
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new glance on R2MGe6 (R = rare earth metal, M = another metal) compounds. An experimental and theoretical study of R2PdGe6 germanides†
Riccardo
Freccero
 a,
Pavlo
Solokha
*a,
Davide M.
Proserpio
bc,
Adriana
Saccone
a and
Serena
De Negri
a
a,
Pavlo
Solokha
*a,
Davide M.
Proserpio
bc,
Adriana
Saccone
a and
Serena
De Negri
a
aUniversità di Genova, Dipartimento di Chimica e Chimica Industriale, Via Dodecaneso 31, 16146 Genova, Italy. E-mail: pavlo.solokha@unige.it; Fax: +39-0103536163; Tel: +39-0103536159
bUniversità degli Studi di Milano, Dipartimento di Chimica, Via Golgi 19, 20133 Milano, Italy
cSamara Center for Theoretical Materials Science (SCTMS), Samara State University, Ac. Pavlov Str. 1, Samara 443011, Russia
First published on 18th September 2017
Abstract
The R2PdGe6 series (R = rare earth metal) was structurally characterized, and the results achieved were extended for a comprehensive study on R2MGe6 (M = another metal) compounds, employing symmetry-based structural rationalization and energy calculations. Directly synthesized R2PdGe6 exists for almost all R-components (R = Y, La–Nd, Sm and Gd–Lu) and even if with La is probably metastable. Several single crystal X-ray analyses (R = Y, Ce, Pr, Nd, Er and Lu) indicated oS72-Ce2(Ga0.1Ge0.9)7 as the correct structure. The alternative In-flux method, once optimized, produced three good quality R2PdGe6 single crystals: La2PdGe6 and Pr2PdGe6 turned out to be mS36-La2AlGe6-type non-merohedrally twinned crystals and Yb2PdGe6 is of oS72-Ce2(Ga0.1Ge0.9)7-type. The vacancy ordering phenomenon was considered as a possible cause of the symmetry reduction relations connecting the most frequently reported 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:6 structural models (oS18, oS72 and mS36) with the oS20-SmNiGe3 aristotype. The detected twin formation is consistent with the symmetry relations, which are discussed even considering the validity of the different structural models. DFT total energy calculations were performed for R2PdGe6 (R = Y and La) in the three abovementioned structural models, and for La2MGe6 (M = Pt, Cu, Ag and Au) in the oS18 and oS72 modifications. The results indicate that the oS18-Ce2CuGe6 structure, prevalently proposed in the literature, is associated with the highest energy and thus it is not likely to be realized in these series. The oS72 and mS36 polytypes are energetically equivalent, and small changes in the synthetic conditions could easily stabilize any of them, in agreement with experimental results obtained by direct and flux syntheses.
:1:6 structural models (oS18, oS72 and mS36) with the oS20-SmNiGe3 aristotype. The detected twin formation is consistent with the symmetry relations, which are discussed even considering the validity of the different structural models. DFT total energy calculations were performed for R2PdGe6 (R = Y and La) in the three abovementioned structural models, and for La2MGe6 (M = Pt, Cu, Ag and Au) in the oS18 and oS72 modifications. The results indicate that the oS18-Ce2CuGe6 structure, prevalently proposed in the literature, is associated with the highest energy and thus it is not likely to be realized in these series. The oS72 and mS36 polytypes are energetically equivalent, and small changes in the synthetic conditions could easily stabilize any of them, in agreement with experimental results obtained by direct and flux syntheses.
1. Introduction
Numerous germanium-rich compounds of idealized general formula R2MGe6 (R = rare earth metal, M = another metal) have been investigated since the eighties of the last century up to the present.1 Some R2MGe6 are off-stoichiometric due to the M/Ge statistical mixture (as in Ce2(Ga0.1Ge0.9)7) or to partially occupied M sites (as in Dy2Zn1−xGe6 (x ∼ 0.5)2). Scientific interest in these phases arises from the fact that they exist with many different metals M, ranging from the s-block (M = Li and Mg) to the p-block (M = Al and Ga) of the periodic table, including the major part of late transition elements. For this reason, title compounds are well suited for systematic studies on crystal structure–electronic structure–property relationships, aiming to evidence for example the role of both R and M properties (size, electronegativity, valence electrons, etc.) in the chemical bonding scenario, set up by characteristic Ge-based covalent fragments.A variety of physical properties, including electrical resistivity, magnetic susceptibility, specific heat and thermoelectric power, have already been measured on several R2MGe6 representative samples, mostly with transition elements, such as Co, Ni, Pd, Pt and Cu, as M components.3–9
Many of the investigated compounds exhibit an antiferromagnetic ordering below ∼30 K;3,4,8 for some Yb2MGe6 phases a behaviour characteristic of intermediate valence systems has been reported.5,9,10
Despite the great amount of experimental work, controversial data exist both on the interpretation of the physical/magnetic properties and on the crystal structures of the R2MGe6 compounds. They are generally reported to crystallize in orthorhombic or monoclinic space groups, distributing among the following prototypes: oS18-Ce2CuGe6 (space group: Amm2), which is the most represented, oS72-Ce2(Ga0.1Ge0.9)7 (space group: Cmce) and mS36-La2AlGe6 (C2/m). A different orthorhombic structure (space group: Cmmm) has been assigned to a few R2LiGe6 (R = La, Ce and Pr) phases, referred to as the oS18-Pr2LiGe6 prototype11,12 and to Yb2Pd1.075(1)Ge6, referred to as the oS20-SmNiGe3 prototype.13
The oS72-Ce2(Ga0.1Ge0.9)7 structure was found to be the correct one for a number of compounds previously reported as oS18, such as La2PdGe6, Dy2PdGe67,14 and Yb2PdGe6,9,15 and for several new 2:1:6 representative samples, such as La2MgGe616 and R2ZnGe6.2 Interestingly, along the Zn-containing series of compounds, a structural change was observed from the orthorhombic oS72-Ce2(Ga0.1Ge0.9)7 structure (R = La–Nd, Sm, Gd and Tb) to the new monoclinic structure mP34-Dy2Zn1−xGe6 (x ∼ 0.5, R = Dy and Ho), which is an ordered superstructure of the La2AlGe6 prototype.
Recently, the alternative synthesis in metal flux was employed for Yb2CuGe6, and the mS36-La2AlGe6 crystal structure was proposed for this compound, instead of the previously reported oS18-Ce2CuGe6 type.17
The abovementioned structural models oS18, oS72 and mS36 are strictly related, as can be pointed out both by means of group–subgroup relations2 and by a description based on the linear intergrowth concept.18,19 As a consequence, the correct model can be assigned only by single crystal X-ray diffraction analysis; powder X-ray diffraction could be sufficient only in the case of high quality patterns of single phase samples, generally quite difficult to obtain.
A correct structural description is of fundamental importance not only to evidence differences, similarities and regularities among the numerous 2:1:6 compounds in view of further chemical bonding studies, but also to appropriately interpret the results of magnetic characterization.
Taking into account the fact that the R2PdGe6 phases are reported to exist for many rare earth metals and that the structures of some of them were recently revised, this 2:1:6 series of compounds were targeted to perform an accurate structural investigation, mostly based on single crystal analysis.
The challenging experimental work necessary for this achievement was complemented both by a comprehensive crystal structure analysis based on group–subgroup relations and by DFT total energy calculations, in order to generalize the obtained results to the whole R2MGe6 family and guide future investigations on new compounds.
2. Experimental section
2.1 Synthesis, microstructure and phase analysis
Different synthetic routes were followed, all starting from the pure components (rare earth metals and In used as a metal solvent were supplied by Newmet Koch, Waltham Abbey, England, palladium and germanium by MaTecK, Jülich, Germany, nominal purities of all metals >99.9 mass %).Samples of about 0.8 g with R22.2Pd11.1Ge66.7 (R = Sc; Y; La–Nd; and Sm–Lu) nominal compositions were prepared by direct synthesis in a resistance furnace. Stoichiometric amounts of components were placed in an arc-sealed Ta crucible in order to prevent oxidation; the constituents of the Eu22.2Pd11.1Ge66.7 sample were weighed and sealed in the crucible inside an inert atmosphere glove box. The Ta crucible was then closed in an evacuated quartz phial to prevent oxidation at high temperature, and finally placed in a resistance furnace, where the following thermal cycle was employed:
 | (I) |
A continuous rotation, at a speed of 100 rpm, was applied to the phial during the thermal cycle. These synthetic conditions were chosen with the aim to obtain samples containing crystals of good quality and size, suitable for further structural studies. The synthesized alloys are very brittle and mostly stable in air, except for the Eu22.2Pd11.1Ge66.7 sample. No tantalum contamination of the samples was detected.
After direct synthesis in the resistance furnace, some pieces of the La22.2Pd11.1Ge66.7 sample were annealed at different temperatures. Further attempts to synthesize the La2PdGe6 compound were made, by arc melting of components, followed by annealing treatments (see the “Results and discussion” section).
Flux synthesis was also performed for samples containing La, Pr and Yb, using In as a metal solvent. Stoichiometric amounts of R, Pd and Ge, giving the nominal composition of R21Pd7Ge72, were placed in an arc-sealed Ta crucible with a 1:45 molar excess of indium, to obtain a total mass of about 3 g. Then, the Ta crucible, closed in an evacuated quartz phial, was placed in a resistance furnace, and the following thermal cycles were employed:
 | (II) |
 | (III) |
For the La-containing sample both thermal cycles were tested, but only cycle III was employed for the Pr and Yb-containing samples.
A continuous rotation of the quartz phial during the thermal cycle was employed to favour better dissolution of the constituting elements inside the flux. In all cases, a vertical section of the obtained ingots revealed the presence of large shining crystals, visible to the naked eye, randomly distributed within the flux solidified matrix.
In order to perform metallographic analysis, samples were embedded in a phenolic resin with a carbon filler by using the automatic hot compression mounting press Opal 410 (ATM GmbH, Germany). The samples synthesized by the flux method were analysed prior to product separation: to avoid the metal solvent melting, they were embedded in a cold-curing resin, conductive due to the presence of a copper filler.
The automatic grinding and polishing machine Saphir 520 (ATM GmbH, Germany) was used to obtain smooth alloy surfaces suitable for microscopic examinations. Grinding was performed using SiC papers with grain size decreasing from 600 to 1200 mesh, using running water as a lubricant; for polishing, diamond paste with particle size decreasing from 6 to 1 μm was used with an alcohol based lubricant. Petroleum ether was employed to clean samples ultrasonically after each polishing step.
In the case of flux prepared samples only the 1200 mesh SiC paper was used applying a low pressure, due to In ductility.
After SEM-EDXS analysis, crystals of R2PdGe6 compounds (R = La, Pr and Yb) obtained by flux synthesis were extracted from the flux by immersion and sonication of the ingot in glacial acetic acid for about 24 h, allowing indium dissolution. The obtained crystalline product was rinsed with water and dried with acetone. After that, another SEM-EDXS analysis was performed in order to check the goodness of the separation procedure and the quality/composition of the isolated crystals.
Microstructure examination as well as qualitative and quantitative analyses were performed using a scanning electron microscope (SEM) Zeiss Evo 40 (Carl Zeiss SMT Ltd, Cambridge, England) equipped with a Pentafet Link Energy Dispersive X-ray Spectroscopy (EDXS) system managed using INCA Energy software (Oxford Instruments, Analytical Ltd, Bucks, UK). Cobalt standard was used for calibration.
2.2 X-ray diffraction (XRD) analysis on single crystal and powder samples
Single crystals of R2PdGe6 compounds prepared by both direct synthesis (R = Y, Ce, Pr, Nd, Er and Lu) and flux synthesis (R = La, Pr and Yb) were selected with the aid of a light optical microscope (Leica DM4000 M, Leica Microsystems Wetzlar GmbH, Wetzlar, Germany) operated in the dark field mode.A full-sphere dataset was obtained in a routine fashion under ambient conditions on a four-circle Bruker Kappa APEXII CCD area-detector diffractometer equipped with graphite monochromatized Mo Kα (λ = 0.71073 Å) radiation, operating in ω-scan mode. Crystals exhibiting metallic luster, glued on glass fibres, were mounted in a goniometric head and then in the goniostat inside the diffractometer camera. Intensity data were collected over the reciprocal space up to ∼30° in θ with exposures of 20–30 s per frame. Semiempirical absorption corrections based on a multipolar spherical harmonic expansion of equivalent intensities were employed for all data using SADABS/TWINABS software.20
The details of the crystal structure solution and refinement are reported in the “Results and discussion” section. The corresponding CIF files are available in the ESI† and they have also been deposited at Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany, with the following depository numbers: CSD 433026 (Y2PdGe6), CSD 433022 (La2PdGe6, flux), CSD 433081 (Ce2PdGe6), CSD 433025 (Pr2PdGe6), CSD 433205(Pr2PdGe6, flux), CSD 433024 (Nd2PdGe6), CSD 433021 (Er2PdGe6), CSD 433151 (Yb2PdGe6, flux), CSD 433023 (Lu2PdGe6).
Selected crystallographic data and structure refinement parameters for the studied single crystals are listed in Tables 1 and 2.
| Empirical formula | Y2PdGe6 | Ce2PdGe6 | Pr2PdGe6 | Nd2PdGe6 | Er2PdGe6 | Lu2PdGe6 |
|---|---|---|---|---|---|---|
| EDXS data | Y21.0Pd10.8Ge68.2 | Ce22.2Pd10.8Ge67.0 | Pr20.2Pd11.4Ge68.4 | Nd21.8Pd11.2Ge67.0 | Er22.7Pd11.0Ge66.3 | Lu23.8Pd10.6Ge65.6 |
| M w, [g mol−1] | 719.76 | 822.18 | 823.76 | 830.42 | 876.46 | 891.88 |
| a [Å] | 8.1703(5) | 8.3548(4) | 8.3129(5) | 8.2831(4) | 8.1122(6) | 8.0725(8) |
| b [Å] | 8.0451(5) | 8.1774(4) | 8.1540(5) | 8.1323(4) | 8.0068(6) | 7.9791(8) |
| c [Å] | 21.558(1) | 22.0272(9) | 21.996(1) | 21.915(1) | 21.399(2) | 21.317(2) |
| V [Å3] | 1417.1(2) | 1504.9(1) | 1491.0(2) | 1476.2(1) | 1389.9(2) | 1373.0(2) |
| Calc. density [g cm−3] | 6.748 | 7.258 | 7.340 | 7.473 | 8.377 | 8.629 |
| Abs. coeff. (μ), mm−1 | 43.6 | 37.7 | 38.9 | 40.1 | 51.8 | 56.8 |
| Unique reflections | 1028 | 1308 | 1296 | 1307 | 1206 | 1225 |
| Reflections I > 2σ(I) | 884 | 1072 | 870 | 848 | 1020 | 874 |
| R sigma | 0.0233 | 0.0084 | 0.0270 | 0.0263 | 0.0133 | 0.0284 |
| Data/parameters | 884/50 | 1072/50 | 870/50 | 848/50 | 1020/50 | 874/50 |
| GOF on F2 (S) | 1.166 | 1.543 | 1.059 | 1.002 | 1.326 | 0.912 |
| R indices [I > 2σ(I)] | R 1 = 0.0232; wR2 = 0.0642 | R 1 = 0.0208; wR2 = 0.0364 | R 1 = 0.0197; wR2 = 0.0372 | R 1 = 0.0165; wR2 = 0.0299 | R 1 = 0.0202; wR2 = 0.0331 | R 1 = 0.0224; wR2 = 0.0557 |
| R indices [all data] | R 1 = 0.0282; wR2 = 0.0667 | R 1 = 0.0260; wR2 = 0.0379 | R 1 = 0.0366; wR2 = 0.0421 | R 1 = 0.0351; wR2 = 0.0354 | R 1 = 0.0264; wR2 = 0.0343 | R 1 = 0.0399; wR2 = 0.0634 |
| Δρfin (max/min), [e Å−3] | 1.31/−1.36 | 1.03/−1.29 | 1.01/−1.68 | 1.26/−1.05 | 1.09/−1.25 | 1.77/−1.63 |
| Empirical formula | La2PdGe6 | Pr2PdGe6 | Yb2PdGe6 |
|---|---|---|---|
| EDXS data | La22.4Pd10.5Ge67.1 | Pr20.6Pd11.3Ge68.1 | Yb23.2Pd10.9Ge65.9 |
| M w, [g mol−1] | 819.76 | 823.76 | 888.02 |
| Space group (no.) | C2/m (12) | C2/m (12) | Cmce (64) |
| Pearson symbol-prototype, Z | mS36-La2AlGe6, 4 | mS36-La2AlGe6, 4 | oS72-Ce2(Ga0.1Ge0.9)7, 8 |
| a [Å] | 8.2163(8) | 8.157(2) | 8.1428(3) |
| b [Å] | 8.4161(9) | 8.328(2) | 7.9807(3) |
| c [Å] | 11.294(1) | 11.195(2) | 21.8331(9) |
| β (°) | 100.477(1) | 100.391(3) | 90 |
| V [Å3] | 768.0(1) | 748.1(3) | 1418.83(9) |
| Calc. density [g cm−3] | 7.090 | 7.314 | 8.314 |
| abs coeff. (μ), mm−1 | 36.2 | 38.7 | 53.5 |
| Twin law | [1 0 0 0 −1 0 −1/2 0 −1] | [1 0 0 0 −1 0 −1/2 0 −1] | — |
| k(BASF) | 0.414(5) | 0.423(5) | — |
| Unique reflections | 1083 | 1262 | 1242 |
| Reflections I > 2σ(I) | 1030 | 1062 | 1144 |
| R sigma | 0.0218 | 0.0298 | 0.0149 |
| Data/parameters | 1030/50 | 1062/50 | 1144/50 |
| GOF on F2 (S) | 1.200 | 1.453 | 1.428 |
| R indices [I > 2σ(I)] | R 1 = 0.0242; wR2 = 0.0773 | R 1 = 0.0357; wR2 = 0.1166 | R 1 = 0.0231; wR2 = 0.0496 |
| R indices [all data] | R 1 = 0.0254; wR2 = 0.0778 | R 1 = 0.0501 wR2 = 0.1209 | R 1 = 0.0255; wR2 = 0.0506 |
| Δρfin (max/min), [e Å−3] | 2.33/−2.61 | 4.35/−5.29 | 1.92/−1.78 |
X-ray powder diffraction (XRPD) was performed on all samples by using a diffractometer Philips X'Pert MPD (Cu Kα radiation, λ = 1.5406 Å, graphite crystal monochromator, scintillation detector, step mode of scanning), with a θ:2θ Bragg–Brentano geometry. Measured powder patterns were collected in the 10°–100°2θ range, with a scanning step of ca. 0.02° with a time per step varying from 10 to 20 s. Phase identification was performed with the help of the software PowderCell.21
2.3 Differential thermal analysis (DTA)
Differential thermal analysis (DTA) was performed on a few La–Pd–Ge samples, in order to gain insights into the formation conditions of the La2PdGe6 compound. Measurements were carried out using a LABSYS EVO (SETARAM Instrumentation, Caluire, France) equipped with type S (Pt–PtRh 10%) thermocouples, in the temperature range of 25–1100 °C using custom-made tantalum crucibles. The sample crucible was loaded with about 200 mg of the alloy to analyze, closed with a cap, and subsequently arc-sealed under an inert atmosphere after cooling it in liquid nitrogen, so as to avoid undesired reactions. A crucible of the same weight as the sample container was used as the reference. The DTA curves were recorded under a continuous flow of argon (20 mL min−1) to avoid oxidation of the crucibles at high temperatures. Different thermal cycles were employed depending on the sample. The obtained thermograms were evaluated with the software Calisto, supplied by SETARAM with the DTA equipment. Prior and after the DTA experiments the samples were characterized by SEM/EDXS and/or XRPD.2.4 Computational details
DFT total energy calculations were performed for R2PdGe6 (R = Y and La) in the three structural modifications oS18, oS72 and mS36, and for La2MGe6 (M = Pt, Cu, Ag and Au) in the two orthorhombic modifications, by means of the plane wave pseudopotential code QUANTUM-ESPRESSO.22 The PBE functional23 for the exchange and correlation energy was used. Ultrasoft pseudopotentials,24 available in the “GBRV” open-source library,25 were employed for M, La and Y, whereas for Ge a norm-conserving pseudopotential, including the 4s and 4p valence orbitals, was used. The semicore states 4p for Pd, 5p for Pt, 3s and 3p for Cu, 4s and 4p for Ag and Y and 5s and 5p for La were treated as valence electrons. The Brillouin zone was sampled within uniform grids generated with different k-points for the three polymorphs: 12 × 12 × 2 for the oS18 modification, 6 × 6 × 2 for oS72 and 6 × 6 × 4 for mS36. The plane-wave and density cut-off were set to 45 Ry and 450 Ry, respectively. The orbital occupancies at the Fermi level were treated with a Gaussian smearing of 0.01 Ry.3. Results and discussion
3.1 The formation of R2PdGe6 germanides along the R series
The results of SEM/EDXS characterization confirm the presence of the R2PdGe6 phase in most of the samples prepared by direct synthesis (see Table 3).| Sample code (R) | R2PdGe6 phase composition [EDXS, at%] R; Pd; Ge | Other known detected phases | ||
|---|---|---|---|---|
| a Samples where single crystals were taken. | ||||
| Sc | – | Sc(Pd,Ge)2−x | Ge | ScPdGe |
| Ya | 21.0; 10.8; 68.2 | YPdGe2 | ||
| La | 22.4; 9.9; 67.7 | La(Pd,Ge)2−x | Ge | LaPdGe3 |
| La,af | 22.4; 10.5; 67.1 | La(Pd,Ge)2−x | Ge | |
| Cea | 22.5; 10.8; 66.7 | Ge | Ce2Pd3Ge5 | |
| Pra | 20.2; 11.4; 68.4 | Ge | Pr2Pd3Ge5 | |
| Pr,af | 20.7; 11.0; 68.3 | Pr(Pd,Ge)2−x | Ge | |
| Nda | 21.8; 11.2; 67.0 | NdPdGe2 | Ge | Nd2Pd3Ge5 |
| Sm | 21.5; 11.1; 67.4 | Ge | Sm2Pd3Ge5 | |
| Eu | – | Eu(Pd,Ge)2−x | Ge | EuPdGe3 |
| Gd | 22.2; 11.4; 66.4 | Gd(Pd,Ge)2−x | ||
| Tb | 22.2; 11.1; 66.7 | TbPdGe2 | ||
| Dy | 22.7; 10.9; 66.4 | DyPdGe2 | ||
| Ho | 22.3; 11.3; 66.4 | Ho(Pd,Ge)2−x | ||
| Era | 22.7; 11.0; 66.3 | Er(Pd,Ge)2−x | ||
| Tm | 23.0; 11.1; 65.9 | Tm(Pd,Ge)2−x | ||
| Yb | 23.1; 10.7; 66.2 | Yb(Pd,Ge)2−x | ||
| Yb,af | 23.2; 10.9; 65.9 | YbGe2−x | ||
| Lua | 23.8; 10.6; 65.6 | Lu(Pd,Ge)2−x | Ge | |
According to the SEM micrographs, under the applied experimental conditions for direct synthesis, R2PdGe6 is generally the highest yield phase. As it is common for alloys prepared by slow cooling from liquid, many secondary phases were detected, among which R(Pd,Ge)2−x, R2Pd3Ge5, RPdGe2 and Ge are the most common. Representative microstructures are available in the ESI.†
In samples with R = Sc and Eu no traces of the R2PdGe6 phase were detected: in both cases three-phase alloys were obtained, in which an already known ternary compound (ScPdGe or EuPdGe3) coexists with Ge and the binary RGe2−x phase dissolving a small amount of Pd.
In the La22.2Pd11.1Ge66.7 sample prepared by direct synthesis employing cycle I (sample code “La” in Table 3), only a small amount of La2PdGe6 was found, in the form of a thin border around big LaGe2−x crystals (see Fig. 1a). From such a sample, it was not possible to isolate suitable single crystals. In the literature, this phase has been reported to crystallize in the oS18-Ce2CuGe63 or in the oS72-Ce2(Ga0.1Ge0.9)7 model,4 the latter with no complete structural data available. For this reason, further attempts were made to synthesize a La2PdGe6 sample convenient for structural resolution, which are briefly accounted for in the following.
 | ||
| Fig. 1 Micrographs (SEM-BSE mode) of representative samples of La22.2Pd11.1Ge66.7 nominal composition (a–d) and of La21Pd7Ge72 nominal composition (e, f) obtained under the following experimental conditions: (a) synthesis in the resistance furnace, cycle I; (b) arc melting; (c, d) arc melting followed by one DTA cycle; (e) synthesis in the resistance furnace with In flux, cycle II; (f) synthesis in the resistance furnace with In flux, cycle III. | ||
The as-cast samples with nominal composition La22.2Pd11.1Ge66.7 did not show any trace of La2PdGe6, independently using the melting method (arc or induction furnace). These samples are characterized by a clear microstructure, where crystals of La(Pd,Ge)2−x and LaPdGe3 coexist with an eutectic structure containing Ge (see Fig. 1b). Annealing at 700 °C of an as-cast sample for 2 weeks, performed on the basis of literature data,4 did not succeed in obtaining the desired compound; what is more, the same annealing treatment carried out on the sample (La) prepared using cycle I caused the thin La2PdGe6 border to disappear.
At this point, it became clear that La2PdGe6 behaves somewhat differently from the other rare earth homologues, and DTA measurements were performed in order to gain insights into its temperature formation.
The DTA curve obtained at a heating rate of 5 °C min−1 (Tmax = 1100 °C), is shown in Fig. S2 (ESI†). Four thermal effects were detected at the following temperatures (onset): 800 °C, 886 °C, 927 °C and 1027 °C. The same thermal effects are recorded on cooling. The peak at 800 °C is in good agreement with the temperature of the binary eutectic equilibrium L → (Ge) + LaGe2−x reported at T = 810 °C.26 However, the other peaks are not easily interpretable. After this thermal cycle the presence of a small amount of La2PdGe6 was indeed detected by SEM-EDXS analysis in the form of a border between La(Pd,Ge)2−x and LaPdGe3 (see Fig. 1c). In a restricted region of the sample after DTA, bigger crystals of La2PdGe6 are present enclosing a brighter core of LaPdGe3 (see Fig. 1d). The measured global composition of this region is La21Pd7Ge72. Aiming to obtain a higher yield of the desired compound, different annealing treatments (lasting for one month each) were performed at 830 °C, 890 °C and 1000 °C: temperatures were chosen slightly below the recorded thermal effects. Unfortunately, no traces of 2:1:6 were found after all these treatments. From the gathered results, it was concluded that La2PdGe6 is probably a metastable phase which is likely to form only in small amounts during relatively slow cooling.
It is known that alternative synthetic routes may help to produce metastable compounds; among them, the flux method was targeted, taking into account both literature data27 and our previous results on related R2Pd3Ge5 compounds.28 As a metal solvent In was chosen considering its ability to dissolve Ge, rare earth and transition metals, without forming In–Ge binary compounds, allowing the formation of ternary phases.27,29 Thus, several attempts were made varying both the nominal composition and the thermal cycle. Good results were obtained starting from the nominal composition La21Pd7Ge72: the 2:1:6 phase was obtained using both cycle II and cycle III (see Fig. 1e and f). After cycle II, however, La2PdGe6 is almost always found as a grey border of big La(Pd,Ge)2−x crystals and only a few single phase crystals were detected. On the contrary, after cycle III, characterized by a lower maximum temperature (750 °C instead of 1000 °C), many 2:1:6 single crystals were obtained, allowing further crystal structure determination after their separation from the In-flux (sample indicated by code “La,af” in Table 3). More details on all samples prepared with La can be found in the ESI.†
Unexpectedly, the flux synthesized La2PdGe6 turned out to be monoclinic, differently from all the other series of samples prepared by direct synthesis (see section 3.2): for this reason the flux method (cycle III) was tested also on Pr21Pd7Ge72 and Yb21Pd7Ge72 samples. In both cases R2PdGe6 phases were detected and analysed by X-ray diffraction (see Table 3); particularly good quality big crystals were obtained in the case of R = Yb (see Fig. 2).
 | ||
| Fig. 2 SEM-BSE micrographs for the Yb2PdGe6 single crystal before (a) and after (b) the selective In oxidation. | ||
3.2 Crystal structure of R2PdGe6 (R = Y, La (flux), Ce, Pr, Pr (flux), Nd, Er, Yb(flux) and Lu)
To definitively establish the crystal structure of R2PdGe6 only the X-ray powder diffraction data are not sufficient. In fact, differences are hardly visible in the calculated diffraction patterns of the orthorhombic and the monoclinic models (see the ESI†). Additionally, the recognition of the correct model is hampered by the fact that samples are usually multiphase giving strong peak overlaps. Hence, single crystal X-ray diffraction is the fundamental method used for the structural investigations conducted here.For single crystals of R2PdGe6 (R = Y, Ce, Pr, Nd, Er, Yb(f) and Lu) compounds (see. Table 1) the cell indexation was straightforward giving an orthorhombic C-centered cell (only h + k = 2n reflections were observed). The analysis of systematic extinctions suggested Cc2e (no. 41) and Cmce (no. 64) as the possible space groups. An almost complete structural model was obtained in the Cmce space group in a few iteration cycles by using the charge-flipping algorithm implemented in JANA2006.30 In this model the rare earth atoms are situated in a 16g general site and palladium occupies the 8f site, while all the other positions are assigned to the lighter germanium atoms. The obtained models have the oS72 Pearson symbol and correspond to the R2PdGe6 stoichiometry, satisfactorily matching with the EDXS microprobe analysis data.
The further structure refinements were carried out by full-matrix least-squares methods on |F2| using the SHELX programs.31 The site occupancy factors of all species were checked for deficiency, in separate cycles of refinement, obtaining values very close to unity. At this point neither the deficiency nor statistical mixture was considered and stoichiometric R2PdGe6 models were further anisotropically refined giving acceptable residuals and flat difference Fourier maps. The results indicate that the Y, Ce, Pr, Er and Lu containing compounds prepared by direct synthesis are isopointal with the Ce2(Ga0.1Ge0.9)7 prototype. The same is true also for the Yb containing compound prepared by flux synthesis (the image of this crystal is reported in Fig. 2).
The atomic positions of the R2PdGe6 compounds are listed in Table 4 together with the equivalent isotropic displacement parameters. The positions were standardized with the Structure Tidy program.32 The interatomic distances are available in the ESI.†
| Atom (site) | Atomic param. | R = Y | R = Ce | R = Pr | R = Nd | R = Er | R = Yb (f) | R = Lu |
|---|---|---|---|---|---|---|---|---|
| R | x/a | 0.25102(5) | 0.25042(2) | 0.25041(2) | 0.25049(2) | 0.25120(2) | 0.24952(3) | 0.25159(3) |
| (16g) | y/b | 0.37584(5) | 0.37513(5) | 0.3753(1) | 0.3751(1) | 0.37605(3) | 0.37604(3) | 0.37592(4) |
| z/c | 0.08125(2) | 0.08270(2) | 0.082540(9) | 0.082326(8) | 0.080953(8) | 0.08257(2) | 0.08075(1) | |
| U eq (Å2) | 0.0065(1) | 0.00492(6) | 0.00671(6) | 0.00527(5) | 0.00541(6) | 0.00529(8) | 0.0072(1) | |
| Ge1 | x/a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (8f) | y/b | 0.13181(8) | 0.1285(1) | 0.1286(2) | 0.1291(3) | 0.1332(1) | 0.1304(1) | 0.1345(1) |
| z/c | 0.02868(3) | 0.03066(2) | 0.03042(3) | 0.03023(2) | 0.02798(3) | 0.02917(4) | 0.02720(5) | |
| U eq (Å2) | 0.0073(2) | 0.0063(1) | 0.0084(1) | 0.0065(1) | 0.0063(1) | 0.0062(1) | 0.0086(2) | |
| Ge2 | x/a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (8f) | y/b | 0.11933(8) | 0.1217(1) | 0.1213(2) | 0.1213(3) | 0.1185(1) | 0.1205(1) | 0.1177(1) |
| z/c | 0.45753(3) | 0.46255(2) | 0.46216(3) | 0.46146(2) | 0.45619(3) | 0.46051(4) | 0.45459(5) | |
| U eq (Å2) | 0.0083(2) | 0.0075(1) | 0.0089(1) | 0.0077(1) | 0.0076(1) | 0.0082(2) | 0.0097(2) | |
| Ge3 | x/a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (8f) | y/b | 0.40519(8) | 0.40516(7) | 0.40475(8) | 0.40492(8) | 0.40524(8) | 0.4047(1) | 0.4052(1) |
| z/c | 0.19344(3) | 0.19462(3) | 0.19441(3) | 0.19422(3) | 0.19305(3) | 0.19403(4) | 0.19299(5) | |
| U eq (Å2) | 0.0073(2) | 0.0076(1) | 0.0089(2) | 0.0072(2) | 0.0062(1) | 0.0048(1) | 0.0085(2) | |
| Ge4 | x/a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (8f) | y/b | 0.34733(8) | 0.34524(7) | 0.34608(8) | 0.34617(8) | 0.34763(8) | 0.3476(1) | 0.3484(1) |
| z/c | 0.30752(3) | 0.30553(3) | 0.30572(3) | 0.30599(3) | 0.30810(3) | 0.30685(4) | 0.30862(5) | |
| U eq (Å2) | 0.0075(2) | 0.0077(1) | 0.0086(2) | 0.0069(2) | 0.0067(1) | 0.0060(2) | 0.0079(2) | |
| Ge5 | x/a | 0.27688(6) | 0.27767(5) | 0.27735(4) | 0.27729(4) | 0.27700(5) | 0.27568(7) | 0.27669(8) |
| (16g) | y/b | 0.12631(5) | 0.12531(8) | 0.12540(1) | 0.1254(2) | 0.12658(7) | 0.12614(7) | 0.1267(1) |
| z/c | 0.19308(2) | 0.19461(2) | 0.19442(2) | 0.19416(2) | 0.19259(2) | 0.19379(2) | 0.19237(3) | |
| U eq (Å2) | 0.0073(1) | 0.00764(8) | 0.00864(9) | 0.00686(8) | 0.00625(9) | 0.0055(1) | 0.0082(2) | |
| Pd | x/a | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| (8f) | y/b | 0.12736(5) | 0.12526(6) | 0.12555(6) | 0.12572(6) | 0.12762(6) | 0.12666(7) | 0.12826(9) |
| z/c | 0.14223(2) | 0.14568(2) | 0.14496(2) | 0.14453(2) | 0.14153(2) | 0.14132(2) | 0.14062(3) | |
| U eq (Å2) | 0.0060(1) | 0.00664(8) | 0.00765(9) | 0.00585(8) | 0.00504(8) | 0.0039(1) | 0.0068(2) | |
The La2PdGe6 crystal selected for X-ray analysis, taken from a flux synthesized sample, is an example of a non-merohedral twin composed of two domains of comparable dimensions. Normally, twins of such a type have problems in the preliminary stages of cell indexing (leading to unexpectedly high values of unit cell parameters) and space group determination (showing inconsistency with any known space group systematic absence).33,34 Instead, in our case the unit cell indexing was straightforward and the following possible space groups were suggested for the C-centered monoclinic cell: C2 (no. 5), Cm (no. 8) and C2/m (no. 12). The lowest combined figure of merit was associated with the only centrosymmetric C2/m space group. These data strongly hint that the studied crystal is isostructural with the monoclinic mS36-La2AlGe6 prototype. A preliminary structural model, obtained by direct methods as implemented in WinGx,35 contained 1 La, 5 Ge and 1 Pd, giving the correct La2PdGe6 composition. Nevertheless, the isotropic thermal parameter values were not coherent for the different types of atoms and three additional intense peaks close to some of the Ge-positions were present in the difference Fourier maps at a distance of ∼0.05 nm. The latter three sites have no physical sense if completely occupied and therefore, the sum of occupations for each pair of very close Ge-sites was restrained to be unity in further cycles of least squares refinement. However, the refinement sticks at R1/wR2 of 0.05/0.17 with a noisy Fourier map give unreasonable thermal parameters when anisotropically refined. Even less chemically sound structural models were obtained with the possible non-centrosymmetric C2 and Cm space groups. At this point, more careful analysis of diffraction spots in reciprocal space was performed with RLATT and CELL_NOW.36 In fact, a regular spatial distribution of extra-peaks was revealed with respect to those associated with the mS36-like monoclinic cell. All of them could be satisfactorily indexed with a twice as big monoclinic base centered unit cell with a ∼ 8.2 Å, b ∼ 8.4 Å, c ∼ 22.6 Å, and β ∼ 100.5°. The structural model deduced in the C2/m space group by the charge-flipping algorithm30 became less disordered, since it contains only one partially occupied Ge position capping the distorted corrugated Ge fragment.2 Even if Ge-rich compounds are frequently off-stoichiometric or disordered,1 residuals remain unsatisfactory (0.09/0.14) with senseless anisotropic thermal parameters. For this reason, more attention was dedicated to the indexing procedure. According to a detailed output of CELL_NOW, one of the possible interpretations of the observed diffraction peak distribution is considering them originating from different domains of a non-merohedrically twinned crystal. In this case, the metric of the single domain remains the same as for the mS36 model. All the extra-peaks, instead, are due to the presence of the second domain, related to the first one by a 180° rotation twin law 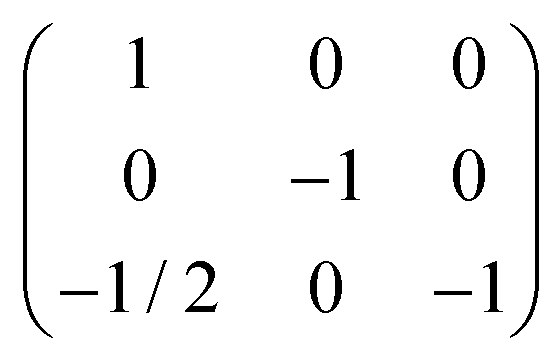 .
.
Consequently, the collected dataset was re-integrated assuming the presence of two domains and the hkl5 file for refinement was prepared by TWINABS.36 After that, excellent residuals were obtained (see Table 5) for the ordered La2PdGe6 model with the mS36-type structure. At the final cycles, this model was refined with anisotropic thermal displacement parameters for all atom sites. The refined fractional contribution k of the second domain is ca. 0.4.
| R2PdGe6 analogue | Atomic param. | R (8j) | Ge1 (4i) | Ge2 (4i) | Ge3 (4i) | Ge4 (4i) | Ge5 (8j) | Pd (4i) |
|---|---|---|---|---|---|---|---|---|
| R = La | x/a | 0.0840(1) | 0.1438(3) | 0.3585(3) | 0.0577(2) | 0.4970(2) | 0.2773(3) | 0.1985(2) |
| y/b | 0.24980(6) | 0 | 0 | 0 | 0 | 0.22206(8) | 0 | |
| z/c | 0.33354(3) | 0.5622(1) | 0.4281(1) | 0.1097(1) | 0.1093(1) | 0.10943(6) | 0.79356(6) | |
| U eq (Å2) | 0.0045(1) | 0.0058(2) | 0.0071(2) | 0.0092(4) | 0.0090(4) | 0.0083(2) | 0.0064(2) | |
| R = Pr | x/a | 0.0832(3) | 0.1453(6) | 0.3580(6) | 0.0573(3) | 0.4981(3) | 0.2778(4) | 0.1979(2) |
| y/b | 0.24962(7) | 0 | 0 | 0 | 0 | 0.2227(1) | 0 | |
| z/c | 0.33491(4) | 0.5609(2) | 0.4243(1) | 0.1115(2) | 0.1112(2) | 0.11130(9) | 0.7899(1) | |
| U eq (Å2) | 0.0047(2) | 0.0057(3) | 0.0075(3) | 0.0068(5) | 0.0070(5) | 0.0066(2) | 0.0056(2) | |
The orientation of twin domains and the corresponding reciprocal plots of the completely overlapped and non-overlapped hkl zones are shown in Fig. 3. Keeping in mind the twin law, it becomes clear that only reflections with h = 2n are affected by the twinning (i.e. they are completely overlapped). Considering also the presence of the only h + k = 2n reflections for the C-centered lattice, it is revealed that half of all measured intensities are affected by twinning. The intensity difference between overlapped/non-overlapped reflections is evident from the corresponding precession photos of the h2l and h1l zones of reciprocal space shown in Fig. 3.
 | ||
| Fig. 3 Upper part: Reciprocal orientation of twin domains together with theoretical reciprocal plots of h2l (totally overlapped) and h1l (non-overlapped) zones generated by XPREP.20 Colors of domains I (red) and II (green) are the same as those of the corresponding hkl reflections. For clarity, relationships between direct/reciprocal lattice vector lengths are not considered. Lower part: Experimental precession photos of h2l and h1l zones. | ||
The crystal structure of Pr2PdGe6 isolated from In flux was solved in the same way as for La2PdGe6, just described. Its structure turned out to be monoclinic mS36, with the same twinning law and with a similar volume ratio to the twinned domains (see Table 2 for more details).
The unit cell volumes of the studied compounds were plotted as a function of the trivalent rare earth metal radii37 (see Fig. 4). The monoclinic cell volume was doubled in order to compare the different related structures.2 Only the lattice parameters obtained from single crystal X-ray diffraction were considered.
 | ||
| Fig. 4 Normalized cell volumes (from single crystal data) of R2PdGe6 compounds as a function of the R3+ ionic radius. The cell volume of Dy2PdGe6 was taken as per ref. 14. | ||
In agreement with the lanthanide contraction, a linear decreasing trend is observed, suggesting a similar chemical bonding scenario for all the studied germanides. The literature data on Dy2PdGe614 fit quite well with the general trend. The most significant deviation is observed for Yb2PdGe6; this result is in good agreement with a recent magnetic investigation8 revealing for Yb a behavior typical of dynamic intermediate valence systems.
The crystal structure of the R2PdGe6 (R = Sm, Gd, Tb, Ho and Tm) compounds was not studied by single crystal X-ray diffraction. The powder diffraction patterns, recorded on the corresponding samples, can be satisfactorily indexed to both oS72 and mS36 structures. However, considering the results obtained on the directly synthesized samples with early and late rare earth metals, it is reasonable to suggest that under the same conditions even the abovementioned R2PdGe6 phases are of the oS72-Ce2(Ga0.1Ge0.9)7 type.
A similar rationalization could not be made for the crystal structure of the 2:1:6 germanides synthesized by the flux method since for Pr2PdGe6 a different structure was stabilized, instead for Yb2PdGe6 the same orthorhombic modification is formed.
3.3 From R2PdGe6 to R2MGe6 compounds: crystallochemical and DFT analyses targeting the correct structural model
From the results described above, it is clear that the oS18-Ce2CuGe6 structure is never realized in the R2PdGe6 series. Within this family, direct synthesis always produces compounds belonging to the oS72-Ce2(Ga0.1Ge0.9)7 structure type, except for La2PdGe6, whose structure under these conditions was not definitely confirmed. The In-flux synthetic route used in this study gave origin to mS36-La2AlGe6 twinned crystals of La2PdGe6 and Pr2PdGe6, whereas Yb2PdGe6 crystals grown from the same metal solvent are orthorhombic oS72. These results are coherent with recent structural studies on 2:1:6 germanides assigning to them the oS727,9,14,15 or the mS36 model17 instead of the previously proposed oS18.
In view of the following discussion it is convenient to distribute all the known 2:1:6 models within a concise scheme (Bärnighausen tree) starting from the oS20-SmNiGe3 aristotype. When considering the vacancy ordering phenomenon16,38,39 causing symmetry reduction, each structural model finds its location on a separate branch. To capture the changes taking place, it is sufficient to follow the M–Ge sub-lattice distortions as depicted in Fig. 5 (R atom positions are negligibly affected by the symmetry reduction).
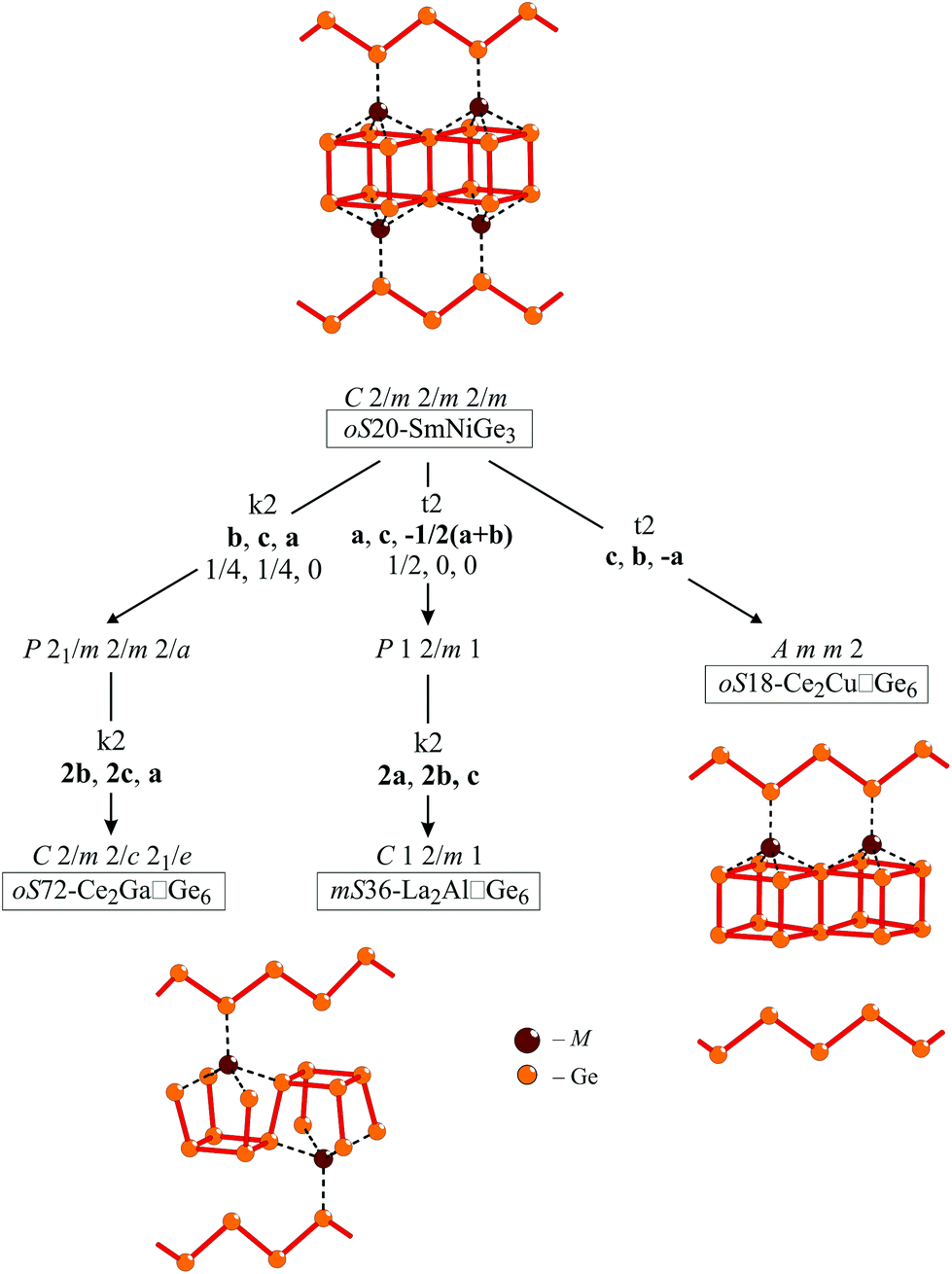 | ||
| Fig. 5 Bärnighausen tree relating to the SmNiGe3 aristotype and its orthorhombic and monoclinic vacancy variants. The type and indexes of the symmetry reductions are indicated. For clarity, only M–Ge frameworks are shown for each structural model; Ge–Ge contacts are shown in red, M–Ge interactions by a dotted line. | ||
From the crystal chemical point of view, the oS18 model is somewhat suspicious:
- it contains many independent crystallographic Ge sites, not in agreement with the symmetry principle;40
- the inhomogeneous vacancy distribution implies that corrugated Ge layers are linked to the zig-zag germanium chains through bridging M atoms only on one side (see Fig. 5).
To corroborate this idea arising from symmetry considerations, the DFT total energy values were calculated for La2PdGe6 and Y2PdGe6 with the oS18, oS72 and mS36 structures. The structural data available in the literature or obtained during this work were used as the starting point for geometric relaxation. In the case of Y2PdGe6, for which the mS36 model has never been reported, the cell dimensions and atomic positions of the La-analogue were chosen. The choice of these rare earths allows avoiding the presence of highly correlated electrons lying in partially filled f states. The relaxed structures, obtained at the end of the variable-cell calculations, showed lattice constants about 2% larger with respect to the literature data and results presented in this work, as expected when using the PBE functional (see Table 6).
| Compound | Experimental | Calculated | |||||
|---|---|---|---|---|---|---|---|
| oS18 | oS72 | mS36 | oS18 | oS72 | mS36 | ||
| Y2PdGe6 | a (Å) | 4.0790(4) | 8.1703(5) | 4.1502 | 8.3117 | 8.2181 | |
| b (Å) | 4.0168(5) | 8.0451(5) | 4.0802 | 8.2239 | 8.3140 | ||
| c (Å) | 21.525(2) | 21.558(2) | 22.212 | 21.857 | 11.123 | ||
| β (°) | 100.5 | ||||||
| V (Å3) | 352.7(1) | 1417.1(2) | 376.14 | 1494.0 | 747.16 | ||
| Energy (eV per atom) | −691.647 | −691.713 | −691.713 | ||||
| Ref. | 3 | ||||||
| La2PdGe6 | a (Å) | 4.2117(3) | 8.430(1) | 8.2163(8) | 4.2914 | 8.5552 | 8.3910 |
| b (Å) | 4.1100(3) | 8.2180(7) | 8.4161(9) | 4.1893 | 8.3902 | 8.5554 | |
| c (Å) | 22.265(5) | 22.192(3) | 11.294(1) | 22.913 | 22.642 | 11.515 | |
| β (°) | 100.5(1) | 100.5 | |||||
| V (Å3) | 385.4(1) | 1537.4(4) | 768.0(1) | 411.92 | 1625.2 | 812.81 | |
| Energy (eV per atom) | −721.181 | −721.244 | −721.244 | ||||
| Ref. | 3 | 4 | |||||
On the basis of the obtained results the oS18 structural model is the worst: its total energy is higher by 0.063 eV per atom for La2PdGe6 and by 0.066 eV per atom in the case of Y2PdGe6 with respect to both oS72 and mS36 models.
Calculations for the oS18 and oS72 models were performed on other La2MGe6 analogues (M = Pt, Cu, Ag and Au). As can be seen from the plot shown in Fig. 6, the highest energy value corresponds always to the oS18 model (ΔE(eV per at) = EoS72 − EoS18, is always negative).
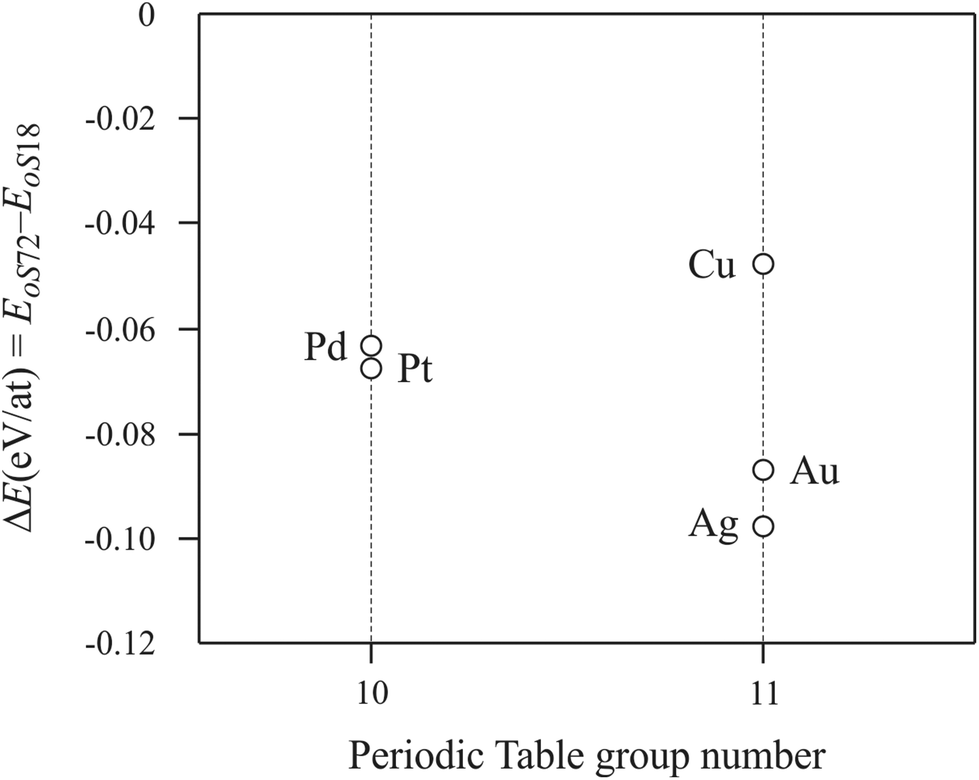 | ||
| Fig. 6 ΔE vs. nature of late transition element M for La2MGe6. | ||
The mS36 model was not considered for energy calculations of La2MGe6 because the results on La2PdGe6 and Y2PdGe6 (Table 6) do not show any energy difference between oS72 and mS36. In fact, they can be considered as two different polytypes of 2:1:6 composition. They both consist of geometrically equivalent layers of defective BaAl4 (of R□MGe2 composition), AlB2 and α-Po type slabs stacked linearly along the c direction.2,41 These layers are also energetically equivalent, and consequently no strong preference for one of the polytypes can be envisaged. Frequently, the structure of such compounds is sensitive to the crystallization conditions and small fluctuations of these may reverse the energetic preferences and at the same time give origin to stacking faults, twinnings or non-periodic structures; as an example the families of SiC, ZnS, CdI2, micas, etc. can be cited.42
These findings do not preclude the possibility that new polytypes exist for this numerous group of compounds. One can suppose that modulated structures may form within this family. The non-periodicity could arise from a vacancy ordering or Ge covalent fragment distortions with a periodicity different from that of lattice. This phenomenon was addressed in a recent paper on structurally related germanides.43
Some more aspects associated with the symmetry reduction scheme can be highlighted. The symmetry reduction path conducive to the mS36 model contains a translationengleiche transformation of index 2 that is in perfect agreement with the fact that non-merohedral two domain twins are formed. The presence of klassengleiche relations, instead, should be at the origin of antiphase domains in both oS72 and mS36 polytypes. These domains are not detectable by conventional X-ray diffraction techniques; however, the quality and dimensions of single crystals obtained by the flux method are well promising for further transmission electron microscopic investigations to achieve this goal.
It remains, however, unclear if the oS20 aristotype has any physical meaning. Further investigations should be performed aiming to clarify if elevated temperature/pressure conditions may stabilize the hypothetical oS20-RPdGe3. If yes, the found non-merohedral twins have been developed by a phase transition in the solid state. Otherwise, their formation took place during the growth of the crystal.
4. Conclusions
The R2PdGe6 series of compounds was targeted with the aim to elucidate more on the formation/crystal structure of the R2MGe6 family (R = rare earth metal, M = another metal), combining the experimental results with structure analysis and energy calculations. This study was motivated by the controversial and sometimes erroneous structural data available for the title compounds, possibly also affecting the interpretation of the numerous magnetic measurements performed by several authors.The R2PdGe6 phase was detected in all samples prepared by direct synthesis, except for R = Sc and Eu.
Single crystal X-ray analyses conducted on several representative samples (R = Y, Ce, Pr, Nd, Er and Lu) indicated that oS72-Ce2(Ga0.1Ge0.9)7 is the correct structural model, as previously reported for the Dy and Yb analogues. The same crystal structure is suggested for R = Sm, Gd, Tb, Ho and Tm on the basis of powder X-ray diffraction measurements and behavior regularities along the rare-earth series.
Different annealing treatments along with thermal analysis investigations were performed with the aim to obtain good crystals or a sufficiently high yield of La2PdGe6; the obtained results suggest that this is a metastable phase likely to form in a small amount during slow cooling treatments.
Therefore, the flux synthesis, able to stabilize metastable phases, was explored, using In as a metal solvent. The good quality La2PdGe6 crystals obtained after optimizing this method are non-merohedrally twins of the mS36-La2AlGe6 structure. Monoclinic twins of two equally big domains of the same morphology were obtained for the flux-synthesized Pr2PdGe6 compound. This result suggests that also La2PdGe6 obtained from direct synthesis might be of the oS72 orthorhombic structure. Instead, for the heavy rare earth representative Yb2PdGe6 the flux does not stabilize the monoclinic structure, and the oS72 model remains the correct one.
Taking into account the considerable amount of data on 2:1:6 germanides, some structure-rationalizing idea was pursued. Considering the vacancy ordering phenomenon to be the key-factor for structural changes, a compact Bärnighausen tree was constructed, with rigorous group–subgroup relations between the oS20-SmNiGe3 aristotype and the three possible derivatives oS72, mS36 and oS18: their structural models are localized on separate branches of the tree, representing different symmetry reduction paths. The presence of a t2 reduction step on the path bringing to the mS36 model is coherent with the formation of twinned crystals.
Both from our experience and literature data,44,45 binary and ternary germanides are particularly prone to geminate, and this possibility should be carefully investigated during structural solution. For this reason these phases are also suitable for studies targeted to better understand the origin, formation conditions and type of twinning in intermetallics. At present, we are further studying twinned crystals of germanides, where twinning seems to be related to vacancy ordering phenomena.
The same phenomena can lead to modulated structures, as already found for binary RGe2−x46 and ternary germanides,46 representing another reason for interest in the studied compounds.
From the presented symmetry reduction scheme the symmetry discrepancy became obvious between the oS18 (2nd order derivative of the aristotype) and both the oS72 and mS36 models (4th order derivatives), highlighting the poor crystal chemical reliability of the structure mostly reported in the literature and already corrected for several R2MGe6 compounds.
Aiming to confirm this idea and to discard energetically less probable modifications, DFT total energy calculations were performed for R2PdGe6 (R = Y and La) in the three abovementioned structural models, and for La2MGe6 (M = Pt, Cu, Ag and Au) in the oS18 and oS72 modifications. Structure optimization was performed for all calculations. Considering the total energy highest values as well as the crystallochemical factors, the oS18-Ce2CuGe6 model is less probable. It was also concluded that the oS72 and mS36 models, containing geometrically equivalent fragments, are energetically equivalent polytypes. In fact, they are sensitive to the crystallization conditions, as it became clear from the results of the flux synthesis. This approach turned out to be an effective alternative method to prepare these compounds.
The mostly structural study presented in this paper constitutes a good and essential basis for further investigations concerning the chemical bonding for R2MGe6, which is already one of the objects of our research work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank D. Ceresoli (CNR-ISTM, Milano) for support in DFT calculations. DMP acknowledges the Ministry of Education and Science of Russia (grant 14.B25.31.0005).References
-
P. Villars and K. Cenzual, Pearson's Crystal Data, ASM International, Ohio, USA, Release 2016/17 Search PubMed
.
- P. Solokha, S. De Negri, D. M. Proserpio, V. A. Blatov and A. Saccone, Inorg. Chem., 2015, 54, 2411 CrossRef CAS PubMed
- O. L. Sologub, K. Hiebl, P. Rogl and O. I. Bodak, J. Alloys Compd., 1995, 227, 37 CrossRef CAS
- M. Konyk, L. Romaka, D. Gignoux, D. Fruchart, O. Bodaka and Yu. Gorelenko, J. Alloys Compd., 2005, 398, 8 CrossRef CAS
- K. Shigetoh, D. Hirata, M. A. Avila and T. Takabatake, J. Alloys Compd., 2005, 403, 15 CrossRef CAS
- D. Kaczorowski, M. Konyk, A. Szytu1a, L. Romaka and O. Bodak, Solid State Sci., 2008, 10, 1891 CrossRef CAS
- R. Wawryk, R. Tróc and A. V. Gribanov, J. Alloys Compd., 2012, 520, 255 CrossRef CAS
- D. Kaczorowski, M. Konyk and L. Romaka, J. Alloys Compd., 2012, 526, 22 CrossRef CAS
- D. Kaczorowski, A. V. Gribanov, P. Rogl and S. F. Dunaev, J. Alloys Compd., 2016, 685, 957 CrossRef CAS
- M. B. Konyk, L. P. Romaka, Yu. K. Gorelenko and O. I. Bodak, J. Alloys Compd., 2000, 311, 120 CrossRef CAS
- V. V. Pavlyuk, V. K. Pecharskii and O. I. Bodak, Kristallografiya, 1988, 33, 43 CAS
- A. Stetskiv, R. Misztal and V. V. Pavlyuk, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, i60 CAS
- C. Rizzoli, O. Sologub and P. Salamakha, J. Alloys Compd., 2003, 351, L10 CrossRef CAS
- A. Gribanov, S. Safronov, E. Murashova and Y. Seropegin, J. Alloys Compd., 2012, 542, 28 CrossRef CAS
- M. L. Fornasini, P. Manfrinetti and A. Palenzona, Z. Kristallogr., 2002, 217 NCS, 173 Search PubMed
- S. De Negri, P. Solokha, M. Skrobańska, D. M. Proserpio and A. Saccone, J. Solid State Chem., 2014, 218, 184 CrossRef CAS
- S. C. Peter, U. Subbarao, S. Sarkar, G. Vaitheeswaran, A. Svane and M. G. Kanatzidis, J. Alloys Compd., 2014, 589, 405 CrossRef CAS
- J. T. Zhao, K. Cenzual and E. Parthè, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1777 CrossRef
-
Yu. Grin, in Modern Perspectives in Inorganic Crystal Chemistry, ed. E. Parthè, Kluwer Academic Publishers, Dordrecht, the Netherlands, 1992, p. 77 Search PubMed
-
Bruker, APEX2, SAINT-Plus, XPREP, SADABS and TWINABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed
- W. Kraus and G. Nolze, J. Appl. Crystallogr., 1996, 29, 301 CrossRef CAS
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892 CrossRef
- GBRV high-throughput pseudopotential, http://physics.rutgers.edu/gbrv, (accessed May 2017).
-
T. B. Massalski, Binary Alloy Phase Diagrams, American Society for Metals, Metals Park, Oh 44073, USA, 1990, vol. 2 Search PubMed
- M. G. Kanatzidis, R. Pöttgen and W. Jeitschko, Angew. Chem., Int. Ed., 2005, 44, 6996 CrossRef CAS PubMed
- P. Solokha, R. Freccero, S. De Negri, D. M. Proserpio and A. Saccone, Struct. Chem., 2016, 27, 1693 CrossRef CAS
- J. R. Salvador, J. R. Gour, D. Bilc, S. D. Mahanti and M. G. Kanatzidis, Inorg. Chem., 2004, 43, 1403 CrossRef CAS PubMed
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr., 2014, 229, 345 CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- L. M. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139 CrossRef
-
P. Müller, Crystal structure refinement: a crystallographer`s guide to SHELXL, Oxford University Press, Oxford, UK, 2006 Search PubMed
-
W. Clegg, Crystal structure analysis: principle and practice, Oxford University Press, Oxford, UK, 2nd edn, 2009 Search PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS
-
Bruker, APEX2, SAINT-Plus, XPREP, SADABS, CELL_NOW and TWINABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2014 Search PubMed
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751 CrossRef
- S. C. Peter, M. Chondroudi, C. D. Malliakas, M. Balasubramanian and M. G. Kanatdidis, J. Am. Chem. Soc., 2011, 133, 13840 CrossRef CAS PubMed
- J. Christensen, S. Lidin, B. Malaman and G. Venturini, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 272 CAS
-
U. Müller, Symmetry Relationships between Crystal Structures. Applications of Crystallographic Group Theory in Crystal Chemistry, Oxford University Press, Oxford, UK, 2013 Search PubMed
- Y. O. Tokaychuk, Y. E. Filinchuk, A. O. Fedorchuk, A. Yu. Kozlov and I. R. Mokra, J. Solid State Chem., 2006, 179, 1323 CrossRef CAS
-
E. Prince, International Tables for Crystallography, Vol. C, Mathematical, physical and chemical tables, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed
- J. Zhang, Y. Liu, C. H. Shek, Y. Wang and S. Bobev, Dalton Trans., 2017, 46, 9253 RSC
- S. Budnyk, F. Weitzer, C. Kubata, Y. Prots, L. G. Akselrud, W. Schnelle, K. Hiebl, R. Nesper, F. R. Wagner and Y. Grin, J. Solid State Chem., 2006, 179, 2329 CrossRef CAS
- O. Shcherban, I. Savysyuk, N. Semuso, R. Gladyshevskii and K. Cenzual, Chem. Met. Alloys, 2009, 2, 115 Search PubMed
- P. H. Tobash, D. Lins and S. Bobev, Inorg. Chem., 2006, 45, 7286 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format; back scattered electron images of alloy surfaces; summary of attempts to synthesize the La2PdGe6 compound; calculated X-ray powder patterns; interatomic distances obtained by single crystal X-ray analysis. See DOI: 10.1039/c7dt02686b |
| This journal is © The Royal Society of Chemistry 2017 |