
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rotaxane synthesis exploiting the M(I)/M(III) redox couple†
Jack
Emerson-King
 ,
Richard C.
Knighton
,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
,
Richard C.
Knighton
,
Matthew R.
Gyton
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 24th August 2017
Abstract
In the context of advancing the use of metal-based building blocks for the construction of mechanically interlocked molecules, we herein describe the preparation of late transition metal containing [2]rotaxanes (1). Capture and subsequent retention of the interlocked assemblies are achieved by the formation of robust and bulky complexes of rhodium(III) and iridium(III) through hydrogenation of readily accessible rhodium(I) and iridium(I) complexes [M(COD)(PPh3)2][BArF4] (M = Rh, 2a; Ir, 2b) and reaction with a bipyridyl terminated [2]pseudorotaxane (3·db24c8). This work was underpinned by detailed mechanistic studies examining the hydrogenation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of 2 and bipy in CH2Cl2, which proceeds with disparate rates to afford [M(bipy)H2(PPh3)2][BArF4] (M = Rh, 4a[BArF4], t = 18 h @ 50 °C; Ir, 4b[BArF4], t < 5 min @ RT) in CH2Cl2 (1 atm H2). These rates are reconciled by (a) the inherently slower reaction of 2a with H2 compared to that of the third row congener 2b, and (b) the competing and irreversible reaction of 2a with bipy, leading to a very slow hydrogenation pathway, involving rate-limiting substitution of COD by PPh3. On the basis of this information, operationally convenient and mild conditions (CH2Cl2, RT, 1 atm H2, t ≤ 2 h) were developed for the preparation of 1, involving in the case of rhodium-based 1a pre-hydrogenation of 2a to form [Rh(PPh3)2]2[BArF4]2 (8) before reaction with 3·db24c8. In addition to comprehensive spectroscopic characterisation of 1, the structure of iridium-based 1b was elucidated in the solid-state using X-ray diffraction.
:1 mixtures of 2 and bipy in CH2Cl2, which proceeds with disparate rates to afford [M(bipy)H2(PPh3)2][BArF4] (M = Rh, 4a[BArF4], t = 18 h @ 50 °C; Ir, 4b[BArF4], t < 5 min @ RT) in CH2Cl2 (1 atm H2). These rates are reconciled by (a) the inherently slower reaction of 2a with H2 compared to that of the third row congener 2b, and (b) the competing and irreversible reaction of 2a with bipy, leading to a very slow hydrogenation pathway, involving rate-limiting substitution of COD by PPh3. On the basis of this information, operationally convenient and mild conditions (CH2Cl2, RT, 1 atm H2, t ≤ 2 h) were developed for the preparation of 1, involving in the case of rhodium-based 1a pre-hydrogenation of 2a to form [Rh(PPh3)2]2[BArF4]2 (8) before reaction with 3·db24c8. In addition to comprehensive spectroscopic characterisation of 1, the structure of iridium-based 1b was elucidated in the solid-state using X-ray diffraction.
Introduction
Coordination chemistry is an increasingly prominent feature of contemporary methods for preparing mechanically interlocked molecules.1,2 In most cases metal ions are employed as well-defined templates, pre-organising the fusion of heteroatom-based molecular building blocks, but thereafter jettisoned to confer an interwoven organic product. An alternative but comparatively underdeveloped approach, resulting instead in retention of the metal complex within the final framework, involves capture of the entangled topology itself through formation of a persistent metal–ligand bond. Such an approach represents a potentially versatile means for preparing new and interesting metal-containing interlocked systems, as exemplified through the recent emergence of polyrotaxane metal–organic frameworks.3In the context of archetypical [2]rotaxane systems, a straight forward scheme employing metal-based building blocks involves coordination of a bulky metal fragment to a donor functionalised “axle” interpenetrated within a macrocycle, viz. metal complex capping of a [2]pseudorotaxane (Scheme 1).1 Indeed, the successful application of such a synthesis was demonstrated as early as 1981, with Ogino using cobalt(III) fragments to capture [2]rotaxanes comprised of a cyclodextrin macrocycle and diaminoalkane axle (A).4 With the notable exception of Sauvage's bis(terpyridine)-ruthenium(II) system B,5 the overwhelming majority of subsequent [2]rotaxanes prepared through metal complex capping (e.g.C–E) have, however, involved relatively weak dative bonding of mono-dentate pyridine donors.6
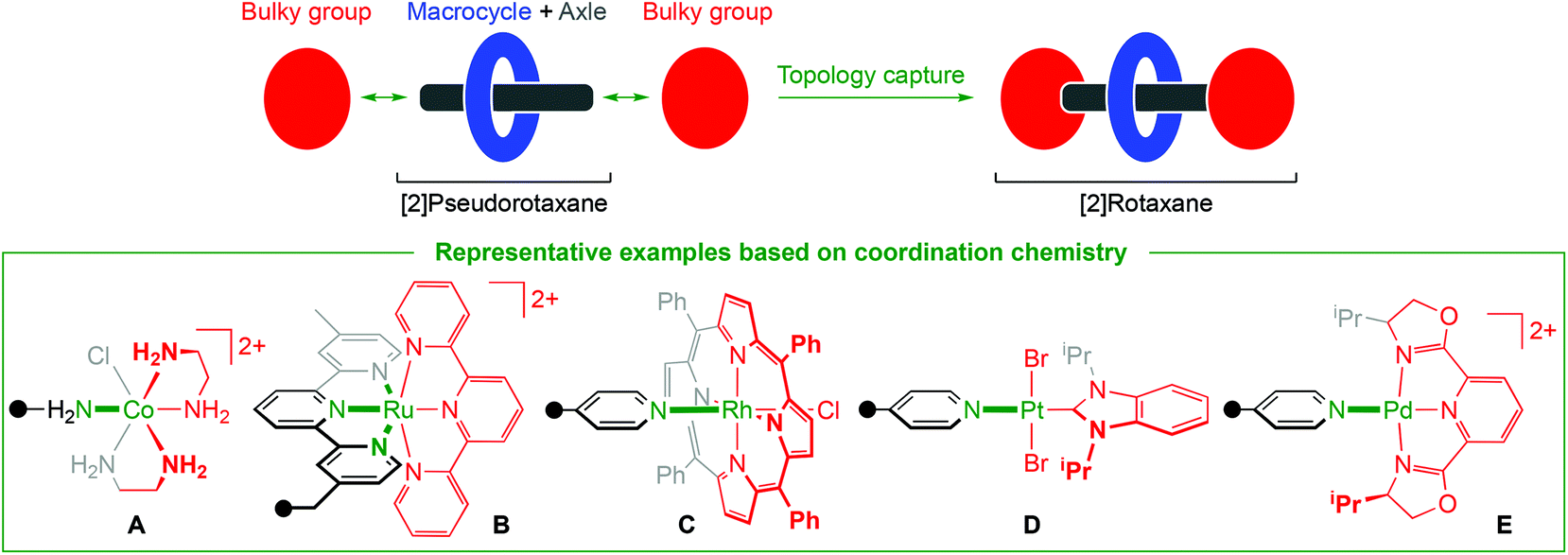 | ||
| Scheme 1 Synthesis of [2]rotaxanes through capping methodology. | ||
Seeking to further develop synthetic methods involving metal complex capping, we targeted the construction of [2]rotaxanes though formation of robust complexes of chelating ligands with bulky late transition metal fragments. To this end, we herein report the preparation of 1 though installation of rhodium(III) and iridium(III) fragments {M(PPh3)2H2}+, generated in situ by hydrogenation of readily accessible rhodium(I) and iridium(I) complexes [M(COD)(PPh3)2][BArF4] (M = Rh, 2a; Ir, 2b; COD = 1,5-cyclooctadiene; ArF = 3,5-(CF3)2C6H3), upon bipyridyl terminated [2]pseudorotaxane 3·db24c8 (Chart 1; db24c8 = dibenzo-24-crown-8). Mechanistic and structural aspects relevant to this process are first detailed using 2,2′-bipyridine (bipy) as a model.
 | ||
| Chart 1 Components of target [2]rotaxanes 1. | ||
Results and discussion
The hydrogenation of [M(diene)(PPh3)2]+ (M = Rh, Ir) to afford metal fragments of the type {M(PPh3)2H2}+ is well established and evidenced through characterisation as adducts of solvent, e.g. [M(PPh3)2H2(OCMe2)2]+ and [Ir(PPh3)2H2(ClCH2CH2Cl)]+,7,8 or the counter anion, e.g. [Ir(PPh3)2H2(1-closo-CB11H6X6)] (X = Cl, I).9 Acetone adducts in particular are well-defined M(III) synthons and have enabled isolation of [M(bipy)H2(PPh3)2]+ (M = Rh, 4a+; Ir, 4b+) through subsequent reaction with bipy in acetone solution.10 In the context of our envisioned synthesis of 1, where robust retention of 3·db24c8 in solution is critical, we sought instead to adapt a procedure reported by Crabtree for the preparation of 4b+ by hydrogenation of a mixture of [Ir(COD)(PPh3)2]+ and bipy in dichloromethane solution (for a range of anions).11 As such we first studied hydrogenation reactions (1 atm) of 1:1 mixtures of 2 (20 mM) and bipy in CD2Cl2, using NMR spectroscopy, to gauge the viability of this procedure for enacting metal complex capping of 3·db24c8 (Scheme 2 and vide infra). Satisfyingly both the rhodium(I) and iridium(I) complexes were hydrogenated quantitatively in the presence of bipy to afford 4[BArF4], although curiously the former required significantly more forcing conditions (t = 18 h @ 50 °C) than the latter (t < 5 min @ RT). In each case the identity of the dihydride products was confirmed by isolation on a preparative scale and full structural characterisation. Spectroscopic data for 4[BArF4] are unsurprisingly in close agreement with precedents bearing different counter anions;10,11 useful markers include low frequency hydride signals at δ −15.66 (app. q, 1JRhH ≈ 2JPH = 14 Hz; M = Rh)/−19.48 (t, 2JPH = 16.6 Hz; M = Ir) and 31P resonances at δ 47.1 (d, 1JRhP = 115 Hz; M = Rh)/20.1 (M = Ir).
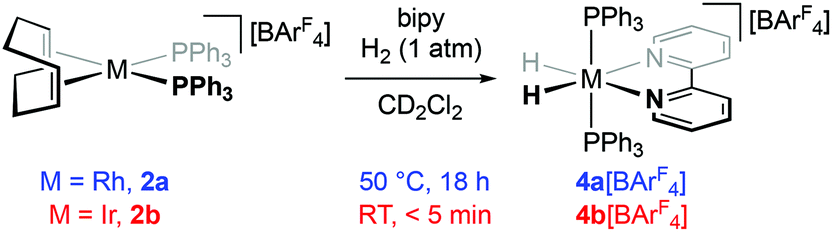 | ||
| Scheme 2 Preparation of 4[BArF4]. | ||
Crystallographic data was also obtained in both cases for 4a[BArF4] and 4b[BArF4]. For the iridium(III) complex two polymorphs were identified and studied, one of which features two independent cations in the asymmetric unit (Z′ = 2). All cations bear the expected pseudo-octahedral metal geometries with trans-phosphine ligands (Fig. 1).12–14 Illustrating the subtle effects of crystal packing, originating though variation of the anion and in some cases presence of solvent molecules, a wide range of geometries are observed for 4+ in the solid-state: including both staggered and eclipsed conformations of the phosphine substituents (cf.4a[BArF4] vs.4b[OTf]), and alternative orientations of the phosphines about the metal–phosphine vector (cf.4a[BArF4] vs.4b[BArF4] (Z′ = 1)).
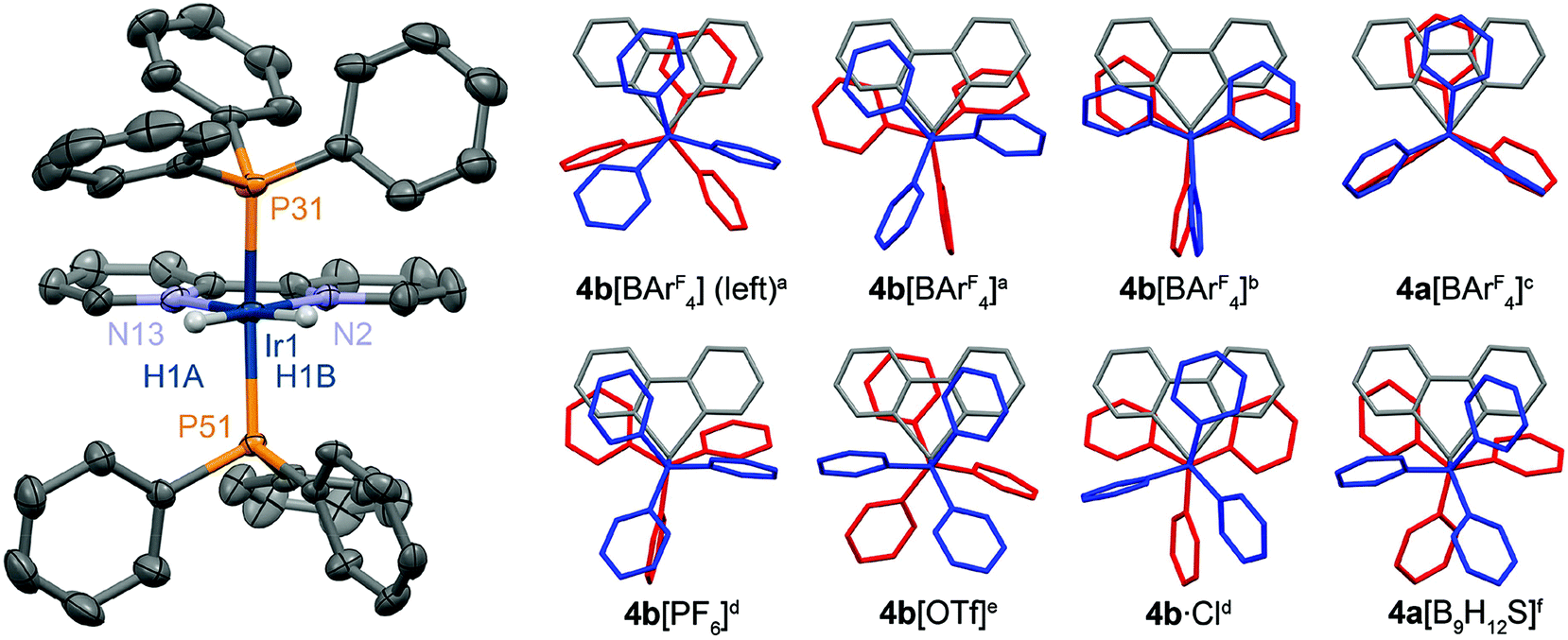 | ||
| Fig. 1 Solid-state structures of 4+. Left: Selected independent cation from the X-ray structure of 4b[BArF4] (Z′ = 2) with thermal ellipsoids drawn at the 50% probability level; non-hydridic hydrogen atoms omitted for clarity. Right: Conformations of 4+ viewed along the P–M–P vector, with phosphine ligands differentially coloured to aid comparison. aThis work, polymorph with Z′ = 2; bthis work, polymorph with Z′ = 1; cthis work; dref. 12; eref. 13; fref. 14. Selected bond lengths (Å) and angles (°): 4a[BArF4], Rh1–P31, 2.3119(6); Rh1–N2, 2.154(2); P31–Rh1–P31*, 174.05(4); *1 − x, +y, 1 − z; 4b[BArF4] (Z′ = 2), Ir1–P31, 2.2924(8); Ir1–P51, 2.3009(7); Ir1–N2, 2.118(2); Ir1–N13, 2.151(2); P31–Ir1–P51, 166.53(3); Ir11–P131, 2.3048(7); Ir11–P151, 2.3035(7); Ir11–N102, 2.115(2); Ir11–N113, 2.145(2); P131–Ir11–P151, 166.40(3); 4b[BArF4] (Z′ = 1), Ir1–P31, 2.3000(8); Ir1–P51, 2.2945(8); Ir1–N2, 2.138(2); Ir1–N13, 2.128(3); P31–Ir1–P51, 164.08(3). | ||
Despite both resulting in formation of the desired products, the disparate rates prompted us to interrogate the mechanism of 2 + bipy hydrogenation in dichloromethane. To this end the kinetics of reactions of 2 (20 mM) with bipy and H2 (1 atm) were examined individually in CD2Cl2 at RT using 1H and 31P NMR spectroscopy (Scheme 3). Presumably driven by the chelate effect, reaction between 2 and bipy resulted in irreversible formation of five coordinate [M(bipy)(COD)(PPh3)][BArF4] (M = Rh, 5a; Ir, 5b) alongside concomitant liberation of PPh3, although under vastly different kinetic regimes (t1/2 = 1.3 h, 2a; 34 h, 2b). Moreover dynamic exchange between bound/free phosphine is observed on the NMR timescale in the rhodium system, leading to a single broad 31P signal at δ 10.6 (fwhm = 55 Hz) at 298 K that decoalesced into two sharp resonances at δ 33.3 (d, 1JRhP = 129 Hz) and −8.3 in a 1:1 ratio on cooling to 200 K. Equivalent reaction mixtures are obtained from reactions between [M(bipy)(COD)][BArF4] (M = Rh, 6a; Ir, 6b) and two equivalents of PPh3 in CD2Cl2, but on a much faster timescale (t < 5 min). Indeed, 5 were subsequently isolated from reactions between 6 and one equivalent of PPh3 for independent structural verification in solution and the solid-state (see Experimental section, Fig. 2 and CIF). While no further reaction of 5b with PPh3 was observed in solution, COD is slowly and reversibly displaced from the lighter congener by PPh3 resulting in equilibrium formation of [Rh(bipy)(PPh3)2][BArF4] 7 (t1/2 = 107 h; K = 2.4) at RT.15
 | ||
| Scheme 3 Hydrogenation of 2 in the presence of bipy. Conditions: 20 mM, CD2Cl2, RT and 1 atm H2 (where relevant). Counter anion = [BArF4]− throughout. COE = cyclooctene. | ||
 | ||
| Fig. 2 Solid-state structure of 5b. Thermal ellipsoids drawn at the 50% probability level; anion and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): 5a, Rh1–P31, 2.3306(7); Rh1–N2, 2.262(2); Rh1–N13, 2.161(2); Rh1–Cnt(C14,C15), 1.996(3); Rh1–Cnt(C18,C19), 2.072(3); P31–Rh1–C18, 179.36(9); 5b, Ir1–P31, 2.3426(6); Ir1–N2, 2.213(2); Ir1–N13, 2.121(2); Ir1–Cnt(C14,C15), 2.013(2); Ir1–Cnt(C18,C19), 2.017(2); P31–Ir1–C18, 171.56(8). | ||
Hydrogenation of 2a in CD2Cl2 proceeded at a moderate rate (t1/2 = 0.8 h) to produce Rh(I) dimer [Rh(PPh3)2]2[BArF4]2 (8) as the exclusive organometallic product alongside cyclooctane (COA).16 Complex 8 was subsequently isolated on a preparative scale and reacted with bipy (under an argon atmosphere) to afford 7 (t < 5 min).15 Placing isolated 7 under hydrogen (1 atm) thereafter quantitatively produced 4a[BArF4] (t < 5 min).15 Reaction of 2b with hydrogen rapidly (t < 5 min) gave rise to a mixture of Ir(III) complexes of the formulation [Ir(PPh3)2H2L2][BArF4] (9; L = H2, CD2Cl2), with concomitant generation of COA, which then afforded 4b[BArF4] quantitatively upon addition of bipy under hydrogen (t < 5 min).8,17
Together these mechanistic experiments allow the disparate rates of 2 + bipy hydrogenation in dichloromethane to be reconciled by (a) the inherently slower reaction of 2a with H2 compared to that of third row congener 2b,18 and (b) competing and irreversible reaction of 2a with bipy, leading to a very slow hydrogenation pathway, involving rate-limiting substitution of COD by PPh3. Consistent with these proposals intermediate presence of 5a is observed in situ during the formation of 4a[BArF4] from hydrogenation of 2a + bipy in dichloromethane at 50 °C (Schemes 2 and 3). Under these conditions a (COA + COE):COD ratio of 42:58 was established at reaction completion by GC analysis,19,20 broadly in line with the relative rates of reaction of 2a with H2 (t1/2 = 0.8 h) and bipy (t1/2 = 1.3 h) determined in dichloromethane at RT. In contrast, no COD was detected by GC analysis on hydrogenation of 2b + bipy in dichloromethane at RT (Schemes 2 and 3).
Moving on from the mechanistic studies associated with metal complex capping reaction, the construction of 1 itself began with preparation of amine 10, which was readily obtained following a straightforward four step synthesis starting from previously reported 5-chloromethylbipyridine (Scheme 4; 66% yield over 4 steps).21 Subsequent protonation and halide exchange, using citric acid and Na[BArF4] under biphasic conditions (CH2Cl2/H2O), enabled isolation of the corresponding ammonium derivative 3 in high yield (84%). Under our chosen conditions, employing anhydrous dichloromethane and strategic incorporation of the weakly coordinating [BArF4]− counter anion,22 analysis by 1H NMR spectroscopy indicated [2]pseudorotaxane 3·db24c8 was assembled rapidly (t < 5 min) and quantitatively on dissolution of a 1:1 mixture of 3 and db24c8 in CD2Cl2 at RT. Distinctive features in the 1H NMR spectrum associated with the formation of 3·db24c8 include methylene resonances at δ 4.82 (bipyC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2) and 4.71 (ArC2) coupled to a partially obscured ammonium resonance at δ 7.70, and significantly perturbed db24c8 resonances (see ESI† for stack plots). Additional experiments involving variation of the components (2:1, 1:2) confirmed that the system is under slow exchange on the NMR timescale (500 MHz, 298 K) consistent with robust retention of 3·db24c8 in solution.
2) and 4.71 (ArC2) coupled to a partially obscured ammonium resonance at δ 7.70, and significantly perturbed db24c8 resonances (see ESI† for stack plots). Additional experiments involving variation of the components (2:1, 1:2) confirmed that the system is under slow exchange on the NMR timescale (500 MHz, 298 K) consistent with robust retention of 3·db24c8 in solution.
 | ||
| Scheme 4 Synthesis of [2]rotaxanes 1. Reagents and conditions: i. Potassium phthalimide, DMF (40 °C); ii. hydrazine monohydrate, EtOH (reflux); iii. 3,5-di-tert-butylbenzaldehyde, 3 Å molecular sieves, MeOH (RT); iv. Na[BH4] (0 °C to reflux); v. Na[BArF4], citric acid monohyrate, CH2Cl2/H2O (RT). | ||
Guided by the aforementioned mechanistic studies, rhodium-based 1a was prepared in high isolated yield (75%) by reaction between 3·db24c8 and 8, individually prepared in situ from 3 + db24c8 and 2a + H2 respectively, under hydrogen (1 atm) in dichloromethane at RT (t = 2 h). A more operationally straightforward procedure was possible for iridium-based 1b, involving addition of 2b to a solution 3·db24c8 and subsequently placing the reaction mixture under H2 (1 atm, t = 1 h), enabling isolation of the analytically pure product in high yield (78%). Employing this hydrogenation procedure, but heating at elevated temperature (50 °C, t = 27 h; cf. Scheme 2), with 2a and 3·db24c8 did result in the formation of 1a, but as the major component of an intractable mixture (ca. 60%). Such an observation is not unexpected given the greater complexity associated with the hydrogenation of 2a + bipy.
The capture of 1 was conviently established by ESI-MS, with strong [M]2+ signals observed at 732.7929 (1a, calcd 732.7937) and 777.8218 (1b, calcd 777.8228) m/z with the expected half integer spacing. The structures of 1 were additionally fully corroborated in solution using by a combination of 1H, 13C and 31P NMR spectroscopy. With the exception of the expected loss of symmetry, the metrics associated with the metal fragment are directly comparable to those of the model systems 4[BArF4]. For instance the hydride resonances of 1a/1b are observed at δ −15.50 and −15.85 (cf. −15.66)/−19.30 and −19.64 (cf. −19.48) with associated 2JHH (11.8/7.4 Hz) coupling, while the 31P resonances of 1a/1b are observed at δ 46.2 (1JRhP = 115 Hz; cf. 47.1, 1JRhP = 115 Hz)/19.8 (cf. 20.1). Likewise the spectroscopic indicators associated with interpenetration of the ammonium axle within db24c8 in 1 are very similar to those seen in the [2]pseudorotaxane 3·db24c8: for example, the methylene 1H resonances bipyC2 (1a, 4.31; 1b, 4.30; cf. 4.82) and ArCH2 (1a, 4.68; 1b, 4.69; cf. 4.71). Gratifyingly, we were also been able to grow single crystals of 1b and elucidate its solid-state structure using X-ray diffraction. [2]Rotaxane 1b crystallises from dichloromethane/hexane in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one full molecule in the asymmetric unit (Fig. 3). The iridium centre adopts the expected distorted octahedral geometry, with comparable metrics to those of 4b[BArF4] and analogues thereof bearing different counter anions (Fig. 1).12,13 Notably there is no meaningful steric buttressing apparent from close proximity of the interlocked db24c8 to the metal fragment (Ir1⋯N15, 6.540(2) Å), evident for example by the similarity of P31–Ir1–P51 bond angles in 1a (165.76(3)°) to those observed in 4b[BArF4] (166.53(3)°, 166.40(3)°, 164.08(3)°). Other salient features include the location of the hydride ligands off the Fourier difference map, and the presence of two strong hydrogen bonds between the ammonium cation and ether linkages of db24c8 (N15–O71, 3.057(3); N15–O77, 3.030(3) Å).
with one full molecule in the asymmetric unit (Fig. 3). The iridium centre adopts the expected distorted octahedral geometry, with comparable metrics to those of 4b[BArF4] and analogues thereof bearing different counter anions (Fig. 1).12,13 Notably there is no meaningful steric buttressing apparent from close proximity of the interlocked db24c8 to the metal fragment (Ir1⋯N15, 6.540(2) Å), evident for example by the similarity of P31–Ir1–P51 bond angles in 1a (165.76(3)°) to those observed in 4b[BArF4] (166.53(3)°, 166.40(3)°, 164.08(3)°). Other salient features include the location of the hydride ligands off the Fourier difference map, and the presence of two strong hydrogen bonds between the ammonium cation and ether linkages of db24c8 (N15–O71, 3.057(3); N15–O77, 3.030(3) Å).
 | ||
| Fig. 3 Solid-state structure of 1b. Thermal ellipsoids drawn at the 50% probability level; anions, most hydrogen atoms, and minor disordered components omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1–P31, 2.3103(5); Ir1–P51, 2.2950(5); Ir1–N2, 2.151(2); Ir1–N13, 2.137(2); Ir1⋯N15, 6.540(2); N15–O71, 3.057(3); N15–O77, 3.030(3); P31–Ir1–P51, 165.76(2). | ||
Summary and perspectives
We have presented a new reaction manifold for preparing [2]rotaxanes (1) using coordination chemistry. Our scheme involves topology capture through construction of bulky and robust rhodium(III) and iridium(III) complex stoppering groups from a bipyridyl terminated [2]pseudorotaxane (3·db24c8) and synthetically convenient metal precursors [M(COD)(PPh3)2][BArF4] (M = Rh, 2a; Ir, 2b) in CH2Cl2 under hydrogen (1 atm). Guided by detailed mechanistic studies, examining the hydrogenation of 1:1 mixtures of 2 and bipy, operationally convenient and mild conditions were developed for both metal-based systems. In the case of rhodium-based 1a, pre-hydrogenation of 2a to form [Rh(PPh3)2]2[BArF4]2 (8, t = 4 h, RT, 1 atm H2) before reaction with 3·db24c8 (t = 2 h, RT, 1 atm H2) is necessary to avoid an exceptionally slow hydrogenation pathway involving substitution of COD by PPh3. The chemistry associated with the iridium(I)/iridium(III) redox couple is much more attractive; in comparison to 2a the iridium precursor 2b shows sluggish reaction with bipy at RT (t1/2 = 34 h), whilst introduction of hydrogenation results in rapid and quantitative stoppering of 3·db24c8 (t < 1 h). Together this work showcases a facile route to the construction of new metal-containing interlocked molecules via formation of robust metal–ligand bonds. In addition to synthetic ease, the installation of {M(PPh3)2H2}+ metal fragments in entangled architectures proffers a number of advantages, including (a) useful spectroscopic handles (distinctive low frequently hydride and 31P signals); (b) large steric profile (trans-phosphine ligands); (c) the presence of heavy transition metals amenable for routine analysis by X-ray diffraction (e.g.Fig. 3); and perhaps more importantly, (d) the possibly to exploit interesting reactivity and spectroscopic properties of the metal fragment. We are particularly interested in exploring the latter as part of our on-going research in this area.
Experimental
General methods
Manipulations were performed under an argon atmosphere using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves (3 Å) were activated by heating at 300 °C in vacuo overnight. Anhydrous solvents (Et2O, CH2Cl2, CHCl3, pentane, hexane, THF and toluene) were purchased from Acros Organics or Sigma-Aldrich and stored over molecular sieves (3 Å). CD2Cl2 was dried over molecular sieves (3 Å) and stored under argon. 5-Chloromethylbipyridine,21 Na[BArF4]23 and [M(COD)2Cl]2 (M = Rh, Ir)24 were synthesised using established procedures. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers at 298 K unless otherwise stated. Chemical shirts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on Bruker Maxis Plus (HR) or Agilent 6130B single Quad (LR) instruments. Gas chromatography analyses were performed on an Agilent 7820A GC system fitted with a 7693A auto-injector. Microanalyses were performed at the London Metropolitan University by Mr Stephen Boyer.Preparation of 1a
A solution of 2a (50.8 mg, 32.0 μmol) in CH2Cl2 (0.9 mL) was freeze–pump–thaw degassed and placed under dihydrogen (1 atm). After stirring at RT for 4 h, the resulting deep red solution was added under dihydrogen to a solution of 3 (40.0 mg, 32.0 μmol) and db24c8 (14.4 mg, 32.1 μmol) in CH2Cl2 (0.9 mL). The solution was stirred at RT for 2 h and then added to pentane (ca. 30 mL) under hydrogen with stirring, whereupon an orange-red gum precipitated. The supernatant was decanted away and the product dried in vacuo for 5 min to afford the product as a dark-yellow foam. Yield = 76.7 mg (75%).
1
H NMR (500 MHz, CD2Cl2): δ 8.17 (s, 1H, bipy), 7.98 (d, 3JHH = 5.0, 1H, bipy), 7.80 (d, 3JHH = 9.0, 1H, bipy), 7.71–7.76 (m, 16H, ArF), 7.67 (vbr, fwhm = 26 Hz, 2H, N2), 7.50–7.61 (obscured, 3H, bipy), 7.55 (br, 8H, ArF), 7.45 (br, 1H, C6H3), 7.23–7.32 (m, 18H, Ph), 7.14–7.22 (m, 14H, C6H3 + Ph), 6.84–6.96 (m, 9H, bipy + C6H4), 4.65–4.72 (m, 2H, ArC2), 4.28–4.36 (m, 2H, bipyC2), 4.05–4.12 (m, 4H, OCH2), 3.94–4.02 (m, 4H, OCH2), 3.52–3.63 (m, 8H, OCH2), 3.33–3.47 (m, 8H, OCH2), 1.17 (s, 18H, tBu), −15.50 (app. p, J = 14 (1JRhH = 15.2, 2JHH = 11.8), 1H, RhH), −15.85 (app. p, J = 14 (1JRhH = 15.6, 2JHH = 11.8), 1H, RhH). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.4 (q, 1JBC = 50, ArF), 155.2 (s, bipy), 154.2 (s, bipy), 153.7 (s, bipy), 153.3 (s, bipy), 152.8 (s, C6H3), 147.8 (s, C6H4), 138.2 (s, bipy), 136.3 (s, bipy), 135.4 (s, ArF), 133.6 (app. t, JPC = 6, Ph), 132.5 (app. t, JPC = 24, Ph), 131.1 (s, Ph), 131.0 (s, bipy), 130.2 (s, C6H3), 129.4 (qq, 2JCF = 32, 2JCB = 3, ArF), 129.2 (app. t, JPC = 5, Ph), 127.1 (s, bipy), 125.4 (s, C6H3), 125.2 (q, 1JFC = 272, ArF), 124.8 (s, C6H3), 123.21 (s, bipy), 123.16 (s, C6H4), 122.7 (s, bipy), 118.1 (sept., 3JFC = 4, ArF), 113.8 (s, C6H4), 71.1 (s, OCH2), 70.7 (s, OCH2), 68.8 (s, OCH2), 55.1 (s, Ar![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 49.3 (s, bipyH2), 35.3 (s, tBu), 31.6 (s, tBu). 31P{1H} NMR (202 MHz, CD2Cl2): δ 46.2 (d, 1JRhP = 115). HR ESI-MS (positive ion, 4 kV): 732.7929 [M]2+ (calcd 732.7937) m/z. Anal. calcd for C150H122B2F48P2Rh (3193.04 g mol−1): C, 56.42; H, 3.85; N, 1.32. Found: C, 56.50; H, 4.02; N, 1.34.
H2), 49.3 (s, bipyH2), 35.3 (s, tBu), 31.6 (s, tBu). 31P{1H} NMR (202 MHz, CD2Cl2): δ 46.2 (d, 1JRhP = 115). HR ESI-MS (positive ion, 4 kV): 732.7929 [M]2+ (calcd 732.7937) m/z. Anal. calcd for C150H122B2F48P2Rh (3193.04 g mol−1): C, 56.42; H, 3.85; N, 1.32. Found: C, 56.50; H, 4.02; N, 1.34.
Attempted preparation of 1a by hydrogenation of 2a + 3·db24c8
To a solution of 3 (20.2 mg, 16.1 μmol) and db24c8 (7.2 mg, 16.1 μmol) in CH2Cl2 (0.8 mL) was added 2a (25.7 mg, 16.1 μmol). The resulting solution was freeze–pump–thaw degassed, placed under dihydrogen (1 atm), and stirred at 50 °C for 27 h. Volatiles were removed in vacuo and the solid redissolved in CH2Cl2 (ca. 2 mL) and layered with hexane (ca. 15 mL) to afford the crude product as a dark yellow gum, which was isolated through decantation of the supernatant and dried in vacuo. Analysis by 1H NMR spectroscopy in CD2Cl2 indicated a mixture of products with the major constituent being 1a in ca. 60% purity.Preparation of 1b
To a solution of 3 (40.0 mg, 32.0 μmol) and db24c8 (14.4 mg, 32.1 μmol) in CH2Cl2 (1.8 mL) was added 2b (54.1 mg, 32.0 μmol). The resulting solution was freeze–pump–thaw degassed and placed under dihydrogen (1 atm), and stirred at RT for 1 h. Volatiles were removed in vacuo and the solid redissolved in Et2O (ca. 2 mL) and layered with hexane (ca. 15 mL). Storage at ambient temperature overnight afforded a yellow oil, which was isolated through decantation of the supernatant and dried in vacuo overnight to afford the product as a bright yellow foam. Yield = 82.2 mg (78%).
1
H NMR (500 MHz, CD2Cl2): δ 8.26 (s, 1H, bipy), 8.16 (d, 3JHH = 5.4, 1H, bipy), 7.82 (d, 3JHH = 9.7, 1H, bipy), 7.71–7.75 (m, 16H, ArF), 7.67 (vbr, fwhm = 43 Hz, 2H, N2), 7.54–7.59 (obscured, 1H, bipy), 7.55 (br, 8H, ArF), 7.51 (d, 3JHH = 8.6, 1H, bipy), 7.47 (d, 3JHH = 8.2, 1H, bipy), 7.45 (s, 1H, C3H3), 7.22–7.31 (m, 18H, Ph), 7.13–7.22 (m, 14H, C6H3 + Ph), 6.86–6.97 (m, 8H, C6H4), 6.81–6.86 (m, 1H, bipy), 4.66–4.73 (m, 2H, ArC2), 4.26–4.34 (m, 2H, bipyC2), 4.05–4.14 (m, 4H, OCH2), 3.95–4.04 (m, 4H, OCH2), 3.52–3.64 (m, 8H, OCH2), 3.35–3.46 (m, 8H, OCH2), 1.17 (s, 18H, tBu), −19.30 (td, 2JPH = 16.9, 2JHH = 7.4, 1H, IrH), −19.64 (td, 2JPH = 16.1, 2JHH = 7.4, 1H, IrH). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JBC = 50, ArF), 156.9 (s, bipy), 155.3 (s, bipy), 154.9 (s, bipy), 154.8 (s, bipy), 152.7 (s, C6H3), 147.8 (s, C6H4), 137.6 (s, bipy), 135.5 (s, bipy), 135.4 (s, ArF), 133.5 (app. t, JPC = 6, Ph), 131.9 (s, bipy), 131.8 (t, 1JPC = 27, Ph), 131.1 (s, Ph), 130.1 (s, C6H3), 129.4 (qq, 2JCF = 32, 2JCB = 3, ArF), 129.1 (app. t, JPC = 5, Ph), 127.9 (s, bipy), 125.4 (s, C6H3), 125.2 (q, 1JFC = 272, ArF), 124.9 (s, C6H3), 123.7 (s, bipy), 123.20 (s, bipy), 123.17 (s, C6H4), 118.0 (sept., 3JFC = 4, ArF), 113.8 (s, C6H4), 71.0 (s, OCH2), 70.7 (s, OCH2), 68.7 (s, OCH2), 55.1 (s, ArH2), 49.1 (s, bipyH2), 35.3 (s, tBu), 31.5 (s, tBu). 31P{1H} NMR (202 MHz, CD2Cl2): δ 19.8 (vbr, fwhm = 43 Hz). HR ESI-MS (positive ion, 4 kV): 777.8218 [M]2+ (calcd 777.8228), 1554.6401 [M − H]+ (calcd 1554.6384) m/z. Anal. calcd for C150H122B2F48P2Ir (3285.14 g mol−1): C, 54.89; H, 3.75; N, 1.28. Found: C, 54.83; H, 3.74; N, 1.31.
Preparation of 2
2a
Yield = 69.9 mg (87%, orange solid).
1
H NMR (500 MHz, CD2Cl2): δ 7.70–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.42 (t, 3JHH = 7.4, 6H, Ph), 7.35–7.40 (m, 12H, Ph), 7.27 (t, 3JHH = 7.2, 12H, Ph), 4.55 (br, 4H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.54–2.42 (m, 4H, CH2), 2.28–2.20 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 135.4 (s, ArF), 134.5 (app. t, JPC = 6, Ph), 131.7 (s, Ph), 130.5–131.3 (m, Ph), 129.4 (qq, 2JCF = 32, 2JCB = 3, ArF), 129.2 (app. t, JPC = 5, Ph), 125.2 (q, 1JFC = 272, ArF), 118.1 (sept., 3JFC = 4, ArF), 99.6 (app. dt, J = 8, J = 5, CHCH), 31.2 (s, CH2). 31P{1H} NMR (162 MHz, CD2Cl2): δ 26.5 (d, 1JRhP = 145). HR ESI-MS (positive ion, 4 kV): 735.1812 [M]+ (calcd 735.1811) m/z. Anal. calcd for C78H54BF24P2Rh (1589.89 g mol−1): C, 57.09; H, 3.40; N, 0.00. Found: C, 57.18; H, 3.56; N, 0.00.
CH), 2.54–2.42 (m, 4H, CH2), 2.28–2.20 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 135.4 (s, ArF), 134.5 (app. t, JPC = 6, Ph), 131.7 (s, Ph), 130.5–131.3 (m, Ph), 129.4 (qq, 2JCF = 32, 2JCB = 3, ArF), 129.2 (app. t, JPC = 5, Ph), 125.2 (q, 1JFC = 272, ArF), 118.1 (sept., 3JFC = 4, ArF), 99.6 (app. dt, J = 8, J = 5, CHCH), 31.2 (s, CH2). 31P{1H} NMR (162 MHz, CD2Cl2): δ 26.5 (d, 1JRhP = 145). HR ESI-MS (positive ion, 4 kV): 735.1812 [M]+ (calcd 735.1811) m/z. Anal. calcd for C78H54BF24P2Rh (1589.89 g mol−1): C, 57.09; H, 3.40; N, 0.00. Found: C, 57.18; H, 3.56; N, 0.00.
2b
Yield = 49.0 mg (58%, red solid). Spectroscopic data are in agreement with the literature values.25
1
H NMR (400 MHz, CD2Cl2): δ 7.70–7.76 (m, 8H, ArF), 7.56 (s, 4H, ArF), 7.42 (t, 3JHH = 7.4, 6H, Ph), 7.35–7.40 (m, 12H, Ph), 7.27 (t, 3JHH = 7.2, 12H, Ph), 4.20 (br, 4H, CHCH), 2.40–2.20 (m, 4H, CH2), 2.06–1.85 (m, 4H, CH2). 31P{1H} NMR (162 MHz, CD2Cl2): δ 17.6 (s). LR ESI-MS (positive ion): 825.2 [M]+ (calcd 825.2) m/z.
NMR scale reactions of 2
The hydrogenation of 2a + bipy was monitored in situ by 1H and 31P NMR spectroscopy at 50 °C. After 1 h, 2a was consumed with concomitant formation of 4a[BArF4] (ca. 58% [Rh]) and 5a (ca. 42% [Rh]). Complete conversion to 4a[BArF4] was observed after 18 h. Analysis by GC gave a hydrocarbon distribution of: COA (6%), COE (36%), COD (58%).
Immediate analysis of the hydrogenation of 2b + bipy at RT (<5 min) by 1H and 31P NMR spectroscopy indicated quantitative conversion to 4b[BArF4]. Analysis by GC gave a hydrocarbon distribution of: COA (14%), COE (86%), COD (0%).
Reaction between 2a and bipy resulted in formation of 5a (t1/2 = 1.3 h) and subsequently slow equilibrium formation of 7, alongside liberation of COD. Modelling the kinetics of the approach to equilibrium over 8 days,26 enabled determination of the equilibrium constant (K = 2.4) and rate of forward reaction (t1/2 = 107 h).
Reaction between 2b and bipy resulted in the slow and quantitative formation of 5b (t1/2 = 34 h). No subsequent reaction was observed.
The smooth conversion of 2a to 8 was observed over 4 h with a t1/2 = 0.8 h, alongside concomitant generation of COA. No COE or COD was evident by 1H NMR spectroscopy.
Hydrogenation of 2b resulted in the immediate colour change from red to colourless within 5 min. Analysis by 1H and 31P NMR spectroscopy indicated the formation of a mixture of hydride species 9, alongside concomitant generation of COA. No COE or COD was evident by 1H NMR spectroscopy. Subsequent transfer of this solution into a J. Young's NMR tube charged with bipy (1.6 mg, 10.0 μmol) resulted in the quantitative formation of 4b[BArF4] within 5 min as gauged by 1H and 31P NMR spectroscopy.
Selected data for 9.
1
H NMR (500 MHz, CD2Cl2): δ 7.71–7.76 (m, 8H, ArF), 7.56 (s, 4H, ArF), 7.47–7.58 (m, 30H, Ph), −26.24 (vbr, fwhm = 190 Hz). 31P{1H} NMR (202 MHz, CD2Cl2): δ 23.4 (vbr, fwhm = 130 Hz). 1H NMR (500 MHz, CD2Cl2, 185 K): δ 7.70–7.76 (m, 8H, ArF), 7.52 (br, 4H, ArF), 7.34–7.53 (m, 30H, Ph), −1.75 (br, T1 = 54 ms, IrH2(2)2), −12.43 (br, T1 = 51 ms, IrH2(2)(CD2Cl2)2), −23.59 (br, T1 = 49 ms, Ir2(H2)2),* −25.11 (t, 2JPH = 14.6, T1 = 401 ms, IrH2(CD2Cl2)2), −26.72 (br, T1 = 361 ms, Ir2(H2)(CD2Cl2)2), −28.13 (br, T1 = 358 ms, Ir2(H2)(CD2Cl2)2). * This resonance is assigned to a hydride despite the short T1 value, which we attribute to chemical exchange on the NMR timescale. 31P{1H} NMR (202 MHz, CD2Cl2, 185 K): δ 27.0 (s, IrH2(H2)(CD2Cl2)2), 23.2 (s, IrH2(H2)2), 15.7 (s, IrH2(CD2Cl2)2).
Preparation of 3
A suspension of 10 (100 mg, 258 μmol), Na[BArF4] (229 mg, 258 μmol) and citric acid monohydrate (54.2 mg, 258 μmol) in CH2Cl2/H2O 1:1 (6 mL) was stirred vigorously at RT for 10 min (air). The organic phase was separated, washed with H2O (5 × 5 mL) and dried over MgSO4. The solvent was removed in vacuo to give the product as an off-white solid. Yield = 270 mg (84%).
1 H NMR (500 MHz, CD2Cl2): δ 8.64 (s, 1H, bipy), 8.57 (d, 3JHH = 5.0, 1H, bipy), 8.08 (app. t, 3JHH = 8, 1H, bipy), 7.97 (d, 3JHH = 7.6, 1H, bipy), 7.93 (d, 3JHH = 7.9, 1H, bipy), 7.81 (d, 3JHH = 7.6, 1H, bipy), 7.69–7.75 (m, 8H, ArF), 7.62 (dd, 3JHH = 7.6, 5.1, 1H, bipy), 7.54 (br, 4H, ArF), 7.43 (t, 4JHH = 1.8, 1H, C6H3), 7.22 (d, 4JHH = 1.8, 2H, C6H3), 4.26 (br, 2H, CH2), 4.24 (br, 2H, CH2), 1.24 (s, 18H, tBu). The NH2 resonance was not unambiguously located. 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 152.8 (s, C6H3), 148.4 (s, bipy), 142.9 (s, bipy), 141.8 (s, bipy), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JBC = 3, ArF), 127.5 (s, bipy), 125.1 (q, 1JFC = 272, ArF), 124.0 (s, bipy), 123.7 (s, C6H3), 123.5 (s, bipy), 123.1 (s, C6H3), 118.0 (sept., 3JFC = 4, ArF), 54.9 (s, CH2), 50.2 (s, CH2), 35.3 (s, tBu), 31.6 (s, tBu). Not all resonances unambiguously located. HR ESI-MS (positive ion, 4 kV): 1252.3484 [M + H]+ (calcd 1252.3484) m/z.
Reaction of 3 with db24c8: preparation of 3·db24c8
A solution of 3 (6.3 mg, 5.0 μmol) and db24c8 (2.3 mg, 5.1 μmol) in CD2Cl2 (0.5 mL) was prepared within a J. Young's NMR tube. Analysis in situ by NMR spectroscopy indicated the quantitative formation of 3·db24c8 (slow exchange at 500 MHz).Repeating the reaction using 0.5 and 2 equiv. db24c8 resulted in the formation of a 1:1 mixture of 3 and 3·db24c8, and 1:1 mixture of 3·db24c8 and db24c8, respectively (both slow exchange at 500 MHz).
1
H NMR (500 MHz, CD2Cl2): δ 8.60 (d, 3JHH = 4.6, 1H, bipy), 8.48 (d, 4JHH = 1.6, 1H, bipy), 8.18 (d, 3JHH = 7.9, 1H), 8.04 (d, 3JHH = 8.2, 1H, bipy), 7.79 (app. td, 3JHH = 8, 4JHH = 1.8, 1H, bipy), 7.70–7.76 (m, 8H, ArF), 7.70 (obscured, 2H, NH2), 7.63 (dd, 3JHH = 8.2, 4JHH = 2.0, 1H, bipy), 7.56 (br, 4H, ArF), 7.47 (t, 4JHH = 1.8, 1H, C6H3), 7.30–7.34 (m, 3H, C6H3 + bipy), 6.69–6.79 (m, 8H, C6H4), 4.79–4.85 (m, 4H, bipyC2), 4.67–4.74 (m, 4H, ArC2), 4.01–4.19 (m, 8H, OCH2), 3.71–3.87 (m, 8H, OCH2), 3.58–3.68 (m, 4H, OCH2), 3.45–3.56 (m, 4H, OCH2), 1.23 (s, 18H, tBu).13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JBC = 50, ArF), 157.2 (s, bipy), 155.5 (s, bipy), 152.5 (s, C6H3), 150.4 (s, bipy), 149.6 (s, bipy), 147.6 (s, C6H4), 138.0 (s, bipy), 137.3 (s, bipy), 135.4 (s, ArF), 131.4 (s, C6H3), 129.4 (qq, 2JCF = 32, 2JCB = 3, ArF), 127.8 (s, bipy), 125.2 (q, 1JFC = 272, ArF), 124.6 (s, bipy), 124.4 (s, C6H3), 124.0 (s, C6H3), 122.3 (s, C6H4), 121.6 (s, bipy), 120.5 (s, bipy), 118.0 (sept. 3JFC = 4, ArF), 113.0 (s, C6H4), 71.2 (s, OCH2), 70.9 (s, OCH2), 68.4 (s, OCH2), 54.0 (s, ArH2), 50.6 (s, bipyH2), 35.4 (s, tBu), 31.6 (s, tBu).
Preparation of 4[BArF4]
4a[BArF4]
Yield = 17.6 mg (53%, off white solid).1 H NMR (500 MHz, CD2Cl2): δ 8.06 (d, 3JHH = 5.2, 2H, bipy), 7.71–7.75 (m, 8H, ArF), 7.71 (obscured, 2H, bipy), 7.64 (t, 3JHH = 7.5, 2H, bipy), 7.56 (br, 4H, ArF), 7.30–7.38 (m, 18H, Ph), 7.23 (t, 3JHH = 7.4, 12H, Ph), 6.82 (ddd, 3JHH = 7.6, 5.5, 4JHH = 1.0, 2H, bipy), −15.66 (app. q, J = 14 (1JRhH = 15.5), 2H, RhH). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 154.5 (s, bipy), 154.3 (s, bipy), 137.8 (s, bipy), 135.4 (s, ArF), 133.7 (app. t, JPC = 7, Ph), 132.5 (app. t, JPC = 24, Ph), 130.8 (s, Ph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 129.0 (app. t, JPC = 5, Ph), 126.4 (s, bipy), 125.2 (q, 3JFC = 272, ArF), 122.6 (s, bipy), 118.0 (sept., 3JFC = 4, CH, ArF). 31P{1H} NMR (162 MHz, CD2Cl2): δ 47.1 (d, 1JRhP = 115). HR ESI-MS (positive ion, 4 kV): 785.1723 [M]+ (calcd 785.1716) m/z. Anal. calcd for C78H52BF24N2P2Rh (1648.91 g mol−1): C, 56.82; H, 3.18; N, 1.70. Found: C, 57.04; H, 2.91; N, 1.80.
4b[BArF4]
Yield = 11.2 mg (34%, yellow solid).1 H NMR (500 MHz, CD2Cl2): δ 8.16 (d, 3JHH = 5.3, 2H, bipy), 7.71–7.56 (m, 8H, ArF), 7.71 (obscured, 2H, bipy), 7.64 (t, 3JHH = 7.8, 2H, bipy), 7.56 (br, 4H, ArF), 7.28–7.36 (m, 18H, Ph), 7.22 (t, 3JHH = 7.4, 12H, Ph), 6.75 (ddd, 3JHH = 7.5, 5.5, 4JHH = 1.0, 2H, bipy), −19.48 (t, 2JPH = 16.6, 2H, IrH). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 156.0 (s, bipy), 155.8 (s, bipy), 137.2 (s, bipy), 135.4 (s, ArF), 133.6 (app. t, JPC = 6, Ph), 131.8 (app. t, JPC = 27, Ph), 130.9 (s, Ph), 129.4 (qq, 2JFC = 31, 3JCB = 3, ArF), 128.9 (app. t, JPC = 5, Ph), 127.3 (s, CH, bipy), 125.2 (q, 1JFC = 272, ArF), 123.2 (s, bipy), 118.0 (sept., 3JFC = 4, ArF). 31P{1H} NMR (162 MHz, CD2Cl2): δ 20.1 (s). HR ESI-MS (positive ion, 4 kV): 875.2286 [M]+ (calcd 875.2293) m/z. Anal. calcd for C78H52BF24IrN2P2 (1738.22 g mol−1): C, 53.90; H, 3.02; N, 1.61. Found: C, 54.03; H, 2.85; N, 1.69.
Attempted hydrogenation of COD mediated by 4a[BArF4]
A solution of 4a[BArF4] (16.5 mg, 10.0 μmol) in CD2Cl2 (0.5 mL) was freeze–pump–thaw degassed, placed under dihydrogen (1 atm), COD (1.2 μL, 10 μmol) added under a hydrogen atmosphere, and finally heated at 50 °C for 18 h. The solution was passed through a short plug of SiO2 (CH2Cl2). Analysis by GC gave a hydrocarbon distribution of: COA (0%), COE (1%), COD (99%).Preparation of 5a
A solution of 6a (24.6 mg, 20.0 μmol) and PPh3 (5.2 mg, 20 μmol) in CH2Cl2 (5 mL) was stirred for 30 min at RT. The solvent was removed in vacuo to give an orange solid, which was washed with hexane (10 mL). Yield = 12.0 mg (40%, orange powder).
1
H NMR (500 MHz, CD2Cl2): δ 9.00 (d, 3JHH = 5.1, 2H, bipy), 7.90 (t, 3JHH = 7.4, 2H, bipy), 7.77 (d, 3JHH = 8.1, 2H, bipy), 7.70–7.75 (m, 8H, ArF), 7.56 (br, 4H, ArF), 7.55 (obscured, 2H, bipy), 7.38 (t, 3JHH = 7.3, 3H, Ph), 7.26 (br app. t, J = 8, 6H, Ph), 7.21 (t, 3JHH = 8.4, 6H, Ph), 3.80 (s, 4H, CHCH), 2.55–2.67 (m, 4H, CH2), 2.03–2.15 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 154.5 (s, bipy), 151.7 (s, bipy), 139.6 (s, bipy), 135.4 (s, ArF), 133.9 (d, 2JPC = 12, Ph), 131.3 (d, 1JPC = 28, Ph), 130.8 (s, Ph), 129.5 (qq, 3JFC = 32, 3JCB = 3, ArF), 129.1 (d, 3JPC = 9, Ph), 127.1 (s, bipy), 125.2 (q, 1JFC = 272, ArF), 123.4 (s, bipy), 118.0 (sept., 3JFC = 4, ArF), 82.8 (d, 1JRhC = 11, CHCH), 31.8 (s, CH2). 31P{1H} NMR (162 MHz, CD2Cl2): δ 22.31 (vbr, fwhm = 45 Hz). HR ESI-MS (positive ion, 4 kV): 367.0679 [M − PPh3]+ (calcd 367.0676) m/z. Anal. calcd for C68H47BF24N2PRh·CH2Cl2 (1577.72 g mol−1): C, 52.53; H, 3.13; N, 1.78. Found: C, 52.91; H, 3.17; N, 2.15.
5b
A solution of 6b (10.0 mg, 7.58 μmol) and PPh3 (2.2 mg, 8.4 μmol) in CH2Cl2 (1 mL) was agitated for 10 min at RT then layered with excess hexane (ca. 20 mL) to afford the product as red crystals on diffusion. Yield = 6.7 mg (56%).
1
H NMR (500 MHz, CD2Cl2): δ 9.13 (br, 2H, bipy), 7.89 (t, 3JHH = 7.6, 2H, bipy), 7.81 (d, 3JHH = 7.9, 2H, bipy), 7.70–7.76 (s, 8H, ArF), 7.56 (br, 4H, ArF), 7.50 (t, 3JHH = 6.4, 2H, bipy), 7.38 (t, 3JHH = 7.3, 3H, Ph), 7.25 (br app t, J = 8, 6H, Ph), 7.13 (t, 3JHH = 8.9, 6H, Ph), 3.23 (br, 4H, CHCH), 2.37–2.51 (m, 4H, CH2), 1.81–1.94 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 155.6 (HMBC, bipy)152.5 (s, bipy), 138.9 (s, bipy), 135.4 (s, ArF), 133.6 (d, 2JPC = 10, Ph), 131.1 (s, Ph), 130.8 (HMBC, Ph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 129.1 (d, 3JPC = 9, Ph), 127.7 (s, bipy), 125.2 (q, 1JFC = 272, ArF), 123.9 (s, bipy), 118.0 (sept., 3JFC = 5, ArF), 65.2 (br, CHCH), 33.0 (s, CH2). 31P{1H} NMR (162 MHz, CD2Cl2): δ 10.1 (s). HR ESI-MS (positive ion, 4 kV): 719.2168 [M]+ (calcd 719.2163) m/z. Anal. calcd for C68H47BF24N2PIr (1582.10 g mol−1): C, 51.62; H, 2.99; N, 1.77. Found: C, 51.43; H, 2.87; N, 1.84.
Preparation of 6
6a
Yield = 91.9 mg (75%, red solid).
1
H NMR (500 MHz, CD2Cl2): δ 8.11 (app. td, 3JHH = 8, 4JHH = 1.5, 2H, bipy), 8.07 (d, 3JHH = 8.2, 2H, bipy), 7.79 (d, 3JHH = 5.5, 2H, bipy), 7.70–7.75 (m, 8H, ArF), 7.59 (ddd, 3JHH = 7.2, 5.5, 4JHH = 1.5, 2H, bipy), 7.55 (br, 4H, ArF), 4.57 (br, 4H, CHCH), 2.54–2.65 (m, 4H, CH2), 2.14–2.23 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 156.8 (s, bipy), 149.1 (s, bipy), 141.7 (s, bipy), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 128.1 (s, bipy), 125.2 (q, 1JFC = 272, ArF), 123.4 (s, bipy), 118.0 (sept., 3JFC = 4, ArF), 86.5 (d, 1JRhC = 12, CHCH), 30.7 (s, CH2). HR ESI-MS (positive ion, 4 kV): 367.0677 [M]+ (calcd 367.0676) m/z. Anal. calcd for C50H32BF24N2Rh (1230.50 g mol−1): C, 48.81; H, 2.62; N, 2.28. Found: C, 48.89; H, 2.52; N, 2.46.
6b
Yield = 91.5 mg (67%, dark green solid).
1
H NMR (500 MHz, CD2Cl2): δ 8.20 (app. td, 3JHH = 8, 4JHH = 1.2, 2H, bipy), 8.14 (d, 3JHH = 8.1, 2H, bipy), 8.11 (d, 3JHH = 5.6, 2H, bipy), 7.71–7.75 (m, 8H, ArF), 7.69 (ddd, 3JHH = 7.8, 5.6, 4JHH = 1.0, 2H, bipy), 7.56 (s, 4H, ArF), 4.40 (br, 4H, CHCH), 2.36–2.48 (m, 4H, CH2), 2.00–2.12 (m, 4H, CH2). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 158.4 (s, bipy), 149.6 (s, bipy), 142.6 (s, bipy), 135.4 (s, ArF), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 128.9 (s, bipy), 125.2 (q, 1JFC = 272, ArF), 123.7 (s, bipy), 118.0 (sept., 3JFC = 4, ArF), 72.2 (s, CHCH), 31.5 (s, CH2). HR ESI-MS (positive ion, 4 kV): 457.1250 [M]+ (calcd 457.1251) m/z. Anal. calcd for C50H32BF24IrN2 (1319.81 g mol−1): C, 45.50; H, 2.44; N, 2.12. Found: C, 45.83; H, 2.56; N, 2.19.
NMR scale reactions of 6
31P{1H} NMR (202 MHz, CD2Cl2): δ 10.6 (vbr, fwhm = 55 Hz).31P{1H} NMR (202 MHz, CD2Cl2, 200 K): δ 33.3 (d, 1JRhP = 129, 5a), −8.3 (s, PPh3).
Preparation of 7
A solution of 8 (14.9 mg, 5.00 μmol) and bipy (1.6 mg, 10 μmol) in CH2Cl2 (1 mL) was agitated for 10 min at RT. Addition of excess hexane (ca. 20 mL) afforded the product as an orange-red oil that solidified on extended drying in vacuo. Yield = 2.4 mg (15%).1 H NMR (500 MHz, CD2Cl2): δ 7.97 (d, 3JHH = 8.1, 2H, bipy), 7.89 (br d, 3JHH = 5, 2H, bipy), 7.82 (app. t, 3JHH = 8, 2H, bipy), 7.71–7.75 (m, 8H, ArF), 7.62–7.68 (m, 12H, Ph), 7.56 (br, 4H, ArF), 7.30 (t, 3JHH = 7.4, 6H, Ph), 7.15 (t, 3JHH = 7.5, 12H, Ph), 6.82 (dd, 3JHH = 7.5, 5.8, 2H, bipy). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 156.5 (s, bipy), 153.8 (s, bipy), 138.6 (s, bipy), 135.4 (s, ArF), 135.2 (app. t, JPC = 6, Ph), 133.0–134.1 (m, Ph), 130.8 (s, Ph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 128.6 (app. t, JPC = 5, Ph), 126.3 (s, bipy), 125.2 (q, 1JCF = 272, ArF), 122.5 (s, bipy), 118.0 (sept., 3JFC = 4, ArF). 31P{1H} NMR (162 MHz, CD2Cl2): δ 46.6 (d, 1JRhP = 182). HR ESI-MS (positive ion, 4 kV): 815.1458 [M + MeOH]+ (calcd 815.1458) m/z.
Hydrogenation of 7
A solution of 7 (16.5 mg, 10.0 μmol) in CD2Cl2 (0.5 mL) within a J. Young's NMR tube was freeze–pump–thaw degassed three times and placed under dihydrogen (1 atm) resulting in quantitative formation of 4a[BArF4] within 5 min as gauged by 1H and 31P NMR spectroscopy.Preparation of 8
A solution of 2a (64.0 mg, 40.0 μmol) in CH2Cl2 (5 mL) was freeze–pump–thaw degassed three times and placed under dihydrogen (1 atm). After stirring for 5 h the solution was freeze–pump–thaw degassed three times and placed under argon. The solution was layered with excess hexane (ca. 45 mL) to afford the product as a dark orange crystalline solid on diffusion. Yield = 33.7 mg (57%).1 H NMR (500 MHz, CD2Cl2): δ 7.72–7.77 (m, 16H, ArF), 7.56 (s, 8H, ArF), 7.43–7.50 (m, 4H, Ph), 7.32–7.40 (m, 6H, Ph), 7.15–7.26 (m, 40H, Ph), 6.87 (t, 3JHH = 6.9, 4H, η-Ph), 6.37 (t, 3JHH = 6.4, 4H, η-Ph), 5.51 (t, 3JHH = 6.5, 2H, η-Ph). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.3 (q, 1JCB = 50, ArF), 135.4 (s, ArF), 135.1 (d, 2JPC = 12, Ph), 134.1 (d, 2JPC = 11, Ph), 132.9 (s, Ph), 132.1 (d, 4JPC = 3, Ph), 129.5 (d, 3JPC = 8, C, Ph), 129.4 (qq, 2JFC = 32, 3JCB = 3, ArF), 129.2 (d, 3JPC = 11, Ph), 129 (obscured, Ph), 125.2 (q, 1JCF = 272, ArF), 123.1 (dd, 1JPC = 33, 2JRhC = 7, η-Ph), 118.0 (sept., 3JFC = 4, ArF), 106.5 (br d, 2JPC = 10, η-Ph), 105.4 (br, η-Ph), 102.4–102.6 (m, η-Ph). 31P{1H} NMR (162 MHz, CD2Cl2): δ 45.9 (ddd, 1JRhP = 217, 2JPP = 38, 2JRhP = 5), 43.2 (dd, 1JRhP = 198, 2JPP = 38). HR ESI-MS (positive ion, 4 kV): 627.0869 [½M]+ (calcd 627.0872) m/z.
Reaction of 8 with bipy
In a J. Young's NMR tube, 8 (14.9 mg, 5.0 μmol) and bipy (1.6 mg, 10.0 μmol) was dissolved in CD2Cl2 (0.5 mL) resulting in quantitative formation of 7 within 5 minutes as gauged by 1H and 31P NMR spectroscopy.Preparation of 5-phthalimidomethylbipyridine
A stirred solution of 5-chloromethylbipyridine (3.00 g, 14.7 mmol) and potassium phthalimide (4.07 g, 21.9 mmol) in DMF (70 mL) under N2 was heated at 40 °C for 18 h. The solvent was removed in vacuo and the mixture dissolved in CH2Cl2 (100 mL) and washed with H2O (2 × 100 mL), brine (100 mL), and then dried over MgSO4. The solvent was removed in vacuo and the product recrystallised from hot EtOH (ca. 450 mL). Yield = 4.19 g (94%, white crystals).1 H NMR (400 MHz, CDCl3): δ 8.76 (dd, 4JHH = 2.3, 5JHH = 0.8, 1H, bipy), 8.65 (ddd, 3JHH = 4.8, 3JHH = 1.8, 5JHH = 0.9, 1H, bipy), 8.36 (app. dt, 3JHH = 7.9, J = 1, bipy), 8.35 (dd, 3JHH = 8.2, 5JHH = 0.8, 1H, bipy), 7.88 (dd, 3JHH = 8.2, 4JHH = 2.3, 1H, bipy), 7.83–7.88 (m, 2H, Phth), 7.79 (app. td, 3JHH = 8, 4JHH = 1.8, 1H, bipy), 7.69–7.74 (m, 2H, Phth), 7.28 (ddd, 3JHH = 7.6, 4.8, 4JHH = 1.2, 1H, bipy), 4.91 (s, 2H, CH2). 13C{1H} NMR (101 MHz, CDCl3): δ 168.0, 155.9, 155.9, 149.6, 149.3, 137.5, 137.0, 134.3, 132.1, 132.1, 123.9, 123.6, 121.3, 121.1, 39.1. HR ESI-MS (positive ion, 4 kV): 338.0903 [M + Na]+ (calcd 338.0900) m/z.
Preparation of 5-aminomethylbipyridine
A solution of 5-phthalimidomethylbipyridine (1.50 g, 4.76 mmol) and hydrazine monohydrate (2.31 mL, 47.6 mmol) in EtOH (100 mL) under N2 was heated at reflux for 18 h. An aqueous solution of NaOH (2 M, 50 mL) was added and the mixture extracted with CH2Cl2 (3 × 100 mL). The organic phase was dried over MgSO4 and the solvent removed in vacuo to give the compound as a waxy off-white solid. Yield = 0.794 g (90%).1 H NMR (400 MHz, (CD3)2SO): δ 8.67 (ddd, 3JHH = 4.8, 4JHH = 1.9, 5JHH = 0.9, 1H, bipy), 8.63 (d, 4JHH = 2.3, 1H, bipy), 8.37 (app. dt, 3JHH = 8.0, J = 1, 1H, bipy), 8.33 (d, 3JHH = 8.1, 1H, bipy), 7.92 (app. td, 3JHH = 8, 4JHH = 1.9, 1H, bipy), 7.90 (dd, 3JHH = 8.1, 4JHH = 2.3, 1H, bipy), 7.42 (ddd, 3JHH = 7.5, 4.8, 4JHH = 1.2, 1H, bipy), 3.82 (s, 2H, CH2). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 155.3, 153.6, 149.2, 148.4, 139.3, 137.2, 136.0, 123.9, 120.2, 119.9, 42.8. HR ESI-MS (positive ion, 4 kV): 208.0846 [M + Na]+ (calcd 208.0845) m/z.
Preparation of 10
A solution of 5-aminomethylbipyridine (730 mg, 3.94 mmol) and 3,5-di-tert-butylbenzaldehyde (860 mg, 3.94 mmol) in MeOH (30 mL) was stirred at RT for 18 h over 3 Å molecular sieves under N2. The resulting mixture was cooled to 0 °C and NaBH4 (298 mg, 7.88 mmol) added in portions. The mixture was warmed to ambient temperature and stirred for 30 min, and then heated at reflux for 2 h. The solvent was removed in vacuo and the crude extracted with CH2Cl2 (30 mL). The organic phase was washed with H2O (3 × 30 mL) and dried over MgSO4. The solvent was removed in vacuo and the product purified by column chromatography (SiO2, CH2Cl2/MeOH 95:5) to give a colourless oil. The product was freeze-dried in vacuo to give an off-white solid. Yield = 1.185 g (78%).
1 H NMR (500 MHz, CD2Cl2): δ 8.63–8.67 (obscured, 1H, bipy), 8.65 (br, 1H, bipy), 8.44 (d, 3JHH = 8.0, 1H, bipy), 8.41 (d, 3JHH = 8.2, 1H, bipy), 7.85 (dd, 3JHH = 8.2, 4JHH = 2.2, 1H, bipy), 7.82 (app. td, 3JHH = 8, 4JHH = 1.8, 1H, bipy), 7.35 (t, 4JHH = 1.9, 1H, C6H3), 7.31 (ddd, 3JHH = 7.5, 4.8, 4JHH = 1.2, 1H, bipy), 7.22 (d, 4JHH = 1.9, 2H, C6H3), 3.90 (s, 2H, CH2), 3.82 (s, 2H, CH2), 1.35 (s, 18H, tBu). The NH resonance was not unambiguously located. 13C{1H} NMR (126 MHz, CD2Cl2): δ 156.7, 155.4, 151.4, 149.7, 149.7, 140.0, 137.3, 137.2, 136.9, 124.1, 122.9, 121.5, 121.2, 121.0, 54.4, 51.0, 35.3, 31.8. HR ESI-MS (positive ion, 4 kV): 388.2747 [M + H]+ (calcd 388.2747) m/z.
Crystallography
Full details about the collection, solution and refinement are documented in the CIF, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 1563158 (1b), 1563159 (4a[BArF4]), 1563160 (4b[BArF4]; Z′ = 1), 1563161 (4b[BArF4]; Z′ = 2), 1563162 (5a), 1563163 (5b) and 1563164 (8).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the EPSRC (J. E.-K.), Leverhulme Trust (RPG-2012-718, R. C. K), ERC (M. R. G., grant agreement 637313), and Royal Society (A. B. C.) for financial support. Crystallographic data for 4a[BArF4] and high-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data for 1b, 4b[BArF4], 5, and 8 were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 637313).References
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC; Y. Suzaki, T. Taira, K. Osakada and M. Horie, Dalton Trans., 2008, 4823–4811 RSC.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef CAS PubMed; J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef PubMed; D.-H. Qu and H. Tian, Chem. Sci., 2011, 2, 1011–1015 RSC; J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; F. Aricó, J. D. Badjić, S. J. Cantrill, A. H. Flood, K. C. F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 203–259 Search PubMed.
- J. Yang, J.-F. Ma and S. R. Batten, Chem. Commun., 2012, 48, 7899–7814 RSC; V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed; S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226–235 RSC.
- H. Ogino and K. Ohata, Inorg. Chem., 1984, 23, 3312–3316 CrossRef CAS; H. Ogino, J. Am. Chem. Soc., 1981, 103, 1303–1304 CrossRef.
- D. J. Cárdenas, P. Gaviña and J.-P. Sauvage, Chem. Commun., 1996, 17, 1915–1916 RSC.
- K. M. Mullen, K. D. Johnstone, D. Nath, N. Bampos, J. K. M. Sanders and M. J. Gunter, Org. Biomol. Chem., 2009, 7, 293–303 Search PubMed; H. V. Huynh, W. Sim and C. F. Chin, Dalton Trans., 2011, 40, 11690–11692 RSC; S. Shinoda, T. Maeda, H. Miyake and H. Tsukube, Supramol. Chem., 2011, 23, 244–248 CrossRef CAS.
- J. A. Osborn and R. R. Schrock, J. Am. Chem. Soc., 1971, 93, 2397–2407 CrossRef CAS; J. R. Shapley, R. R. Schrock and J. A. Osborn, J. Am. Chem. Soc., 1969, 91, 2816–2817 CrossRef.
- G. L. Moxham, S. K. Brayshaw and A. S. Weller, Dalton Trans., 2007, 1759–1761 RSC.
- G. L. Moxham, T. M. Douglas, S. K. Brayshaw, G. Kociok-Köhn, J. P. Lowe and A. S. Weller, Dalton Trans., 2006, 88, 5492–5505 RSC.
- B. D. Alexander, B. J. Johnson, S. M. Johnson, A. L. Casalnuovo and L. H. Pignolet, J. Am. Chem. Soc., 1986, 108, 4409–4417 CrossRef CAS; R. R. Schrock and J. A. Osborn, J. Am. Chem. Soc., 1976, 98, 2134–2143 CrossRef.
- A. Macchioni, C. Zuccaccia, E. Clot, K. Gruet and R. H. Crabtree, Organometallics, 2001, 20, 2367–2373 CrossRef CAS.
- P. Alam, C. U. Climent, G. Kaur, D. Casanova, A. Roy Choudhury, A. Gupta, P. Alemany and I. R. Laskar, Cryst. Growth Des., 2016, 16, 5738–5752 CAS.
- I. A. Guzei, A. L. Rheingold, D. H. Lee and R. H. Crabtree, Z. Kristallogr. – New Cryst. Struct., 1998, 213, 585–587 CAS.
- Á. Álvarez, R. Macías, M. J. Fabra, M. L. Martín, F. J. Lahoz and L. A. Oro, Inorg. Chem., 2007, 46, 6811–6826 CrossRef PubMed.
- C. Cocevar, G. Mestroni and A. Camus, J. Organomet. Chem., 1972, 35, 389–395 CrossRef CAS.
- A. Rifat, N. J. Patmore, M. F. Mahon and A. S. Weller, Organometallics, 2002, 21, 2856–2865 CrossRef CAS; P. Marcazzan, M. B. Ezhova, B. O. Patrick and B. R. James, C. R. Chim., 2002, 5, 373–378 CrossRef . See CIF for solid-state structure.
- M. J. Ingleson, S. K. Brayshaw, M. F. Mahon, G. D. Ruggiero and A. S. Weller, Inorg. Chem., 2005, 44, 3162–3171 CrossRef CAS PubMed.
- J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, California, 2010 Search PubMed.
- COE = cyclooctene.
- Hydrogenation of COD mediated by isolated 4a[BArF4] proceeds only very slowly at 50 °C. After 18 h, 1% conversion to COE was determined by GC analysis.
- A. D. Faulkner, PhD thesis, Univeristy of Warwick, 2014.
- N.-C. Chen, C.-J. Chuang, L.-Y. Wang, C.-C. Lai and S.-H. Chiu, Chem. – Eur. J., 2012, 18, 1896–1900 CrossRef CAS PubMed.
- W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
- G. Giordano, R. H. Crabtree, R. M. Heintz, D. Forster and D. E. Morris, Inorg. Synth., 1990, 28, 88–90 CrossRef CAS; J. L. Herde, J. C. Lambert, C. V. Senoff and M. A. Cushing, Inorg. Synth., 1974, 15, 18–20 Search PubMed.
- A. Rifat, G. Kociok-Köhn, J. W. Steed and A. S. Weller, Organometallics, 2004, 23, 428–432 CrossRef CAS.
- J. W. Moore and R. G. Pearson, Kinetics and Mechanism, John Wiley & Sons, New York, 3rd edn, 1981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 31P{1H} NMR and ESI-MS spectra of new compounds and selected reactions. CCDC 1563158–1563164. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02648j |
| This journal is © The Royal Society of Chemistry 2017 |