
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel stibacarbaborane cluster with adjacent antimony atoms exhibiting unique pnictogen bond formation that dominates its crystal packing†
Josef
Holub
a,
Petr
Melichar
b,
Zdeňka
Růžičková
c,
Jan
Vrána
c,
Derek A.
Wann
 d,
Jindřich
Fanfrlík
*b,
Drahomír
Hnyk
*a and
Aleš
Růžička
*c
d,
Jindřich
Fanfrlík
*b,
Drahomír
Hnyk
*a and
Aleš
Růžička
*c
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, v.v.i., 250 68 Husinec-Řež, Czech Republic. E-mail: hnyk@iic.cas.cz
bInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic. E-mail: fanfrlik@uochb.cas.cz
cDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210 Pardubice, Czech Republic. E-mail: ales.ruzicka@upce.cz
dDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK
First published on 12th September 2017
Abstract
We have prepared nido-7,8,9,11-Sb2C2B7H9, the first cluster with simultaneous Sb–B, Sb–C and Sb–Sb atom pairs with interatomic separations with magnitudes that approach the respective sums of covalent radii. However, the length of the Sb–Sb separation in this cluster is slightly less than the sum of the covalent radii. Quantum chemical analysis has revealed that the crystal packing of nido-7,8,9,11-Sb2C2B7H9 is predominantly dictated by pnictogen (Pn) bonding, an unconventional σ-hole interaction. Indeed, the interaction energy of a very strong Sb2⋯H–B Pn-bond in the nido-7,8,9,11-Sb2C2B7H9 dimer exceeds −6.0 kcal mol−1. This is a very large value and is comparable to the strengths of known Pn-bonds in Cl3Pn⋯π complexes (Pn = As, Sb).
Introduction
Boron atoms have a remarkable ability to form 3-center-2-electron (3c2e) bonds, which results in an astonishing variety of stable three-dimensional molecular structures composed of triangular B–B–B and B–H–B faces.1,2 An important class of boranes comprises closo-BnHn2− and nido-BnHn4−. BH vertices can be formally replaced by CH+, S2+, Se2+, P+, As+ and Sb+ units which results in a variety of heteroboranes. While carboranes and thiaboranes are common, heteroboranes with heavier main group elements are rare. The first Sb-containing boron clusters to be synthesized were closo-1,2-AsSbB10H10, closo-1,2-Sb2-B10H10 and nido-7,8-SbCB9H11− although no crystal structures could be determined.3 The closo-1,2-Sb2B10H10 compound subsequently allowed a bis(phosphine)metalladistibaborane to be synthesized and crystallized.4 However, the Sb2B3 pentagonal belt was stabilized by Pd in this cluster. It is often the case that slightly unstable arrangements are capped with a metal atom to help their stabilization.5 Inspired by 3-Cl-nido-7,8,9,11-P2C2B7H9 (1) and 3-I-nido-7,8,9,11-As2C2B7H9 (2) compounds,6,7 we have succeeded for the first time in generating a pentagonal belt containing two Sb atoms without having to cap it with a metal atom nido-7,8,9,11-Sb2C2B7H9 (3, see Fig. 1).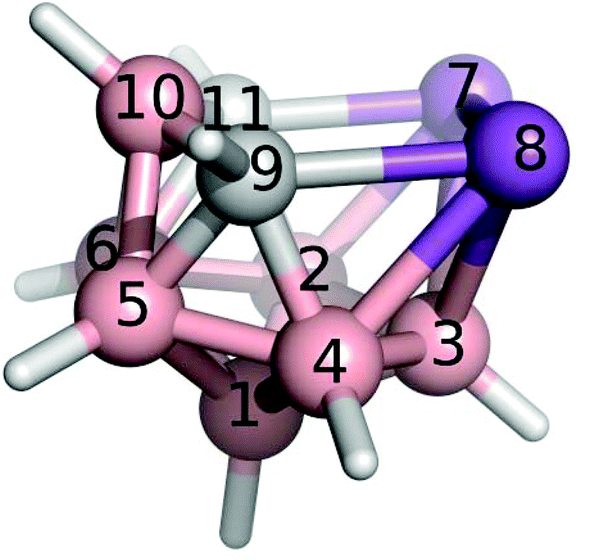 | ||
| Fig. 1 The molecular structure and atomic numbering of nido-7,8,9,11-Sb2C2B7H9, 3. | ||
Nowadays, particular attention is paid to σ-hole noncovalent interactions.8 The σ-holes are defined as positive electrostatic potential (ESP) areas capable of interacting with electron-rich regions.9 They are most often located along the vector of a covalent bond to a halogen (X), chalcogen (Y), or pnictogen (Pn) atom. Consequently, σ-hole interactions are often called X-, Y- or Pn-bonds. These interactions are well known in organic chemistry. A σ-hole is most often characterized by its magnitude (Vs,max); the higher the Vs,max value, the stronger the X-bond. The Vs,max value can be increased by increasing the atomic number of the X atom or by changing the chemical environment. Recently, σ-hole interactions within heteroboranes with positive σ-holes on heteroatoms incorporated have received attention due to the remarkably strong σ-hole interactions.10 This is counterintuitive when one considers the very low electronegativity of B atoms. It was a phenyl-exo-substituted thiaborane molecule where the σ-hole interactions were first noticed with the resulting formation of an Y-bond that was responsible for the overall packing motif in the crystal structure.10a A strong X-bond has been observed in the crystal packing of Br-exo-substituted carbaborane.11 Although a series of heteroboranes containing Pn atoms is already known experimentally such as 1 and 2,6 no experimental evidence for Pn-bonding in heteroboranes has been reported. Therefore, we used 3 to validate the ability of heteroboranes to form noncovalent Pn-bonds.
Results and discussion
Structural characterization by NMR
The properties of 3 can be inferred from NMR experiments and QM calculations. Table 1 compares experimental and computed 11B NMR chemical shifts, showing excellent agreement between the experimental and computed NMR values with the exception of B(5,6), which are antipodally coupled12 to the Sb atoms. The discrepancy, which is not unusual for the third row and other heavier elements,12 indicates that B(5,6) atoms are strongly influenced by Sb(7,8).Aromatic ring currents are observed in 2D and 3D aromatic molecules such as benzene and closo-B12H122−. The nucleus-independent chemical shift (NICS) is a computational method that expresses the absolute magnetic shielding at the center of a 2D or 3D molecule. The NICS values13 for various parts of molecule 3 are computed to be −41.7, −44.5, −41.7, −44.9, −22.5 and −17.4 ppm for the Sb–B–B, B–B–B, Sb–Sb–B, and Sb–B–C triangles, center of the molecule, and open pentagonal belt, respectively. These results indicate that the cluster as a whole is of aromatic nature. The fact that the triangles that are closest to antimony are highly aromatic indicates that the Sb atoms donate valence electrons to the corresponding atomic triangles. Even the NICS values that are less negative than −40 ppm are comparable to the aromaticity displayed by the 1,3-dehydro-5,7-adamantanediyl dication and closo-B12H122− (NICS values of −49.6 and −34.4 ppm, respectively).13 The NICS value for the Sb–Sb–C–B–C pentagon of −17.4 ppm is comparable to the values associated with pyrrole and thiophene (NICS values of −17.3 and −14.7 ppm, respectively; more details in ref. 13). The 7,8,9,11-Pn2C2B7H9 (Pn = P, As) compounds display similar trends of NICS values.
Crystal structure of 3
In the crystal structure of 3 (see Fig. 2), four molecules were found in the chiral unit cell. A striking feature of this structure is the well-determined Sb–Sb separation (2.7546(6) Å), which seems to be in line with analogous distances in reduced or over-reduced Sb compounds containing two or more metal atoms. There are several different classes of compounds that could be used for comparison where the Sb–Sb distances are a bit longer than the sum of the covalent radii of 2.8 Å.14a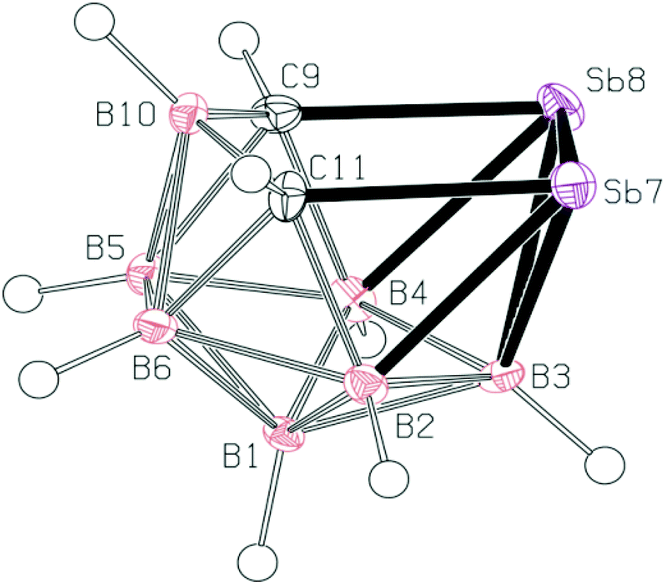 | ||
| Fig. 2 The molecular structure (ORTEP 50% probability level) and atomic numbering of 3. Selected interatomic distances [Å] and angles [°]: Sb7–Sb8 2.7546(6), Sb7–C11 2.225(8), Sb8–C9 2.217(8), C9–B10 1.611(11), B10–C11 1.603(11), B2–Sb7 2.326(9), B3–Sb7 2.458(8), B3–Sb8 2.446(7), Sb8–B4 2.313(8); C9–Sb8–Sb7 89.8(2), C11–Sb7–Sb8 90.61(19), B10–C11–Sb7 115.7(5), B10–C9–Sb8 116.9(5), C11–B10–C9 119.2(6), C9–Sb8–B4 44.5(3), C9–Sb8–B3 77.0(3), B4–Sb8–B3 45.8(3), B4–Sb8–Sb7 92.17(19), B3–Sb8–Sb7 56.03(19), C11–Sb7–B2 44.0(3), C11–Sb7–B3 76.8(3), B2–Sb7–B3 46.1(3), B2–Sb7–Sb8 92.4(2), B3–Sb7–Sb8 55.62(17). | ||
Complexes with Sb–B and Sb–C bonds in a single molecule are very rare.14b,c The reported Sb–C separation of 3 is elongated by ca. 0.07 Å when compared to the typical C(aromatic)–SbIII distance. Similarly, the Sb–B distances in 3 (range from 2.312 to 2.458 Å) approach the sum of the covalent radii of the constituent atoms, i.e. 2.25 Å.14a
Electrostatic potential (ESP) analysis
In order to understand Pn-bonding more generally, we analyzed the charge distributions for a series of PnCl3 and nido-Pn2C2B7H9 (Pn = P, As, and Sb) molecules. The computed Vs,max and dipole moment (μ) values are summarized in Table 2. Sb has very low electronegativity comparable to that of B but considerably smaller than those values for C and Cl. In accordance with the concept of electronegativity, the Sb atom has highly positive Vs,max values in SbCl3 of 48.6 kcal mol−1. However, the Sb atoms in 3 also have a very high Vs,max value of 42.7 kcal mol−1 (the σ-hole in between the adjacent Sb atoms; see Fig. 3) despite being bonded to B and C atoms, which are considerably less electronegative than Cl atoms. Interestingly, the Sb vertices have more positive ESP surfaces than the CH vertices (see Fig. 3) and the Sb atoms lie at the center of a partial positive charge within 3. A very large μ of 3 (μ of 4.0 D) also confirms that the center of the Sb–Sb vector is positively charged.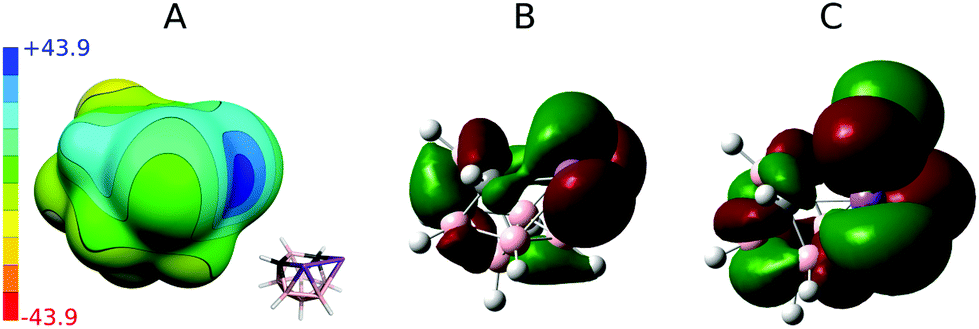 | ||
| Fig. 3 Computed electrostatic potential (ESP) surface for compound 3 (A). The color range of the ESP in kcal mol−1. The HOMO (B) and LUMO (C) of 3 were derived at the same level of theory as ESP, see computational modeling. | ||
| V s,max/kcal mol−1 | μ/D | ||
|---|---|---|---|
| Group V atoms | Group VII atoms | ||
| PCl3 | 3 × 29.2 | 3 × 6.7 | 1.1 |
| AsCl3 | 3 × 38.6 | 3 × 2.9 | 2.1 |
| SbCl3 | 3 × 48.6 | 3 × −3.3 | 3.3 |
| nido-7,8,9,11-P2C2B7H9 (1) | 24.8 | — | 2.0 |
| 3-Cl-nido-7,8,9,11-P2C2B7H9 | 25.4 | −3.1 | 2.8 |
| nido-7,8,9,11-As2C2B7H9 | 31.1 and 2 × 24.4 | — | 2.8 |
| 3-I-nido-7,8,9,11-As2C2B7H9 (2) | 32.2 and 2 × 29.0 | 8.2 | 3.5 |
| nido-7,8,9,11-Sb2C2B7H9 (3) | 1 × 42.7 and 2 × 25.7 | — | 4.0 |
Crystal packing and noncovalent interactions
We have studied pairwise interactions in the crystal structure of 3. It was not possible to compare these interactions to the parent nido-7,8,9,11-Pn2C2B7H9 (Pn = P, As) complexes, as it reveals poor quality crystals, which prevents them from performing reliable X-ray structure determination. Hence, we opted for the halogenated analogues of 1 and 2, for which the crystal structures had already been determined.6,7 We performed highly accurate MP2.5/CBS calculations for all of the bonding motifs as well as benchmark CCSD(T)/CBS calculations for selected motifs. All the computed interaction energies (ΔE) are summarized in Table 3 (see also Fig. 4). Each molecule of 1 makes two Pn-bonds (P2⋯H–B and P2⋯Cl–B). These two Pn-bonds form the A⋯B binding motif which has ΔE of −6.1 kcal mol−1 and was the dominant binding motif for 1. Besides Pn-bonding, molecule 1 also forms a CH⋯Cl–B H-bond and a C–H⋯B–H HH-bond. The H-bonding of 1 is, however, weaker than its Pn-bonding and the ΔE of the A⋯C motif is less negative (−4.4 kcal mol−1).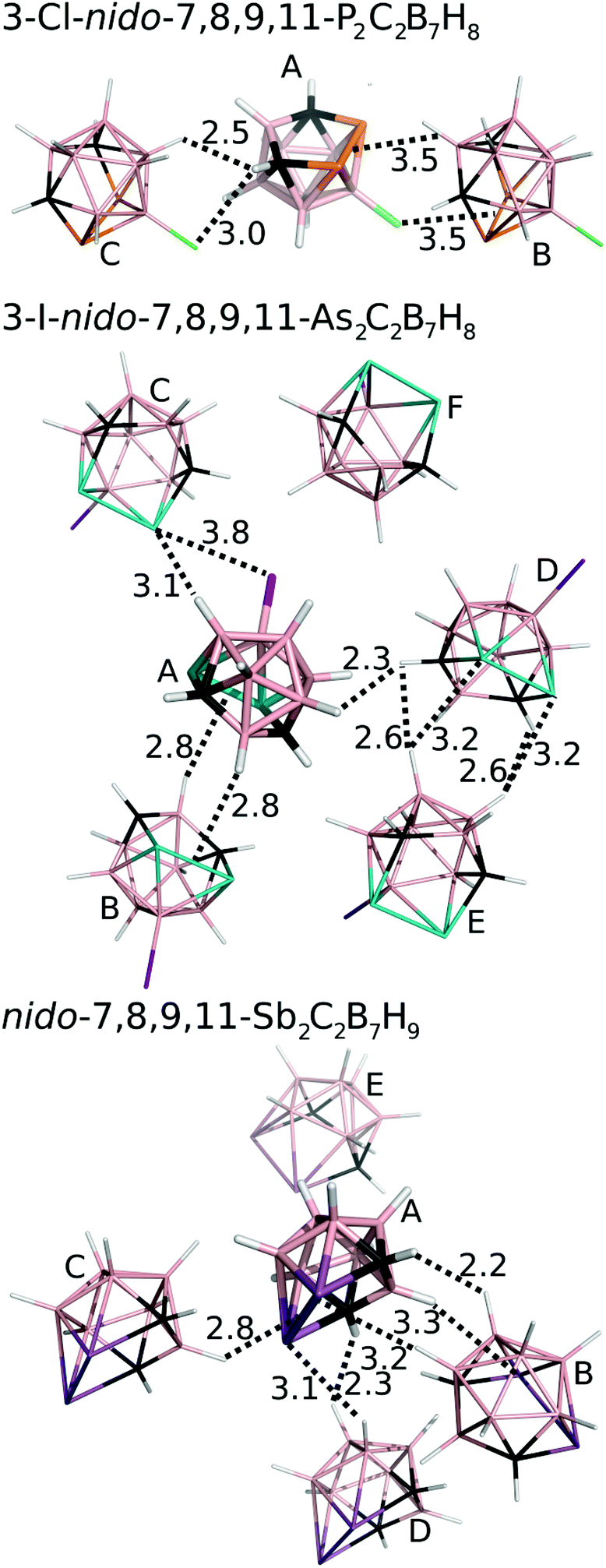 | ||
| Fig. 4 The most significant/stable interaction motifs from the crystalline structures of 3-Cl-nido-7,8,9,11-P2C2B7H8 (1), 3-I-nido-7,8,9,11-As2C2B7H8 (2), and nido-7,8,9,11-Sb2C2B7H9 (3). Distances are in Å. The color coding as follows: pink – B; black – C; orange – P; green – Cl; white – H; cyan – As; purple – I; magenta – Sb. Positions of H atoms were optimized at the DFT-D3/BLYP/DZVP level.11 | ||
| Interaction | MP2.5/CCSD(T) | |
|---|---|---|
| 3-Cl-nido-7,8,9,11-P 2 C 2 B 7 H 8 | ||
| A⋯B | 2 × Pn-bonds | −5.93/−6.10 |
| A⋯C | H- and HH-bonds | −4.19/−4.39 |
| 3-I-nido-7,8,9,11-As 2 C 2 B 7 H 8 | ||
| A⋯B | 2 × Pn-bonds | −5.98 |
| A⋯C | 2 × Pn-bonds | −5.67 |
| D⋯E | Pn- and HH-bonds | −4.52 |
| A⋯D | HH-bond | −3.52 |
| A⋯F | Stacking | −2.66 |
| nido-7,8,9,11-Sb 2 C 2 B 7 H 9 | ||
| A⋯B | 2 × Pn-bond, HH-bond | −6.80/−7.05 |
| A⋯C | Single Pn-bond | −6.22/−6.46 |
| A⋯D | HH-bond, Pn-bond | −4.79 |
| A⋯E | Stacking | −4.10 |
The crystal packing of 2 is also dominated by Pn-bonding, as the binding motifs with most negative ΔE values are stabilized by Pn-bonding. The A⋯B motif of 2 is stabilized by two symmetrical As2⋯H–B Pn-bonds and has ΔE of −6.0 kcal mol−1. An estimated ΔE of this isolated Pn-bond would thus be about −3 kcal mol−1. The A⋯C motif of 2 has a comparable ΔE of −5.7 kcal mol−1 and is stabilized by As⋯H–B and As⋯I–B Pn-bonds. The other motifs have less negative ΔE values. Among them, the most negative is the D⋯E motif with two As⋯H–B Pn-bonds and ΔE of −4.5 kcal mol−1.
The crystal structure of 3 has two binding motifs with ΔE exceeding −6 kcal mol−1. The A⋯B motif has ΔE of −7.1 kcal mol−1 and is stabilized by multiple interactions, specifically by two Sb⋯H–B Pn-bonds and one C–H⋯H–B HH-bond. On the other side, the A⋯C motif is exclusively formed by a single interaction, namely a Sb2⋯H–B Pn-bond. ΔE of −6.5 kcal mol−1 computed for this binding motif can thus be considered as ΔE of the isolated Pn-bond. This is a very large value, exceeding ΔE of isolated Pn-bonding of 2 by about 100%. The Pn-bonding of 3 is even slightly more favorable than Pn-bonding in Cl3Sb⋯N(Me)3 and Cl3Sb⋯benzene complexes (ΔE of −5.8 and −6.1 kcal mol−1, respectively).15
Methods
Synthesis
A solution of arachno-4,6-C2B7H13 (0.226 g, 2 mmol, see also Fig. 5) in dichloromethane (20 mL) was treated with proton-sponge (1.5 g, 7 mmol). SbCl3 (2.0 g, 16 mmol) was added under stirring and cooling to 0 °C. Stirring was continued at room temperature for 4 h and the mixture was then cooled to 0 °C and decomposed by the dropwise addition of water (20 mL). Column chromatography was carried out on a silica gel as the stationary phase (3 g). The solids were mounted onto a silica gel column and the column was eluted with dichloromethane. The chromatography led to the separation of a major yellow fraction of Rf = 0.73 (CH2Cl2), which was evaporated to dryness. 0.255 g of yellowish nido-7,8,9,11-Sb2C2B7H9 was obtained (yield 36%, with respect to the starting material 4,6-C2B7H13).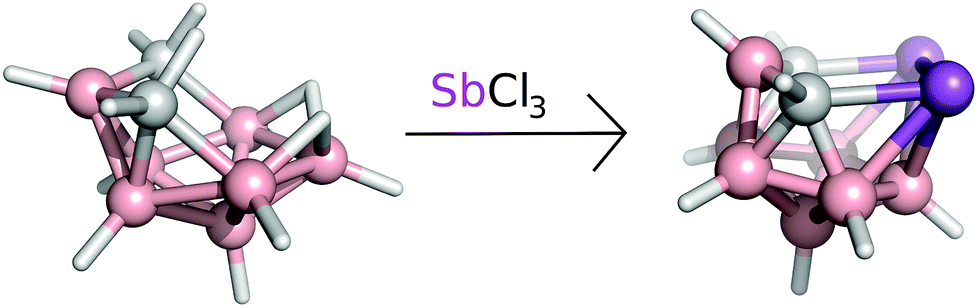 | ||
| Fig. 5 The reaction scheme of arachno-4,6-C2B7H13 with SbCl3 yielding the nido-7,8,9,11-Sb2C2B7H9, compound. | ||
Computational modeling
Magnetic shielding was calculated using the GIAO-MP2 method incorporated into Gaussian0916 utilizing the IGLO-II basis with the MP2/cc-pVTZ geometry (with the TZVP basis set on Sb by Weigend17 with ECP by Metz18) and frozen core electrons. Electrostatic potentials were computed at the HF/cc-pVDZ level (for I basis set in ref. 19) using Gaussian09 and Molekel4.320 programs. It has recently been shown that this basis set size is sufficient for these purposes.21Interaction energy (ΔE) values were calculated for all pairwise interactions of the crystal structures of 1, 2 and 3. All hydrogen atoms were optimized using the DFT-D3/BLYP/DZVP method prior to the energy calculations.11 ΔE for the central molecule with each surrounding molecule was evaluated at MP2.5/CBS using the Turbomole 6.622 and Cuby423 programs. MP2.5/CBS was calculated as the sum of MP2/CBS energy and MP2.5 correction. MP2/CBS was approximated by RI-MP2-F12/cc-pVTZ-F12 for 1.24 In the case of 2 and 3, we used MP2 extrapolation to the CBS from cc-pVTZ to cc-pVQZ (for I and Sb atoms cc-pVTZ-PP and cc-pVQZ-PP pseudopotentials were used).25 The MP2.5 correction term was calculated using the aug-cc-pVDZ basis set. MP2.5/CBS interaction energies were compared to benchmark CCSD(T)/CBS for the interaction motifs of 1 and the most favourable motifs of 3. The CCSD(T) correction term was also determined using the aug-cc-pVDZ basis sets. Counterpoise corrections for basis set superposition error (BSSE) are used for all energy calculations.
Crystallography
Full sets of diffraction data for 3 were collected at 150(2) K using a Bruker D8-Venture diffractometer equipped with a Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) source, a Photon CMOS detector and an Oxford Cryosystems cooling device.The frames were integrated using the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS). Data were treated by using XT-version 2014/5 and SHELXL-2014/7 software implemented in an APEX3 v2016.5-0 (Bruker AXS) system.26 Hydrogen atoms were mostly localized on a difference Fourier map; however, to ensure uniformity of treatment of the crystal, all hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom). H atoms in were placed with C–H distances of 1.1 Å for B–H and C–H bonds in the carborane cage. Hydrogen atoms in the upper rim of the cage were placed according to the appropriate maxima found on the Fourier difference electron density map.
R int = ∑|Fo2 – Fo,mean2|/∑Fo2, S = [∑(w(Fo2 – Fc2)2)/(Ndiffrs – Nparams)]1/2 for all data, R(F) = ∑||Fo| – |Fc||/∑|Fo| for observed data, wR(F2) = [∑(w(Fo2 – Fc2)2)/(∑w(Fo2)2)]1/2 for all data.
Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 1553208 for 3.†
Crystallographic data and structural refinement parameters for colorless single crystals of 3: C2H9B7Sb2, M = 352.26 g mol−1, orthorhombic, Pna21, a = 20.4799(12), b = 6.6471(4), c = 6.5803(4) Å, β = 90°, Z = 4, V = 895.79(9) Å3, Dc = 2.612 g cm−3, μ = 5.946 mm−1, Tmin/Tmax = 0.583/0.745; −27 ≤ h ≤ 27, −8 ≤ k ≤ 8, −8 ≤ l ≤ 8; 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 233 reflections measured (θmax = 28.35°), 2223 independent (Rint = 0.1199), 1796 with I > 2σ(I), 106 parameters, S = 1.049, R1 (obs. data) = 0.0271, wR2 (all data) = 0.0398; max., min. residual electron density = 1.091, −1.107 e Å−3.
233 reflections measured (θmax = 28.35°), 2223 independent (Rint = 0.1199), 1796 with I > 2σ(I), 106 parameters, S = 1.049, R1 (obs. data) = 0.0271, wR2 (all data) = 0.0398; max., min. residual electron density = 1.091, −1.107 e Å−3.
Conclusions
To sum up, in compound 3 (nido-7,8,9,11-Sb2C2B7H9) we have synthesized and crystallized the very first reported cluster containing simultaneously Sb–B, Sb–C and Sb–Sb covalent bonds. In addition to that, the distance between two Sb atoms in the open pentagonal belt of 3 is less than the sum of Sb covalent radii. Further quantum chemical analysis revealed that the most stable crystal motifs in the crystal structure of 3 were directed by Pn-bonding. ΔE of a very strong Sb2⋯H–B Pn-bond would exceed −6.0 kcal mol−1. For comparison, the Pn-bonding in 3 is more favorable than the reported Pn-bonding in Cl3Pn⋯N(Me)3 and is comparable to the Pn-bonding in Cl3Pn⋯π complexes (Pn = As, Sb). Phosphorus and arsenic can occupy the same positions as Sb in 3. However, in contrast to 3, crystallization of the parent nido-7,8,9,11-Pn2C2B7H8 (Pn = P, As) has not been possible. Indeed, crystal packing of halogenated 3-X-nido-7,8,9,11-Pn2C2B7H8 (X = Cl or I; Pn = P, As) derivatives is also dominated by Pn-bonding. We believe that Pn-bonding of nido-7,8,9,11-Pn2C2B7H9-type compounds offers a tantalizing possibility to further examine the Pn-bonding in a very detailed manner.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research project RVO 61388963 of the Czech Academy of Sciences. We acknowledge the financial support of the Czech Science Foundation (17-08045S).References
- (a) W. N. Lipscomb, Boron Hydrides, Benjamin, New York, 1963 Search PubMed; (b) D. Hnyk and D. A. Wann, Challenges and Advances in Computational Chemistry and Physics, in Boron – the Fifth Element, ed. D. Hnyk and M. McKee, Springer, Heidelberg, New York, Dordrecht and London, 2015, ch. 2, vol. 20 Search PubMed.
- K. Wade, Nat. Chem., 2009, 1, 92 CrossRef CAS PubMed.
- (a) J. L. Little, Inorg. Chem., 1979, 18, 1598 CrossRef CAS; (b) L. J. Todd, A. R. Burke, A. R. Garber, H. T. Silverstein and B. N. Storhoff, Inorg. Chem., 1970, 9, 2175 CrossRef CAS . Note that HOMO of (Me2PPh)2Pd(2+) and SLUMO (the second lowest unoccupied orbital) of the B9H9Sb2(2−) accounts for the stabilization.
- S. A. Jasper Jr., S. Roach, J. N. Stipp, J. C. Huffman and L. J. Todd, Inorg. Chem., 1993, 32, 3072 CrossRef.
- R. N. Grimes, Angew. Chem., Int. Ed., 2003, 42, 1198 CrossRef CAS PubMed . The interaction of the suitable symmetrical d orbitals of a metal with HOMO of the borane-cluster fragment is a driving force for the origination of the metallacarbaboranes.
- J. Holub, T. Jelínek, D. Hnyk, Z. Plzák, I. Císařová, M. Bakardjiev and B. Štíbr, Chem. – Eur. J., 2001, 7, 1546 CrossRef CAS.
- L. Mikulášek, B. Grüner, I. Císařová and B. Štíbr, Dalton Trans., 2003, 7, 1332 RSC.
- (a) W. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072 CrossRef PubMed; (b) A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS PubMed.
- (a) J. Fanfrlík, A. Přáda, Z. Padělková, A. Pecina, J. Macháček, M. Lepšík, J. Holub, A. Růžička, D. Hnyk and P. Hobza, Angew. Chem., Int. Ed., 2014, 53, 10139 CrossRef PubMed; (b) J. Fanfrlík and D. Hnyk, CrystEngComm, 2016, 18, 8973 RSC.
- J. Fanfrlík, J. Holub, Z. Růžičková, J. Řezáč, P. D. Lane, D. A. Wann, D. Hnyk, A. Růžička and P. Hobza, ChemPhysChem, 2016, 17, 3373 CrossRef PubMed.
- D. Hnyk, E. Vajda, M. Bühl and P. v. R. Schleyer, Inorg. Chem., 1992, 31, 2464 CrossRef CAS . For the mechanism of the antipodal effect, see. D. Hnyk, D. A. Wann, J. Holub, S. Samdal and D. W. H. Rankin, Dalton Trans., 2011, 40, 5734 RSC.
- P. v. R. Schleyer, Ch. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed.
- (a) P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed; (b) W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650 CrossRef CAS PubMed; (c) C. R. Wade, M. R. Saber and F. P. Gabbai, Heteroat. Chem., 2011, 22, 500 CrossRef CAS.
- A. Bauza, D. Quinonero, P. M. Deya and A. Frontera, CrystEngComm, 2013, 15, 3137 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- In-Sb(ecp-28): B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431 CrossRef CAS.
- (a) P. Flükiger, H. P. Lüthi, S. Portmann and J. Weber, MOLEKEL 4.3, Swiss Center for Scientific Computing, Manno, Switzerland, 2000 Search PubMed; (b) S. Portmann and H. P. Lüthi, MOLEKEL: CHIMIA, 2007, vol. 28, p. 555 Search PubMed.
- K. E. Riley, K.-A. Tran, P. Lane, J. S. Murray and P. Politzer, J. Comput. Sci., 2016, 17, 273 CrossRef.
- R. Ahlrichs, et al., TURBOMOLE V6.2 2010, a development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com, Turbomole V6, Karlsruhe, Deutschland, 2008 Search PubMed.
- J. Řezáč, J. Comput. Chem., 2016, 37, 1230 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed.
Footnote |
| † CCDC 1553208. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02845h |
| This journal is © The Royal Society of Chemistry 2017 |