
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin-state-correlated optical properties of copper(II)–nitroxide based molecular magnets†
Irina Yu.
Barskaya
a,
Sergey L.
Veber
 *ab,
Elizaveta A.
Suturina
b,
Peter S.
Sherin
ab,
Kseniya Yu.
Maryunina
c,
Natalia A.
Artiukhova
a,
Evgeny V.
Tretyakov
a,
Renad Z.
Sagdeev
a,
Victor I.
Ovcharenko
a,
Nina P.
Gritsan
bd and
Matvey V.
Fedin
*ab
*ab,
Elizaveta A.
Suturina
b,
Peter S.
Sherin
ab,
Kseniya Yu.
Maryunina
c,
Natalia A.
Artiukhova
a,
Evgeny V.
Tretyakov
a,
Renad Z.
Sagdeev
a,
Victor I.
Ovcharenko
a,
Nina P.
Gritsan
bd and
Matvey V.
Fedin
*ab
aInternational Tomography Center SB RAS, 630090 Novosibirsk, Russia. E-mail: sergey.veber@tomo.nsc.ru; mfedin@tomo.nsc.ru
bNovosibirsk State University, 630090 Novosibirsk, Russia
cGraduate School of Science and Center for Chiral Science, Hiroshima University, 1-3-1 Kagamiyama, 739-8526, Higashi-Hiroshima, Japan
dInstitute of Chemical Kinetics and Combustion SB RAS, 630090 Novosibirsk, Russia
First published on 5th September 2017
Abstract
Molecular magnets based on copper(II) ions and stable nitroxide radicals exhibit promising switchable behavior triggered by a number of external stimuli; however, their spin-state-correlated optical properties vital for photoinduced switching have not been profoundly investigated to date. Herein, the electronic absorption spectra of single crystals of three representatives of this unique family are studied experimentally and theoretically in the visible and near-IR regions. We established that the color of the complexes is mainly determined by optical properties of the nitroxide radicals, whereas the Cu(hfac)2 fragment contributes to the near-IR range with the intensity smaller by an order of magnitude. The thermochromism of these complexes evident upon thermal spin state switching is mainly caused by a spectral shift of the absorption bands of the nitroxides. The vibrational progression observed in the visible range for single crystals as well as for solutions of pure nitroxides is well reproduced by DFT calculations, where the C–C stretching mode governs the observed progression. The analysis of the spectra of single crystals in the near-IR region reveals changes in the energy and in the intensity of the copper(II) d–d transitions, which are well reproduced by SOC-NEVPT2 calculations and owe to the flip of the Jahn–Teller axis in the coordination environment of copper. Further strategies for designing bidirectional magnetic photoswitches using these appealing compounds are discussed.
1. Introduction
The development of functional stimuli-responsive molecular materials is a hot topic in modern science. Bringing electronic devices and computer technologies to a new level requires principally new building blocks, and switchable magnetoactive compounds are among the promising candidates in this regard. To date, a large number of such compounds that exhibit spin crossover (SCO) or valence tautomerism and are sensitive to various external stimuli (temperature, pressure, light, etc.) have been developed.1–4 Special attention was drawn to materials with magnetic states operated by light because this type of manipulation is the most suitable for applications in spintronics and nanoelectronics. Therefore, the understanding of optical properties of such materials and their light-driven changes is of crucial importance.Functional properties of SCO compounds have been intensively investigated over the past few decades.5–21 In particular, thermochromism was found in most of the compounds: e.g., iron-based compounds, which as a rule are intensively colored at low temperatures but switch to a colorless state at high temperatures upon spin transition. More importantly, a Light-Induced Excited Spin State Trapping (LIESST) phenomenon has been found, which involves (i) photoswitching between two spin states of the system and (ii) metastability of the photoinduced (low-spin) state at cryogenic temperatures.5,18,22,23 Later, more LIESST-related phenomena have been found and exploited for the development of photosensitive magnetoactive materials.24–28
In particular, SCO-like and LIESST-like phenomena were found in a relatively new family of polymer-chain compounds based on copper(II) ions and stable nitroxide radicals.28–37 The spin state of the whole copper(II)–nitroxide cluster can be changed depending on temperature, applied pressure or irradiation with light. The spins of copper(II) and nitroxides can either be strongly-coupled by antiferromagnetic exchange interaction (strongly-coupled spin state, SS), or they can be weakly-coupled by ferromagnetic exchange (weakly-coupled spin state, WS). As a rule, the SS state is found at low temperatures and corresponds to shorter distances of the copper(II)–nitroxide (nitroxide(s) are in equatorial coordination positions of the copper(II) ion) bond. In contrast, the WS state is typically found at high temperatures where the Jahn–Teller axis of the coordination octahedron flips and nitroxide(s) occupy axial positions. It is remarkable that despite the principal differences between SCO compounds and copper(II)–nitroxide based molecular magnets (often called “breathing crystals” for brevity), the manifestation of thermal transitions and LIESST-like phenomena is quite similar.11,12,28,32 However, contrary to the SCO compounds, optical properties of breathing crystals are significantly less understood. The UV/Vis absorption spectra are dominated by nitroxide radicals, whose absorption bands are more intense compared to d–d transitions of copper and metal-to-ligand charge transfer (MLCT) bands, complicating the interpretation of spectra and their spin-state dependent changes. In the case of SCO compounds, a suitable choice of the excitation light wavelength allows both direct and reverse photoswitching.27 Similar functionality, unfortunately, has not yet been found for breathing crystals, and only SS → WS photoswitching has been achieved to date. One of the reasons could be more complicated UV/Vis spectra with significant overlap of the bands responsible for the direct and reverse switching. Although breathing crystals also exhibit pronounced thermochromism upon spin transitions,36,37 a deep understanding of optical properties and the corresponding electronic structure is currently missing.
In this work we report the first detailed experimental and theoretical study of UV/Vis–near-IR absorption spectra of breathing crystals, with the primary focus on the electronic structure of the copper(II)–nitroxide clusters and temperature dependence of their spectra upon magnetostructural transitions. Three representative compounds Cu(hfac)2LMe, Cu(hfac)2LEt-CP and Cu(hfac)2LPr showing either abrupt (former two) or gradual (latter one) magnetostructural transition were selected. The recording of the absorption spectra in the broad spectral range (200 to 2500 nm or 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 4000 cm−1) allowed experimental detection of all types of transitions from the high-energy π–π* and MLCT to low-energy d–d types. The assignment of these transitions was supported by quantum chemical calculations. The obtained data are of high importance for the following optimization of the direct and reverse switching of breathing crystals by light in order to control their spin states.
000 to 4000 cm−1) allowed experimental detection of all types of transitions from the high-energy π–π* and MLCT to low-energy d–d types. The assignment of these transitions was supported by quantum chemical calculations. The obtained data are of high importance for the following optimization of the direct and reverse switching of breathing crystals by light in order to control their spin states.
2. Experimental and computational details
2.1. Synthesis and sample preparation
Pyrazolyl-substituted nitronyl nitroxide radicals LMe, LEt-CP, and LPr and complexes Cu(hfac)2LMe, Cu(hfac)2LEt-CP and Cu(hfac)2LPr studied in this work were synthesized according to the previously developed procedures.37,38 Their structure and magnetic36–39 and optical (mid-IR)40 properties have also been investigated previously. The thin single crystals were obtained by boosting of the crystallization process to aid rapid precipitation. Note that, as was shown previously,40 the obtained thin crystals undergo magnetostructural transitions at the same temperature as polycrystalline powders and bigger single crystals. Benzene or ethanol was used as a solvent to prepare solutions for the UV/Vis spectra measurements. Cu(hfac)2 was preliminarily dried and stored in a desiccator; benzene was dried using molecular sieves.2.2. UV/Vis spectroscopy
000–10000 cm−1 using an infrared microscope HYPERION 2000 (Bruker Optics, Germany) equipped with an extension for the VIS spectral region. The microscope is coupled to the FTIR spectrometer Bruker Vertex 80v. A Q428/7 tungsten halogen lamp, T602/8 CaF2 NIR/Vis/UV beam splitter, and D510/3 Si diode detector were used to measure the spectra. The spectral resolution was 8 cm−1. The thickness of the crystals was ∼2–6 μm (according to the Vis–near-IR transparency level compared to the pellet sample with measured compound quantity), and the probed area of the crystals was ∼0.2 × 0.2 mm2, which is slightly smaller than the crystal size.
000–4000 cm−1 using the same configuration of the microscope and FTIR spectrometer except for the detector: a D316 liquid N2-cooled MCT detector was used instead of the D510/3 Si diode detector. The spectral resolution was 4 cm−1. The thickness of the crystals was ∼20–80 μm. A sample stage Linkam FTIR600 (Linkam Scientific Instruments, United Kingdom) equipped with BaF2 windows was used to control the temperature of single crystals in both Vis and near-IR experiments.
2.3. Fluorescence spectroscopy
Fluorescence emission and excitation spectra were measured with a FLSP920 (Edinburgh Instruments, UK) spectrofluorimeter equipped with a low-noise micro-channel plate photomultiplier detecting in the range of 250–850 nm. Liquid or solid samples were placed in a quartz tube (1.8/2.8 inner/outer diameter) and all fluorescence experiments were performed in the reflective mode with a cylindrical quartz Dewar vessel at room temperature or at 77 K. All fluorescence spectra were corrected for the wavelength-dependent sensitivity of the detection.2.4. Computational details
Geometry optimizations were performed using the pure BP8641,42 or hybrid B3LYP functionals with the Ahlrichs polarized def2-TZVP43–45 basis set and Grimme's dispersion correction D3.46,47 The auxiliary basis set def2-TZVP/J was used in conjunction with the resolution of identity approximation.48The time-dependent (TD) DFT49 at the UB3LYP/def2-TZVP level was employed to calculate the electronic absorption spectrum of nitroxide LMe. The vibrational progression in the absorption spectrum was computed using the Franck–Condon and Heller approximations with the “orca asa” module50 as implemented in the ORCA51 program package. In this approach the energy of a vertical electron excitation (E0) is computed by time-dependent DFT, and the excited state energy gradient is estimated numerically with respect to each totally symmetric vibrational mode. The Huang–Rhys dimensionless parameter S (S = α(ΔQ)2/2 with ΔQ being dimensionless displacement of the excited state minimum along the normal mode, α = Mω/ħ and the reduced mass of the molecule M) is computed from the gradient under the assumption that the excited state has the same Hessian as the ground state (Fig. 1). The Huang–Rhys parameter defines the relative intensities of the transitions in the progression at zero-temperature with the corresponding Franck–Condon factors for a particular vibration:
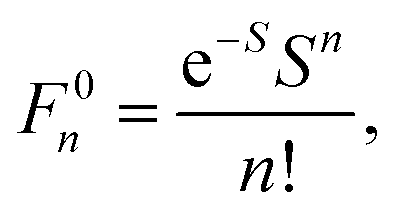 | (1) |
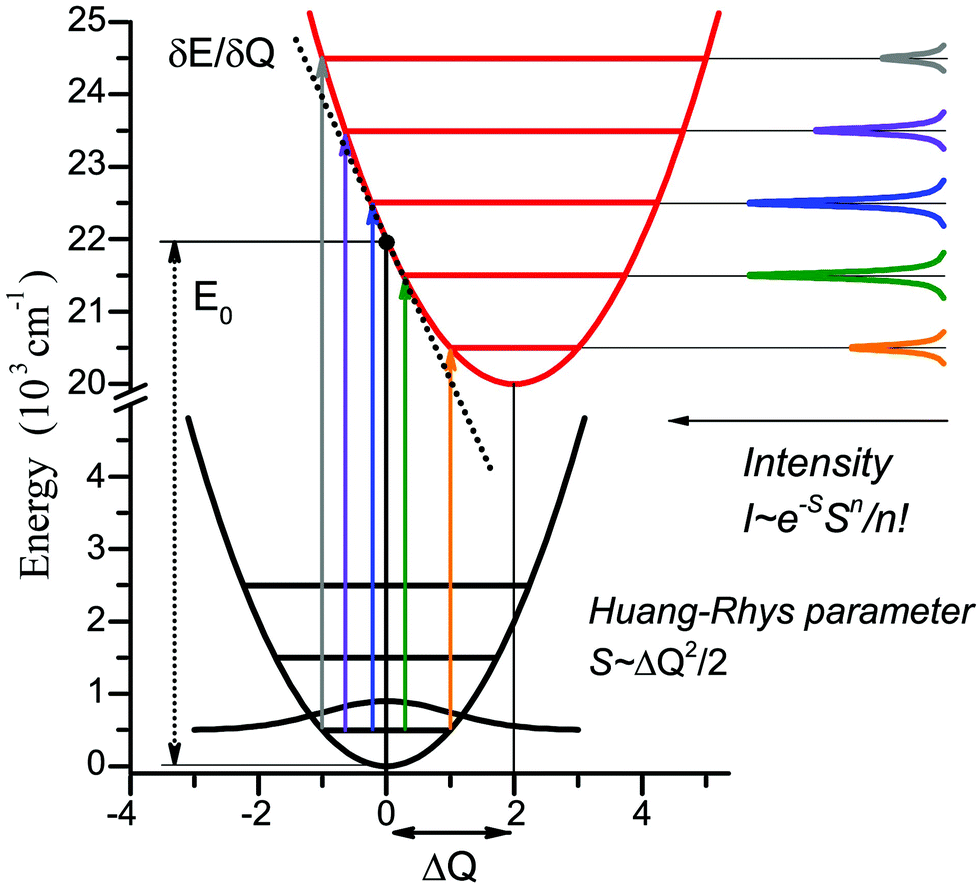 | ||
| Fig. 1 Illustration of the theoretical approach to the vibrational progression calculation for the vertical excitation of 22000 cm−1, normal mode frequency 1000 cm−1, and Huang–Rhys parameter S = 2. | ||
The TD-DFT approach with several functionals such as double-hybrid B2PLYP,54 long range corrected hybrid functional wB97x55,56 was employed to analyze charge transfer (CT) transitions in the electronic absorption spectrum of Cu(hfac)2.
To calculate positions and oscillator strengths of the d–d transitions in the Cu(hfac)2LMe complex, the state-averaged complete active space self-consisted field (SA-CASSCF)57–59 approach together with the second order N-electron valence perturbation theory (NEVPT2)60–63 were also used. The scalar relativistic effects were taken into account using a standard second-order Douglas–Kroll–Hess (DKH2) procedure.64 Relativistically reconstructed versions of the def2-TZVP basis sets65 were used in the calculations. Spin–orbit coupling was taken into account using the spin–orbit mean-field approximation (SOMF)66 as implemented in the ORCA package. The active space was chosen to contain five 3d orbitals. To simplify calculations, the LMe radicals were substituted by the corresponding hydroxylamines. The X-ray geometries of the complex at low (110 K) and high (240 K) temperatures were used in the calculations. The ORCA program package was employed for all calculations.
The NBO (Natural Bond Orbitals) analysis67,68 of the bonding interaction between Cu(II) and nitroxide has been performed using the NBO6 program.69 The strength of donor–acceptor interactions in the case of NBO analysis is determined by the second-order perturbation energy.
3. Results and discussion
3.1. Chemical structure and magnetic properties of compounds under study
We investigated three polymer-chain complexes Cu(hfac)2LPr, Cu(hfac)2LMe and Cu(hfac)2LEt-CP where the structures of Cu(hfac)2, LPr, LMe and LEt-CP fragments are shown in Fig. 2a.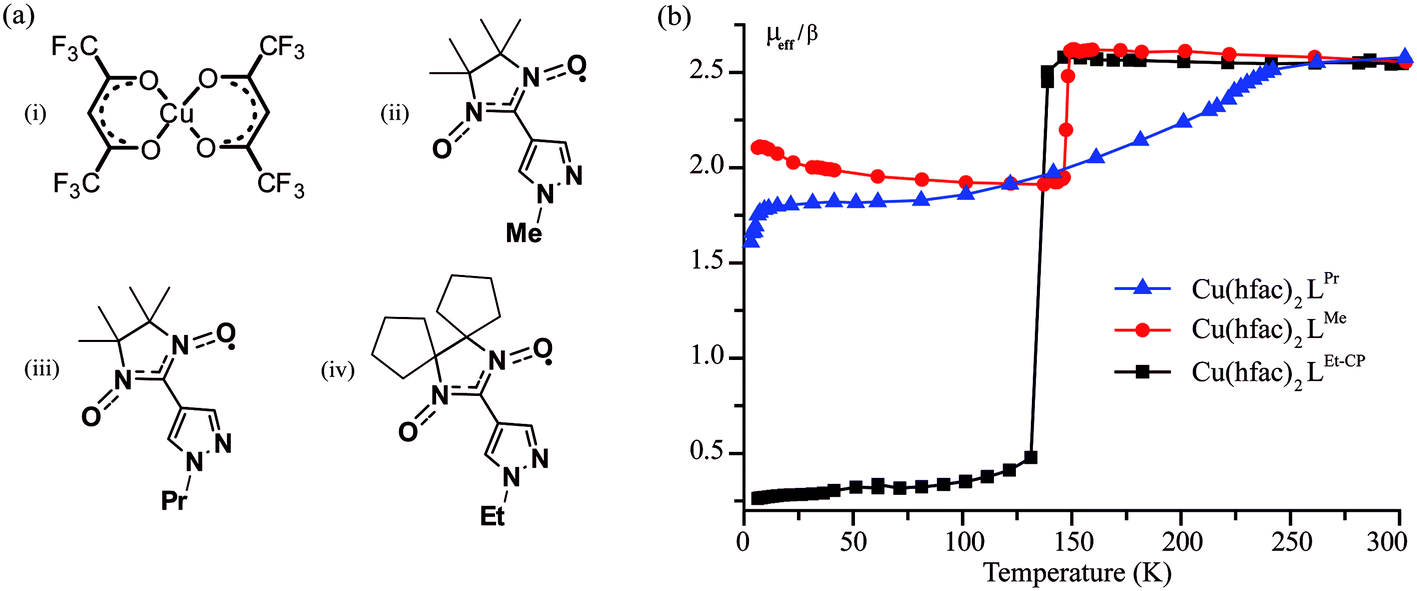 | ||
| Fig. 2 (a) Chemical structures of Cu(hfac)2 (i), LMe (ii), LPr (iii) and LEt-CP (iv). Hydrogen atoms are omitted for clarity. (b) The temperature dependence of the effective magnetic moment of studied compounds. | ||
The complex Cu(hfac)2LPr has a “head-to-head” polymer chain motif, i.e. the chains are formed by two alternating units: three-spin exchange-coupled nitroxide-copper(II)–nitroxide clusters and one-spin magnetically-isolated copper(II) unit. The complexes Cu(hfac)2LMe and Cu(hfac)2LEt-CP have a “head-to-tail” polymer chain motif, i.e. their chains are formed by two-spin exchange-coupled clusters of copper(II)–nitroxide. Structural and magnetic properties of these compounds, including their spin-crossover-like behavior, were studied in detail in our previous works.24,36–38 Temperature-induced magnetostructural transition in Cu(hfac)2LPr occurs in a gradual manner (Fig. 2b) and involves only three-spin exchange-coupled nitroxide-copper(II)–nitroxide clusters.39 In Cu(hfac)2LMe36,38,39 and Cu(hfac)2LEt-CP37 the transition is abrupt (T↓ = 141 K and 125 K, respectively) and occurs in two-spin exchange-coupled clusters. The main difference between Cu(hfac)2LMe and Cu(hfac)2LEt-CP is that only a half of the copper(II)–nitroxide clusters undergo the magnetostructural transition in Cu(hfac)2LMe, whereas in Cu(hfac)2LEt-CP all the clusters change their spin state (Fig. 2b).
Before we focus on the spectroscopic properties of studied polymer-chain complexes, it is useful at first to analyze the properties of their building blocks.
3.2. UV/Vis, fluorescence emission and excitation spectra of LMe
The room temperature absorption UV/Vis spectrum of the LMe ligand in ethanol features three distinct bands (Fig. 3a), which are well reproduced by the band-shaped TD-DFT calculations (Fig. 3b). The corresponding spectra of two other ligands LPr and LEt-CP are very similar to LMe and are shown in the ESI (Fig. S1†).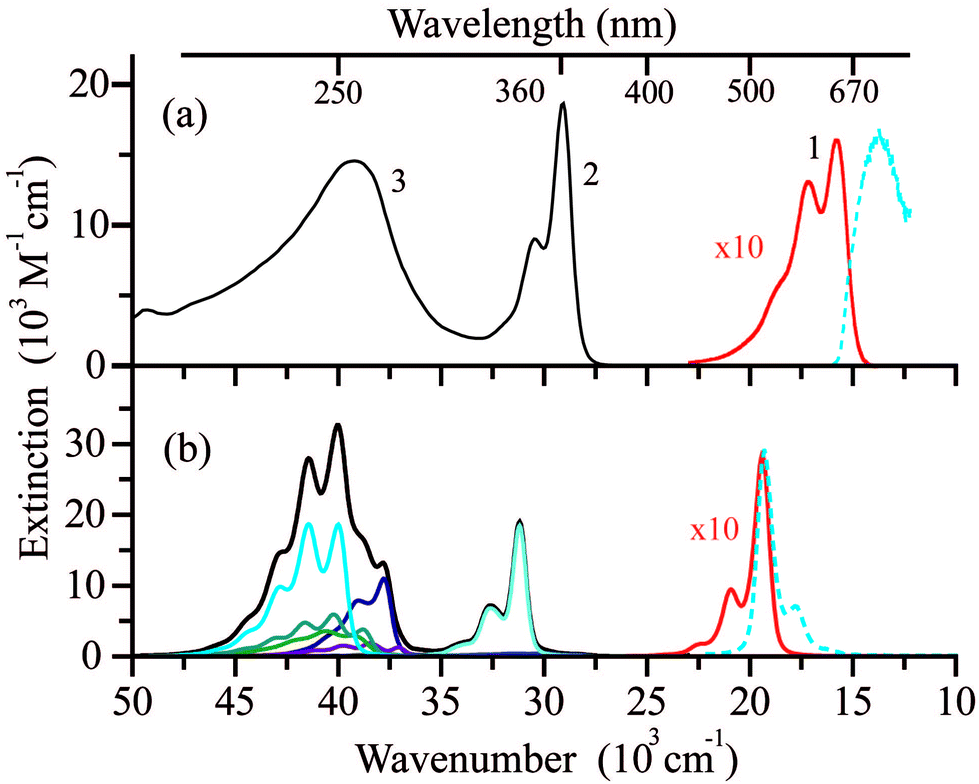 | ||
| Fig. 3 (a) The electronic absorption spectrum of LMe in ethanol at room temperature (black) with an intensity of band (1) increased by a factor of 10 (red) and fluorescence emission spectrum (dashed blue) of LMe in a 1:1 methanol/ethanol (v/v) mixture at 77 K and λex = 580 nm. (b) Electronic absorption/emission spectra of LMe (black curve) computed using the results of TD-UB3LYP/def2-TZVP calculations (colored curves represent individual electronic transitions) with the intensity of the lowest-energy band increased by a factor of 10 (red). | ||
The lowest-energy band (1) in the spectral range (15–20) × 103 cm−1 (εmax ∼ 1600 M−1 cm−1) is responsible for the blue color of the radical LMe and characterized by the structure with three maxima at ∼15800, 17200 and 18800 cm−1. According to the calculations, the band (1) corresponds to a single electronic transition that involves a sum of the electron promotions (a) from the highest occupied MO (HOMO) to the singly occupied MO (SOMO) and (b) from the SOMO to the lowest unoccupied MO (LUMO) as presented in Fig. 4a. The coupling of this electronic transition with the molecular vibration visualized in Fig. 4b leads to the observed structure of band (1). Note that the vibration presented in Fig. 4b corresponds mainly to the C–C stretching mode. The computed dimensionless displacement of the excited state minimum relative to the ground state along the mode ΔQ = −0.63 gives the Huang–Rhys parameter S = 0.2. According to eqn (1), such S value corresponds to the monotonous decrease of the intensity in the vibrational progression, where the second line for n = 1 is five times less intense than the n = 0 line. The visual comparison with experiment indicates that the Huang–Rhys parameter is underestimated by the theory. Other vibrations coupled to this electronic transition including vibrations of the O–N–C–N–O fragment are characterized by a smaller ΔQ parameter (see ESI Tables S1 and S2†) and their contribution to the observed vibronic progression is minor. The absorption band (1) (Fig. 3a) is similar to those observed for other nitroxyl radicals, which also feature vibrational progressions.70 However, for the unsubstituted nitronyl nitroxide radical there is an overlap of two electronic transitions with less resolved vibrational progression, whereas substituted radicals feature more pronounced vibrational progression due to the vibronic coupling with the C–C vibration of a linker.
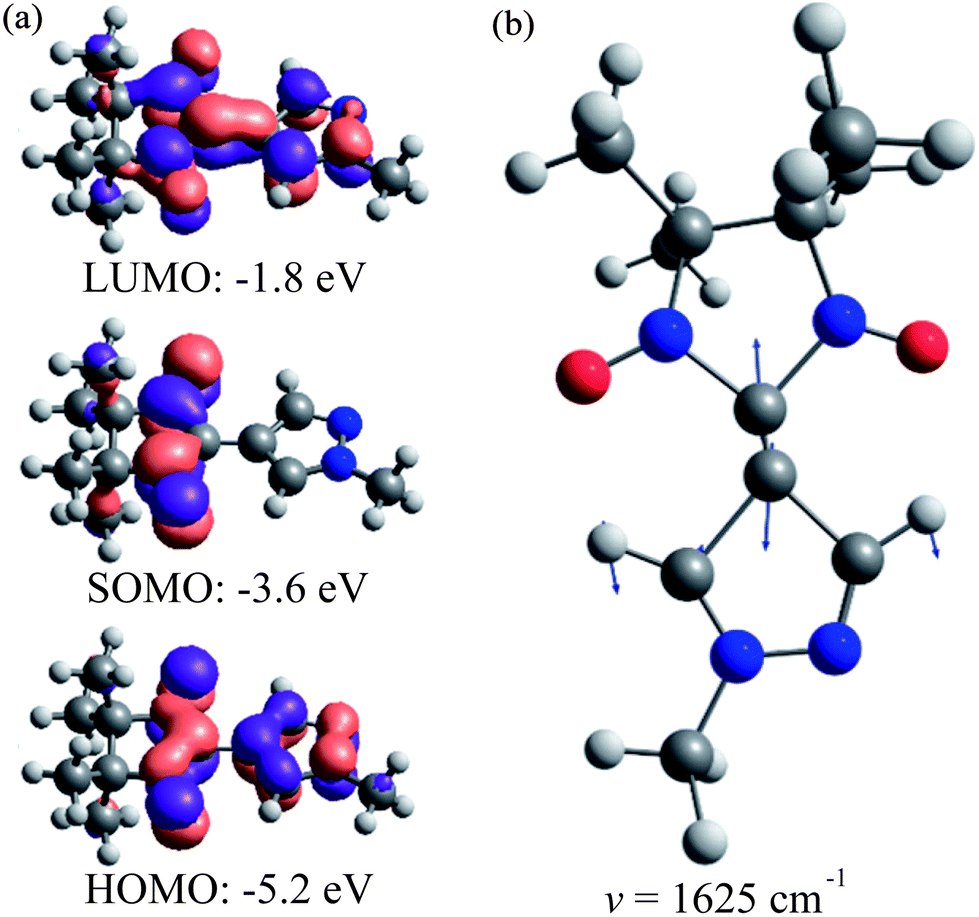 | ||
| Fig. 4 (a) LMe natural molecular orbitals involved in the electronic transition of band (1) calculated at the UB3LYP/def2-TZVP level and (b) LMe C–C stretching mode vibration (hν = 1625 cm−1) responsible for the observed vibrational progression (arrows’ lengths are proportional to the displacement amplitude). | ||
Absorption band (2) in the spectral range (27.5–32.5) × 103 cm−1 (εmax ∼ 18700 M−1 cm−1) also corresponds to a single electronic transition coupled to the same vibration (Fig. 4b). The nature of this electronic transition is similar to that of band (1), viz. a negative combination of (a) and (b) with some contributions of electron promotions from the lower-lying occupied MOs to the SOMO. In contrast to bands (1) and (2), the broad band (3) in the spectral range (35–45) × 103 cm−1 is composed of several overlapping bands with different shapes corresponding to individual electronic transitions (Fig. 3b).
The emission from the LMe solution could be observed only at 77 K (in a methanol/ethanol mixture, Fig. 3a). The same fluorescence emission spectra with maximum at ca. 730 nm were recorded upon excitation of LMe at different wavelengths over the whole absorption spectrum (Fig. S2, ESI†). Excitation spectra coincide with the absorption spectrum of LMe, regardless of the emission wavelength (Fig. S2, ESI†). There is a breakdown of the mirror image symmetry of absorption and fluorescence spectra of LMe (Fig. 3 and S2, ESI†). This effect is not related to the different experimental conditions as the low-temperature spectra are usually characterized by a more pronounced structure. Moreover, analogous spectral features were observed for the nitronyl nitroxide radical covalently linked with 2-pyrazolylquinoline,71 which exhibits detectable fluorescence at room temperature due to energy transfer from the pyrazolylquinoline moiety. Most likely, the structure-less fluorescence band (Fig. 3a) indicates a strongly anharmonic potential for some low-energy vibrational modes in the excited state.72 For the polycrystalline samples of the radicals LMe, LPr and LEt-CP, the broad fluorescence emission spectra with maxima at 750–850 nm (13.2–12.5 × 103 cm−1) were recorded at room temperature (Fig. S3, ESI†). In contrast, the blue-shifted (by about ∼1000 cm−1) and structured emission bands with characteristic intervals of about 200 cm−1 were detected at 77 K (Fig. S3, ESI†).
3.3. UV/Vis spectrum of Cu(hfac)2
Fig. 5a displays the electronic absorption spectrum of Cu(hfac)2 measured at 300 K in benzene (red). This spectrum consists of one intense band with the maximum at 32.5 × 103 cm−1 (313 nm) and a shoulder at 30 × 103 cm−1 (325 nm). In order to detect less intense d–d transitions expected for Cu(hfac)2, additional measurements were performed for the concentrated Cu(hfac)2 solution. As a result, a set of overlapping absorption bands covering the range from 500 to 700 nm was detected (Fig. 5a, red curve).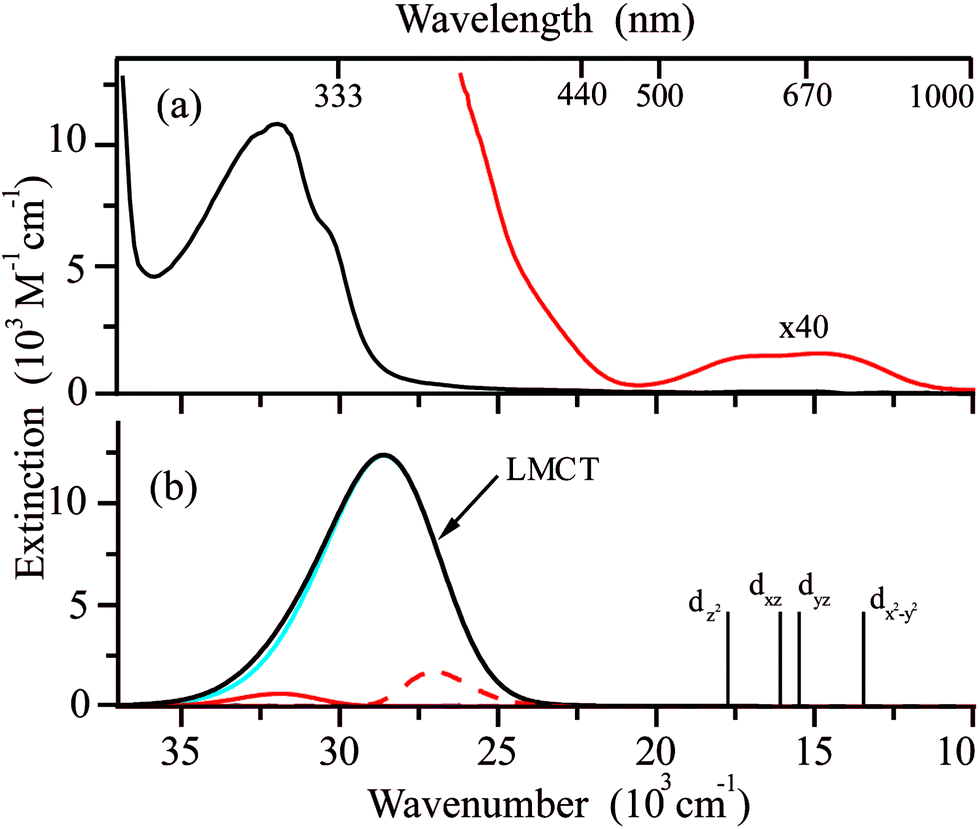 | ||
| Fig. 5 (a) The electronic absorption spectrum of Cu(hfac)2 in benzene at room temperature with the intensity of low energy bands increased by a factor of 40 (red). (b) The electronic absorption spectra of Cu(hfac)2 (black curve) computed using results of TD-B3LYP/def2-TZVP calculations (see section 2.4 for details). Blue and red curves represent individual electronic transitions. Positions of the d–d transitions (SOC-NEVPT2) are shown by vertical bars together with the SOMO orbitals of the corresponding excited states. | ||
According to the B3LYP calculations, the intense UV band at 28.6 × 103 cm−1 (353 nm) in the absorption spectrum of Cu(hfac)2 corresponds mostly to the single electronic transition involving electron promotion from the in-plane p-orbital of oxygen atoms onto the singly-occupied dxy orbital of the Cu ion (Fig. 5b, blue line). Thus, this band is a ligand-to-metal charge transfer (LMCT) band. The pure DFT functional usually significantly underestimates the energy of charge transfer transitions.49 Indeed, the calculated maximum of this band is significantly red shifted at the BP86 level (22.9 × 103 cm−1) as compared with the experimentally observed band. The use of a more advanced functional leads to the overestimated energy of this CT band (34.8 × 103 cm−1 and 37.7 × 103 cm−1 for calculations at the wB97x and B2PLYP levels, respectively). The band shape calculations were performed using TD-B3LYP results (Fig. 5b). Fig. 5 demonstrates that calculations reproduce well the shape of the LMCT band. The band is very broad due to large displacements along several modes (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 169 cm−1, ΔQ = 5.2 and = 88 cm−1, ΔQ = −5.5, see Fig. S4, ESI†). The linewidth for simulation was set to 100 cm−1 for Lorentzian shape and 200 cm−1 for Gaussian shape, and the broadening due to the population of the excited vibrational levels is also considered at T = 300 K. As the vibrational quanta for these modes are smaller than the line broadening, the progression is not resolved.
= 169 cm−1, ΔQ = 5.2 and = 88 cm−1, ΔQ = −5.5, see Fig. S4, ESI†). The linewidth for simulation was set to 100 cm−1 for Lorentzian shape and 200 cm−1 for Gaussian shape, and the broadening due to the population of the excited vibrational levels is also considered at T = 300 K. As the vibrational quanta for these modes are smaller than the line broadening, the progression is not resolved.
The Cu(hfac)2 complex dissolved in benzene has very weak d–d absorption bands which are characterized by extinction coefficients of ∼30 M−1 cm−1 that is typical for Cu(II) chelates in both the solid state73 and solution.74 Positions of the d–d electronic transitions predicted at the SOC-CASSCF(9,5)/NEVPT2 level agree very well with the broad low-energy experimental absorption bands. The ab initio calculations predict the following order of the d-orbitals: dz2 < dxz < dyz < dx2−y2 < dxy (see Table S3, ESI†). The Cu(hfac)2 complex dissolved in benzene exhibits very low emission, which could not be detected even with the highly sensitive photomultiplier used in our experiments. This indicates highly efficient internal conversion from the LMCT state to the lower energy d–d excited states.
3.4. Electronic absorption spectra of Cu(hfac)2 complexes with nitroxide radicals. Vis–near-IR spectroscopy of single crystals
The Vis–near-IR spectra of single crystals of Cu(hfac)2LR complexes (LPr, LMe and LEt-CP) measured at high temperatures (in the WS state) closely resemble the spectra of the ligands: a set of the observed bands is attributed to the vibrational progression with the most intense band centered at ∼650 nm (Fig. 6). The d–d transitions of Cu(II) ions, which are expected in the near-IR range, are almost not manifested at high temperatures. Thus, the color of the complexes at high temperatures is mainly determined by the color of the corresponding ligands. The lowering of the temperature does not influence the spectra significantly until the temperature of the magnetostructural transition is reached. At this point, the absorption spectra change drastically in agreement with thermochromism that is visible to the naked eye. In all three cases, the band with vibrational progression, associated with the radicals, shifts to a higher energy at lower temperature. This might occur due to the stronger polarization of the electron density by Cu(II) in the SS-state with the shortened Cu–O bond. The spectral changes upon decreasing temperature differ for these complexes reflecting the differences in the magnetostructural transitions. For example, in the case of Cu(hfac)2LMe only a half of the clusters undergo the magnetostructural transition, whereas in the case of Cu(hfac)2LEt-CP all the clusters convert to the SS state.37 Therefore, the low temperature spectrum of Cu(hfac)2LMe consists of two overlapping bands, viz. those of SS- and WS-states, with similar vibrational progression, where the SS-state is shifted to higher energy by ∼1500 cm−1. In the case of Cu(hfac)2LEt-CP, only the shifted band of the SS-state is observed. In the case of Cu(hfac)2LPr, the magneto-structural transition takes place in a wide temperature range (∼90–240 K),31,39 and the temperature lowering leads to simultaneous displacements of the structured band and the appearance of a broad structure-less band centered at ∼500 nm, which could be tentatively assigned to a charge-transfer band (ligand-to-metal or metal-to-ligand).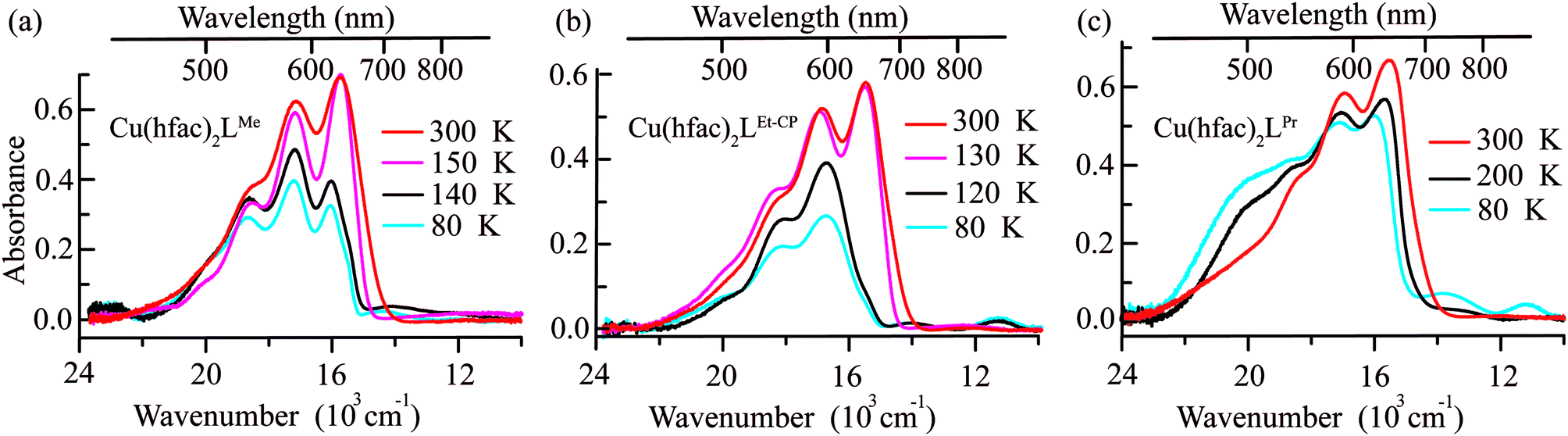 | ||
| Fig. 6 Vis–near-IR absorption spectra of (a) Cu(hfac)2LMe, (b) Cu(hfac)2LEt-CP, and (c) Cu(hfac)2LPr crystals before and after the magnetostructural transition, measured for single crystals. Temperatures are indicated on the right side of each figure. | ||
In addition to the above-mentioned spectral changes upon cooling, Fig. 6 demonstrates also an appearance of weak absorption bands in the near-IR range. To investigate the changes occurring at λ > 700 nm in more detail, near-IR absorption spectra were recorded for optically dense crystals, which were too dark for the 400–700 nm region but suitable for >700 nm region (Fig. 7). It is clearly visible that at low temperature the spectra are more intense and better resolved than at high temperatures: for each compound three bands can be recognized.
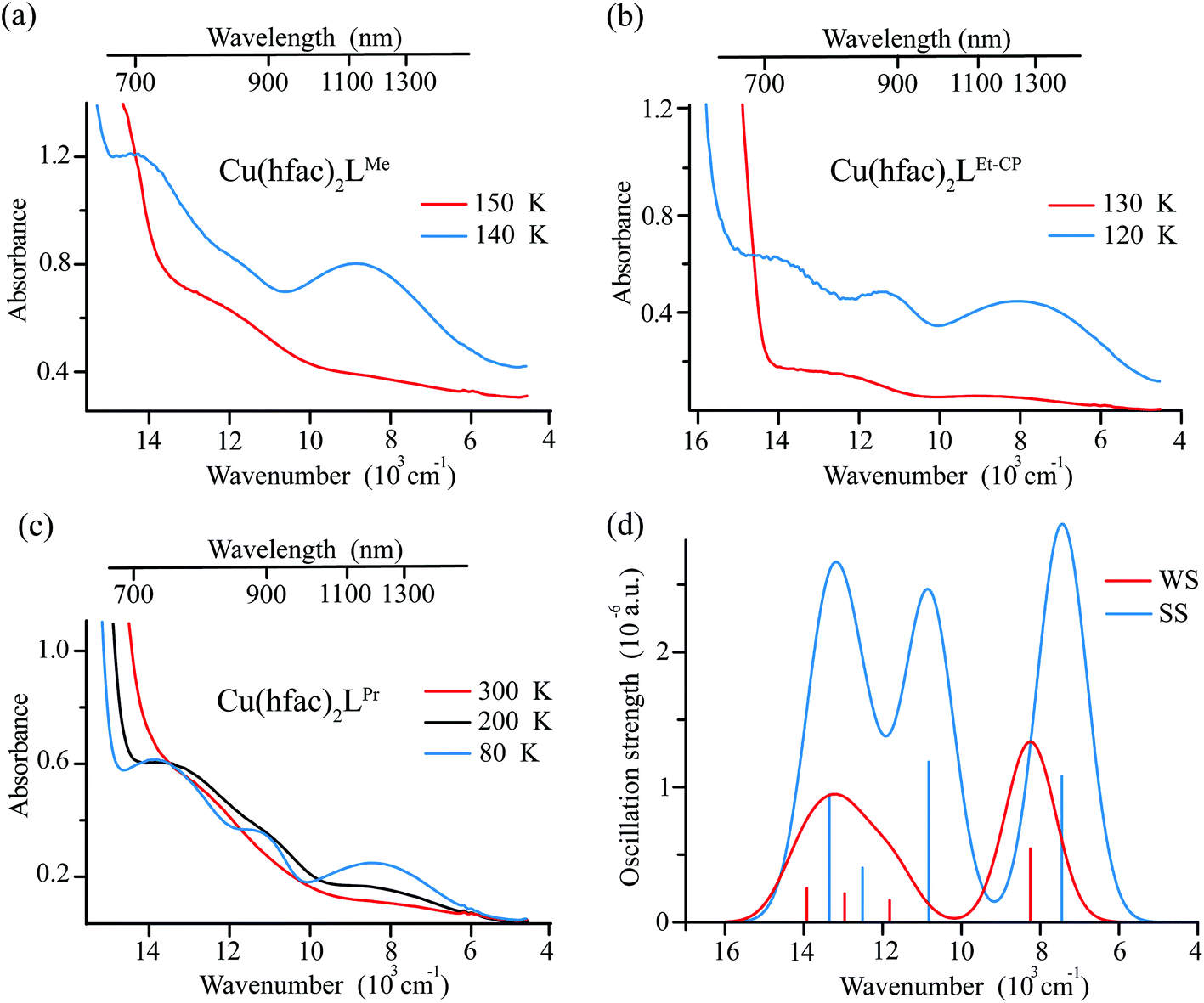 | ||
| Fig. 7 The near-IR absorption spectra recorded for optically dense single crystals of complexes Cu(hfac)2LMe (a), Cu(hfac)2LEt-CP (b) and Cu(hfac)2LPr (c) before and after the magnetostructural transition. Temperatures are indicated on the right side of each figure. (d) The d–d spectrum computed at the SOC-CASSCF(9,5)/NEVPT2 level of theory (applying 1500 cm−1 Gaussian broadening of the individual transitions) for the Cu(hfac)2LMe complex with the XRD structures at 295 K and 140 K. The SOMO orbitals of the corresponding excited states are shown. | ||
Ab initio calculations suggest that these bands correspond to d–d transitions of Cu(II) (Fig. 7). The change in the first coordination sphere of the Cu(II) ion caused by the magnetostructural transition leads to the change of both the intensity and positions of the d–d transitions. Analysis of the active space (five d-orbitals) orbitals also demonstrates the switch of the z-axis from the Cu-radical to the Cu-hfac direction and the change in the interaction of the Cu ion with the radical (see Table S4, ESI†). In agreement with the experiment, results of calculations show that the complex with more distorted low-temperature structure has more intense transitions with three distinguishable maxima. The high temperature structure is more symmetric and the d–d spectrum has only two well-separated bands with almost twice smaller intensities compared to the low-temperature structure.
NBO analysis for both the low- and high-temperature structures (SS and WS states, correspondingly) of Cu(hfac)2LMe shows that four interactions give the main contributions to the bonding between Cu(II) and nitroxide (ESI, Tables S5 and S6†). Two interactions delocalize electrons from the d-orbitals of Cu(II) onto orbitals combined from 3s and 3p AO of oxygen. Two other interactions delocalize the lone pairs of oxygen onto the 4s orbital of Cu(II). The population of the 4s orbital was estimated to be about 0.26 in both the SS and WS states of Cu(hfac)2LMe. The bonding between Cu(hfac)2 and nitroxides is quite weak and leads to dissociation of the complexes in solution at room temperature.
We were unable to detect any fluorescence for polycrystalline samples of complexes Cu(hfac)2LR (LR = LMe, LPr, LEt-CP) at both room temperature and 77 K. This might be due to the presence of fast internal conversion to the d–d excited states. Moreover, the LIESST phenomenon is most likely associated with the d–d excited states of Cu(hfac)2LR complexes. Indeed, it was shown11,12,29,33 that LIESST can be initiated by excitation at λ ≥ 900 nm (d–d absorption region, Fig. 6 and 7).
3.5. Comparison with iron-based SCO compounds and implications for photoswitching
The above observations allow us to conclude on the following trends in UV/Vis–near-IR spectroscopy of breathing crystals Cu(hfac)2LR:(i) UV/Vis–near-IR spectra are dominated by the absorption bands of nitroxides in both WS (high-temperature) and SS (low-temperature) states.
(ii) The spectra are changed moderately depending on the magnetostructural state – the nitroxide spectrum shifts to the higher energies (lower wavelengths) upon the transition from the WS to SS state. In addition, three-spin nitroxide-copper(II)–nitroxide clusters manifest a new band centered at ∼500 nm in the SS state, tentatively assigned to the charge transfer transition.
(iii) The d–d transitions of copper(II) are relatively weak in intensity; the band positions and shapes do change upon WS ↔ SS conversion, but cover the same spectral region ∼700–2000 nm.
As we noticed already, breathing crystals Cu(hfac)2LR manifest LIESST-like phenomena similar to iron(II) SCO compounds. The WS state can be photogenerated from the ground SS state at low temperatures (typically <30 K) and remain metastable on the scale of hours (referred to as mWS). However, the reverse photoswitching mWS → SS (analogue of reverse LIESST, i.e. photoinduced high-spin (HS) to low-spin (LS) conversion in SCO compounds) has not yet been found for breathing crystals, which, in particular, was a strong motivation for the present study.
In contrast to breathing crystals, typical iron(II) SCO compounds have easier interpretable UV/Vis–near-IR spectra dominated by d–d and MLCT transitions (see Fig. 8 for example). The distinct difference between this spectrum (Fig. 8) and the spectra of breathing crystals (Fig. 6 and 7) is that there are almost no absorption bands of the low-temperature LS state at λ > 670 nm (15000 cm−1), whereas the photo/thermo-induced high-temperature HS state features a well-resolved band centered around ∼830 nm (12000 cm−1). Thus, one needs higher energy quanta (green light) to induce LS → HS conversion (direct LIESST) and lower energy quanta (red light) to induce HS → LS conversion (reverse LIESST). Qualitatively, upon irradiation of the metastable HS state by red light for reverse LIESST one does not reach necessary energy to induce simultaneously direct LIESST; therefore the reverse LIESST is feasible.
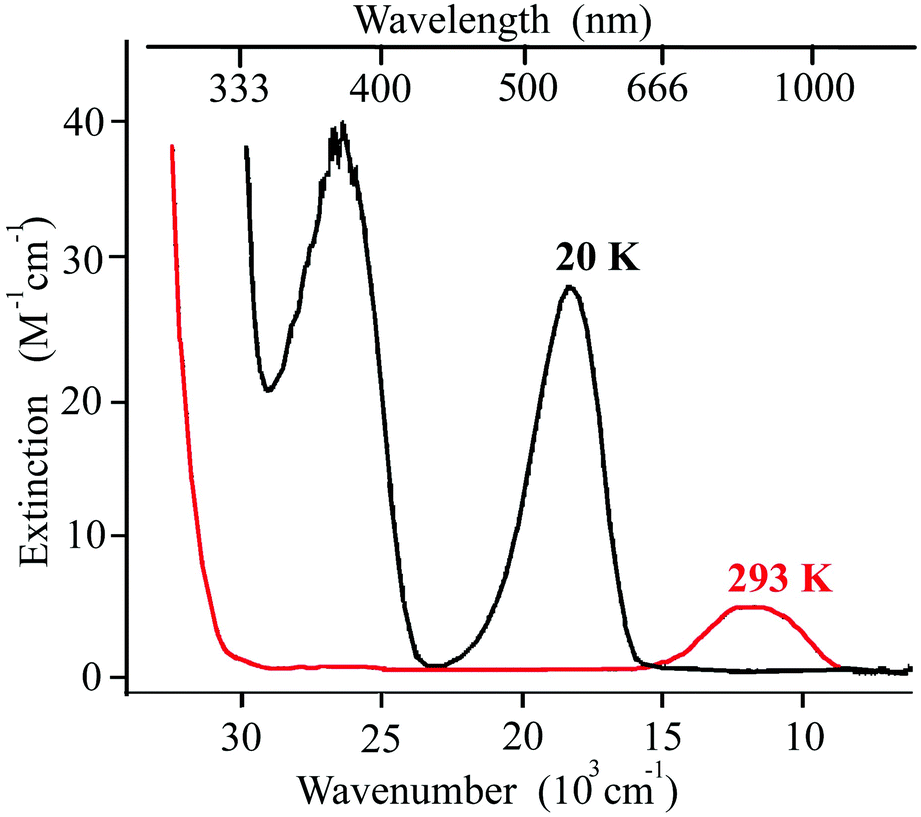 | ||
| Fig. 8 An example of UV/Vis spectra of the iron(II) SCO complex [Fe(ptz)6](BF4)2 at 293 and at 20 K. Adapted from ref. 75. | ||
In contrast, for breathing crystals d–d transitions of copper(II) are present at λ ∼ 700–2000 nm in both SS and WS states (Fig. 7). They do not noticeably shift to the higher energy in the WS state, most likely because the switch of the Jahn–Teller axis mainly interchanges the axes of crystal field and does not change significantly the corresponding energy values, in contrast to SCO compounds where the mean metal-to-ligand distance does change upon the transition leading to a significant change of the crystal field. Therefore, the low-energy route between the SS and mWS states via d–d transitions is feasible when irradiating either ground SS or photoinduced mWS states. Most likely, this is the main obstacle for inducing the reverse LIESST in breathing crystals.
All synthetic approaches applied to date in order to influence the SCO-like behavior in breathing crystals have relied on structural modification of either radical or solvent molecules included in the crystal. Both factors influenced crystal packing, viz. bond lengths and inter-bond angles, and eventually the exchange interactions between paramagnetic centers. However, in all cases Cu(hfac)2 was the indispensable component of the complexes, and the switch of the elongated Jahn–Teller axis in copper(II) units coupled with the change of copper(II)–nitroxide exchange interactions was the core of the observed SCO-like phenomena. Therefore, it is difficult to anticipate that the situation with d–d transitions covering the range of ∼700–2000 nm in both SS and WS states can be remedied synthetically. The absorption in the visible region λ ∼ 400–700 nm can be reduced by adjusting the structure of the nitroxide radical, as was shown earlier by replacing the nitronyl nitroxide by tert-butyl pyrazolyl nitroxide;76 however, this should not make the reverse mWS → SS photoswitching better isolated from simultaneous direct SS → mWS one. We suppose that a more feasible strategy for inducing the reverse LIESST in breathing crystals could be the use of excitation energies that are so low that direct LIESST cannot be induced, whereas reverse LIESST is still feasible. This requires the adjustment of copper(II) coordination environments in order to provide simultaneously a high enough energy barrier for SS → mWS switching and the presence of low-lying d–d transitions.
Finally, it is worth mentioning that the existence of low-energy d–d bands in both SS and WS states of breathing crystals (λ ∼ 700–2000 nm and beyond) makes an interesting difference compared to iron-based SCO compounds. For instance, the direct photoswitching requires lower energy quanta which potentially means that the other radiation sources such as CO and CO2 infrared lasers can be implemented to control the spin state of the copper(II)–nitroxide photoswitchable compounds.
4. Conclusions
The electronic absorption spectra of single crystals of three thermoswitchable molecular magnets (Cu(hfac)2LMe, Cu(hfac)2LEt-CP, Cu(hfac)2LPr) have been analyzed in the visible and near-IR regions. The color of the complexes is mainly determined by optical properties of the nitroxide radicals, while the Cu(hfac)2 fragment contributes to the near-IR range with the intensity smaller by an order of magnitude. The observed thermochromism of the complexes is mainly caused by the spectral shift of the absorption bands of the nitroxide radicals to higher energy upon the magnetostructural transition to the SS state. The important feature of the absorption band in the visible range is the vibrational progression observed in solutions of nitroxide radicals as well as in the single crystals of the complexes. These progressions differ only slightly in the solution spectrum of radicals and in the WS and SS states of complexes in crystals. UV/Vis absorption spectra of the nitroxide radical LMe in ethanol solution have been well reproduced by DFT calculations.The low-temperature absorption spectra of single crystals manifest distinct differences in the studied complexes upon magnetostructural transitions. In the case of Cu(hfac)2LMe, the low temperature spectrum features two overlapping bands of SS and WS states with maxima separated by ∼1500 cm−1, whereas the spectrum of Cu(hfac)2LEt-CP has only the SS band at low temperature, reflecting 50% (for Cu(hfac)2LMe) and 100% (for Cu(hfac)2LEt-CP) conversion. In contrast, the spectrum of Cu(hfac)2LPr changes gradually with temperature and is characterized by the appearance of both the slightly shifted structured band and the broad structure-less band on the higher energy side, which is tentatively assigned to a charge transfer transition. The analysis of the spectra of single crystals in the near-IR region reveals changes in the energy and the intensity of the Cu(II) d–d transitions, which are well reproduced by SOC-NEVPT2 calculations and are due to a switching of the Jahn–Teller axis and corresponding changes in the coordination sphere distortions.
The rational design of photomagnetic switches relying on breathing crystals requires an in-depth understanding of the structure and the origin of contributing optical absorption bands. In this work we attempted to solve this task using three representative compounds of the family and drawing general conclusions on the corresponding UV/Vis properties. Based on the insights gained, we proposed further strategies for designing bidirectional photoswitches using these appealing compounds, which will be pursued in our future work.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has been supported by RFBR (no. 15-03-07640, 17-33-80025, 16-33-00675) and the Grant Council of the President of Russian Federation (MK-3597.2017.3). M. V. F. thanks FASO Russia (project 0333-2016-0004) and N. P. G. thanks FASO Russia (project 0304-2015-0005).References
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308(Part 2), 395–408 CrossRef.
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, Wiley, 2013 Search PubMed.
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I, Springer, 2004 Search PubMed.
- S. Decurtins, P. Gütlich, C. Köhler, H. Spiering and A. Hauser, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS.
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS PubMed.
- R. Sessoli, Nat. Chem., 2010, 2, 346–347 CrossRef CAS.
- R. Bertoni, M. Lorenc, H. Cailleau, A. Tissot, J. Laisney, M.-L. Boillot, L. Stoleriu, A. Stancu, C. Enachescu and E. Collet, Nat. Mater., 2016, 15, 606–610 CrossRef CAS PubMed.
- M. Fedin, V. Ovcharenko, R. Sagdeev, E. Reijerse, W. Lubitz and E. Bagryanskaya, Angew. Chem., Int. Ed., 2008, 47, 6897–6899 CrossRef CAS PubMed.
- M. V. Fedin, K. Y. Maryunina, R. Z. Sagdeev, V. I. Ovcharenko and E. G. Bagryanskaya, Inorg. Chem., 2012, 51, 709–717 CrossRef CAS PubMed.
- P. Chakraborty, C. Enachescu, C. Walder, R. Bronisz and A. Hauser, Inorg. Chem., 2012, 51, 9714–9722 CrossRef CAS PubMed.
- A. Marino, P. Chakraborty, M. Servol, M. Lorenc, E. Collet and A. Hauser, Angew. Chem., Int. Ed., 2014, 53, 3863–3867 CrossRef CAS PubMed.
- H. Z. Ye, C. Sun and H. Jiang, Phys. Chem. Chem. Phys., 2015, 17, 6801–6808 RSC.
- K. E. Funck, A. V. Prosvirin, C. Mathonière, R. Clérac and K. R. Dunbar, Inorg. Chem., 2011, 50, 2782–2789 CrossRef CAS PubMed.
- N. F. Sciortino, S. M. Neville, J.-F. o. Létard, B. Moubaraki, K. S. Murray and C. J. Kepert, Inorg. Chem., 2014, 53, 7886–7893 CrossRef CAS PubMed.
- B. Rösner, M. Milek, A. Witt, B. Gobaut, P. Torelli, R. H. Fink and M. M. Khusniyarov, Angew. Chem., Int. Ed., 2015, 54, 12976–12980 CrossRef PubMed.
- A. Hauser, in Spin Crossover in Transition Metal Compounds II, Springer, 2004, pp. 155–198 Search PubMed.
- A. Tissot, J.-F. Bardeau, E. Rivière, F. Brisset and M.-L. Boillot, Dalton Trans., 2010, 39, 7806–7812 RSC.
- J. Laisney, A. Tissot, G. Molnár, L. Rechignat, E. Rivière, F. Brisset, A. Bousseksou and M.-L. Boillot, Dalton Trans., 2015, 44, 17302–17311 RSC.
- H. Naggert, A. Bannwarth, S. Chemnitz, T. von Hofe, E. Quandt and F. Tuczek, Dalton Trans., 2011, 40, 6364–6366 RSC.
- D. Unruh, P. Homenya, M. Kumar, R. Sindelar, Y. Garcia and F. Renz, Dalton Trans., 2016, 45, 14008–14018 RSC.
- S. L. Veber, M. V. Fedin, K. Y. Maryunina, G. V. Romanenko, R. Z. Sagdeev, E. G. Bagryanskaya and V. I. Ovcharenko, Inorg. Chim. Acta, 2008, 361, 4148–4152 CrossRef CAS.
- M. M. Khusniyarov, T. Weyhermüller, E. Bill and K. Wieghardt, Angew. Chem., Int. Ed., 2008, 120, 1248–1251 CrossRef.
- M.-L. Boillot, S. Pillet, A. Tissot, E. Riviere, N. Claiser and C. Lecomte, Inorg. Chem., 2009, 48, 4729–4736 CrossRef CAS PubMed.
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds II, Springer, 2004 Search PubMed.
- W. Kaszub, A. Marino, M. Lorenc, E. Collet, E. G. Bagryanskaya, E. V. Tretyakov, V. I. Ovcharenko and M. V. Fedin, Angew. Chem., Int. Ed., 2014, 53, 10636–10640 CrossRef CAS PubMed.
- I. Y. Barskaya, E. V. Tretyakov, R. Z. Sagdeev, V. I. Ovcharenko, E. G. Bagryanskaya, K. Y. Maryunina, T. Takui, K. Sato and M. V. Fedin, J. Am. Chem. Soc., 2014, 136, 10132–10138 CrossRef CAS PubMed.
- V. Ovcharenko and E. Bagryanskaya, Spin-Crossover Materials: Properties and Applications, 2013, pp. 239–280 Search PubMed.
- M. V. Fedin, S. L. Veber, E. G. Bagryanskaya and V. I. Ovcharenko, Coord. Chem. Rev., 2015, 289, 341–356 CrossRef.
- M. V. Fedin, E. G. Bagryanskaya, H. Matsuoka, S. Yamauchi, S. L. Veber, K. Y. Maryunina, E. V. Tretyakov, V. I. Ovcharenko and R. Z. Sagdeev, J. Am. Chem. Soc., 2012, 134, 16319–16326 CrossRef CAS PubMed.
- I. Y. Barskaya, S. Veber, S. Fokin, E. Tretyakov, E. Bagryanskaya, V. Ovcharenko and M. Fedin, Dalton Trans., 2015, 44, 20883–20888 RSC.
- M. V. Fedin, S. L. Veber, E. G. Bagryanskaya, G. V. Romanenko and V. I. Ovcharenko, Dalton Trans., 2015, 44, 18823–18830 RSC.
- I. Y. Barskaya, S. Veber, S. Fokin, E. Tretyakov, E. Bagryanskaya, V. Ovcharenko and M. Fedin, Dalton Trans., 2015, 44, 20883–20888 RSC.
- K. Y. Maryunina, X. Zhang, S. Nishihara, K. Inoue, V. A. Morozov, G. V. Romanenko and V. I. Ovcharenko, J. Mater. Chem. C, 2015, 3, 7788–7791 RSC.
- N. A. Artiukhova, G. V. Romanenko, A. S. Bogomyakov, I. Y. Barskaya, S. L. Veber, M. V. Fedin, K. Y. Maryunina, K. Inoue and V. I. Ovcharenko, J. Mater. Chem. C, 2016, 4, 11157–11163 RSC.
- V. Ovcharenko, S. Fokin, G. Romanenko, Y. G. Shvedenkov, V. Ikorskii, E. Tretyakov and S. Vasilevskii, J. Struct. Chem., 2002, 43, 153–167 CrossRef CAS.
- V. I. Ovcharenko, K. Y. Maryunina, S. V. Fokin, E. V. Tretyakov, G. V. Romanenko and V. N. Ikorskii, Russ. Chem. Bull., 2004, 53, 2406–2427 CrossRef CAS.
- S. L. Veber, E. A. Suturina, M. V. Fedin, K. N. Boldyrev, K. Y. Maryunina, R. Z. Sagdeev, V. I. Ovcharenko, N. P. Gritsan and E. G. Bagryanskaya, Inorg. Chem., 2015, 54, 3446–3455 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- T. Petrenko and F. Neese, J. Chem. Phys., 2007, 127, 164319 CrossRef PubMed.
- F. Neese, Wiley Interdisciplinary Reviews: Computational Molecular Science, 2012, vol. 2, pp. 73–78 Search PubMed.
- M. de Jong, L. Seijo, A. Meijerink and F. T. Rabouw, Phys. Chem. Chem. Phys., 2015, 17, 16959–16969 RSC.
- T. Petrenko and F. Neese, J. Chem. Phys., 2012, 137, 234107 CrossRef PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- S. Per, H. Anders, R. Björn and L. Bernard, Phys. Scr., 1980, 21, 323 CrossRef.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- P. E. M. Siegbahn, J. Almlöf, A. Heiberg and B. O. Roos, J. Chem. Phys., 1981, 74, 2384–2396 CrossRef.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- C. Angeli and R. Cimiraglia, Theor. Chem. Acc., 2002, 107, 313–317 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- B. A. Hess, Phys. Rev. A, 1986, 33, 3742–3748 CrossRef CAS.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- D. Ganyushin and F. Neese, J. Chem. Phys., 2013, 138, 104113 CrossRef PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, 2005 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- G. Bussière, R. Beaulac, H. Bélisle, C. Lescop, D. Luneau, P. Rey and C. Reber, in Transition Metal and Rare Earth Compounds: Excited States, Transitions, Interactions III, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 97–118 Search PubMed.
- E. V. Tretyakov, V. F. Plyusnin, A. O. Suvorova, S. V. Larionov, S. A. Popov, O. V. Antonova, E. M. Zueva, D. V. Stass, A. S. Bogomyakov and G. V. Romanenko, J. Lumin., 2014, 148, 33–38 CrossRef CAS.
- G. Heimel, M. Daghofer, J. Gierschner, E. J. W. List, A. C. Grimsdale, K. Müllen, D. Beljonne, J.-L. Brédas and E. Zojer, J. Chem. Phys., 2005, 122, 054501 CrossRef PubMed.
- J. Ferguson, J. Chem. Phys., 1961, 34, 1609–1613 CrossRef CAS.
- R. L. Belford, M. Calvin and G. Belford, J. Chem. Phys., 1957, 26, 1165–1174 CrossRef CAS.
- A. Hauser, J. Chem. Phys., 1991, 94, 2741–2748 CrossRef CAS.
- I. Y. Drozdyuk, S. E. Tolstikov, E. V. Tretyakov, S. L. Veber, V. I. Ovcharenko, R. Z. Sagdeev, E. G. Bagryanskaya and M. V. Fedin, J. Phys. Chem. A, 2013, 117, 6483–6488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electronic absorption spectra of LMe, LPr and LEt-CP, fluorescence emission and excitation spectra of LMe as well as computed vertical transitions and the dimensionless displacements (ΔQ) of LMe, energies of ab initio ligand field d-orbitals of Cu(hfac)2, NBO analysis of Cu(hfac)2LMe and some other computational details (PDF). See DOI: 10.1039/c7dt02719b |
| This journal is © The Royal Society of Chemistry 2017 |