
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissimilar catalytic behavior of molecular or colloidal palladium systems with a new NHC ligand†
Fernando
Gómez-Villarraga‡
a,
Jonathan
De Tovar‡
 a,
Miguel
Guerrero
bc,
Pau
Nolis
d,
Teodor
Parella
d,
Pierre
Lecante
e,
Nuria
Romero
a,
Lluís
Escriche
a,
Roger
Bofill
a,
Josep
Ros
a,
Xavier
Sala
a,
Karine
Philippot
*bc and
Jordi
García-Antón
*a
a,
Miguel
Guerrero
bc,
Pau
Nolis
d,
Teodor
Parella
d,
Pierre
Lecante
e,
Nuria
Romero
a,
Lluís
Escriche
a,
Roger
Bofill
a,
Josep
Ros
a,
Xavier
Sala
a,
Karine
Philippot
*bc and
Jordi
García-Antón
*a
aDepartament de Química, Unitat de Química Inorgànica, Facultat de Ciències, Universitat Autònoma de Barcelona, 08193-Bellaterra, Barcelona, Spain. E-mail: Jordi.GarciaAnton@uab.es; Fax: (+34) 93 581 3101
bCNRS, LCC (Laboratoire de Chimie de Coordination du CNRS), 205, route de Narbonne, F-31077 Toulouse, France. E-mail: Karine.Philippot@lcc-toulouse.fr; Fax: (+33) 5 61553003
cUniversité de Toulouse, UPS, INPT, LCC, F-31077 Toulouse, France
dServei de Ressonància Magnètica Nuclear, Facultat de Ciències, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Catalonia, Spain
eCNRS, CEMES (Centre d'Elaboration de Matériaux et d'Etudes Structurales), 29 rue J. Marvig, F-31055 Toulouse, France
First published on 8th August 2017
Abstract
In this work, we describe the synthesis of a new N-heterocyclic carbene (NHC) ligand, derived from a hybrid pyrazole-imidazolium scaffold, namely 1-[2-(3,5-dimethylpyrazol-1-yl)ethyl]-3-((S)-1-phenylethyl)-3H-imidazol-2-ylidene (L). This ligand has been used as a stabilizer for the organometallic synthesis of palladium(0) nanoparticles (Pd NPs). L presents a better stabilizing effect than its pre-carbenic HLCl counterpart, allowing the formation of isolated Pd NPs while HLCl yields aggregated ones. Additionally, molecular Pd(II) coordination compounds of L and HLCl were synthesized and characterized to better understand the coordination modes of these ligands. Both molecular and colloidal Pd systems have been further tested in catalytic C–C coupling processes. Three different types of reactions have been observed depending on the catalytic system: (i) the Suzuki–Miyaura reaction takes place with Pd molecular complexes; (ii) a secondary reaction, the dehalogenation of the substrate, is always detected and (iii) the C–C homocoupling between two molecules of bromoarenes is observed with colloidal catalysts.
Introduction
During the last few years, the use of N-heterocyclic carbenes (NHCs) as primary ligands has emerged in catalysis and beyond. Amongst other catalytic processes, organometallic compounds containing these ligands have been applied in the cross-coupling reactions of un-activated substrates.1 This can be explained not only by their remarkably strong σ-binding and steric tunability,2 but also by their ability to stabilize highly unusual and hitherto elusive reactive species such as metal nanoparticles (MNPs) thanks to their stability under oxidative conditions once coordinated.3 Thus, NHCs have become ligands of paramount importance in nanochemistry given their advantageous behavior for the stabilization and functionalization of MNPs.4 Although different families of NHCs have been largely explored in various important organic transformations when combined with metal pre-catalysts, imidazolium salts are the most frequently employed as the core structures for NHCs given their straightforward syntheses and feasible tuning properties.5Different methodologies can be followed to obtain and stabilize NPs in different media while controlling their size, shape and composition, which, in turn, can tune their unique properties.6 The use of coordinating ligands is of particular interest to prepare MNPs because it gives the possibility of modulating their surface properties as known for the synthesis of molecular complexes.7 As a consequence, ligand-stabilized MNPs became very attractive for applications in catalysis.8 MNPs are able to catalyze not only reactions that can also be catalyzed by molecular complexes (i.e. C–C coupling or hydrogenation of olefins) but also reactions that cannot be catalyzed by molecular complexes (i.e. hydrogenation of arenes).
Furthermore, palladium is the most versatile transition metal in chemical catalytic reactions since many of these processes cannot be catalyzed by other transition metals.9 Thus, Pd(0) NPs have shown large catalytic efficiency in C–C coupling reactions specially when non-aggregated homogeneous NPs of 1–4 nm size are used as catalysts.10
The Suzuki–Miyaura reaction is one of the most important cross-coupling processes from an industrial point of view.11 It allows to easily obtain biaryl products, important intermediates in the organic syntheses of various target products ranging from performance materials to pharmaceuticals.12 Although noble metal complexes are usually employed as catalysts for this reaction, MNPs merit study since playing with stabilizing ligands may tune their catalytic performance.13
Our research group has focused on discerning the catalytic behavior of colloidal and molecular palladium systems in C–C coupling reactions. For example, we have recently studied Pd molecular/colloidal systems containing new hybrid pyrazole derived ligands with alkylether, alkylthioether or alkylamino moieties leading to different catalytic outputs depending on the system.14 Here we present improved Pd systems based on a new NHC–pyrazole hybrid ligand. Interestingly, as it will be described hereafter, different behaviors were observed in C–C coupling catalysis that illustrates the possibility of optimizing the catalytic system according to the final aim.
Results and discussion
Synthesis of the ligand (L)
For the synthesis of 1-[2-(3,5-dimethylpyrazol-1-yl)ethyl]-3-((S)-1-phenylethyl)-3H-imidazol-1-ium chloride (HLCl), 1-(2-chloroethyl)-3,5-dimethyl-1H-pyrazole and 1-(S)-(phenylethyl)imidazole were heated solventless at 120 °C for 64 h. The deprotonation of HLCl to yield 1-[2-(3,5-dimethylpyrazol-1-yl)ethyl]-3-((S)-1-phenylethyl)-3H-imidazol-2-ylidene (L) was achieved by reacting HLCl with NaH and a catalytic amount of tBuOK in anhydrous dichloromethane (Scheme 1). Both L and HLCl have been fully characterized by NMR and IR spectroscopies as well as by ESI-MS (Fig. S1–S6 in the ESI†).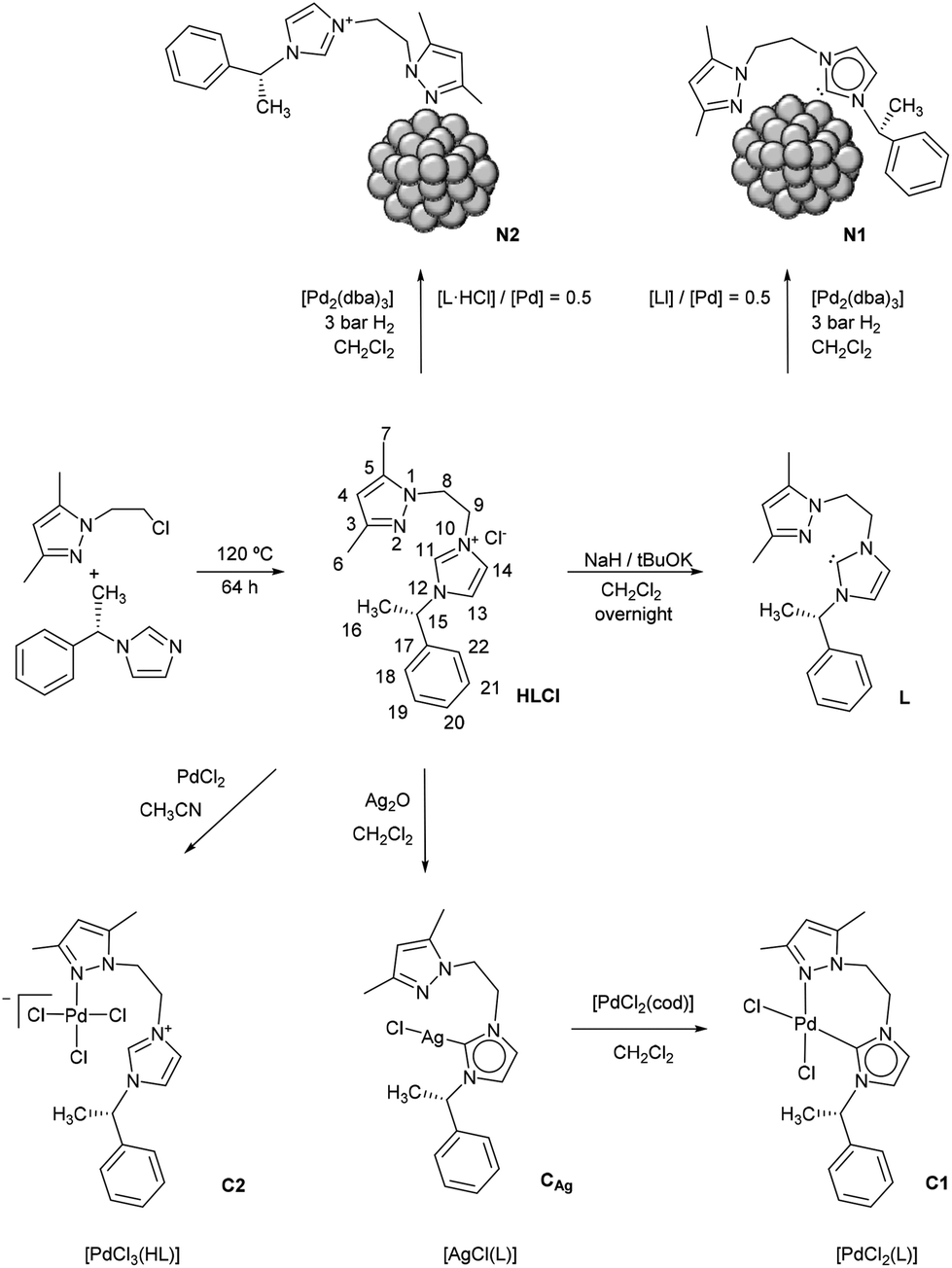 | ||
| Scheme 1 Synthetic preparation of L and HLCl and their corresponding Ag and Pd molecular complexes (CAg, C1 and C2) and Pd nanoparticulate systems (N1 and N2). | ||
Synthesis of Pd(0) nanoparticles (Pd NPs)
Ligand L has been used to prepare Pd NPs according to the organometallic approach under mild reaction conditions.15 The syntheses have been carried out by reacting [Pd2(dba)3] (dba = dibenzylideneacetone) and L ([L]/[Pd] molar ratios varying from 0.1 to 1.0) in anhydrous dichloromethane for 18 h in a Fisher-Porter reactor, under 3 bar of dihydrogen at room temperature. Finally, Pd NPs were precipitated and washed with cold pentane and dried under reduced pressure.TEM (transmission electron microscopy) and SEM (scanning electron microscopy) analyses (after deposition/drying of a drop of crude colloidal solutions onto a holey carbon-covered copper grid) were performed with all the obtained samples. The results showed that the Pd NPs are aggregated into superstructures when a L/Pd ratio of 0.1 is used (HR-TEM images are shown in Fig. 1a and SEM-FEG images are shown in ESI, Fig. S7a†). The single NPs are spherical and have a mean size of 3.6(0.9) nm but the superstructures do not have a preferential shape and their size varies from 50 to 250 nm. For L/Pd ratios of 0.3, 0.5 and 1.0, only isolated NPs were observed (Fig. 1b–d). In all these cases, NPs are spherical and present similar mean sizes in the range of 2.1–2.4 nm. These similar sizes indicate that a minimum L/Pd molar ratio of 0.3 is needed to have stable isolated NPs, but the addition of a higher ligand/metal ratio does not have a significant effect on the size of the NPs.
 | ||
| Fig. 1 HR-TEM micrographs and the corresponding size-histograms of Pd nanoparticles stabilized with L synthesized as follows: (a) [L]/[Pd] = 0.1; (b) [L]/[Pd] = 0.3; (c) [L]/[Pd] = 0.5; (d) [L]/[Pd] = 1.0. | ||
For comparative purposes, the stabilization of the NPs has also been attempted with the protonated ligand HLCl (see HR-TEM images in Fig. 2 and SEM-FEG images in ESI, Fig. S7b–d†). In this case, a molar ratio of 0.1 failed to afford stable NPs. For molar ratios of 0.3 and 0.5 Pd NPs appeared organized into superstructures, and for the molar ratio of 1.0, a mixture of isolated and aggregated NPs was observed. The mean sizes of these NPs are 3.0(0.7), 3.2(0.9) and 2.1(0.9) nm for HLCl/Pd molar ratios of 0.3, 0.5 and 1.0, respectively. Therefore, with HLCl a higher ratio of ligand is needed to successfully stabilize the NPs. From these results and given the main difference of the two ligands (L is a carbene while HLCl is not deprotonated), we believe that the stabilization of the Pd NPs takes place by the coordination of L through the carbene (M–C) and pyrazolic (M–N) groups to the surface of the Pd NPs whereas HLCl would only coordinate through the pyrazolic group (Scheme 1).
 | ||
| Fig. 2 HR-TEM micrographs and the corresponding size-histograms of Pd nanoparticles stabilized with HLCl synthesized as follows: (a) [HLCl]/[Pd] = 0.3; (b) [HLCl]/[Pd] = 0.5; (c) and (d) [HLCl]/[Pd] = 1.0. | ||
The presence of the ligands L or HLCl in N1 or N2 (those prepared with L and HLCl, respectively, in a ligand/Pd molar ratio equal to 0.5) has been demonstrated by IR spectroscopy (Fig. S8† and the Experimental section), and ICP-OES and elemental analyses. The Pd/ligand ratios found are Pd561L37 and Pd1415HLCl93 for N1 and N2, respectively.
Wide-angle X-ray scattering (WAXS) measurements have been performed on nanoparticles N1 and N2. The two radial distribution functions (RDFs) (Fig. 3a) are very similar and in good agreement with the fcc structure of bulk Pd. However, significant distances can be observed of at least up to 4 nm, thus being in disagreement with the sizes obtained from HR-TEM (2.1(0.9) and 3.2(0.9) for N1 and N2, respectively). Indeed, when compared with the RDF computed (Fig. 3b) from a spherical model 2.0 nm in diameter, the agreement does not go beyond 1.5 nm. A much better result can be obtained with a dual size model, including a small amount (10%) of much bigger spherical particles (5 nm in diameter). Actually, this dual size model is likely itself oversimplified and different combinations including a large majority of 2 nm particles and a minority of bigger ones should probably lead to the same level of agreement. This result, however, is not in contradiction with the TEM results taking into account not only the mean sizes but also the relatively large tail on the bigger size side.
 | ||
| Fig. 3 WAXS measurements on Pd nanoparticles for N1 and N2 and comparison with Pd fcc in reciprocal space (a) and real space including simulated from a model including only 2 nm particles based on the Pd fcc structure (lower) and simulated from a model including 2 nm particles plus 10% of 5 nm particles (upper) (b). | ||
Synthesis and characterization of Pd(II) molecular complexes (C1–C2)
Pd(II) molecular complexes of L and HLCl have been synthesized in order to better understand the coordination properties of these ligands (Scheme 1).The silver carbene transmetallation reaction is a general procedure for the preparation of palladium–carbene complexes16 and was considered to obtain the Pd complex with L. The silver complex of L (CAg) was prepared by reacting silver dioxide and HLCl in anhydrous dichloromethane. The 1H NMR spectrum of this complex shows that the pre-carbene proton of the imidazolium salt is no longer present, thus confirming the deprotonation of the imidazolium group and formation of the Ag–NHC bond (Fig. 4 and Fig. S9†). Also, the formation of CAg has been confirmed by ESI-MS and IR spectroscopy (Fig. S10 and S11,† respectively). The next step consisted of the reaction of CAg with [PdCl2(cod)] in anhydrous dichloromethane (Scheme 1) to obtain the carbenic Pd complex [PdCl2L] (C1). The absence of the pre-carbene proton in the 1H-NMR spectrum of C1 confirms the formation of the Pd–NHC bond (Fig. 4 and Fig. S12†). The formation of the complex has been attested by HR-ESI-MS and IR spectroscopy (Fig. S13 and S14,† respectively). Moreover, the NMR data also evidence the chelation of the ligand to the metallic centre through the NHC and pyrazolic groups. The rigidity imposed by this coordination mode causes the two protons of each CH2 in the N–CH2CH2–N moiety (H8 and H9) to become diastereotopic, resulting in four visible signals assigned to a single hydrogen atom each (see high-resolution NMR spectra of C1 including 1H and 13C NMR spectroscopy chemical shift assignments in Fig. S12a and b†). It is also noticeable that some proton signals split, which is a single quote labeled in Fig. S12a.† This splitting is due to conformational exchange between a major and a minor component with a ratio of approximately 70/30, as deduced from signal integration.
 | ||
| Fig. 4 1H-NMR of HLCl, L and CAg registered at 360 MHz (CDCl3, 298 K) and C1 registered at 600 MHz (CDCl3, 298 K). | ||
In order to study further this chemical exchange process, a 2D ROESY (Rotating Frame Overhauser Effect Spectrocopy) spectrum was recorded.17 Briefly explained, in this NMR experiment ROE and occasionally chemical exchange cross peaks are observed.18 The former are indicative of through space distance proximity, and the latter are detected when protons experience distinct chemical environments, which is commonly the case when a conformational equilibrium is present. The distinction of the above mentioned different cross peaks families is possible from the opposite signal phase in the ROESY spectrum. Fig. 5 and S12d† show the 2D ROESY spectrum of C1, where chemical exchange cross peaks are detected in the negative phase (blue color), while ROE cross peaks are positive phased (red color). Thus, protons H8 and H9 in the ethyl chain undergo a pronounced chemical environment exchange, with cross peaks situated notably separated from the spectrum diagonal. This conformational exchange might be explained on the basis of a slow conformational equilibrium of the new seven-membered ring formed. Other protons (H4, H6, H7, H13, H14 and H16) also exhibit chemical exchange cross peaks, which are closer to the spectrum diagonal, indicating that chemical exchange is taking place between less differentiated chemical environments. This latter effect could be explained as a side-effect from the mentioned seven ring conformational exchange. On the other side, neither benzyl ring protons H18–H22 nor H15 show any chemical exchange cross peaks, suggesting that these protons are situated far enough from the conformational exchange described.
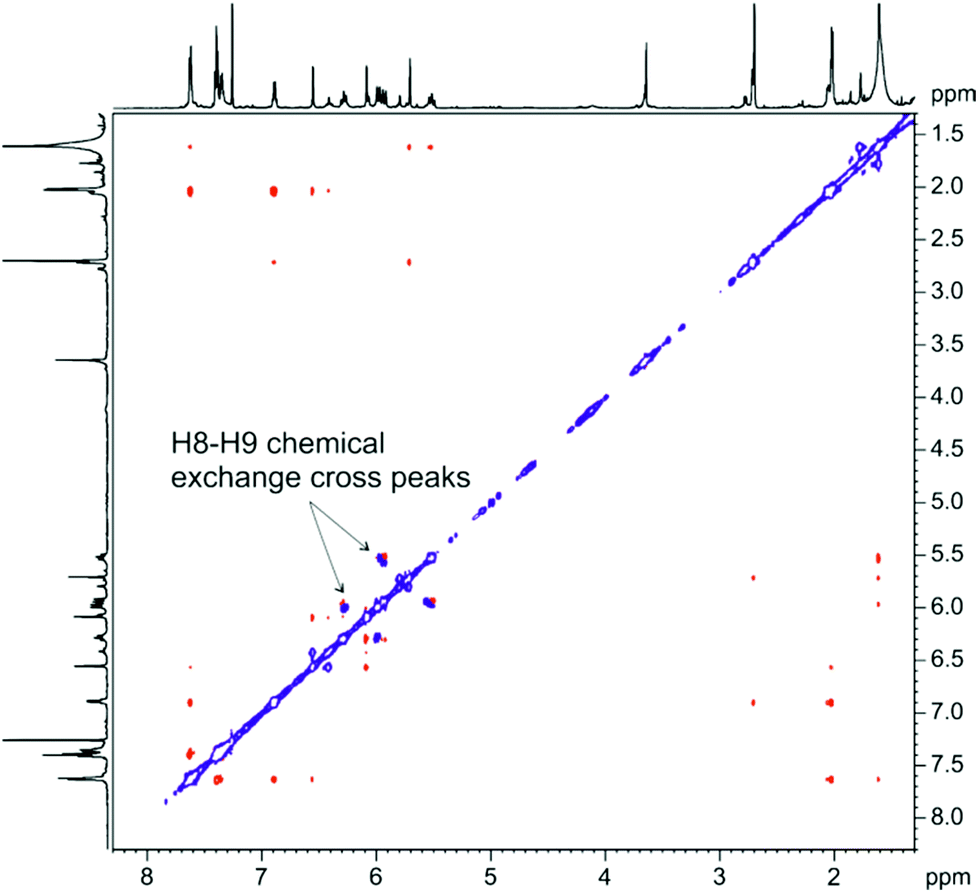 | ||
| Fig. 5 2D ROESY spectrum of C1 registered at 600 MHz (CDCl3, 298 K). Arrows pointing negative cross peaks (blue color) indicate chemical exchange between H8 and H9 protons. | ||
On the other hand, the direct reaction of HLCl with PdCl2 in acetonitrile yielded the complex [PdCl3(HL)] (C2, Scheme 1), which has been characterized by NMR and IR spectroscopies and ESI-MS (Fig. S15–S17†), thus confirming its stoichiometry. Interestingly, the existence of the protonated ligand HL+ is confirmed by the singlet 1H NMR spectroscopy resonance appearing at 9.03 ppm for the H11 pre-carbene proton (Fig. S15†).
Crystal and molecular structure of trichloro[(S)-3-(2-(3,5-dimethyl-1H-pyrazol-1-yl)ethyl)-1-(1-phenylethyl)-1H-imidazol-3-ium-κ1N]palladium(II) (C2)
Single crystals of C2 suitable for X-ray diffraction analysis were obtained by slow evaporation of a concentrated acetonitrile solution of the complex at RT. The structure of C2 consists of monomeric [PdCl3(HL)] units (Fig. 6a), linked by van der Waals forces (Fig. 6b). The palladium center is coordinated by three terminal chloride ligands and one pyrazolic nitrogen from a 1-[2-(3,5-dimethylpyrazol-1-yl)ethyl]-3-((S)-1-phenylethyl)-3H-imidazol-1-ium cation in a slightly distorted square-planar geometry. Crystallographic data are listed in Table S1,† and the data on selected bond lengths and angles are gathered in Table S2.† The positive charge generated by the imidazolium cation is canceled by the negative charge provided by the third chloride ion coordinated to Pd(II), thus forming a zwitterionic structure. | ||
| Fig. 6 ORTEP drawing of C2, showing the atom-numbering scheme; 50% probability amplitude displacement ellipsoids are shown for all non-hydrogen atoms (a) and hydrogen interactions present in the unit cell of C2 (b). | ||
There are three complexes reported in the literature with a PdCl3N core (terminal chloride ligands and pyrazolic nitrogen).19–21 The Pd–N and Pd–Cl bond lengths here observed are comparable to the values found in the literature, and the N–Pd–Cl and Cl–Pd–Cl bond angles slightly deviate from the square planar angles (Table S2†).
In compound C2, chlorine atoms (Cl2 and Cl3, Fig. 6b) form three different hydrogen bonds: C3–H3A⋯Cl2 (2.707 Å, 146.11°) and C15–H15⋯Cl3 (2.935 Å, 154.30°) interacting with hydrogens from the ethylene group of an adjacent C2 unit, and C11–H11⋯Cl2 (2.840 Å, 138.45°) interacting with a hydrogen atom from the imidazole aromatic ring of another adjacent C2 complex. These interactions are responsible for the expansion in the crystallographic a direction.
Catalytic experiments with Pd molecular or colloidal systems
C1 and C2 Pd complexes (with L and HLCl ligands, respectively) as well as N1 and N2 Pd nanoparticles have been evaluated as catalysts for the Suzuki–Miyaura reaction (Scheme 2, Table 1).22,23 | ||
| Scheme 2 Suzuki–Miyaura, homocoupling and dehalogenation reactions performed in the presence of Pd complexes C1 and C2 and Pd NPs N1 and N2. | ||
| Entry | Catalyst | X | [PhB(OH)2]/[Pd] | Conv.b (%) | BTc (%) | TTc (%) | PhMec (%) | Yield BT (%) | Yield TT (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 × 10−3 mmol of Pd, 2.5 mmol of 4-halogenotoluene, 5.0 mmol tBuOK and 0.5 mmol naphthalene as an internal standard in 8.0 mL of DMF and 2.0 mL of H2O. Temperature, 100 °C. b Conversion after 6 h reaction. c Chemoselectivity in 4-methylbiphenyl (BT), 4,4′-dimethylbiphenyl (TT) and toluene (PhMe), respectively. | |||||||||
| 1 | C1 | I | 3125 | 100 | 51 | 0 | 49 | 51 | 0 |
| 2 | C2 | I | 3125 | 82 | 41 | 0 | 59 | 34 | 0 |
| 3 | C1 | Br | 3125 | 28 | 71 | 0 | 29 | 20 | 0 |
| 4 | C2 | Br | 3125 | 46 | 93 | 0 | 7 | 43 | 0 |
| 5 | N1 | I | 3125 | 100 | 81 | 0 | 19 | 81 | 0 |
| 6 | N2 | I | 3125 | 100 | 47 | 0 | 53 | 47 | 0 |
| 7 | N1 | I | 0 | 100 | 0 | 0 | 100 | 0 | 0 |
| 8 | N2 | I | 0 | 56 | 0 | 0 | 56 | 0 | 0 |
| 9 | N1 | Br | 3125 | 66 | 5 | 51 | 44 | 3 | 34 |
| 10 | N2 | Br | 3125 | 36 | 7 | 37 | 56 | 3 | 13 |
| 11 | N1 | Br | 0 | 72 | 0 | 83 | 17 | 0 | 60 |
| 12 | N2 | Br | 0 | 36 | 0 | 72 | 28 | 0 | 26 |
4-Halogenotoluene derivatives (4-chlorotoluene, 4-bromotoluene or 4-iodotoluene) and an excess of phenylboronic acid were chosen as substrates in order to easily distinguish between cross-coupling and homocoupling products. Under the catalytic reaction conditions applied (Scheme 2),14 two different by-products can be formed besides the expected cross-coupled product (4-methylbiphenyl; BT) as follows: (i) toluene (PhMe) arising from the dehalogenation of the substrates and (ii) 4,4′-dimethylbiphenyl (TT), resulting from the 4-bromotoluene homocoupling. Once optimized, 4-halogenotoluene (substrate/Pd = 2500), phenylboronic acid (PhB(OH)2/Pd = 3125 or 0), and tBuOK (base/Pd = 5000) were employed in a DMF/water mixture (4/1) as the solvent. As a general trend, higher conversions were obtained with 4-iodotoluene compared to 4-bromotoluene in both molecular and colloidal systems (Table 1), as expected from the respective C(Ph)–X bond energies (X = Br, 84 kcal mol−1; X = I, 67 kcal mol−1).24 Unfortunately, 4-chlorotoluene could not be activated probably due to the higher C(Ph)–Cl bond energy value (97.1 kcal mol−1)24 and the Suzuki–Miyaura coupling reaction was unsuccessful.
Interestingly, in the presence of the molecular catalysts C1 and C2, toluene was the only side product observed in a significant amount using both 4-bromotoluene and 4-iodotoluene as substrates (Table 1, entries 1–4). The homocoupling reaction does not take place, as otherwise predictable according to the literature on Pd molecular complexes.14 Also, 4-bromotoluene yields BT as the major product, while the amount of the dehalogenation product (toluene) significantly increases when 4-iodotoluene is used.
Recently, V. P. Ananikov et al. have demonstrated the formation of Pd NPs when molecular Pd complexes with NHC ligands are used in the Mizoroki–Heck reaction.25 In our case, even if it is not a definite proof, TEM control analyses of the solutions after catalysis with C1 or C2 did not show the presence of Pd nanoparticles. In any case, Hg poisoning tests carried out using these systems (vide infra) confirm the molecular nature of the real catalyst when C1 or C2 is used.
When 4-iodotoluene was used as a substrate with nanocatalysts N1 and N2, complete conversions were achieved in 6 h in both cases. In the case of N1, the formation of the cross-coupling product (4-methylbiphenyl; BT) was mainly observed together with a small amount of toluene (Table 1, entry 5). The formation of toluene increases to 53% in the case of N2 (Table 1, entry 6). In contrast, N1 and N2 behave similarly against 4-bromotoluene, with the formation of 4,4′-dimethylbiphenyl (TT), arising from the homocoupling reaction, together with toluene and small amounts of BT (Table 1, entries 9 and 10). Furthermore, for the two halogenotoluene substrates, both conversion and chemoselectivities are higher for the Pd NP/L system (N1) than that for the Pd NP/HLCl system (N2). These differences can be explained by the binding of the carbene group at the Pd NP surface in the case of N1 compared to N2. Indeed, the different coordination modes (by C and N in N1 and only by N in N2) may tune both electronic and steric properties at the surface and consequently the catalytic behavior of N1.
In order to determine the influence of phenylboronic acid in C–C coupling reactions catalyzed by the colloidal systems, catalytic experiments were also carried out in the absence of the boron reagent. Besides that BT was not detected, it is remarkable that full conversion to the dehalogenated product (PhMe) is observed in the case of the reaction carried out with 4-iodotoluene catalyzed by N1 (Table 1, entry 7). However, when 4-bromotoluene was employed a significant increase in the yield of the TT homocoupling product is observed for both nanocatalysts (from 34% to 60%; Table 1, entries 9 and 11, respectively, for N1, and from 13% to 26% for N2, entries 10 and 12). When performed with the molecular complexes, the same reaction of 4-halotoluene in the absence of phenylboronic acid reagent did not yield homocoupling products at all.
A recurrent discussion with Pd NPs as catalysts for C–C coupling reactions is whether the real catalysts are the NPs or molecular Pd species leached from them.10 There are several publications that support either a surface-based26 or atom-leaching mechanism.27 In this context, three different tests have been carried out to assess the nature of the active species during the catalytic process. First of all, in the mercury poisoning test,28 100 equivalents of Hg were added to the catalytic mixtures after 10 min of reaction. For molecular systems (C1 and C2) the conversions were not affected, while for the colloidal systems (N1 and N2) the catalytic reactions ceased completely. These data suggest that C1 and C2 act as real molecular catalysts when starting with them, whereas Pd NPs are the active species when N1 and N2 are introduced into the catalysis. This is in accordance with the different results observed in catalysis depending on the molecular or colloidal nature of the introduced catalysts. However, the mercury poisoning test is not definitive, since the amalgam of Hg and Pd from the NPs could prevent potential Pd molecular species from leaching. For this reason, additional experiments have been performed with systems from entries 9 and 10 in Table 1 (Suzuki–Miyaura reaction conditions, p-bromotoluene as the haloderivative and N1 or N2 as the catalyst, respectively). First of all, TEM grids were prepared after the catalytic experiments, observing the existence of Pd NPs (Fig. S18†). The mean size of these NPs (2.7(0.7) and 3.3(0.6) nm for N1 and N2 after catalysis, respectively) is similar to those found for the same NPs prior to the catalytic tests (2.1(0.9) and 3.2(0.9) nm for N1 or N2, respectively). Even if no restructuring of the NPs seems to have taken place (which could indicate a leaching/deposition mechanism through the Ostwald ripening process29), it is important to note that for N2 the NPs appear isolated after catalysis, in contrast with the aggregation observed prior to catalysis.
Secondly, ICP-MS analyses of the solutions after catalysis for entries 9 and 10 in Table 1 were carried out in order to know the amount of Pd leached from the NPs to the solution. The concentrations of Pd in these solutions are 0.17 and 1.8 ppm for N1 or N2, respectively, meaning that 0.7% and 15% of Pd have leached to the solution for N1 or N2, respectively. The differences on the percentage of leached Pd can be explained by the different coordinative properties of the ligands on each system. For N1, the carbene and pyrazolyl groups from L can coordinate to the surface of the NPs, and better stabilize them from Pd leaching than in N2, where only the pyrazolyl group from HLCl can coordinate to the surface of the NPs.
Moreover, the leached molecular Pd species can be responsible for the small amount of C–C heterocoupling reaction observed in entries 9 and 10 of Table 1 (5% and 7%, respectively).
All in all, these experiments indicate that C1 and C2 act as molecular catalysts in Suzuki–Miyaura C–C heterocoupling and N1 and N2 are the real active species for the C–C homocoupling reaction.
Experimental section
General procedure and reagents
All manipulations were carried out under an argon atmosphere using standard Schlenk tubes or Fisher-Porter reactors and vacuum line techniques, or in a glove-box. [Pd2(dba)3] was purchased from Strem Chemicals, and [PdCl2(CH3CN)2]30 and [PdCl2(cod)]31 (cod = 1,5-cyclooctadiene) were prepared as described in the literature. Solvents were purchased from SDS and dried using a purification machine (MBraun MB SPS-800) or distilled prior to use: tetrahydrofuran and diethyl ether over sodium/benzophenone, and pentane, n-hexane, acetonitrile and dichloromethane over calcium hydride.Elemental analyses (C, H, and N) were carried out by the Chemical Analyses Service of the Universitat Autònoma de Barcelona on a Eurovector 3011 instrument. The Pd weight percentages or concentrations were analyzed by the Chemical Analyses Service of the Universitat Autònoma de Barcelona using an inductively coupled plasma mass spectrometry (ICP-MS) Agilent 7500ce model system or an inductively coupled plasma optical emission spectrometry (ICP-OES) PerkinElmer Optima 4300DV model system. Infrared spectra were recorded on a PerkinElmer FT spectrophotometer, series 2000, as KBr pellets or polyethylene films in the range of 4000–150 cm−1. 1H NMR, 13C {1H} NMR, HSQC, COSY, DOSY and NOESY spectra for all compounds but C1 were recorded on Bruker AVANCE 360 and 400 NMR spectrometers in CDCl3 solutions at room temperature. For C1, NMR spectroscopy experiments were performed on a Bruker Avance 600 spectrometer (Bruker Biospin, Rheinstetten, Germany) equipped with TXI HCN z-grad probes. The temperature for all measurements was set to 298 K. The NMR spectroscopy experiments performed for the structural characterization were the standard 1H, 13C, COSY, ROESY, multiplicity-edited HSQC and HMBC experiments (see the ESI†). All chemical shift values (δ) are given in ppm. Electrospray ionization mass spectra (SI-MS) were obtained with an Esquire 3000 ion trap mass spectrometer from Bruker Daltonics.
Specimens for TEM/HR-TEM and SEM-FEG analyses were prepared by slow evaporation of a drop of crude colloidal solution deposited under argon onto holey carbon-covered copper grids. TEM/HR-TEM analyses were performed at the Servei de Microscopia de la UAB with a JEOL JEM 2010 electron microscope working at 200 kV with a resolution point of 2.5 Å. SEM-FEG analyses were performed at the Service Commun de Microscopie Electronique de l′Université Paul Sabatier in Toulouse with an MEB JSM6700F microscope. The size distributions were determined via manual analysis of enlarged micrographs by measuring ca. 200 particles on a given grid to obtain a statistical size distribution and a mean diameter.
Data collection for WAXS was performed at the CEMES-CNRS (Toulouse) on small amounts of powder. All samples were sealed in 1 mm diameter Lindemann glass capillaries. The measurements of the X-ray intensity scattered by the samples irradiated with graphite monochromatized MoKα (0.071069 nm) radiation were performed using a dedicated two-axis diffractometer. The measurement time was 15 h for each sample. Scattering data were corrected for polarization and absorption effects, then normalized to one Pd atom and Fourier transformed to obtain the RDFs. To make comparisons with the crystalline structure in real space, a model was generated from bulk Pd parameters. The classic Debye's function was then used to compute the intensity values, subsequently Fourier transformed under the same conditions as the experimental ones.
Synthesis and characterization of the ligands
Anal. calcd for C18H23N4Cl·1.5H2O: C, 60.41; H, 7.32; N, 15.66. Found: C, 60.54; H, 7.27; N, 16.02. 1H-NMR: (CDCl3, 360 MHz, 298 K) δ: 1.89 [s, 3H, H6 or H7], 1.92 [br, 3H, H16], 2.06 [s, 3H, H6 or H7], 4.44 [br, 2H, H9], 4.87 [br, 2H, H8], 5.61 [s, 1H, H4], 5.76 [q, 1H, 3J = 7.0 Hz, H15], 7.00, 7.23 [2br, 1H each one, H13, H14], 7.30 [br, 5H, H18–H22], 10.32 [s, 1H, H11]. (DMSO, 360 MHz, 298 K) δ: 1.81 [d, 3H, 3J = 7.0 Hz, H16], 1.99 [s, 6H, H6 and H7], 4.42 [t, 2H, 3J = 5.0 Hz, H9], 4.58 [t, 2H, 3J = 5.0 Hz, H8], 5.73 [s, 1H, H4], 5.83 [q, 1H, 3J = 7.0 Hz, H15], 7.36 [br, 5H, H18–H22], 7.72, 7.92 [2br, 1H each one, H13, H14], 9.39 [s, 1H, H11]. 13C{1H}-NMR: (CDCl3, 91 MHz, 298 K) δ: 10.7, 13.4 [C6, C7], 21.1 [C16], 47.9 [C9], 49.2 [C8], 59.9 [C15], 105.5 [C4], 120.4, 123.0 [C13, C14], 126.8, 129.4, 137.8 [C17–C22], 136.6 [C11], 140.5, 148.7 [C3, C5]. (DMSO-d6, 91 MHz, 298 K) δ: 10.2, 13.3 [C6, C7], 20.6 [C16], 47.2 [C9], 48.8 [C8], 58.5 [C15], 105.0 [C4], 121.3, 123.2 [C13, C14], 126.5, 128.7, 129.0, [C17–C22], 136.0 [C11] 139.4, 146.8 [C3, C5]. IR (ATR) cm−1: 3040, 2978 ν(C–H), 1552 (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), ν(CN))ar, 1455 (δ(CC), δ (CN))ar, 1160 δ (C–H)ip, 703 δ (C–H)oop. MS (ESI): m/z (%) 295.2 (100.0%) [C18H23N4Cl − Cl−].
C), ν(CN))ar, 1455 (δ(CC), δ (CN))ar, 1160 δ (C–H)ip, 703 δ (C–H)oop. MS (ESI): m/z (%) 295.2 (100.0%) [C18H23N4Cl − Cl−].
1H-NMR: (CDCl3, 400 MHz, 298 K) δ: 2.00 [s, 3H, H6 or H7], 2.02 [d, 3H, 3J = 7.0 Hz, H16], 2.16 [s, 3H, H6 or H7], 4.55 [t, 2H, 3J = 5.0 Hz, H9], 5.01 [t, 2H, 3J = 5.0 Hz, H8], 5.65 [q, 1H, 3J = 7.0 Hz, H15], 5.71 [s, 1H, H4], 6.77, 6.90 [2d, 1H each one, 3J = 2.0 Hz, H13, H14], 7.31–7.42 [2 m, 5H, H18–H22]. 13C{1H}-NMR: (CDCl3, 100 MHz, 298 K) δ: 10.7, 13.5 [C6, C7], 21.2 [C16], 48.0 [C9], 49.2 [C8], 60.2 [C15], 105.6 [C4], 120.4, 123.0 [C13, C14], 126.8, 129.5, 137.8 [C17–C22], 140.5, 148.8 [C3, C5], signal for [C11] was not observed. IR (ATR) cm−1: 3033, 2960 ν(C–H), 1552 (ν(CC), ν(CN))ar, 1454 (δ(CC), δ(CN))ar, 1015 δ(C–H)ip, 700 δ(C–H)oop.
Synthesis and characterization of the Pd(II) or Ag(I) complexes
Anal. calcd for C18H22AgClN4: C, 49.39; H, 5.07; N, 12.80. Found: C, 49.92; H, 5.14; N, 12.47. 1H-NMR: (CDCl3, 360 MHz, 298 K) δ: 1.77 [s, 3H, H6 or H7], 1.81 [br, 3H, H16], 2.18 [s, 3H, H6 or H7], 4.29 [br, 2H, H9], 4.54 [br, 2H, H8], 5.68 [s, 1H, H4], 5.76 [br, 1H, H15], 6.43, 6.80 [2br, 1H each one, H13, H14], 7.23, 7.32 [2br, 5H, H18–H22]. 13C{1H}-NMR: (CDCl3, 91 MHz, 298 K) δ: 10.6, 13.7 [C6, C7], 21.4 [C16], 49.3 [C9], 51.9 [C8], 60.9 [C15], 105.5 [C4], 118.5, 122.2 [C13, C14], 126.6, 128.7, 129.2, 139.7 [C17–C22], 140.2, 148.9 [C3, C5], signal for [C11] was not observed. IR (ATR) cm−1: 3087, 2977 ν(C–H), 1551 (ν(CC), ν(CN))ar, 1448 (δ(CC), δ(CN))ar, 1219 δ(C–H)ip, 700 δ(C–H)oop. MS (ESI): m/z (%) 401.1 (100.0%) [C18H22N4ClAg − Cl−].
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl ether 4:1 as the eluent and then by second column chromatography (silica gel 60 Å) using dichloromethane:hexane 9:1 as the eluent. The product was obtained as a pale yellow powder. Yield: 133 mg, 51%.
:diethyl ether 4:1 as the eluent and then by second column chromatography (silica gel 60 Å) using dichloromethane:hexane 9:1 as the eluent. The product was obtained as a pale yellow powder. Yield: 133 mg, 51%.
1H-NMR: (CDCl3, 600 MHz, 298 K) δ: 1.61 [s, 3H, H6 or H7], 2.02 [br, 3H, H16], 2.70 [s, 3H, H6 or H7], 5.71 [s, 1H, H4], 5.87 [br, 2H, H9], 6.27 [br, 2H, H8], 6.08, 6.55 [2br, 1H each one, H13, H14], 6.89 [br, H15], 7.39, 7.62 [2br, 5H, H18–H22]. 13C{1H}-NMR: (CDCl3, 150 MHz, 298 K) δ: 10.9, 14.5 [C6, C7], 19.5 [C16], 50.1 [C9], 51.1 [C8], 59.2 [C15], 106.4 [C4], 118.5, 123.4 [C13, C14], 127.4, 128.5, 128.8 [C17–C22], 139.6, 144.1 [C3, C5], signal for [C11] was not observed. IR (ATR) cm−1: 3097, 2978 ν(C–H), 1555 (ν(CC), ν(CN))ar, 1452 (δ(CC), δ(CN))ar, 1180 δ(C–H)ip, 694 δ(C–H)oop. HRMS (ESI): m/z 399.0808 [C18H22N4Cl2Pd − Cl− − HCl].
Anal. calcd for C18H23N4Cl3Pd: C, 42.54; H, 4.56; N, 11.02. Found: C, 42.03; H, 4.62; N, 10.76. 1H-NMR: (DMSO-d6, 360 MHz, 298 K) δ: 1.80 [d, 3H, 3J = 7.0 Hz, H16], 1.98 [s, 3H, H6 or H7], 2.01 [s, 3H, H6 or H7], 4.38 [t, 2H, 3J = 5.0 Hz, H9], 4.54 [t, 2H, 3J = 5.0 Hz, H8], 5.75 [s, 1H, H4], 5.79 [br, 1H, H15], 7.32, 7.40 [2 m, 5H, H18–H22], 7.64, 7.83 [2br, 1H each one, H13, H14], 9.03 [s, 1H, H11]. 13C{1H}-NMR: (DMSO-d6, 91 MHz, 298 K) δ: 10.2, 13.3 [C6, C7], 20.6 [C16], 47.1 [C9], 48.9 [C8], 58.5 [C15], 105.0 [C4], 121.3, 123.2 [C13, C14], 126.4, 128.7, 129.0, 139.4 [C17–C22], 135.8 [C11], 139.4, 146.9 [C3, C5]. IR (ATR) cm−1: 3091, 2984 ν(C–H), 1555 (ν(CC), ν(CN))ar, 1453 (δ(CC), δ(CN))ar, 1152 δ(C–H)ip, 704 δ(C–H)oop. MS (ESI): m/z (%) 399.1 (100.0%) [C18H23N4Cl3Pd − Cl− − 2HCl], 437.1 (26.5%) [C18H23N4Cl3Pd − Cl− − HCl], 473.0 (25.7%) [C18H23N4Cl3Pd − Cl−].
Synthesis of Pd/L and Pd/HLCl nanoparticles
The general procedure for the preparation of palladium nanoparticles is detailed through the case of [L]/[Pd] = 0.5 (Fig. 2a and Scheme 1). The procedure was similar for all other samples.[Pd2(dba)3] (200 mg, 0.22 mmol) and L (28.6 mg, 0.11 mmol) were dissolved in a Fisher-Porter reactor in previously degassed anhydrous dichloromethane (200 mL) under argon at 196 K. The mixture was pressurized under 3 bar of dihydrogen and kept at room temperature under vigorous stirring. The color of the solution turned from purple to black after 1 h. The hydrogen pressure and the temperature were maintained for 18 h. After that period of time, the colloidal solution was black and homogeneous. Hydrogen was evacuated and a drop of the crude colloidal solution was deposited under argon on a holey carbon-covered copper grid using filter paper under the grid for TEM and SEM analysis. Then, the colloidal solution was concentrated to ca. 10 mL. The addition of cold pentane (20 mL) allowed the precipitation of the particles as a black solid, which was washed with pentane (3 × 20 mL) and dried under reduced pressure. The filtered pentane was slightly yellow due to dba elimination. This was corroborated by 1H-NMR experiments of the dried pentane solution. N1 Anal. found (wt%): Pd, 66.0; N, 5.34 for a stoichiometry of Pd561L37. IR (ATR, cm−1): 3026, 2939 ν(C–H), 1564 (ν(CC), ν(CN))ar, 1462 (δ(CC), δ (CN))ar, 700 δ (C–H)oop. N2: anal. found (wt%): Pd, 83.0; N, 2.90 for a stoichiometry of Pd1415HLCl93. IR (KBr, cm−1): 3019, 2960 ν(C–H), 1563 (ν(CC), ν(CN))ar, 1456 (δ(CC), δ(CN))ar, 1137 δ(C–H)ip.
Crystal structure determination of C2
Crystallographic data for compound C2 were collected at low temperature (180 K) on a Bruker Kappa Apex II diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) and equipped with an Oxford Cryosystems Cryostream Cooler device. Crystallographic data for C2 can be gathered in the ESI, Tables S1 and S2.†The structures have been solved by Direct Methods using SIR92,33 and refined by means of least-squares procedures on F2 with the aid of the program SHELXL9734 included in the software package WinGX version 1.63.35 The atomic scattering factors were taken from International tables for X-ray crystallography.36,37 All hydrogen atoms were placed geometrically, and refined by using a riding model. All non-hydrogen atoms were anisotropically refined, and in the last cycles of refinement a weighing scheme was used, where weights are calculated from the following formula: w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2 + 2Fc2)/3.
Crystallographic data for the structural analyses have been deposited at the Cambridge Crystallographic Data Centre, CCDC, with reference number 1432563 (C2).†
Catalytic experiments
The quantification of the catalytic reactions was carried out in an HP5890 Hewlett Packard gas chromatograph equipped with a FID detector and an HP-5 column (5% diphenylpolysiloxane and 95% dimethylpolysiloxane). The products obtained in the catalytic reactions were identified using a G1800A Hewlett Packard gas chromatograph with an electron impact ionization detector and a HP-5 column (5% diphenylpolysiloxane and 95% dimethylpolysiloxane). The mass spectra of the catalytic products are in agreement with those published in the literature.38,39Suzuki–Miyaura reactions
In a two-neck round-bottom flask fitted with a reflux condenser and a septum, 4-halogenotoluene (2.5 mmol), phenylboronic acid (3.125 mmol), tBuOK (5.0 mmol), and naphthalene (0.5 mmol) as internal standards were dissolved in DMF/H2O (10 mL, 4/1). Next, the palladium organometallic complex (1 × 10−3 mmol) or palladium nanoparticles (1 × 10−3 mmol Pd atoms) were added. The solution was vigorously stirred and heated at 100 °C for 6 h under nitrogen. Then, the reaction crude was cooled to room temperature and the products were extracted with a mixture of diethyl ether/brine (20 mL, 1/1). The organic phase was analyzed by GC and GC-MS.Conclusions
A new hybrid pyrazole-imidazol-2-ylidene ligand, 1-[2-(3,5-dimethylpyrazol-1-yl)ethyl]-3-((S)-1-phenylethyl)-3H-imidazol-2-ylidene (L), has been synthesized for the first time and proved to effectively stabilize small and isolated palladium(0) nanoparticles. A comparison with its counterpart HLCl as a stabilizer led to badly stabilized NPs which evidenced that the coordination of the ylidene group is a key factor in the stabilization process of the colloidal system. The successful preparation and complete characterization of molecular Pd(II) coordination compounds with the same ligands confirmed a chelated coordination mode through the pyrazolic nitrogen and the ylidene group for L but a terminal monodentate mode through pyrazolic nitrogen for HLCl. From our previous research with carbene or pyrazole-containing ligands as stabilizers, we can conclude that similar coordination modes of L and HLCl here take place for both the Pd NP particles and Pd complexes respectively. As a consequence, the coordination of L is stronger and leads to nanoparticles with well-controlled size.All these systems have been tested in catalytic C–C coupling reactions. For colloidal systems (N1 and N2), we observed an improving effect, in terms of chemoselectivity and yield, of L containing NPs (N1) with respect to HLCl containing NPs (N2) as a result of the different coordination modes of the carbene group to the surface of the NPs. Interestingly, these systems are the only ones able to achieve the C–C homocoupling reaction between two molecules of bromoarenes or the complete dehalogenation reaction of iodoarenes. The Suzuki–Miyaura reaction is favored with the Pd molecular complexes.
Taking into account the advantages of the organometallic approach and the different attributes of these catalysts, we believe that these materials could find practical uses in C–C coupling reactions in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from MINECO (CTQ2015-64261-R and CTQ2015-64436-P) is gratefully acknowledged. F. G. V. and J. DeT. acknowledge the Universitat Autònoma de Barcelona for their pre-doctoral grants. J. G.-A. acknowledges the Serra Húnter Program. The authors thank the Microscopy Service of the Universitat Autònoma de Barcelona for technical assistance with TEM and HR-TEM. CNRS is also thanked through the LTPMM-LEA 368 action.Notes and references
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) F. Glorius, Top. Organomet. Chem., 2006, 21, 1–20 CrossRef; (c) J. A. M. Lummiss, C. S. Higman, D. L. Fyson, R. McDonald and D. E. Fogg, Chem. Sci., 2015, 6, 6739–6746 RSC.
- N. Hadei, E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Org. Lett., 2005, 7, 1991 CrossRef CAS PubMed.
- (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed; (b) Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- (a) A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed; (b) P. Lara, O. Rivada-Wheelaghan, S. Conejero, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS PubMed; (c) P. Lara, A. Suárez, V. Colliere, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87–90 CrossRef CAS; (d) J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 46, 7230–7232 RSC; (e) E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837–1840 RSC; (f) D. González-Gálvez, P. Lara, O. Rivada-Wheelaghan, S. Conejero, B. Chaudret, K. Philippot and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 99–105 RSC; (g) E. A. Baquero, S. Tricard, J. C. Flores, E. de Jesús and B. Chaudret, Angew. Chem., Int. Ed., 2014, 53, 13220–13224 CrossRef CAS PubMed; (h) A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414–5420 CrossRef CAS; (i) L. M. Martínez-Prieto, A. Ferry, P. Lara, C. Richter, K. Philippot, F. Glorius and B. Chaudret, Chem. – Eur. J., 2015, 21, 17495–17502 CrossRef PubMed; (j) C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204–3207 RSC; (k) L. M. Martínez-Prieto, A. Ferry, C. Richter, P. Lecante, K. Philippot, F. Glorius and B. Chaudret, Chem. Commun., 2016, 52, 4768–4771 RSC; (l) L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Cámpora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647–4650 RSC.
- M. Albrecht, Adv. Organomet. Chem., 2014, 62, 111–158 CrossRef CAS.
- (a) P. Lara, K. Philippot, L. M. Lacroix, S. Lachaize, N. Liakakos, K. Soulantica and B. Chaudret, Organometallic Nanoparticles, in Advances in Organometallic Chemistry: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, Wiley VCH, Weinheim, 2013, ch. 31, pp. 421–436 Search PubMed; (b) S. Kinayyigit and K. Philippot, Organometallic Approach for the Synthesis of Noble Metal Nanoparticles: Towards Application in Colloidal and Supported Nanocatalysis, in Metal Nanoparticles for Catalysis: Advances and Applications, ed. T. Tao, RSC, 2014, ch. 4, pp. 47–82 Search PubMed; (c) K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1442 CrossRef CAS; (d) N. J. Costa, M. Guerrero, V. Colliere, E. Teixeira-Neto, R. Landers, K. Philippot and L. M. Rossi, ACS Catal., 2014, 4, 1735–1742 CrossRef CAS.
- C. Amiens, D. Ciuculescu-Pradines and K. Philippot, Coord. Chem. Rev., 2016, 38, 409–432 CrossRef.
- (a) K. Philippot and P. Serp, Concepts in Nanocatalysis, in Nanomaterials in Catalysis, ed. P. Serp and K. Philippot, Wiley-VCH, Weinheim, 2013, ch. 1, pp. 1–54 Search PubMed; (b) T. Ayvali and K. Philippot, On the use of Organometallic Concepts for the Synthesis of Nanocatalysts, in New Materials for Catalytic Applications, ed. E. Kemnitz and V. Parvulescu, Elsevier, 2016, ch. 3, pp. 41–79 Search PubMed; (c) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed; (d) M. Guerrero, N. T. T. Chau, S. Noel, A. Denicourt-Nowicki, F. Hapiot, A. Roucoux, E. Montflier and K. Philippot, Curr. Org. Chem., 2013, 17, 364–399 CrossRef CAS; (e) B. Chaudret, M. Gómez and K. Philippot, Top. Catal., 2013, 56, 1153 CrossRef CAS; (f) D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- (a) A. Kolmakov, X. Chen and M. J. Moskovits, Nanosci. Nanotechnol., 2008, 8, 111–121 CrossRef CAS; (b) M. Guerrero, N. J. S. Costa, L. L. R. Vono, L. M. Rossi, E. V. Gusevskaya and K. Philippot, J. Mater. Chem. A, 2013, 1, 1441–1449 RSC.
- A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC.
- S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS.
- A. Ganesan, Drug Discovery Today, 2001, 6, 238 CrossRef CAS PubMed.
- D. Astruc, C. Ornelas, A. K. Diallo and J. Ruiz, Molecules, 2010, 15, 4947–4960 CrossRef CAS PubMed.
- D. Peral, F. Gómez-Villarraga, X. Sala, J. Pons, J. C. Bayon, J. Ros, M. Guerrero, L. Vendier, P. Lecante, J. García-Antón and K. Philippot, Catal. Sci. Technol., 2013, 3, 475–489 CAS.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374–3401 RSC.
- J. Deng, H. Gao, F. Zhu and Q. Wu, Organometallics, 2013, 32, 4507–4515 CrossRef CAS.
- T. L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1992, 114, 3157–3159 CrossRef CAS.
- A. D. Bain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef CAS.
- Z. A. Savel'eva, L. A. Glinskaya, S. A. Popov, R. F. Klevtsova, A. V. Tkachev and S. V. Larionov, Russ. J. Coord. Chem., 2009, 35, 668–673 CrossRef.
- H. M. Lee, P. L. Chiu, C.-H. Hu, C.-L. Lai and Y.-C. Chou, J. Organomet. Chem., 2005, 690, 403–414 CrossRef CAS.
- G. Aragay, J. Pons, J. García-Antón, X. Solans, M. Font-Bardia and J. Ros, J. Organomet. Chem., 2008, 693, 3396–3404 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 36, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981–1992 CrossRef CAS.
- (a) P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed; (b) R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2004, 108, 8572–8580 CrossRef CAS; (c) R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed; (d) A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 10473–10482 RSC; (e) C. Sánchez-Sánchez, N. Orozco, J. P. Holgado, S. K. Beumont, G. Kyriakou, D. J. watson, A. R. González-Eripe, L. Feria, J. Fernández-Sanz and R. M. Lambert, J. Am. Chem. Soc., 2014, 137, 940–947 CrossRef PubMed; (f) V. K. Kanuru, G. Kyriakou, S. K. Beaumont, A. C. Papageorgiou, D. J. Watson and R. M. Lambert, J. Am. Chem. Soc., 2010, 132, 8081–8086 CrossRef CAS PubMed; (g) C. Sánchez-Sánchez, F. Yubero, A. R. González-Elipe, L. Feria, J. Fernández-Sanz and R. M. Lambert, J. Phys. Chem. C, 2014, 118, 11677–11684 CrossRef; (h) F. Wang, C. Li, L.-D. Sun, H. Wu, T. Ming, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 1106–1111 CrossRef CAS PubMed; (i) F. Wang, C. Li, H. Chen, R. Jiang, L.-D. Sun, Q. Li, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- (a) Z. Niu, Q. Peng, Z. Zhuang, W. He and Y. Li, Chem. – Eur. J., 2012, 18, 9813–9817 CrossRef CAS PubMed; (b) D. B. Pacardo, J. M. Slocik, K. C. Kirk, R. R. Naik and M. R. Knecht, Nanoscale, 2011, 3, 2194–2201 RSC; (c) A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Chem. – Eur J., 2007, 13, 6908–6913 CrossRef CAS PubMed; (d) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–168 CrossRef CAS; (e) J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J. P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staud, Organometallics, 1985, 4, 1819–1830 CrossRef CAS.
- R. L. Oliveira, W. He, R. J. M. Klein Gebbink and K. P. de Jong, Catal. Sci. Technol., 2015, 5, 1919–1928 CAS.
- S. Komiya, in Synthesis of Organometallic Compounds: A Practice Guide, Board, New York, 1997 Search PubMed.
- D. Drew and J. R. Doyle, in Inorganic Syntheses, 28 (Reagents Transition Met. Complex Organomet. Synth.), 1990, pp. 346–349 Search PubMed.
- H. Konishi, T. Ueda, T. Muto and K. Manabe, Org. Lett., 2012, 14, 4722–4725 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- T. Hahn, in International Tables for Crystallography, Volume A, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- A. J. C. Wilson, in International Tables for Crystallography, Volume C, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- X. Xu, D. Cheng and W. Pei, J. Org. Chem., 2006, 71, 6637–6639 CrossRef CAS PubMed.
- I. J. S. Fairlamb, A. R. Kapdi and A. F. Lee, Org. Lett., 2004, 6, 4435–4438 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1432563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02729j |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |