
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLi3Co1.06(1)TeO6: synthesis, single-crystal structure and physical properties of a new tellurate compound with CoII/CoIII mixed valence and orthogonally oriented Li-ion channels†
Gunter
Heymann
 *a,
Elisabeth
Selb
a,
Michaela
Kogler
b,
Thomas
Götsch
b,
Eva-Maria
Köck
b,
Simon
Penner
b,
Martina
Tribus
c and
Oliver
Janka
d
*a,
Elisabeth
Selb
a,
Michaela
Kogler
b,
Thomas
Götsch
b,
Eva-Maria
Köck
b,
Simon
Penner
b,
Martina
Tribus
c and
Oliver
Janka
d
aInstitut für Allgemeine, Anorganische und Theoretische Chemie, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria. E-mail: Gunter.Heymann@uibk.ac.at; Fax: +43(0)512-507 57099
bInstitut für Physikalische Chemie, Leopold-Franzens-Universität Innsbruck, Innrain 52c, A-6020 Innsbruck, Austria
cInstitut für Mineralogie und Petrographie, Leopold-Franzens-Universität Innsbruck, Innrain 52, A-6020 Innsbruck, Austria
dInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 28/30, D-48149 Münster, Germany
First published on 25th August 2017
Abstract
A tellurate compound with CoII/CoIII mixed valence states and lithium ions within orthogonally oriented channels was realized in Li3Co1.06(1)TeO6. The single-crystal structure determination revealed two independent and interpenetrating Li/O and (Co,Te)/O substructures with octahedral oxygen coordination of the metal atoms. In contrast to other mixed oxides, a honeycomb-like ordering of CoO6 and TeO6 octahedra was not observed. Li3Co1.06(1)TeO6 crystallizes orthorhombically with the following unit cell parameters and refinement results: Fddd, a = 588.6(2), b = 856.7(2), c = 1781.5(4) pm, R1 = 0.0174, wR2 = 0.0462, 608 F2 values, and 33 variables. Additional electron density in tetrahedral voids in combination with neighboring face-linked and under-occupied octahedral lithium sites offers an excellent possible diffusion pathway for lithium ions. According to the symmetry of the crystal structure the diffusion pathways in Li3Co1.06(1)TeO6 were found in two orthogonal orientations. The CoII/CoIII mixed valence was investigated via X-ray photoelectron spectroscopy (XPS), revealing a composition comparable to that derived from single-crystal X-ray diffractometry. Magnetic susceptibility measurements underlined the coexistence of CoII and CoIII, the title compound, however, showed no magnetic ordering down to low temperatures. The ionic conductivity of Li3Co1.06(1)TeO6 was determined via alternating current (AC) electrochemical impedance spectroscopy and was found to be in the range of 1.6 × 10−6 S cm−1 at 573 K.
1 Introduction
Nowadays, there is a great demand for efficient electrical energy storage not only for mobile electric devices but also for regenerative energy resources like solar and wind power. In the case of rechargeable Li-ion batteries, the potentially best chance for improvement lies in the development of new cathode materials. Since 1977, several cathode materials have been used starting with titanium disulphide TiS2, followed by lithium cobalt dioxide LiCoO2 (1991), lithium manganese spinel LiMn2O4, and, finally, lithium iron phosphate LiFePO4 (2006).1 Because of their high stability and easy synthetic accessibility, oxide-based inorganic materials play a major role in the innovation process of electric components.Hitherto, only little research was carried out in the field of lithium transition metal tellurates as potential cathode materials. This can be attributed to difficulties in the crystallization of these materials and the following crystal structure investigation mostly possible only from powder diffraction data. Recently, a new lithium rich material Li4NiTeO6 was synthesized by Sathiya et al., which showed an excellent volume stability and a relatively high capacity of 110 mA h g−1, based on a 2 e− redox mechanism associated with the Ni2+/Ni4+ redox couple.2 These findings were derived from first-principles investigations on the delithiation process during charging.3 McCalla et al. reported on a material in the lithium rich Li–Fe–Te–O system that exhibited no transition metal oxidation during the charging process. Two oxygen-related mechanisms were identified to contribute to the whole capacity change of Li4.27Fe0.57TeO6 during charge and discharge.4 The crystal structures of many of these mixed oxides (including various superstructure variants) can be attributed to the general formula Ax(M,L)O2 consisting of alkali cations A and brucite-like layers of (M,L)O3/6 octahedra. LiCoO2 and its substitution variants with intercalated lithium ions between the octahedral slabs are excellent electrode materials for Li-ion batteries.5 With sodium instead of lithium, NaxCoO2 bronzes are formed, which are efficient thermoelectric materials or even superconductors when hydrated.6,7 Furthermore, these partially complex oxidic materials show very high alkali-cation conductivity. From tellurates, the compounds Na2M2TeO6 (M = Mg, Co, Ni, Zn)8 and Na2LiFeTeO6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 should be mentioned here exhibiting a pure ionic conductivity of 4–11 S m−1 at 573 K. Depending on the different stacking of the brucite-like (M,L)O3/6 octahedral layers and in particular on the different coordination of the interlayer alkali cations, various compounds can be classified into different polytypes named O3, P2, P3, etc. In this notation, according to Fouassier et al.,10 O stands for octahedral and P for trigonal prismatic coordination of the A ions and the digit for the number of brucite-like layers in the unit cell. In many cases, honeycomb-like superlattice ordering of the two heterovalent M and L cations was observed by the surroundings of isolated LO6 octahedra from six MO6 octahedra in a honeycomb-like arrangement. General compositions of these rock-salt type superstructures are Li3MII2LO6 (M = Mg, Co, Ni, Cu; L = Nb, Ta, Sb, Bi)11–17 and Na3MIII2LO6 (M = Mg, Co, Ni, Cu; L = Sb).18–20 Compounds belonging to the family of tellurates are A2MII2TeO6 (A = Li, Na; M = Mg, Co, Ni, Cu, Zn)8,21,22 and the recently reported Li8MII2Te2O12 (M = Co, Ni, Cu, Zn),23 which belongs to the former known series Li4MIITeO6 (M = Ni, Zn).2,24 An elegant method to obtain isostructural honeycomb-like ordered structures at low temperatures, inaccessible by direct solid state high-temperature reactions, is alkali ion-exchange experiments, as was shown for Na2Cu2TeO6.22 Besides the MII/TeVI honeycomb-like ordering, Tarte et al. investigated a possible MIV/TeVI cation ordering and identified two different structure types for the compounds Li2(Zr,Hf)TeO625 and Li2(Ti,Sn)TeO6.26 To the best of our knowledge, in these ordered AxMyTeO6 compounds always a heterovalence of MII/TeVI and MIV/TeVI was observed. MIII ions occur only in combination with the pentavalent LV ions, for example in Li4MIIISbO6 (M = Fe)27 and a possible MIII/TeVI ordering is unknown. Within the P2 and O3 family, all the honeycomb-like ordered phases can be attributed to one of the following space groups; the P2 family with known space groups P63/mmc, P6322, and P212121 as well as the O3 family with the space groups C2/m, C2/c, and P312. Kumar et al. reported on a tellurium containing mixed oxide crystallizing in a disordered Fddd structure type without a MII/TeVI honeycomb-like ordering.22 The given unit cell parameters of this compound (Li2Ni2TeO6: a = 589.21(7), b = 859.9(1), c = 1769.1(2) pm) are nearly the same as those of the here presented Li3Co1.06(1)TeO6, also crystallizing without the honeycomb-like ordering in an orthorhombic unit cell with Fddd symmetry. Other previously known oxides with an orthorhombic Fddd symmetry are Li3MII2LO6 (M = Ni, Co, Mg; L = Nb, Ta, Sb)11–14 with the exception of Li3Co2NbO6. As already mentioned, the general observed valences of the M and L ions of the tellurate compounds are MII and MIV in combination with LVI. However, the here presented compound Li3Co1.06(1)TeO6 is as far as we know the only AxMyTeO6 compound exhibiting CoIII next to TeVI cations. In contrast to the majority of published structure solutions derived from X-ray powder diffraction data, Li3Co1.06(1)TeO6 was examined by single-crystal diffraction methods. This method enabled a distinct site assignment and allowed distinguishing between CoII/CoIII mixed valence states, which are present in Li3Co1.06(1)TeO6 or CoIII/LiI mixed occupied sites. In order to quantify and differentiate between the CoII/CoIII mixed valence states, additional X-ray photoelectron spectroscopy (XPS) as well as magnetic susceptibility measurements were performed. The sample conductivity was determined by two different types of electrochemical impedance experiments.
9 should be mentioned here exhibiting a pure ionic conductivity of 4–11 S m−1 at 573 K. Depending on the different stacking of the brucite-like (M,L)O3/6 octahedral layers and in particular on the different coordination of the interlayer alkali cations, various compounds can be classified into different polytypes named O3, P2, P3, etc. In this notation, according to Fouassier et al.,10 O stands for octahedral and P for trigonal prismatic coordination of the A ions and the digit for the number of brucite-like layers in the unit cell. In many cases, honeycomb-like superlattice ordering of the two heterovalent M and L cations was observed by the surroundings of isolated LO6 octahedra from six MO6 octahedra in a honeycomb-like arrangement. General compositions of these rock-salt type superstructures are Li3MII2LO6 (M = Mg, Co, Ni, Cu; L = Nb, Ta, Sb, Bi)11–17 and Na3MIII2LO6 (M = Mg, Co, Ni, Cu; L = Sb).18–20 Compounds belonging to the family of tellurates are A2MII2TeO6 (A = Li, Na; M = Mg, Co, Ni, Cu, Zn)8,21,22 and the recently reported Li8MII2Te2O12 (M = Co, Ni, Cu, Zn),23 which belongs to the former known series Li4MIITeO6 (M = Ni, Zn).2,24 An elegant method to obtain isostructural honeycomb-like ordered structures at low temperatures, inaccessible by direct solid state high-temperature reactions, is alkali ion-exchange experiments, as was shown for Na2Cu2TeO6.22 Besides the MII/TeVI honeycomb-like ordering, Tarte et al. investigated a possible MIV/TeVI cation ordering and identified two different structure types for the compounds Li2(Zr,Hf)TeO625 and Li2(Ti,Sn)TeO6.26 To the best of our knowledge, in these ordered AxMyTeO6 compounds always a heterovalence of MII/TeVI and MIV/TeVI was observed. MIII ions occur only in combination with the pentavalent LV ions, for example in Li4MIIISbO6 (M = Fe)27 and a possible MIII/TeVI ordering is unknown. Within the P2 and O3 family, all the honeycomb-like ordered phases can be attributed to one of the following space groups; the P2 family with known space groups P63/mmc, P6322, and P212121 as well as the O3 family with the space groups C2/m, C2/c, and P312. Kumar et al. reported on a tellurium containing mixed oxide crystallizing in a disordered Fddd structure type without a MII/TeVI honeycomb-like ordering.22 The given unit cell parameters of this compound (Li2Ni2TeO6: a = 589.21(7), b = 859.9(1), c = 1769.1(2) pm) are nearly the same as those of the here presented Li3Co1.06(1)TeO6, also crystallizing without the honeycomb-like ordering in an orthorhombic unit cell with Fddd symmetry. Other previously known oxides with an orthorhombic Fddd symmetry are Li3MII2LO6 (M = Ni, Co, Mg; L = Nb, Ta, Sb)11–14 with the exception of Li3Co2NbO6. As already mentioned, the general observed valences of the M and L ions of the tellurate compounds are MII and MIV in combination with LVI. However, the here presented compound Li3Co1.06(1)TeO6 is as far as we know the only AxMyTeO6 compound exhibiting CoIII next to TeVI cations. In contrast to the majority of published structure solutions derived from X-ray powder diffraction data, Li3Co1.06(1)TeO6 was examined by single-crystal diffraction methods. This method enabled a distinct site assignment and allowed distinguishing between CoII/CoIII mixed valence states, which are present in Li3Co1.06(1)TeO6 or CoIII/LiI mixed occupied sites. In order to quantify and differentiate between the CoII/CoIII mixed valence states, additional X-ray photoelectron spectroscopy (XPS) as well as magnetic susceptibility measurements were performed. The sample conductivity was determined by two different types of electrochemical impedance experiments.
2 Experimental
2.1 Synthesis
X-ray pure samples with the ideal composition of Li3CoTeO6 were synthesized by conventional solid-state techniques. According to eqn (1), mixtures of lithium carbonate (Li2CO3: purity ≥99.5%, Fluka), cobalt powder (Co: purity ≥99.8%, Strem Chemicals), and tellurium powder (Te: purity ≥99.99%, Alfa Aesar) in the correct stoichiometric ratio were used.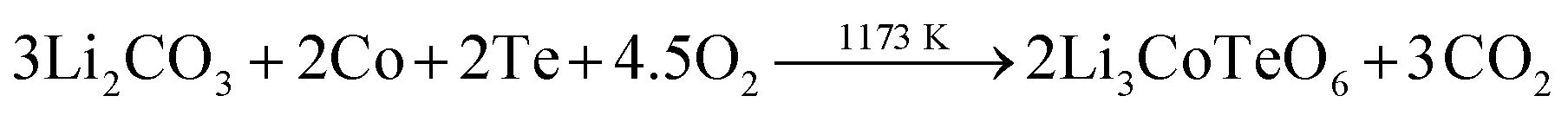 | (1) |
The thoroughly ground mixtures of the starting materials were filled into a corundum crucible and placed in an open silica glass ampoule to ensure sufficient oxygen availability. Subsequently, the silica glass ampoules were placed in a vertical tube furnace regulated by using an eight level PID controller with a type K thermocouple. With a heating rate of 1 K min−1, the mixtures were first heated in air to a temperature of 773 K for 3 h to completely oxidize tellurium to its oxidation stage TeVI.26 Afterwards, the temperature was increased to 1173 K for 36–100 h with a ramp rate of 2 K min−1 and then slowly cooled down to a temperature of 473 K with a ramp rate of 0.1 K min−1 to ensure good crystal quality. The polycrystalline product appears bluish grey with a metallic lustre and is stable in air. Instead of Li2CO3, elemental lithium as a starting material is also possible but more homogeneous results are derived from the syntheses with the carbonate.
2.2 Characterization
EDX and ICP. The Li3Co1.06(1)TeO6 crystals were semi-quantitatively investigated by the use of a Jeol JSM-6010LV scanning electron microscope with a Quantax (Bruker Nano) energy-dispersive system (EDX) for element identification. Small particles of the samples were placed on adhesive carbon platelets and four suitable regions of the crystals were selected as measurement points. The experimentally observed Co to Te ratio in Li3Co1.06(1)TeO6 of Co/Te = 21 ± 3 at% Co/17 ± 3 at% Te was close to the ideal ratio of Co/Te = 13.2 at% Co
:12.4 at% Te. The light element Li was not detectable with this measurement setup and the oxygen determination in this semi-quantitative approach is not reliable. Therefore, only the ratios were compared and the absolute values can be neglected.
The Li/Co ratio was determined by ICP-OES (Inductively Coupled Plasma-Optical Emission Spectrometry) on a Horiba Jobin Yvon ACTIVA high-resolution spectrometer. 100.0 ± 0.1 mg of powdered Li3Co1.06(1)TeO6 sample was completely dissolved in a 50.0 ml nitric acid/hydrogen peroxide mixture (HNO3 65% p.a. (Carl Roth GmbH), H2O (ultrapure, 18 MΩ), H2O2 30% (Carl Roth GmbH)). The actual measurement solution was diluted twice by a ratio of 1:10. Li and Co standard solutions for ICP measurements (Merck) were used to evaluate the concentrations. Calibration curves with a Li and Co content of 0–10–20 mg l−1 and 0–40–60 mg l−1 were recorded, respectively. The used emission wavelengths were 228.616 nm for Co and 670.784 nm for Li. The given relative standard deviations were calculated on four different measurements. The ICP-OES analysis revealed a content of 439 ± 7 mg l−1 Co and 147 ± 4 mg l−1 Li within the solution which corresponds to a Li/Co ratio of 2.84(9). This Li/Co ratio was used for the refinement of the site occupancy parameters of the single-crystal structure data.
| Empirical formula | Li3Co1.06(1)TeO6 |
|---|---|
| Molar mass, g mol−1 | 307.03 |
| Crystal system | Orthorhombic |
| Space group | Fddd (no. 70) |
| Formula units per cell, Z | 8 |
| Powder diffractometer | STOE Stadi P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| Powder data: | |
| a, pm | 587.81(1) |
| b, pm | 857.71(2) |
| c, pm | 1787.72(3) |
| V, Å3 | 896.78(3) |
| Single-crystal diffractometer | Bruker D8 Quest |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| Single-crystal data: | |
| a, pm | 588.6(2) |
| b, pm | 856.7(2) |
| c, pm | 1781.5(4) |
| V, Å3 | 898.4(3) |
| Calculated density, g cm−3 | 4.54 |
| Crystal size, mm3 | 0.04 × 0.03 × 0.03 |
| Temperature, K | 273(2) |
| Absorption coefficient, mm−1 | 10.3 |
| F (000), e | 1102 |
| Detector distance, mm | 40 |
| θ range, ° | 4.4–37.9 |
| Range in hkl | ±10, ±14, ±30 |
| Total reflections | 11929 |
| Data/ref. parameters | 608/33 |
| Reflections with I ≥ 2σ(I) | 535 |
| R int, Rσ | 0.0316, 0.0112 |
| Goodness-of-fit on F2 | 1.378 |
| Absorption correction | Multi-scan28 |
| R 1/wR2 for I ≥ 2σ(I) | 0.0174/0.0462 |
| R 1/wR2 (all data) | 0.0216/0.0477 |
| Largest diff. peak/hole, e Å−3 | 1.89/−0.75 |
| Extinction coefficient | 0.0012(2) |
| Transmission min./max. | 0.695/0.747 |
A couple of crystal fragments of the crushed sample were embedded in polyfluoropolyalkylether (viscosity 1800) and treated under a microscope. Smaller fragments of Li3Co1.06(1)TeO6 appear transparent blue. Selected single-crystal fragments were fixed on the tip of MicroMounts™ (MiTeGen, LLC, Ithaca, NY, USA) with a diameter of 30 μm and a subsequent intensity data collection of the Li3Co1.06(1)TeO6 crystals was carried out using a Bruker D8 Quest diffractometer with a Photon 100 detector system and an Incoatec Microfocus source generator (multi-layered optic, monochromatized Mo-Kα radiation, λ = 71.073 pm). To enhance the collection strategies concerning ω- and φ-scans, the APEX 2 program package28 was used and data sets of complete reciprocal spheres up to high angles with high redundancies were received. Further data processing and data reduction were performed with the program SAINT28 and a correction regarding absorption effects was carried out on the semi-empirical “multi scan” approach with the program SADABS.28
Magnetic properties. The powdered sample of Li3Co1.06(1)TeO6 was packed into polyethylene (PE) capsules and attached to the sample holder rod of a Vibrating Sample Magnetometer (VSM) unit for measuring the magnetization M(T,H) in a Quantum Design Physical Property Measurement System (PPMS). The samples were investigated in the temperature range of 2.5–300 K with magnetic flux densities up to 80 kOe.
X-ray photoelectron spectroscopy (XPS). The oxidation states of the samples were investigated by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific MultiLab 2000 spectrometer. This instrument is equipped with a monochromated Al-Kα X-ray source, an Alpha 110 hemispherical sector analyzer as well as an ion gun for sputter-depth profiling (with 3 keV argon ions), and features a base pressure in the low 10−10 mbar range. In order to avoid charging of the samples upon irradiation, a flood gun, providing electrons of 6 eV, was utilized.
The Li 1s, Te 3d and C 1s spectra were fitted using Gaussian/Lorentzian products (30% Lorentzian character). Because of the complex shape of the Co 2p region, reference spectra had to be employed to determine the amount of CoIII in the samples. Due to the lack of a pure oxidic CoIII reference, references from CoO and Co3O4 were used. To determine the CoIII concentration, a linear combination of the respective sample spectrum and the CoO reference was fitted to the Co3O4 spectrum, which allows for the determination of CoIII amounts larger than that in Co3O4. During the sputter depth profiling, using adventitious carbon as the charge reference was not possible (because it was sputtered away during the first step). The latter was used for the surface spectra, and the other spectra were referenced to the Co 2p region because no peak shift between CoII and CoIII was observed.29
Electrochemical impedance spectroscopy (EIS). The in situ impedance cell consists of an outer quartz tube with two inner quartz tubes to which the sample and the electrodes were attached. Heating is provided by using a tubular Linn furnace and controlled by the use of a thermocouple (K-element) located in the reactor about 5 mm downstream of the sample and a Micromega PID temperature controller.30 The impedance is measured by using an IM6e impedance spectrometer (Zahner Messsysteme, Germany), which provides data on the impedance and the phase angle of the current as a function of voltage. The powder samples were pressed into pellets with a pressure of 1.5 t (5 mm diameter, 0.1 mm thick) and placed between two circular Pt electrodes forming a plate capacitor in mechanically enforced contact with the sample pellet.
For all temperature-programmed impedance measurements, 20 mV signal amplitude and a frequency of 1 Hz were applied to the Pt electrodes. The impedance modulus value |Z| of the pellet was, thus, effectively measured in an electrochemically unpolarized state.
The electrical properties were also determined by AC EIS measurements in a frequency range between 100 mHz and 1 MHz with 20 mV signal amplitude. The real and imaginary parts of the impedance were first measured from 1 kHz up to 1 MHz (within 19 s) and then from 1 MHz down to 100 mHz (within 4 min 37.7 s; total measuring time: 4 min 56.7 s) to check for changes of the system during EIS. A 1 h interval was allowed for thermal stabilization after each temperature change. Curve fitting and resistance calculation were performed with the Zahner ThalesBox and with an adapted equivalent circuit model consisting of two R-CPE elements in series as described in ref. 31 and 32.
3 Results and discussion
3.1 Structure refinements
The diffraction data showed an F-centered orthorhombic lattice and the systematic extinctions were in agreement with the space group Fddd. The starting atomic parameters were derived from the “Intrinsic Phasing” method,33 implemented in the APEX 2 program package.28 Subsequently, full-matrix least squares refinements based on F2 were executed with the program SHELXL-2013.34,35 All atoms were refined with anisotropic displacement parameters. The correct composition of the compound was verified by refining the occupation parameters in a separate series of least-squares cycles. Except the Co and Li atoms, all sites were fully occupied within two standard deviations. The ideal composition of Li3CoTeO6 with all Co atoms in the oxidation state CoIII is formed by one fully occupied tellurium site (8a), two oxygen sites (16f, 32h) as well as two lithium sites (8b, 16g). The Co site (16g) is only half filled resulting in the following refinement residuals: R1 = 0.0190/wR2 = 0.0526, and GooF = 1.340. A possible ordering of Co on the 16g site was checked by symmetry reduction to the space group Fdd2. The two resulting 8a sites were more than 50% occupied without the ordering tendency and the Flack parameter of around 0.5 indicated centro-symmetry. Separate refinements of the occupation of Co and Li sites in the centro-symmetric space group Fddd revealed significant deviations from the ideal composition Li3CoTeO6. Cobalt exhibits a site occupation of 52.7% and the Li2 site seems to be occupied only by 73.6%, leading to a composition of Li2.6(1)Co1.06(1)TeO6 and slightly improved refinement results of: R1 = 0.0172/wR2 = 0.0460, and GooF = 1.372. The Li1 site is completely occupied within two standard deviations. In addition to this, residual electron density in a tetrahedral oxygen environment was left and taking into account this by an occupation with lithium and free refinement of the occupation parameters, the general Li site becomes occupied by 12(2)%. The resulting composition of Li2.97(9)Co1.05(1)TeO6 is in good agreement with the EDX, ICP, and XPS measurements. Unfortunately, lithium located at this tetrahedral position face-linked to the octahedral position is defined as non-positive (NPD). For this reason, the occupation parameters of Li2 were raised manually to achieve the former given composition Li2.97(9)Co1.05(1)TeO6 (Li/Co = 2.83) reflecting the measured Li/Co ratio of 2.84 derived from the ICP measurement. The composition Li3Co1.06(1)TeO6 (Li/Co = 2.83) was achieved by a full occupation of the Li2 site. This increased the thermal anisotropic displacement parameters of Li2. Actually, however, a part of the Li2 atoms will be statistically distributed to the neighboring tetrahedral positions. Possible Li/Co mixed occupations were considered in various refinement cycles but did not improve the structure model nor reflected the results of the additional analyses. Nevertheless, mixed occupations in this highly dynamic system cannot be excluded or are even most likely. The correctness of the space group was checked with the ADDSYM36 routine of the PLATON program package.37 Detailed information about the single-crystal structure determination can be found in Table 1. The positional parameters, anisotropic displacement parameters, interatomic distances, and angles are given in Tables 2–5.| Atom | Wyckoff-position | x | y | z | SOF | U eq |
|---|---|---|---|---|---|---|
| Co | 16g | 3/8 | 3/8 | 0.04298(4) | 0.532(2) | 0.0075(3) |
| Te | 8a | 1/8 | 1/8 | 1/8 | 1 | 0.00457(8) |
| O1 | 16f | 1/8 | 0.3499(3) | 1/8 | 1 | 0.0086(4) |
| O2 | 32h | 0.1076(3) | 0.3742(2) | 0.29817(8) | 1 | 0.0087(3) |
| Li1 | 8b | 3/8 | 3/8 | 3/8 | 1 | 0.017(2) |
| Li2 | 16g | 3/8 | 3/8 | 0.2135(8) | 1 | 0.05(4) |
| Atom | U 11 | U 22 | U 33 | U 23 | U 13 | U 12 |
|---|---|---|---|---|---|---|
| Co | 0.0079(4) | 0.0077(4) | 0.0068(3) | 0 | 0 | −0.0029(3) |
| Te | 0.0043(1) | 0.0050(1) | 0.0044(1) | 0 | 0 | 0 |
| O1 | 0.0088(8) | 0.0060(7) | 0.0111(8) | 0 | 0.0013(8) | 0 |
| O2 | 0.0076(6) | 0.0109(6) | 0.0076(5) | −0.0003(5) | 0.0019(4) | 0.0008(6) |
| Li1 | 0.008(4) | 0.032(5) | 0.010(4) | 0 | 0 | 0 |
| Li2 | 0.054(8) | 0.045(7) | 0.041(6) | 0 | 0 | 0.038(6) |
| Co: | O1 | 208.50(6) | 2× |
| O2 | 212.4(2) | 2× | |
| O2 | 213.9(2) | 2× | |
| Te: | O1 | 192.6(2) | 2× |
| O2 | 193.6(2) | 4× | |
| Li1: | O2 | 208.6(2) | 4× |
| O1 | 235.7(2) | 2× | |
| Li2: | O2 | 216.1(2) | 2× |
| O1 | 216.8(9) | 2× | |
| O2 | 218.0(9) | 2× |
| O2–Co–O2 | 80.29(9) | |
| O1–Co–O2 | 80.38(8) | 2× |
| O2–Co–O2 | 89.94(6) | 2× |
| O1–Co–O1 | 91.01(3) | |
| O2–Co–O2 | 93.85(6) | 2× |
| O1–Co–O2 | 94.61(4) | 2× |
| O1–Co–O2 | 96.11(7) | 2× |
| O1–Co–O2 | 172.36(6) | 2× |
| O2–Co–O2 | 175.05(9) | |
| O1–Te–O2 | 89.79(5) | 4× |
| O2–Te–O2 | 89.98(9) | 2× |
| O2–Te–O2 | 90.02(9) | 2× |
| O1–Te–O2 | 90.21(5) | 4× |
| O2–Te–O2 | 179.58(9) | 2× |
| O1–Te–O1 | 180 | |
| O2–Li1–O2 | 82.03(9) | 2× |
| O2–Li1–O1 | 89.80(4) | 4× |
| O2–Li1–O1 | 90.20(4) | 4× |
| O2–Li1–O2 | 97.97(9) | 2× |
| O2–Li1–O2 | 179.61(9) | 2× |
| O1–Li1–O1 | 180 | |
| O2–Li2–O1 | 78.4(2) | 2× |
| O1–Li2–O1 | 86.6(5) | |
| O1–Li2–O2 | 90.73(5) | 2× |
| O2–Li2–O2 | 91.7(2) | 2× |
| O2–Li2–O2 | 92.4(5) | |
| O2–Li2–O1 | 93.5(3) | 2× |
| O2–Li2–O2 | 96.0(2) | 2× |
| O2–Li2–O2 | 169.0(7) | |
| O1–Li2–O1 | 173.6(2) | 2× |
Further details of the structure refinements may be obtained from the Fachinformationszentrum Karlsruhe, D-76344 Eggenstein-Leopoldshafen, Germany (fax: (+49)7247-808-666; e-mail: crysdata@fiz-karlsruhe.de), on quoting the deposition number CSD 433117 (Li3Co1.06(1)TeO6).
3.2 Crystal chemistry
The here presented compound Li3Co1.06(1)TeO6 crystallizes orthorhombically with the following unit cell parameters and refinement results: Fddd, a = 588.6(2), b = 856.7(2), c = 1781.5(4) pm, R1 = 0.0174, wR2 = 0.0462, 608 F2 values and 33 variables. The unit cell parameters are close to the isopointal compound Li2Ni2TeO6 reported by Kumar et al. but because of the low thermal stability, they did not attempt to grow single crystals. From powder data they derived a disordered structure type without a NiII/TeVI honeycomb-like ordering and LiI/NiII mixed occupation of the 8b and the two 16g sites.22 Our single-crystal investigations confirmed the missing honeycomb-like ordering and furthermore, Li3Co1.06(1)TeO6 exhibits a comparatively high-order three-dimensional lattice of two interpenetrating substructures. These two substructures consist of a lithium–oxygen and a cobalt/tellurium–oxygen structure part. During various refinement cycles, mixed occupations of the cobalt and lithium sites were tested. A simultaneous refinement of both Li sites together with the Co site as Li/Co mixed occupied sites according to Kumar et al.22 was not possible. In addition to this, refinements of the under-occupied Co site as the mixed Li/Co site resulted in a compound with a Li/Co ratio larger than that detected by the ICP measurements. Moreover, the additional Li site remains non-positive definite (NPD) and for a valence balanced compound a combination of CoIII and CoIV would be necessary. This is in conflict with the results of the XPS measurements which specify the CoII/CoIII ratio to 9(3)% CoII and 91(3)% CoIII. For the Co site, an occupation of 53(1)% was found after a free refinement of the occupation parameters. The diffraction data gave no hints of a symmetry reduction and an ordering of the Co site in a larger unit cell. Due to the fact that the Co site is a CoII/CoIII mixed valent site, the occupation must be larger than 50% compared to the ideal formula sum of Li3CoTeO6 with 100% CoIII. For this reason, a statistical occupation of the Co site is assumed. The structural model proposed here is the most suitable model with the joint consideration of all here presented analytical results.
Fig. 1 gives an impression of the crystal structure of Li3Co1.06(1)TeO6 along the [110] direction (bottom) and of the 90° turned view along the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] direction (top). Both sights reveal an identical linking pattern of edge linked TeO6 and CoO6 octahedra. However, it should be noted that the displayed Co octahedra are only about 53% occupied in a statistical manner. This part of the crystal structure belongs to the first substructure of the two interpenetrating networks. Within the so-formed channels, the lithium ions are located building up the second substructure if drawn in their polyhedral view. Fig. 2 shows the crystal structure of Li3Co1.06(1)TeO6 with the only displayed LiO6 octahedra network in identical directions as before. Now the Co and Te atoms are located inside the channels. Each atom in this structure type exhibits an octahedral coordination sphere like it is found in rock-salt type structures. The Te atoms are coordinated in nearly perfect octahedral geometry with typical TeVI–O distances of 193 (2×) and 194 pm (4×) and O–Te–O angles of 89.8–90.2° (see Tables 4 and 5). The octahedral Co coordination sphere is a bit more distorted with Co–O distances from 209–214 pm and O–Co–O angles varying from 80.3 to 96.2° which are comparable to CoII–O distances of 213 pm found in NaCl-type CoO.38 Spinel-type Co2O3 with tetrahedral and octahedral Co coordination exhibits CoII–O (tetrahedral position) and CoIII–O (octahedral position) distances of 198 and 189 pm, respectively.39 Cobalt in its threefold low-spin oxidation state can be found in corundum type high-pressure sesquioxide with Co–O distances of 189 and 193 pm.40 Compared to binary cobalt oxide polymorphs, the Co–O contacts appearing in Li3Co1.06(1)TeO6 are comparatively long for CoIII–O distances. However, in ternary or quaternary Co-containing compounds for example LiCoO2,41 CoLa2Li0.5Co0.5O4,42 and Ba6La2Co4O1543 CoIII–O distances in an octahedral configuration from 205 to 215 pm are documented. The Li1 atoms show four equal distances of 209 pm to the surrounding oxygen atoms and two longer contacts of 236 pm. A more uniform coordination is given for the Li2 atoms, which are encircled by six oxygen atoms with distances of 216–218 pm. All corresponding angles of the octahedral coordination spheres can be found in Table 5. In the literature, the documented range of Li–O distances in lithium-transition metal-tellurates is around 200 pm but can be escalated up to 247 pm and more. In Li8Cu2Te2O12, a coordination comparable to the Li1 atoms occurs with four shorter Li–O distances of 201 pm and two longer distances of 247 pm.23 The anisotropic displacement parameters of the lithium ions inside the octahedral coordination sphere are enlarged, indicating a possible Li ion mobility. As described in the Structure refinement section, the Li1 site is fully occupied within two standard deviations, whereas the Li2 site revealed an under-occupation and was filled up manually until the Li/Co ratio determined by ICP analyses was achieved. This was also legitimated by the fact that additional electron density located in a neighboring tetrahedral position resulted in the same Li/Co ratio if taken into account. However, an occupation of this tetrahedral position with lithium resulted in negative displacement parameters. Nevertheless, the located electron density in a tetrahedral environment in combination with direct face-linked under-occupied octahedral sites offers an excellent diffusion pathway for lithium ions. According to the crystal structure, two of these diffusion pathways in the orthogonal orientation exist in Li3Co1.06(1)TeO6. In Fig. 3 the diffusion pathway along the [110] direction with Li–Li hopping distances of 179–186 pm is emphasized. The second diffusion pathway runs along the [10] direction. Investigations of the physical properties of Li3Co1.06(1)TeO6 are discussed in the following section.
10] direction (top). Both sights reveal an identical linking pattern of edge linked TeO6 and CoO6 octahedra. However, it should be noted that the displayed Co octahedra are only about 53% occupied in a statistical manner. This part of the crystal structure belongs to the first substructure of the two interpenetrating networks. Within the so-formed channels, the lithium ions are located building up the second substructure if drawn in their polyhedral view. Fig. 2 shows the crystal structure of Li3Co1.06(1)TeO6 with the only displayed LiO6 octahedra network in identical directions as before. Now the Co and Te atoms are located inside the channels. Each atom in this structure type exhibits an octahedral coordination sphere like it is found in rock-salt type structures. The Te atoms are coordinated in nearly perfect octahedral geometry with typical TeVI–O distances of 193 (2×) and 194 pm (4×) and O–Te–O angles of 89.8–90.2° (see Tables 4 and 5). The octahedral Co coordination sphere is a bit more distorted with Co–O distances from 209–214 pm and O–Co–O angles varying from 80.3 to 96.2° which are comparable to CoII–O distances of 213 pm found in NaCl-type CoO.38 Spinel-type Co2O3 with tetrahedral and octahedral Co coordination exhibits CoII–O (tetrahedral position) and CoIII–O (octahedral position) distances of 198 and 189 pm, respectively.39 Cobalt in its threefold low-spin oxidation state can be found in corundum type high-pressure sesquioxide with Co–O distances of 189 and 193 pm.40 Compared to binary cobalt oxide polymorphs, the Co–O contacts appearing in Li3Co1.06(1)TeO6 are comparatively long for CoIII–O distances. However, in ternary or quaternary Co-containing compounds for example LiCoO2,41 CoLa2Li0.5Co0.5O4,42 and Ba6La2Co4O1543 CoIII–O distances in an octahedral configuration from 205 to 215 pm are documented. The Li1 atoms show four equal distances of 209 pm to the surrounding oxygen atoms and two longer contacts of 236 pm. A more uniform coordination is given for the Li2 atoms, which are encircled by six oxygen atoms with distances of 216–218 pm. All corresponding angles of the octahedral coordination spheres can be found in Table 5. In the literature, the documented range of Li–O distances in lithium-transition metal-tellurates is around 200 pm but can be escalated up to 247 pm and more. In Li8Cu2Te2O12, a coordination comparable to the Li1 atoms occurs with four shorter Li–O distances of 201 pm and two longer distances of 247 pm.23 The anisotropic displacement parameters of the lithium ions inside the octahedral coordination sphere are enlarged, indicating a possible Li ion mobility. As described in the Structure refinement section, the Li1 site is fully occupied within two standard deviations, whereas the Li2 site revealed an under-occupation and was filled up manually until the Li/Co ratio determined by ICP analyses was achieved. This was also legitimated by the fact that additional electron density located in a neighboring tetrahedral position resulted in the same Li/Co ratio if taken into account. However, an occupation of this tetrahedral position with lithium resulted in negative displacement parameters. Nevertheless, the located electron density in a tetrahedral environment in combination with direct face-linked under-occupied octahedral sites offers an excellent diffusion pathway for lithium ions. According to the crystal structure, two of these diffusion pathways in the orthogonal orientation exist in Li3Co1.06(1)TeO6. In Fig. 3 the diffusion pathway along the [110] direction with Li–Li hopping distances of 179–186 pm is emphasized. The second diffusion pathway runs along the [10] direction. Investigations of the physical properties of Li3Co1.06(1)TeO6 are discussed in the following section.
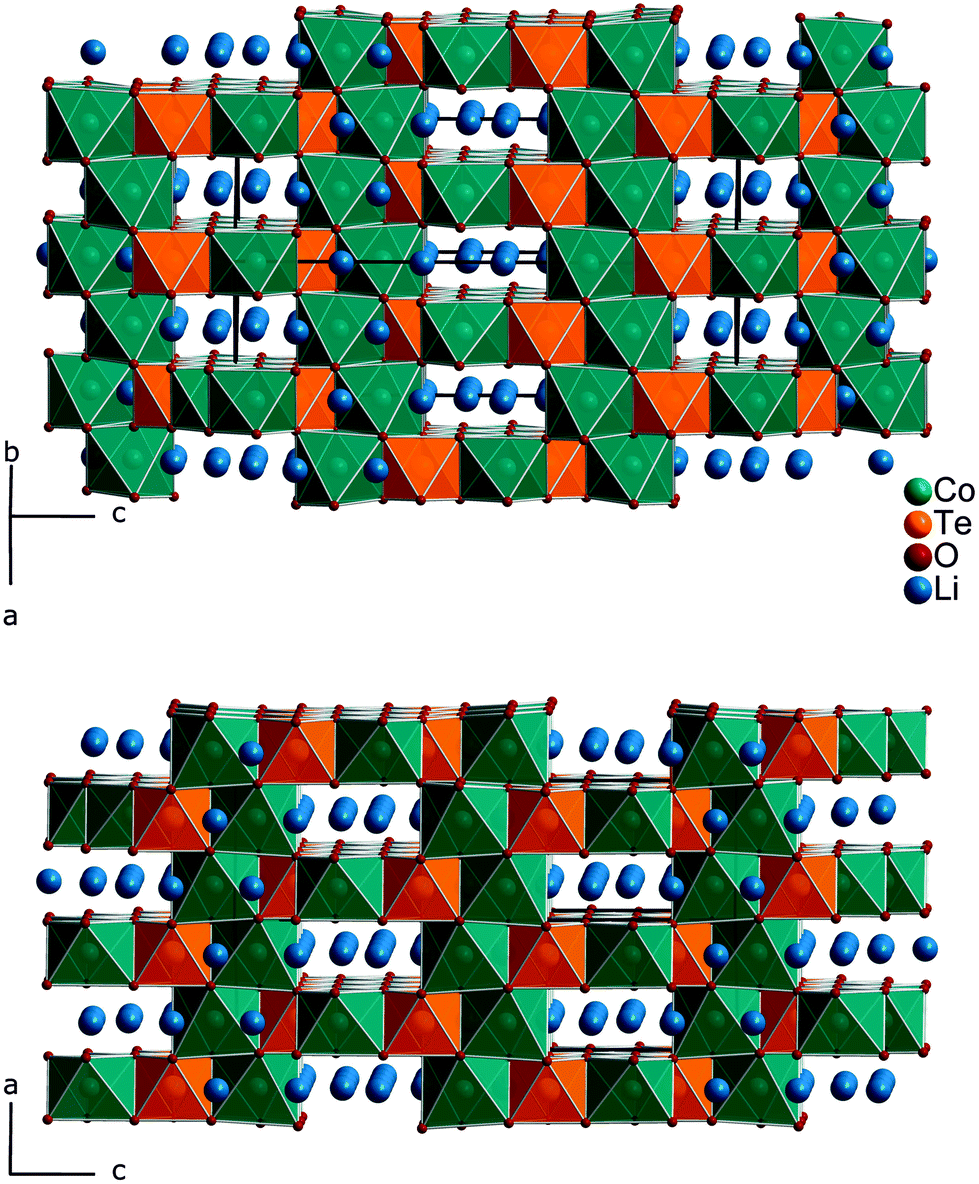 | ||
| Fig. 1 The crystal structure of Li3Co1.06(1)TeO6 with a view along the [10] direction (top) and the 90° turned (rotation around the c-axis) view along the [110] direction (bottom). TeO6 and CoO6 octahedra are drawn in orange and petrol, respectively. The lithium atoms are located inside the channels of the crystal structure. | ||
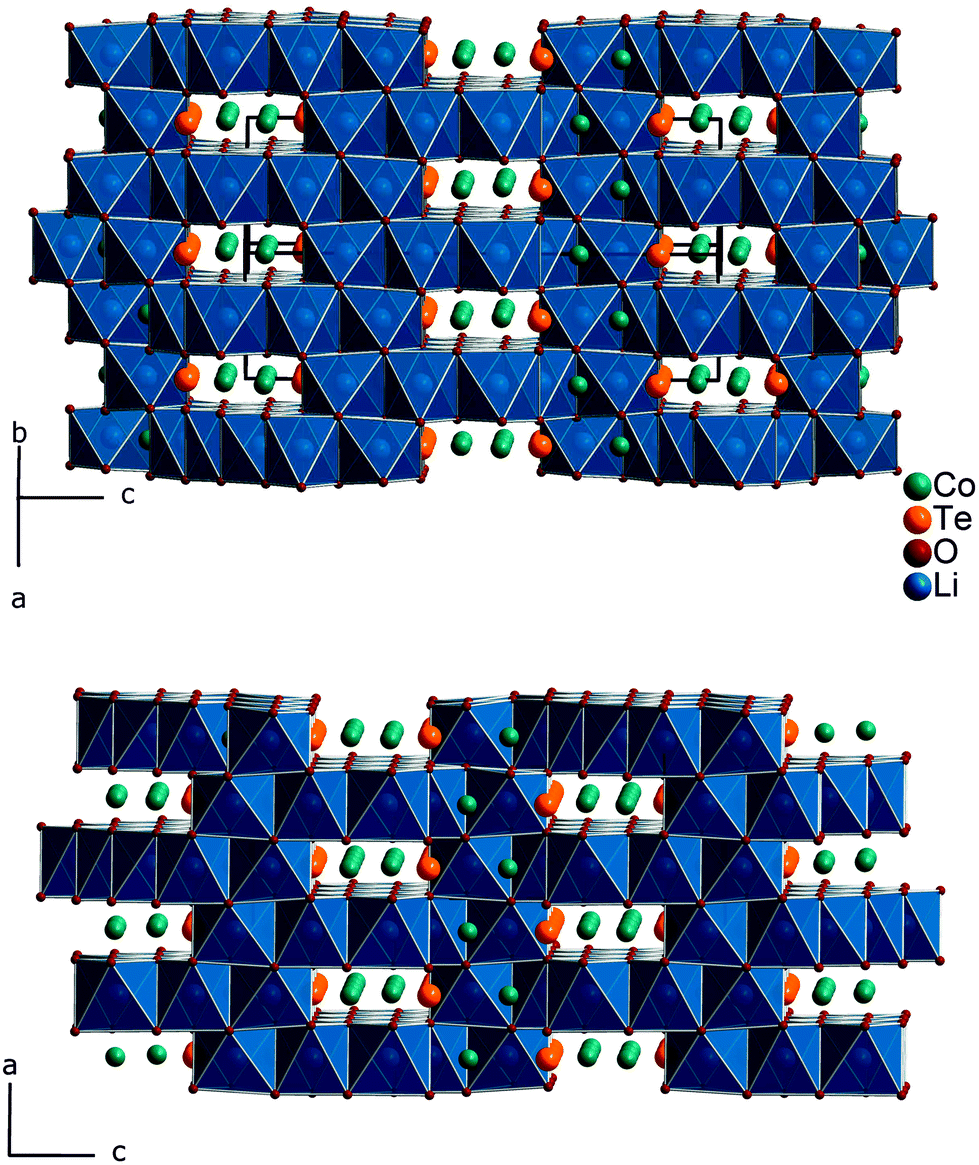 | ||
| Fig. 2 Inverted crystal structure of Li3Co1.06(1)TeO6 with emphasized LiO6 polyhedra and Co and Te located inside the channels. Viewing direction [10] (top) and the 90° turned (rotation around the c-axis) view along the direction [110] (bottom). | ||
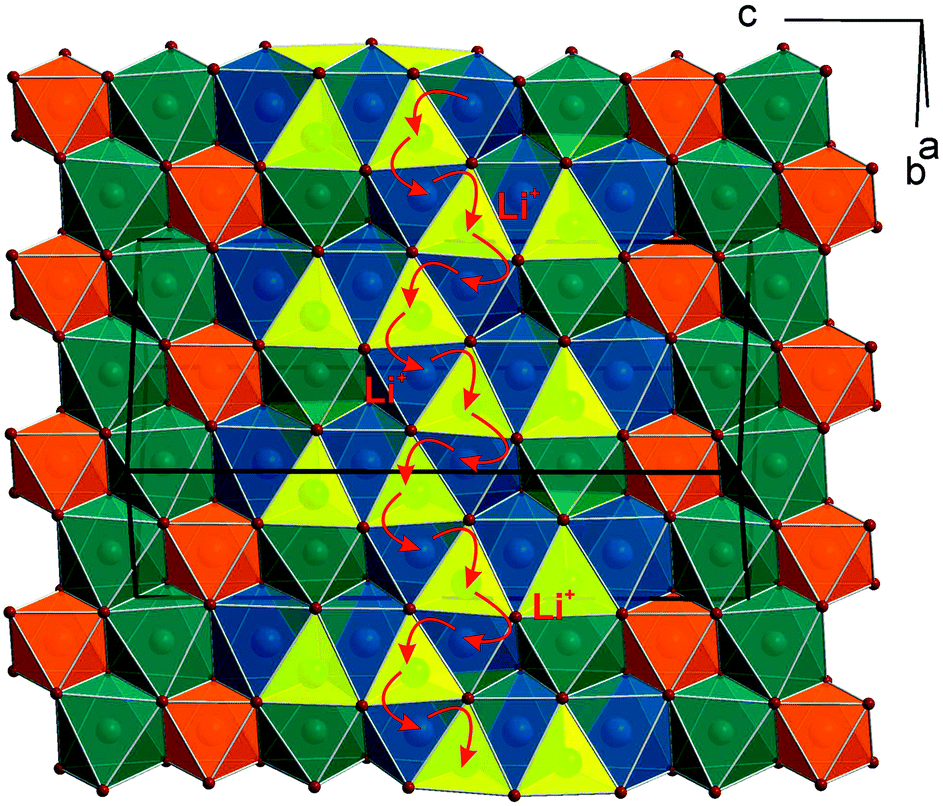 | ||
| Fig. 3 Possible diffusion pathway from octahedral to tetrahedral sites for Li-ions within one octahedral slab. The diffusion pathways run along the [110] direction and a second identical pathway direction is parallel to the [10] direction. | ||
3.3 Physical properties
:9 determined by XPS into account, the expected magnetic moment for Li3Co1.06(1)TeO6 is 4.81μB, considering all Co cations in the high-spin state. When considering Co2+ to be in a low-spin state (μcalc = 1.73μB), the total effective magnetic moment adds up to 4.68μB. Co3+ as the low-spin cation has an effective moment of 0μB; therefore, this case cannot apply. Both cases result in magnetic moments that are higher than the experimentally found value of μeff = 4.32(1)μB; therefore, more detailed information about the magnetic spin-state of the Co cations cannot be given. The experimentally determined value, however, points towards high-spin Co3+ and low-spin Co2+.
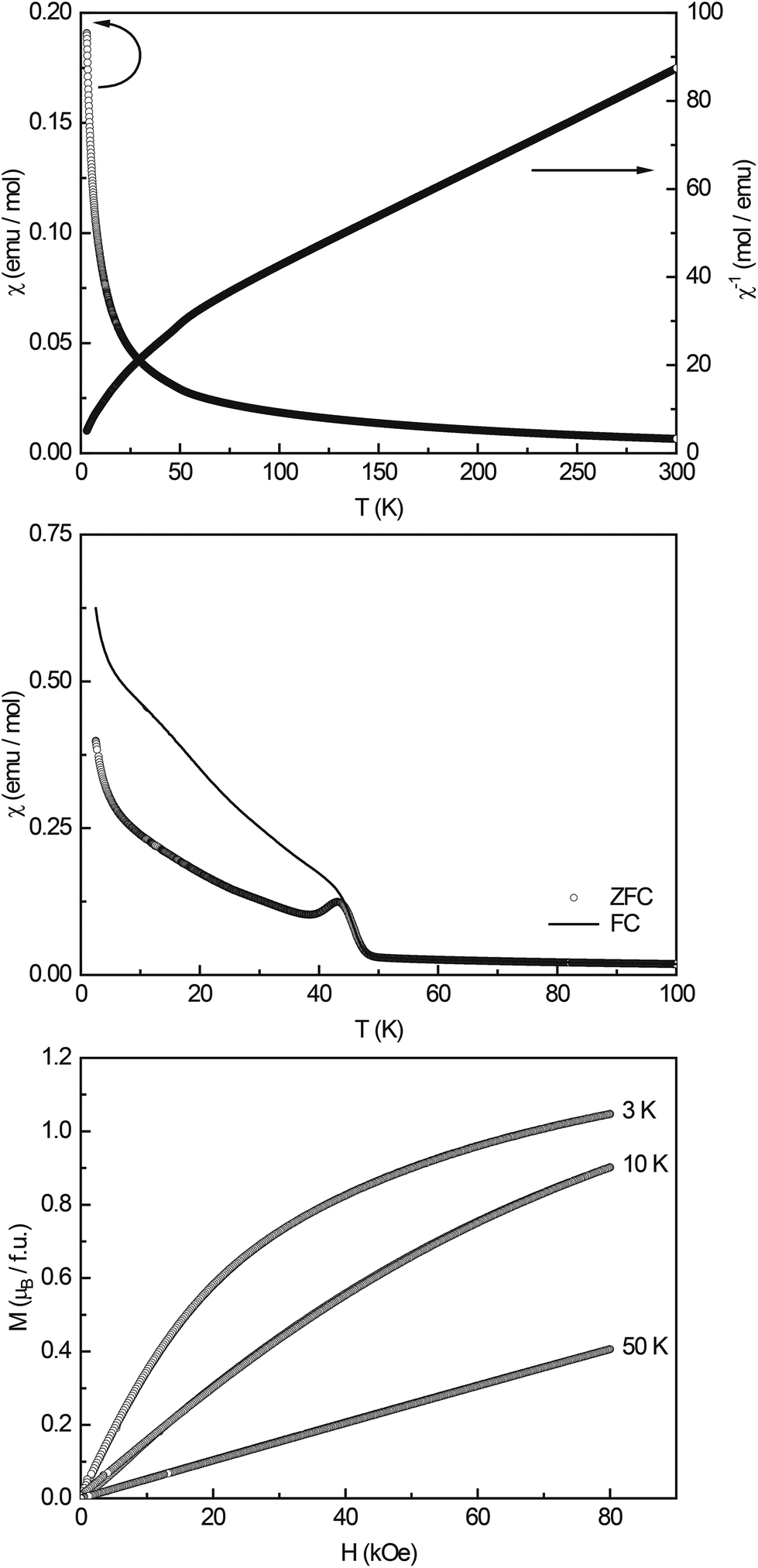 | ||
| Fig. 4 Magnetic properties of Li3Co1.06(1)TeO6: susceptibility and inverse susceptibility (χ and χ−1 data), measured at an external field strength of 10 kOe (top); zero-field-cooled/field-cooled data (ZFC/FC) measured at 100 Oe (middle); magnetization isotherms recorded at 3, 10, and 50 K (bottom). | ||
Low-field measurements between 2.5 and 100 K (Fig. 4, middle) show an antiferromagnetic transition at TN = 43.5(2) K; the Curie-tail towards low temperatures and the magnitude of the transition however indicate that the AFM ordering is not intrinsic. In the literature, several ternary cobalt tellurates and lithium cobalt oxides are found which exhibit magnetic ordering. CoTe6O13 has a Néel temperature of TN = 21 K45 and Co2Te3O8 of TN = 70 K.46 Co3TeO6 was found to exhibit a complex magnetism with transitions T1 = 26 K, T2 = 19.5 K, and T3 = 18 K,47 LiCo2O4 orders antiferromagnetically at TN ∼ 35 K,48 and the solid solution Li1−xCoxO at temperatures >200 K.49 Li6CoO450 and Li8Co2Te2O623 finally only show paramagnetic behavior and no magnetic ordering down to low temperatures. Therefore, no clear indication of the origin of the magnetic ordering at ∼40 K can be found, substitutional effects as e.g. seen in Li1−xCoxO show that the ordering temperature shifts towards lower temperatures upon increasing Li content. Hence, one could speculate that Li-substituted Co2Te3O8 could cause the observed transition.
The magnetization isotherms recorded at 3, 10, and 50 K (Fig. 4, bottom) finally show the typical behavior of a paramagnetic material, with slightly curved isotherms at 3 and 10 K. The saturation magnetization at 3 K and 80 kOe was determined to be μsat = 1.05(1)μB.
:55, i.e. the sampled depth of approximately 2.8 nm for the kinetic energy of the Li 1s electrons52 contained slightly more of the Li2 species (again located at a binding energy of 52.5 eV). The Li content seemed to be highly variable within the bulk sample depending on the ratio of CoII/CoIII valences. This can also be gathered from the results of the quantification, listed in Table 6, as well as from the lithium concentration profile in Fig. 6, with the dashed line representing the expected Li concentration based on the sum formula (27.12 at%). Whereas at 5 nm, the amount of Li was lower than that value, it was within the standard deviation at 25 nm, and at 75 nm, the observed Li concentration was actually significantly higher than the expected value, which corroborated this theory.
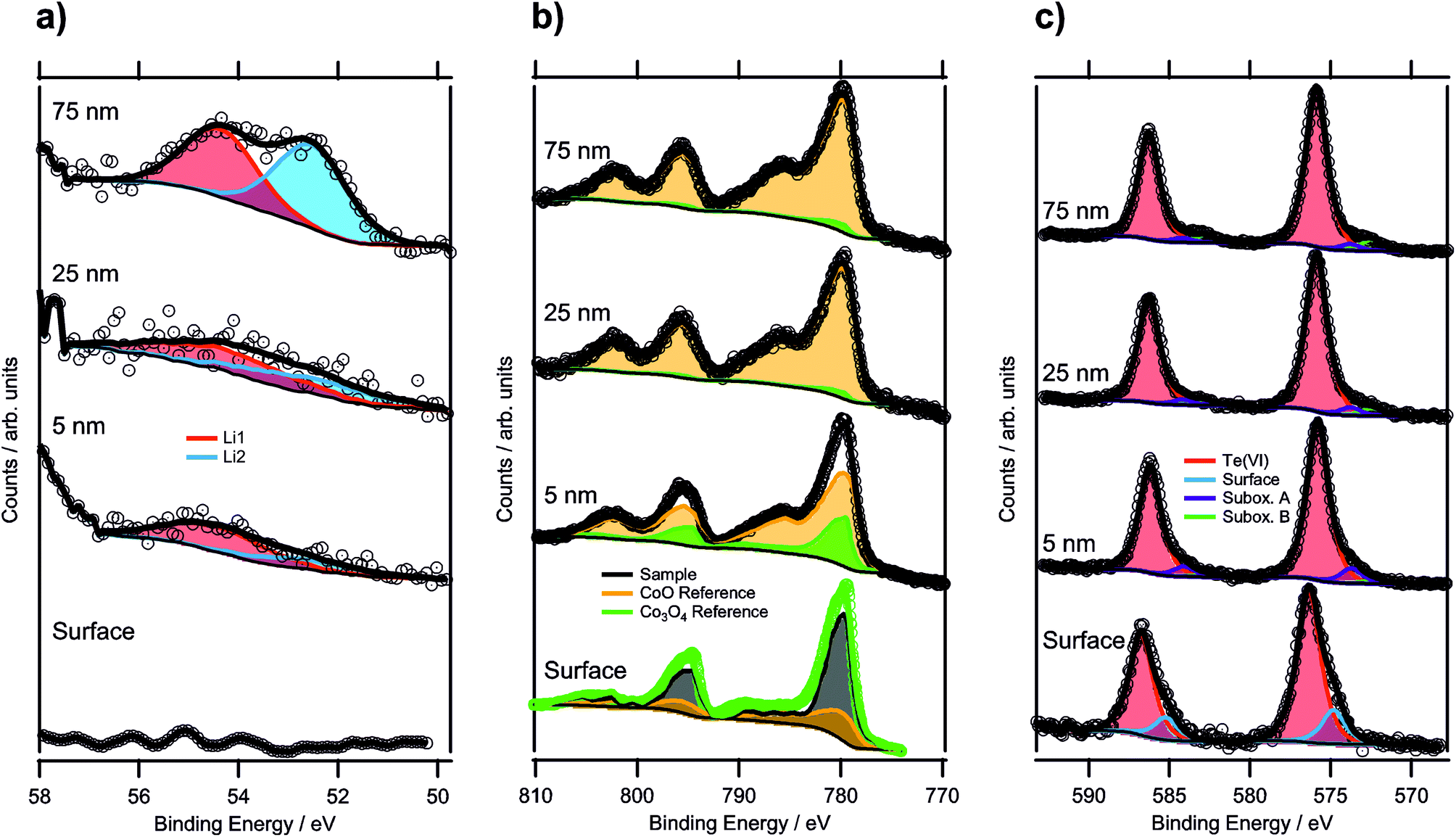 | ||
| Fig. 5 XPS depth profile spectra of a sample annealed for 100 h (up to a maximum depth of 75 nm): (a) Li 1s regions, (b) Co 2p regions and (c) Te 3d regions of the same depth profiling experiment. | ||
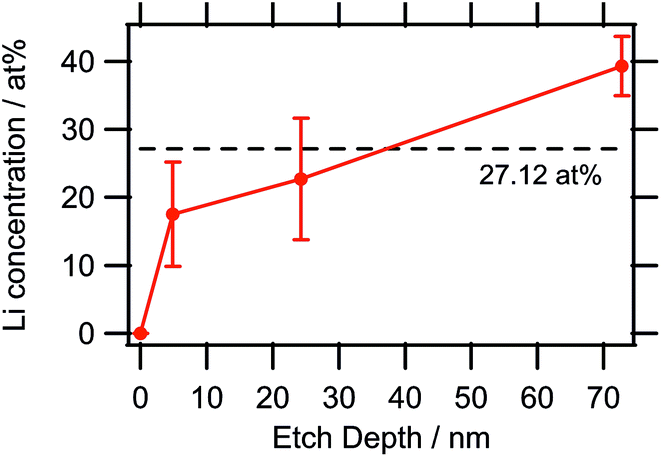 | ||
| Fig. 6 Lithium concentration depth profile as determined by XPS. The large error bars are due to the very low sensitivity factor of Li causing the noise to have a higher impact on the quantification. | ||
| Etch depth/nm | at% | |||
|---|---|---|---|---|
| Li | Co | Te | O | |
| 0 | 0 | 26(2) | 11.0(6) | 63(5) |
| 5 | 18(7) | 30.0(7) | 10.6(2) | 42(2) |
| 25 | 23(9) | 26.2(13) | 13.4(3) | 38(4) |
| 75 | 39(4) | 17.7(6) | 13.3(2) | 30(2) |
In Fig. 5b, the Co 2p regions are displayed for each depth profiling step. In order to determine the oxidation state of the cobalt in the structure, the reference spectra of CoO and Co3O4 were used. However, the surface spectrum (bottom-most spectrum) could not be fitted by a linear combination of these two references due to the higher CoIII concentration as compared to Co3O4, and no reliable pure CoIII reference could be obtained. Therefore, a different fitting procedure had to be applied for this surface spectrum. Instead of using the sample spectrum as the one to be fitted, the Co3O4 spectrum was used. The Co3O4 reference was, thus, fitted by a linear combination of the CoO spectrum and the sample spectrum. The fitting equation is shown by the following formula, with Sx being the spectra of compound x and Ix the respective intensity factors:
| SCo3O4 = ICoO × SCoO + Isample × Ssample |
Since the amount of CoIII is the quantity of interest, and CoO contains only CoII, all the CoIII required for the description of Co3O4 (corresponding to 66.67%) has to come from the sample compound. Thus, the intensity of the sample spectrum can directly be linked to the concentration of CoIII in the sample by rewriting this equation to c(CoIII, Co3O4) = 2/3 = Isample × c(CoIII, sample), with c(CoIII, x) describing the CoIII concentration of compound x. Since the fitting procedure showed that the Co3O4 spectrum can be described by a linear combination of 27% of the CoO spectrum and 73% of the sample spectrum, the CoIII concentration in the sample was 91(3)%. This means that the average oxidation state of cobalt in the oxide was 2.91(9). From the formula sum, the calculated theoretical oxidation state was 2.83, assuming that tellurium was only present as TeVI, which was within the experimental errors of this analysis.
The surface oxidation state of a sample, annealed for 36 hours only, was analyzed too, and showed 89(3)% of CoIII, which is in excellent agreement with the sample annealed for 100 h. This indicated that the oxidation of Co had already been completed after 36 hours heat treatment.
Upon sputtering the specimen annealed for 100 hours, the oxidation state of Co was reduced significantly, shown in the top three spectra in Fig. 5b. For these spectra, the fitting of a linear combination of CoO and Co3O4 was possible, because the CoIII concentration was lower than that for Co3O4. At 5 nm, the sum of 75% CoO and 25% Co3O4 described the spectrum well, resulting in a CoIII concentration of 16.8% in the sample corresponding to an oxidation state of 2.17. This drastic shift in the oxidation state can also be seen from the satellite feature to the high binding energy side of the 2p3/2 component, which was shifted to lower binding energies with increasing CoII concentration. At 25 nm, the Co ions were reduced even further, leading to only 2% of CoIII and a sum oxidation state of 2.02. Upon reaching a depth of 75 nm, no change compared to the 25 nm depth was detected. This strong reduction of the cobalt species most likely resulted from the sputtering process itself by bombardment with highly energetic argon ions (such as the 3 keV ones employed here). It is known that especially transition metals are affected to be reduced by preferential sputtering with argon ions.53,54
The Te 3d components featured a distinct shoulder at the lower binding energy sides (see Fig. 5c). While the majority of the signal could be described by a component with a binding energy of 576.4 eV for 3d5/2, and thus could be attributed to TeVI,55 the asymmetry of the peaks required a second component at 574.8 eV to be added. Based on the binding energy, this could originate from a TeIV species,56 which was expected to exist on the surface exhibiting a lower coordination number for tellurium. Similar surface species are known for various other compounds such as Sr-rich perovskites or yttrium oxide.57,58 Because TeIV accounted for 18% of the total Te 3d signal with an escape depth of 2.02 nm of the corresponding electrons, an overlayer thickness of 0.37 nm could be assumed relating to a single atomic layer. This gives strong evidence that TeIV is a surface species. The presence of TeIV also indicated that the crystal is tellurium-terminated, instead of being, e.g. cobalt-terminated. For confirmation, ion scattering data giving information about the outermost surface composition would be required.
During sputtering, there was a noticeable peak shift in the Te 3d region, as the major component was at a binding energy of 575.8 eV. In the literature, there are reports of mixed TeIV/VI oxides resulting in peaks in this area.56 However, it was still located 1.0 eV higher than the possible TeIV species, attributed to the surface spectrum. The difference in binding energy further confirmed the assignment of TeIV as a surface species. Additionally, small contributions from suboxidic species with binding energies of 573.7 eV and 572.4 eV were identified. The lower binding energy component could even be attributed to elemental Te,55,59 but exact assignment was difficult with a low concentration making up only 2% of the total spectrum. Further sputtering steps did not alter the spectrum significantly.
Frequency-dependent impedance measurements (Nyquist plots). To gain information on the ionic conductivity of the sample, frequency dependent EIS measurements (Nyquist plots) were performed for Li3Co1.06(1)TeO6. The tests were conducted in air between RT and 723 K. The Li3Co1.06(1)TeO6 sample shows the characteristic semicircles as displayed in Fig. 7 indicating the ionic conductivity of the material. At temperatures between 523 and 573 K, two depressed semicircles are apparent: a very large one at high frequencies and a small one at low frequencies. The bulk (b) contribution is usually observed at high frequencies in the Nyquist plot, whereas the grain boundary (gb) contribution is located at mid and low frequencies.30,60–62 The equivalent circuit model that was used in this study to fit the experimental data is composed of constant phase elements (CPEs) instead of ideal Debye's capacitors. This is generally done for polycrystalline samples due to material inhomogeneity, surface defects, ionic transport deviation from Fick's law, and electrode roughness. All these parameters give rise to a certain degree of frequency dispersion and non-uniformity of the current density. Hence, a CPE represents more accurately the capacitive behavior of this kind of cell in the whole studied frequency range.30,63–65 This can also be clearly seen in the depressed semicircles in Fig. 7. It is shown that with an increase in temperature, Rb and Rgb decrease. The determined b and gb resistances were then converted to electrical conductivities (σ) by considering the thickness (0.1 mm) and area (0.196 cm2) of the sample (see Table 7).
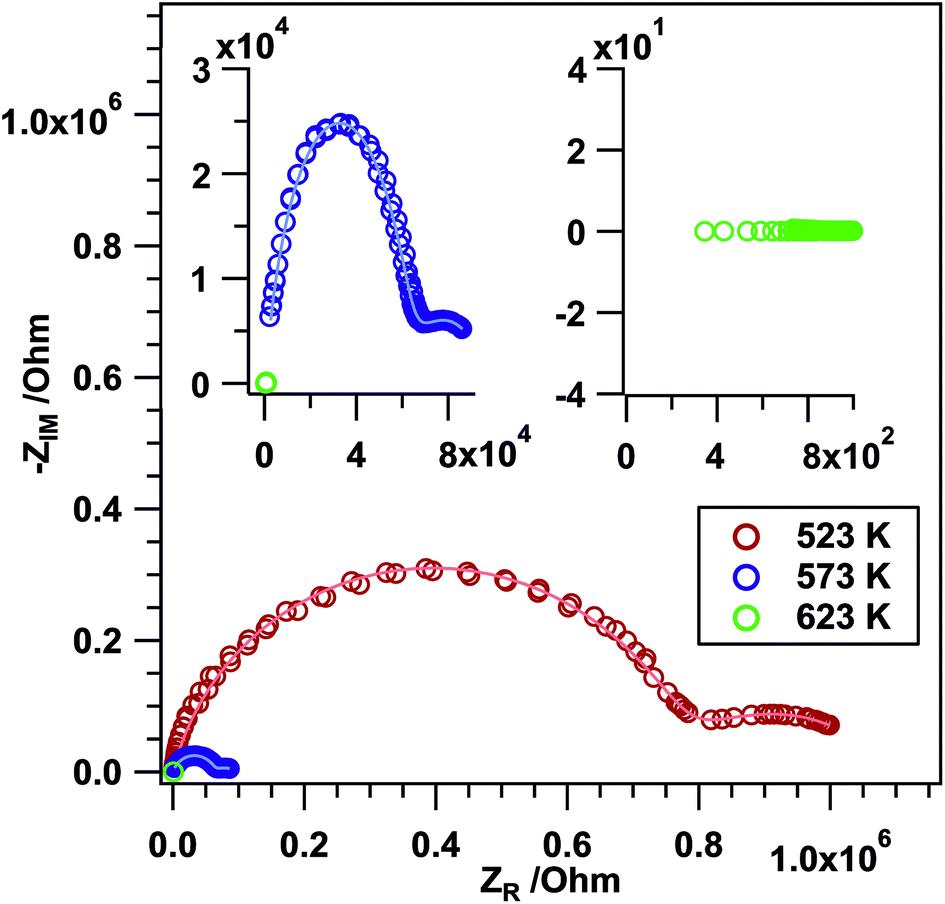 | ||
| Fig. 7 AC impedance spectra (data points) and simulated spectra (continuous lines) of the bulk and grain boundary contribution of the Li3Co1.06(1)TeO6 sample in air at selected temperatures between 523 and 623 K. The lowest frequency of 0.1 Hz is at the right side and the highest one of 1 MHz at the left side of the x-axis. The insets show zoomed regions of the Nyquist plots (NPs) at higher temperatures. | ||
| Temperature/K | R b/Ω | σ b/S cm−1 | C b/F | R gb/Ω | σ gb/S cm−1 | C gb/F |
|---|---|---|---|---|---|---|
| 523 | 6.82 × 105 | 7.47 × 10−8 | 3.56 × 10−11 | 1.87 × 105 | 2.72 × 10−8 | 6.17 × 10−8 |
| 573 | 8.07 × 104 | 6.31 × 10−7 | 3.06 × 10−11 | 3.10 × 104 | 1.64 × 10−6 | 4.88 × 10−9 |
Kumar et al. determined the ionic conductivities for the compound Li2Ni2TeO6 which crystallizes isopointal to our investigated sample Li3Co1.06(1)TeO6 and reported values in the range of 2 × 10−4 S cm−1 at 573 K. This value is about two orders of magnitude higher than the grain boundary conductivity σgb of 1.64 × 10−6 S cm−1 measured for Li3Co1.06(1)TeO6 at 523 K. The differences in conductivity may be explained by changes in the lattice polarization (NiII instead of CoIII), changes in the lithium coordination geometries (significant occupation of tetrahedral voids) and site occupation. Comparable values were found for the lithium-containing garnets Li3Ln3Te2O12 (Ln = Y, Pr, Nd, Sm–Lu) with a maximum value of 1 × 10−5 S cm−1 at 873 K measured for the neodymium compound.66 In crystalline solids, the highest reported Li+ conductivities of about 1 × 10−3 S cm−1 at room temperature have been realized in a perovskite-type lithium lanthanum titanate.67,68
However, starting at a temperature of 623 K and above only a straight line in the corresponding NPs is visible. The imaginary part of the impedance is basically zero and only a “real” resistance (without a phase shift) is observed indicating a significantly increased conductivity with ohmic resistance contributions.
Time-/temperature-dependent impedance measurements. Time- and temperature dependent AC impedance experiments were carried out to check for eventual phase transformations in the studied temperature region. At first, the same temperature region as used for the NPs was chosen. At RT, an impedance value of 2.26 × 107 Ω is apparent. Upon heating to ∼353 K the impedance starts to increase leading to a value of 8.27 × 109 Ω. In the temperature region between 353 and 723 K, semiconductive behavior with a decreasing impedance value upon increasing the temperature is visible. Basically, the same impedance course is visible upon re-cooling to RT with slight differences between 371 K and RT. No directly visible phase transformation processes are apparent during the heating–cooling cycle. Further details as well as plots of the time- and temperature-dependent in situ EIS measurements (Fig. S2†) are given in the ESI.†
4 Conclusion
The compound Li3Co1.06(1)TeO6 synthesized under high-temperature conditions exhibited Li-ion conductivity as well as CoII/CoIII mixed valence states. In contrast to the isopointal compound Li2Ni2TeO6 reported by Kumar et al., the here presented Li3Co1.06(1)TeO6 revealed MIII ions next to TeVI ions for the first time. Magnetic susceptibility measurements and XPS investigations clearly indicated the presence of CoIII in this compound, although the exact quantification is difficult. ICP measurements were supposed to be the most accurate way to quantify the Li and Co content of a 100 mg Li3Co1.06(1)TeO6 sample. The Li/Co ratio thus obtained was used for the single-crystal structure refinement and resulted in completely occupied Li sites after consideration of additional electron density in tetrahedral voids. Consequently, for reasons of electro-neutrality, the cobalt site must be mixed valent, which was proved by magnetic susceptibility and XPS measurements. The facts that XPS measurements even indicated a higher lithium content in the sample, and the effective magnetic moment proposed a lower CoIII content than 91(3)% suggest that Li3Co1.06(1)TeO6 could be a promising electrode material based on the CoII/CoIII redox couple.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Dr. H. Huppertz for continuous support and usage of all the facilities of the Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck. For the execution of the ICP measurements we thank Ass. Prof. Dr. R. Tessadri, Institute of Mineralogy and Petrography. This work was financially supported by the Tiroler Wissenschaftsfond (TWF); project no.: 235863. Dr. G. Heymann was supported by the program Nachwuchsförderung of the University Innsbruck. T. Götsch acknowledges funding by the Austrian Science Fund (FWF) via grant F4503-N16. This work was performed within the framework of the research platform “Material's and Nanoscience” at the University of Innsbruck.References
- M. S. Whittingham, MRS Bull., 2008, 33, 411–419 CrossRef CAS.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, K. Guruprakash, A. S. Prakash, M. L. Doublet and J. M. Tarascon, Chem. Commun., 2013, 49, 11376–11378 RSC.
- J. Bao, D. Wu, Q. Tang, Z. Ma and Z. Zhou, Phys. Chem. Chem. Phys., 2014, 16, 16145–16149 RSC.
- E. McCalla, A. S. Prakash, E. Berg, M. Saubanère, A. M. Abakumov, D. Foix, B. Klobes, M.-T. Sougrati, G. Rousse, F. Lepoivre, S. Mariyappan, M.-L. Doublet, D. Gonbeau, P. Novak, G. Van Tendeloo, R. P. Hermann and J.-M. Tarascon, J. Electrochem. Soc., 2015, 162, A1341–A1351 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- I. Terasaki, Y. Sasago and K. Uchinokura, Phys. Rev. B: Condens. Matter, 1997, 56, R12685–R12687 CrossRef CAS.
- K. Takada, H. Sakurai, E. Takayama-Muromachi, F. Izumi, R. A. Dilanian and T. Sasaki, Nature, 2003, 422, 53–55 CrossRef CAS PubMed.
- M. A. Evstigneeva, V. B. Nalbandyan, A. A. Petrenko, B. S. Medvedev and A. A. Kataev, Chem. Mater., 2011, 23, 1174–1181 CrossRef CAS.
- V. B. Nalbandyan, A. A. Petrenko and M. A. Evstigneeva, Solid State Ionics, 2013, 233, 7–11 CrossRef CAS.
- C. Fouassier, C. Delmas and P. Hagenmuller, Mater. Res. Bull., 1975, 10, 443–449 CrossRef CAS.
- M. Castellanos, J. A. Gard and A. R. West, J. Appl. Crystallogr., 1982, 15, 116–119 CrossRef CAS.
- J. G. Fletcher, G. C. Mather, A. R. West, M. Castellanos and M. P. Gutierrez, J. Mater. Chem., 1994, 4, 1303–1305 RSC.
- G. C. Mather, R. I. Smith, J. M. S. Skakle, J. G. Fletcher, M. A. Castellanos R, M. P. Gutierrez and A. R. West, J. Mater. Chem., 1995, 5, 1177–1182 RSC.
- G. C. Mather and A. R. West, J. Solid State Chem., 1996, 124, 214–219 CrossRef CAS.
- J. M. S. Skakle, M. A. Castellanos R, S. T. Tovar and A. R. West, J. Solid State Chem., 1997, 131, 115–120 CrossRef CAS.
- E. A. Zvereva, M. A. Evstigneeva, V. B. Nalbandyan, O. A. Savelieva, S. A. Ibragimov, O. S. Volkova, L. I. Medvedeva, A. N. Vasiliev, R. Klingeler and B. Buechner, Dalton Trans., 2012, 41, 572–580 RSC.
- R. Berthelot, W. Schmidt, S. Muir, J. Eilertsen, L. Etienne, A. W. Sleight and M. A. Subramanian, Inorg. Chem., 2012, 51, 5377–5385 CrossRef CAS PubMed.
- O. A. Smirnova, V. B. Nalbandyan, A. A. Petrenko and M. Avdeev, J. Solid State Chem., 2005, 178, 1165–1170 CrossRef CAS.
- V. V. Politaev, V. B. Nalbandyan, A. A. Petrenko, I. L. Shukaev, V. A. Volotchaev and B. S. Medvedev, J. Solid State Chem., 2010, 183, 684–691 CrossRef CAS.
- L. Viciu, Q. Huang, E. Morosan, H. W. Zandbergen, N. I. Greenbaum, T. McQueen and R. J. Cava, J. Solid State Chem., 2007, 180, 1060–1067 CrossRef CAS.
- J. Xu, A. Assoud, N. Soheilnia, S. Derakhshan, H. L. Cuthbert, J. E. Greedan, M. H. Whangbo and H. Kleinke, Inorg. Chem., 2005, 44, 5042–5046 CrossRef CAS PubMed.
- V. Kumar, A. Gupta and S. Uma, Dalton Trans., 2013, 42, 14992–14998 RSC.
- V. Kumar, N. Bhardwaj, N. Tomar, V. Thakral and S. Uma, Inorg. Chem., 2012, 51, 10471–10473 CrossRef CAS PubMed.
- V. B. Nalbandyan, M. Avdeev and M. A. Evstigneeva, J. Solid State Chem., 2013, 199, 62–65 CrossRef CAS.
- J. Choisnet, A. Rulmont and P. Tarte, J. Solid State Chem., 1988, 75, 124–135 CrossRef CAS.
- J. Choisnet, A. Rulmont and P. Tarte, J. Solid State Chem., 1989, 82, 272–278 CrossRef CAS.
- E. A. Zvereva, O. A. Savelieva, Y. D. Titov, M. A. Evstigneeva, V. B. Nalbandyan, C. N. Kao, J. Y. Lin, I. A. Presniakov, A. V. Sobolev, S. A. Ibragimov, M. Abdel-Hafiez, Y. Krupskaya, C. Jahne, G. Tan, R. Klingeler, B. Buchner and A. N. Vasiliev, Dalton Trans., 2013, 42, 1550–1566 RSC.
- APEX2 (v. 2014.11-0), CELL_NOW (v. 2008/4), SAINT (v. 8.34A), TWINABS (v. 2012/1), and SADABS (v. 2014/5), Bruker AXS GmbH, Karlsruhe, Germany Search PubMed.
- Thermo Scientific XPS Reference, Thermo Fisher Scientific Inc., http://www.xpssimplified.com, (accessed 23.05.2017, 2017).
- M. Kogler, E. M. Köck, B. Klötzer, L. Perfler and S. Penner, J. Phys. Chem. C, 2016, 120, 3882–3898 CAS.
- H. J. Avila-Paredes, J. Zhao, S. Wang, M. Pietrowski, R. A. De Souza, A. Reinholdt, Z. A. Munir, M. Martin and S. Kim, J. Mater. Chem., 2010, 20, 990–994 RSC.
- N. J. Kidner, N. H. Perry, T. O. Mason and E. J. Garboczi, J. Am. Ceram. Soc., 2008, 91, 1733–1746 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, ShelXL – Crystal Structure Refinement – Multi-CPU Version, University of Göttingen Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- Y. Le Page, J. Appl. Crystallogr., 1988, 21, 983–984 CrossRef.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- P. Schmidt, Eur. J. Inorg. Chem., 2008, 2847–2855, DOI:10.1002/ejic.200800218.
- W. L. Smith and A. D. Hobson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 362–363 CrossRef CAS.
- J. Chenavas, J. C. Joubert and M. Marezio, Solid State Commun., 1971, 9, 1057–1060 CrossRef CAS.
- H. J. Orman and P. J. Wiseman, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 12–14 CrossRef.
- S. Abou-Warda, W. Pietzuch, G. Berghöfer, U. Kesper, W. Massa and D. Reinen, J. Solid State Chem., 1998, 138, 18–31 CrossRef CAS.
- H. Mevs and H. Müller-Buschbaum, Z. Anorg. Allg. Chem., 1990, 584, 114–118 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- J. T. S. Irvine, M. G. Johnston and W. T. A. Harrison, Dalton Trans., 2003, 2641–2645, 10.1039/B300573A.
- C. R. Feger, G. L. Schimek and J. W. Kolis, J. Solid State Chem., 1999, 143, 246–253 CrossRef CAS.
- C.-W. Wang, C.-H. Lee, C.-Y. Li, C.-M. Wu, W.-H. Li, C.-C. Chou, H.-D. Yang, J. W. Lynn, Q. Huang, A. B. Harris and H. Berger, Phys. Rev. B: Condens. Matter, 2013, 88, 184427 CrossRef.
- O. Peña, V. Bodenez, T. Guizouarn, E. Meza and J. L. Gautier, J. Magn. Magn. Mater., 2004, 272–276(Supplement), E1579–E1580 CrossRef.
- W. D. Johnston, R. R. Heikes and D. Sestrich, J. Phys. Chem. Solids, 1958, 7, 1–13 CrossRef CAS.
- A. Möller, Chem. Mater., 1998, 10, 3196–3201 CrossRef.
- CasaXPS: Processing Software for XPS, AES, SIMS and More, Casa Software Ltd Search PubMed.
- W. H. Gries, Surf. Interface Anal., 1996, 24, 38–50 CrossRef CAS.
- T. Götsch, W. Wallisch, M. Stöger-Pollach, B. Klötzer and S. Penner, AIP Adv., 2016, 6, 025119 CrossRef.
- T. Götsch, T. Schachinger, M. Stöger-Pollach, R. Kaindl and S. Penner, Appl. Surf. Sci., 2017, 402, 1–11 CrossRef.
- W. E. Sartz, K. J. Wynne and D. M. Hercules, Anal. Chem., 1971, 43, 1884–1887 CrossRef CAS.
- B. V. R. Chowdari and P. Pramoda Kumari, J. Non-Cryst. Solids, 1996, 197, 31–40 CrossRef CAS.
- R. Thalinger, T. Götsch, C. Zhuo, W. Hetaba, W. Wallisch, M. Stöger-Pollach, D. Schmidmair, B. Klötzer and S. Penner, ChemCatChem, 2016, 8, 2057–2067 CrossRef CAS.
- E.-M. Köck, M. Kogler, T. Götsch, B. Klötzer and S. Penner, Phys. Chem. Chem. Phys., 2016, 18, 14333–14349 RSC.
- J. M. Thomas, I. Adams, R. H. Williams and M. Barber, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 755–764 RSC.
- L. Rojo, G. Ga Mandayo and E. Castano, Thin film YSZ solid state electrolyte characterization performed by electrochemical impedance spectroscopy, 9th Spanish Conference on Electron Devices (CDE), Valladolid, Spain, 2013.
- M. Wierzbicka, P. Pasierb and M. Rekas, Physica B, 2007, 387, 302–312 CrossRef CAS.
- E. M. Köck, M. Kogler, B. Klötzer, M. F. Noisternig and S. Penner, ACS Appl. Mater. Interfaces, 2016, 8, 16428–16443 Search PubMed.
- K. Obal, Z. Pedzich, T. Brylewski and M. Rekas, Int. J. Electrochem. Sci., 2012, 7, 6831–6845 CAS.
- S. Vishweswaraiah, Masters Thesis, Central Florida, 2004.
- J. R. MacDonald, Impedance Spectroscopy - Theory, Experiment and Applications, Wiley, New York, 1987 Search PubMed.
- M. P. O'Callaghan, D. R. Lynham, E. J. Cussen and G. Z. Chen, Chem. Mater., 2006, 18, 4681–4689 CrossRef.
- S. Stramare, V. Thangadurai and W. Weppner, Chem. Mater., 2003, 15, 3974–3990 CrossRef CAS.
- A. D. Robertson, A. R. West and A. G. Ritchie, Solid State Ionics, 1997, 104, 1–11 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt02663c |
| This journal is © The Royal Society of Chemistry 2017 |