeld surfaces for the analysis of molecular crystal structures encourages the adoption of a whole structure view of intermolecular interactions, rather than concentrating exclusively on assumed interactions. While the discussion of crystal structures in terms of individual interatomic contacts is unavoidable and certainly valuable, a broader picture of intermolecular interactions in the crystal is increasingly desirable; such a picture is available from the Hirshfeld surface. The size and shape of the Hirshfeld surface are intimately related to the chemical environment surrounding the molecule, making it ideal for use when comparing different crystal structures.
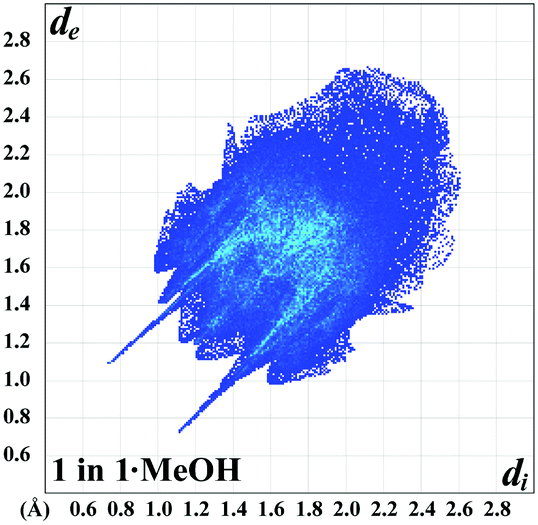
According to Hirshfeld surface analysis, for the molecule of 1 in the structure of 1·MeOH the intermolecular H⋯H, H⋯C and H⋯N contacts, comprising 42.3%, 24.1% and 27.4% of the total number of contacts, are the major contributors to the crystal packing (Table 1). The same intermolecular contacts are also the major contributors on the Hirshfeld surface of the ligand L in the structure of 2. However, the intermolecular H⋯H contacts are remarkably less and occupy 29.5%, while a proportion of the H⋯N contacts is more, comprising 35.1% (Table 1). The intermolecular H⋯C contacts on the surface of L in 2 occupy a similar proportion on the surface of 1 and are of 21.3%. The shortest H⋯H contacts are shown in the fingerprint plots of both species as characteristic broad spikes at de + di ≈ 2.2–2.4 Å (Fig. S1 and S2 in the ESI†). Furthermore, a subtle feature is evident in the fingerprint plot of 1. There is a splitting of the short H⋯H fingerprint. This splitting occurs when the shortest contact is between three atoms, rather than for a direct two-atom contact.37 The H⋯C contacts in the fingerprint plots of both 1 and L in 2 are shown in the form of “wings” (Fig. S1 and S2 in the ESI†) with the shortest de + di ≈ 2.6 Å. These contacts are recognized as characteristic of C–H⋯π nature.37 It is worth adding that the fingerprint plot of L in 2 exhibits a significant number of points at large de and di, shown as tails at the top right of the plot (Table 1). These points, similar to those observed in the fingerprint plot of benzene37 and phenyl-containing compounds,42–47 correspond to the regions on the Hirshfeld surface without any close contacts with nuclei in the adjacent molecules. Notably, the shortest H⋯N contacts in the fingerprint plot of 1 are shown as a sharp spike at de + di ≈ 1.9 Å (Fig. S1 in the ESI†). These contacts correspond to the O–H⋯N hydrogen bonds (Fig. 2 and 4, Table S2 in the ESI†). The structure of 1 is also characterized by intermolecular H⋯O contacts of 2.1% of which the shortest are shown in the fingerprint plot as a sharp spike at de + di ≈ 1.9 Å (Fig. S1 in the ESI†). These contacts correspond to the O–H⋯O hydrogen bonds (Fig. 2 and 4, Table S2 in the ESI†). The structures of both species are also described by C⋯C and are of 3.2% and 4.5%, respectively (Table 1), and are shown in the fingerprint plots as the symmetric area on the diagonal at de = di ≈ 1.7–2.1 Å (Fig. S1 and S2 in the ESI†). These contacts are evidence for π⋯π stacking interactions (Table S3 in the ESI†). Moreover, the Hirshfeld surface of 1 also contains a negligible proportion of the C⋯N contacts of 0.9%, which are shown in the fingerprint plot as the symmetric area on the same diagonal as C⋯C contacts at de = di ≈ 1.8–2.0 Å (Fig. S1 in the ESI†). Thus, the C⋯N contacts on the molecular surface of 1 are also due to π⋯π stacking between pyridyl rings (Table S3 in the ESI†). Notably, the C⋯N contacts occupy a significantly higher proportion (3.9%) of the Hirshfeld surface area of L in the structure of 2. This is not only due to π⋯π stacking between pyridyl rings (Table S3 in the ESI†) but rather due to N–N⋯π interactions (Table S4 in the ESI†). This is further supported by the shortest C⋯N contacts in the fingerprint plot of L in 2 shown at de + di ≈ 3.3 Å (Fig. S1 in the ESI†). Close inspection of other intermolecular contacts in the structure of L in 2 also revealed a negligible proportion of the N⋯N contacts (1.6%) and a proportion of the N⋯Cd contacts (3.9%). However, the latter contacts originate from the covalent bonding of L towards the chelated CdII center.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| H | C | N | O | H | C | N | Cd | |
| a Values are obtained from CrystalExplorer 3.1.39 b The enrichment ratios were not computed when the “random contacts” were lower than 0.9%, as they are not meaningful.48 | ||||||||
| Contacts (C, %)a | ||||||||
| H | 42.3 | — | — | — | 29.5 | — | — | — |
| C | 24.1 | 3.2 | — | — | 21.3 | 4.5 | — | — |
| N | 27.4 | 0.9 | 0.0 | — | 35.1 | 3.9 | 1.6 | — |
| O/Cd | 2.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.9 | 0.0 |
| Surface (S, %) | ||||||||
| 69.1 | 15.7 | 14.2 | 1.1 | 57.7 | 17.1 | 23.1 | 2.0 | |
| Random contacts (R, %) | ||||||||
| H | 47.7 | — | — | — | 33.3 | — | — | — |
| C | 21.7 | 2.5 | — | — | 19.7 | 2.9 | — | — |
| N | 19.6 | 0.7 | 2.0 | — | 26.7 | 1.4 | 5.3 | — |
| O/Cd | 1.5 | 0.3 | 0.3 | 0.0 | 2.3 | 0.7 | 0.9 | 0.0 |
| Enrichment (E)b | ||||||||
| H | 0.89 | — | — | — | 0.89 | — | — | — |
| C | 1.11 | 1.28 | — | — | 1.08 | 1.55 | — | — |
| N | 1.40 | — | 0.0 | — | 1.31 | 2.79 | 0.30 | — |
| O/Cd | 1.40 | — | — | — | 0.0 | — | 4.33 | — |


We have also determined the enrichment ratios (E)48 of the intermolecular contacts for both structures to study the propensity of two chemical species to be in contact. The enrichment ratio, derived from Hirshfeld surface analysis, is defined as the ratio between the proportion of actual contacts in the crystal and the theoretical proportion of random contacts. E is larger than unity for a pair of elements with a higher propensity to form contacts, while pairs which tend to avoid contacts yield an E value lower than unity.
The H⋯C, H⋯N and C⋯C contacts as well as H⋯O contacts in 1 are highly favoured in the structure of 1 and L in the structure of 2 since the corresponding enrichment ratios EHH, EHN and ECC are larger than unity (1.08–1.55) (Table 1). This is explained by a relatively higher proportion of these contacts on the total Hirshfeld surface area over a corresponding proportion of random contacts RHH, RHN, RCC and RHO, respectively (Table 1). Contrarily, the H⋯H contacts on the surface of both species are less favoured (EHH = 0.89). This is due to a relatively higher amount of random contacts RHH, despite both molecules being characterised by a high amount of SH (Table 1). Interestingly, the C⋯N contacts in the structure of L in 2 are remarkably enriched (ECN = 2.79). This is explained by a significantly higher amount of the C⋯N contacts on the molecular surface together with a lower amount of random contacts RCN (Table 1). Finally, the structures of L in 2 are characterized by very impoverished N⋯N (ENN = 0.30) contacts, which are due to a significantly higher proportion of random contacts RNN compared to a proportion of N⋯N contacts on the molecular surface (Table 1).
Conclusions
In summary, we have obtained and fully structurally characterized a heteroleptic mononuclear discrete complex [Cd(N3)2(L)(MeOH)]·MeOH (1·MeOH) and a one-dimensional coordination polymer of the composition [Cd3(N3)6(L)]n (2), fabricated from Cd(NO3)2·4H2O and the helical organic ligand benzilbis((pyridin-2-yl)methylidenehydrazone) (L) in the presence of two equivalents of NaN3. The formation of two significantly different structures is exclusively dictated by the solvent nature. Particularly, the former complex is formed in the presence of MeOH as a reaction medium, while the latter complex is formed in EtOH. Thus, different solvent template molecules (MeOH and EtOH) act as structure-directing agents, adjusted by their size and shape, for the Cd(N3)2L-system.According to the single crystal diffraction data the CdII centre in 1·MeOH is bound to the tetradentate Schiff-base ligand L, two azide ligands and one methanol ligand with the CdN6O coordination polyhedron. The asymmetric unit of 2 consists of three symmetrically independent atoms of CdII, six azide anions and one molecule of L. The polymeric structure of 2 is achieved through the chains of Cd(N3)2 units along the crystallographic a axis which are further decorated with Cd(N3)2L units. In both complexes, the 12π electron chelate ring of the CdL fragment is shown to be aromatic by establishing it as a Möbius object.
Experimental
Materials
L was prepared following the reported method as described elsewhere.22 All other reagents and solvents were commercially available and used as received without further purification.Physical measurements
FTIR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer. Microanalyses were performed using an ElementarVario EL III analyzer.Hirshfeld surface analysis
The Hirshfeld molecular surfaces37 and their corresponding 2D fingerprint plots38 were generated using the CrystalExplorer 3.1 software39 on the basis of crystal structures. The dnorm (normalized contact distance) surface and the breakdown of the 2D fingerprint plots were used for decoding and quantifying the intermolecular interactions in the crystal lattice. The dnorm is a symmetric function of distances to the surface from the nuclei inside (di) and outside (de) the Hirshfeld surface, relative to their respective van der Waals radii. 2D fingerprint plots were generated using di and de in the translated 0.4–3.0 Å range and including reciprocal contacts as a pair of coordinates in 2D histograms. A colour gradient in the fingerprint plots ranging from blue to red is used to visualize the proportional contribution of contact pairs on the global surface.Enrichment ratio
The enrichment ratio (E)48 of a pair of elements (X,Y) is the ratio between the proportion of actual contacts in the crystal and the theoretical proportion of random contacts. E is larger than unity for pairs of elements which have a high propensity to form contacts in crystals, while pairs which tend to avoid contacts with each other yield an E value lower than unity. The E values are calculated from the percentage of contacts, which, in turn, are given by CrystalExplorer 3.1 software,39 between one type or two types of chemical elements in crystal packing.Synthesis of 1·MeOH and 2
Cd(NO3)2·4H2O (0.154 g, 0.5 mmol), NaN3 (0.065 g, 1 mmol) and L (0.222 g, 0.5 mmol) were placed in the main arm of a branched tube. MeOH or EtOH (15 mL) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. X-ray suitable crystals were formed during the next days in the cooler arm and were filtered off, washed with acetone and diethyl ether, and dried in air.X-ray powder diffraction
X-ray powder diffraction for bulk samples was carried out using a Rigaku Ultima IV X-ray powder diffractometer. The parallel beam mode was used to collect the data (λ = 1.54184 Å).Single-crystal X-ray diffraction of 1·MeOH
The X-ray data were collected on an Agilent Technologies SuperNova-Atlas CCD single crystal diffractometer. Data were processed using the CrysAlisPro49 program and corrected for absorption using SADABS.50 The structure was solved by direct methods,51 which revealed the positions of all non-hydrogen atoms. These were refined on F2 by a full-matrix least-squares procedure using anisotropic displacement parameters.51 Hydrogen atoms were inserted at calculated positions, except for the hydrogen atoms of the hydroxyl groups, which were located from a difference Fourier map. All hydrogen atoms were included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 times those of the respective atom. Certain crystal geometry parameters referred in the discussion were calculated using PLATON.52, a = 9.8020(1), b = 10.3207(1), c = 15.5967(2) Å, α = 94.002(3), β = 94.287(4), γ = 91.342(4)°, V = 1568.92(3) Å3, Z = 2, ρ = 1.492 g cm−3, μ(Mo-Kα) = 5.979 mm−1, reflections: 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 281 collected, 5582 unique, Rint = 0.032, R1(all) = 0.0271, wR2(all) = 0.0654.
281 collected, 5582 unique, Rint = 0.032, R1(all) = 0.0271, wR2(all) = 0.0654.
Single-crystal X-ray diffraction of 2
The X-ray data were collected on a Bruker Apex2 CCD single crystal diffractometer. Data were processed with the Saint53 program and corrected for absorption using SADABS.50 The space group was assigned using XPREP of the Bruker ShelXTL54 package, solved with ShelXT54 and refined with ShelXL54 and graphical interface ShelXle.55 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms attached to carbon were positioned geometrically and constrained to ride on their parent atoms., a = 8.9122(3), b = 14.7168(5), c = 15.1730(5) Å, α = 111.392(1), β = 92.184(1), γ = 94.028(1)°, V = 1844.04(11) Å3, Z = 2, ρ = 1.862 g cm−3, μ(Mo-Kα) = 1.773 mm−1, reflections: 70672 collected, 10789 unique, Rint = 0.025, R1(all) = 0.0204, wR2(all) = 0.0455.
Figures were generated using the program Mercury.56
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. This publication was supported by the “RUDN University Program 5-100”.References
- V. G. Machado, P. N. W. Baxter and J.-M. Lehn, J. Braz. Chem. Soc., 2001, 12, 431–462 CrossRef CAS.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS.
- G. Struckmeier, U. Thewalt and J.-H. Fuhrhop, J. Am. Chem. Soc., 1976, 98, 278–279 CrossRef CAS.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS PubMed.
- M. G. B. Drew, D. Parui, S. De, J. P. Naskar and D. Datta, Eur. J. Inorg. Chem., 2006, 4026–4028 CrossRef CAS.
- T. Riss-Johannessen, L. P. Harding, J. C. Jeffery, R. Moon and R. C. Rice, Dalton Trans., 2007, 1577–1587 RSC.
- Y. Furusho and E. Yashima, Chem. Rec., 2007, 7, 1–11 CrossRef CAS PubMed.
- A. M. Stadler, N. Kyritsakas, G. Vanghan and J.-M. Lehn, Chem. – Eur. J., 2007, 13, 59–68 CrossRef CAS PubMed.
- Q. Sun, Y. Bai, G. He, C. Duan, Z. Lin and Q. Meng, Chem. Commun., 2006, 2777–2779 RSC.
- M. G. B. Drew, S. De and D. Datta, Inorg. Chim. Acta, 2009, 362, 2487–2291 CrossRef CAS.
- S. De, M. G. B. Drew and D. Datta, Inorg. Chim. Acta, 2010, 363, 4123–4126 CrossRef CAS.
- E. Heilbronner, Tetrahedron Lett., 1964, 29, 1923–1928 CrossRef.
- D. Ajami, O. Oeckler, A. Simon and R. Herges, Nature, 2003, 426, 819–821 CrossRef CAS PubMed.
- R. P. John, M. Park, D. Moon, K. Lee, S. Hong, Y. Zou, C. S. Hong and M. S. Lah, J. Am. Chem. Soc., 2007, 129, 14142–14143 CrossRef CAS PubMed.
- S. De, M. G. B. Drew, H. S. Rzepa and D. Datta, New J. Chem., 2008, 32, 1831–1834 RSC.
- H. S. Rzepa, Inorg. Chem., 2008, 47, 8932–8934 CrossRef CAS PubMed.
- J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J. Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568–13579 CrossRef CAS PubMed.
- Y. M. Sung, M. C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418–422 CrossRef CAS PubMed.
- A. Masoumi, M. S. Gargari, G. Mahmoudi, B. Machura, V. Lynch, G. Giester, M. Abedi and P. Hazendonk, Z. Anorg. Allg. Chem., 2015, 641, 1176–1181 CrossRef CAS.
- G. Mahmoudi, V. Stilinović, M. S. Gargari, A. Bauzá, G. Zaragoza, W. Kaminsky, V. Lynch, D. Choquesillo-Lazarte, K. Sivakumar, A. A. Khandari and A. Frontera, CrystEngComm, 2015, 17, 3493–3502 RSC.
- M. G. B. Drew, S. De and D. Datta, Inorg. Chim. Acta, 2008, 361, 2967–2972 CrossRef CAS.
- N. K. Shee and D. Datta, Inorg. Chim. Acta, 2016, 453, 339–344 CrossRef CAS.
- W. Beck, K. Feldl and E. Schuierer, Angew. Chem., Int. Ed. Engl., 1965, 4, 439–440 CrossRef.
- W. Beck, E. Schuierer and K. Feldl, Angew. Chem., Int. Ed. Engl., 1966, 5, 249 CrossRef CAS.
- W. Beck, E. Schuierer, P. Pöllmann and W. P. Fehlhammer, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1966, 21, 811–812 CAS.
- W. Beck, W. P. Fehlhammer, P. Pöllmann, E. Schuierer and K. Feldl, Chem. Ber., 1967, 100, 2335–2361 CrossRef CAS.
- W. Beck, M. Bauder, W. P. Fehlhammer, P. Pöllmann and H. Schächl, Inorg. Nucl. Chem. Lett., 1968, 4, 143–146 CrossRef CAS.
- W. Beck, W. P. Fehlhammer, P. Pöllmann and H. Schächl, Chem. Ber., 1969, 102, 1976–1987 CrossRef CAS.
- W. P. Fehlhammer and W. Z. Beck, Anorg. Allg. Chem., 2013, 639, 1053–1082 CrossRef CAS.
- J. A. R. P. Sarma and G. R. Desiraju, in Crystal Engineering: Design and Application of Functional Solids, ed. K. R. Seddon and M. Zaworotko, Kluwer, Norwell, MA, 1999, pp. 325–356 Search PubMed.
- S. A. Barnett, D. A. Tocher and M. Vickers, CrystEngComm, 2006, 8, 313–319 RSC.
- G. Mahmoudi, A. A. Khandar, J. White, M. P. Mitoraj, H. S. Jena, P. Van Der Voort, N. Qureshi, A. M. Kirillov, K. Robeyns and D. A. Safin, CrystEngComm, 2017, 19, 3075–3025 Search PubMed.
- T. H. Noh, J. H. Kim, Y.-A. Lee, H. Suh and O.-S. Jung, J. Mol. Struct., 2004, 691, 165–169 CrossRef CAS.
- F. A. Mautner, F. R. Louka, J. Hofer, M. Spell, A. Lefèvre, A. E. Guilbeau and S. S. Massoud, Cryst. Growth Des., 2013, 13, 4518–4525 CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer 3.1, University of Western Australia, 2012 Search PubMed.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem. – Eur. J., 1998, 4, 2136 CrossRef CAS.
- A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- D. A. Safin, M. P. Mitoraj, K. Robeyns, Y. Filinchuk and C. M. L. Vande Velde, Dalton Trans., 2015, 44, 16824–16832 RSC.
- M. G. Babashkina, K. Robeyns, Y. Filinchuk and D. A. Safin, New J. Chem., 2016, 40, 1230–1236 RSC.
- D. A. Safin, C. M. L. Vande Velde, M. G. Babashkina, K. Robeyns and Y. Filinchuk, New J. Chem., 2016, 40, 6156–6163 RSC.
- D. A. Safin, K. Robeyns, M. G. Babashkina, Y. Filinchuk, A. Rotaru, C. Jureschi, M. P. Mitoraj, J. Hooper, M. Brela and Y. Garcia, CrystEngComm, 2016, 18, 7249–7259 RSC.
- D. A. Safin, K. Robeyns and Y. Garcia, CrystEngComm, 2016, 18, 7284–7296 RSC.
- D. A. Safin, M. G. Babashkina, M. P. Mitoraj, P. Kubisiak, K. Robeyns, M. Bolte and Y. Garcia, Inorg. Chem. Front., 2016, 3, 1419–1431 RSC.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CAS.
- Oxford Diffraction, CrysAlis CCD, Oxford Diffraction Ltd, Abingdon, Oxfordshire, England, 2006 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- Bruker Saint Plus, Saint Plus 8.34A, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2, Tables S1–S4 and cif files. CCDC 1564162 and 1564163. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02952g |
| This journal is © The Royal Society of Chemistry 2017 |