DOI:
10.1039/C7DT02631E
(Paper)
Dalton Trans., 2017,
46, 13590-13596
Nickel–ruthenium-based complexes as biomimetic models of [NiFe] and [NiFeSe] hydrogenases for dihydrogen evolution†
Received
19th July 2017
, Accepted 19th September 2017
First published on 21st September 2017
Abstract
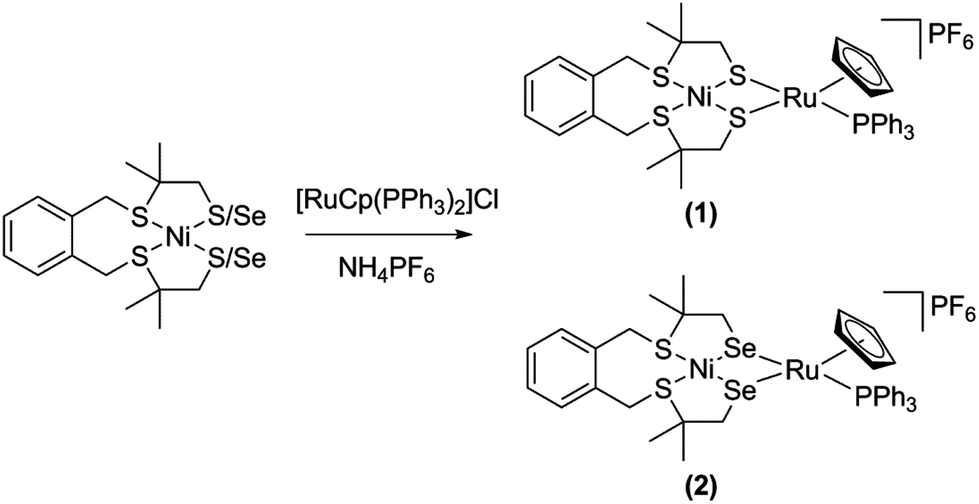
The two heterodinuclear nickel–ruthenium complexes [Ni(xbSmS)RuCp(PPh3)]PF6 and [Ni(xbSmSe)RuCp(PPh3)]PF6 (H2xbSmS = 1,2-bis(4-mercapto-3,3-dimethyl-2-thiabutyl)benzene, H2xbSmSe = 1,2,-bis(2-thiabutyl-3,3-dimethyl-4-selenol)benzene, Cp = cyclopentadienyl) were synthesized as biomimetic models of [NiFe] and [NiFeSe] hydrogenases. The X-ray structural analyses of the complexes show that the two NiRu complexes are isomorphous; in both NiRu complexes the nickel(II) centers are coordinated in a square-planar environment with two thioether donor atoms and two thiolate or selenolate donors that are bridging to the ruthenium(II) center. The Ru(II) ion is further coordinated to a η5-cyclopentadienyl group and a triphenylphosphane ligand. These complexes catalyze hydrogen evolution in the presence of acetic acid in acetonitrile solution at around −2.20 V vs. Fc+/Fc with overpotentials of 810 and 830 mV, thus they can be regarded as functional models of the [NiFe] and [NiFeSe] hydrogenases.
Introduction
Hydrogenases are enzymes that have a catalytic role in the oxidation of molecular hydrogen (H2) and the reduction of protons; this catalytic interconversion plays an important role in the metabolism of a number of algae and bacteria.1 The hydrogenase enzymes are relevant for future energy applications since dihydrogen is a clean source of energy.2 Researchers are looking for new and cleaner ways for the production of dihydrogen gas and the development of functional mimics of the hydrogenases might aid in finding a solution for our energy problem.3 In nature these enzymes are highly efficient catalysts for proton reduction with turnover frequencies ranging between 1500–9000 per second at 30 °C. Unfortunately, it is difficult to isolate these enzymes in a pure form, and they are very fragile and air-sensitive.4,5 With a biomimetic approach the active site of the enzyme can be mimicked by way of the synthesis and characterization of low-molecular mass compounds.5 Ample research has been done on [NiFe] hydrogenases to unravel its catalytic activity and mechanism in the oxidation of dihydrogen and reduction of protons.6 A significant amount of data has been gathered over the years concerning the enzyme redox states and the reaction mechanism for the reversible heterolytic splitting of dihydrogen at the [NiFe] hydrogenase active site.7 The knowledge thus gathered has led to progress in the design, synthesis and characterization of models of the active site of [NiFe] and [FeFe] hydrogenases. A variety of interesting structural models has been published over the past decades and many of these have been investigated for their electrocatalytic activity.8–11 Reported complexes include NiS4 compounds,6,12 mononuclear Ni/Co/Fe complexes with phosphane ligands,13 thiolate-bridged [NiFe] carbonyl complexes,14,15 and a number of [NiRu] heterobimetallic complexes.9,10,16,17 The choice of substituting iron by ruthenium in mimicking the active site is based on the fact that ruthenium complexes are active catalysts in hydrogenation and hydrogen transfer reactions and generally form more stable compounds. Most importantly Ru(II) ions are able to accept both hard and soft ligands such as hydride and dihydrogen, which makes ruthenium a suitable replacement of the Fe center in models of the [NiFe] hydrogenases.18 In some [NiFe] hydrogenase mimics a Cp− or Cp*− ligand has been used instead of the CO ligands coordinated to the iron center; it was shown that this created lower overpotentials for proton reduction.7,15,19 So far, mostly models for the active site of [NiFe] hydrogenases have been studied, but recently a number of reports appeared describing the first [NiFe] models for the active site in [NiFeSe] hydrogenase containing an S2Se2 coordination environment around the nickel center instead of S4.20–22 Furthermore, new dithiolato and diselenolato bridged models for the active site in [NiFe(Se)] hydrogenases have been published comprising Ni(P2S2) or Ni(P2Se2) environments in order to compare their properties and also a cobalt selenolate electrocatalyst has been reported as a functional mimic of [NiFeSe] hydrogenase.23,24 Generally, the Sec-containing redox proteins show higher catalytic activities than their Cys-containing homologues. The relevant properties of selenium that could explain this difference in activity are the higher nucleophilicity of selenium, the lower redox potentials of the Sec-containing homologues and the higher acidity of selenocysteine; the pKa of Sec is 5.3 whereas that of Cys is 8.3. The increased acidity of Sec allows the selenol groups to be active at lower pH ranges. Selenium is also a softer donor atom than sulfur, the polarizable volume of selenium is 3.8 Å3vs. 2.9 Å3 of sulfur.25
So far no heterodimetallic nickel–ruthenium complexes have been reported comprising a NiS2Se2 unit as mimics of the [NiFeSe] hydrogenase active site. In this paper we describe the synthesis and characterization of the two nickel–ruthenium complexes [Ni(xbSmS)RuCp(PPh3)]PF6 and [Ni(xbSmSe)RuCp(PPh3)]PF6 as mimics of the active site of the [NiFe] and [NiFeSe] hydrogenases. The compound [Ni(xbSmS)RuCp(PPh3)]PF6 has been previously reported without crystallographic information.10 Herein, we report the detailed structural and electrochemical analysis of the compounds [Ni(xbSmS)RuCp(PPh3)]PF6 and [Ni(xbSmSe)RuCp(PPh3)]PF6 and their electrocatalytic properties in proton reduction.
Results
Synthesis and characterization
The two heterodinuclear compounds [Ni(xbSmS)RuCp(PPh3)]PF6 and [Ni(xbSmSe)RuCp(PPh3)]PF6 were synthesized following the procedure shown in Scheme 1. The mononuclear nickel compounds and [RuCp(PPh3)2Cl] have been reported earlier and were synthesized according to the published methods.12,21,26 Reaction of the mononuclear nickel complexes with one equivalent of [RuCp(PPh3)2Cl] in dichloromethane provided the compounds [Ni(xbSmS)RuCp(PPh3)]Cl and [Ni(xbSmSe)RuCp(PPh3)]Cl. The counter ion was exchanged by the addition of NH4PF6 to a solution of the chloride compounds in acetonitrile resulting in the compounds[Ni(xbSmS)RuCp(PPh3)]PF6 (1) and [Ni(xbSmSe)RuCp(PPh3)]PF6 (2) in 20% and 29% yield, respectively. The [NiRu] complexes were characterized by using 1H, 31P, 13C NMR spectroscopy, mass spectrometry, elemental analysis and single crystal X-ray crystallography. Both [NiRu] complexes give rise to sharp, clear resonances in the 1H NMR, 31P NMR and 13C NMR spectra. In the 1H NMR spectra of both compounds the resonances of the four methyl groups are observed as two singlets and the four methylene groups are observed as four doublets.
 |
| | Scheme 1 Synthesis scheme of the heterodinuclear NiRu complexes (1) and (2). | |
Description of the structures
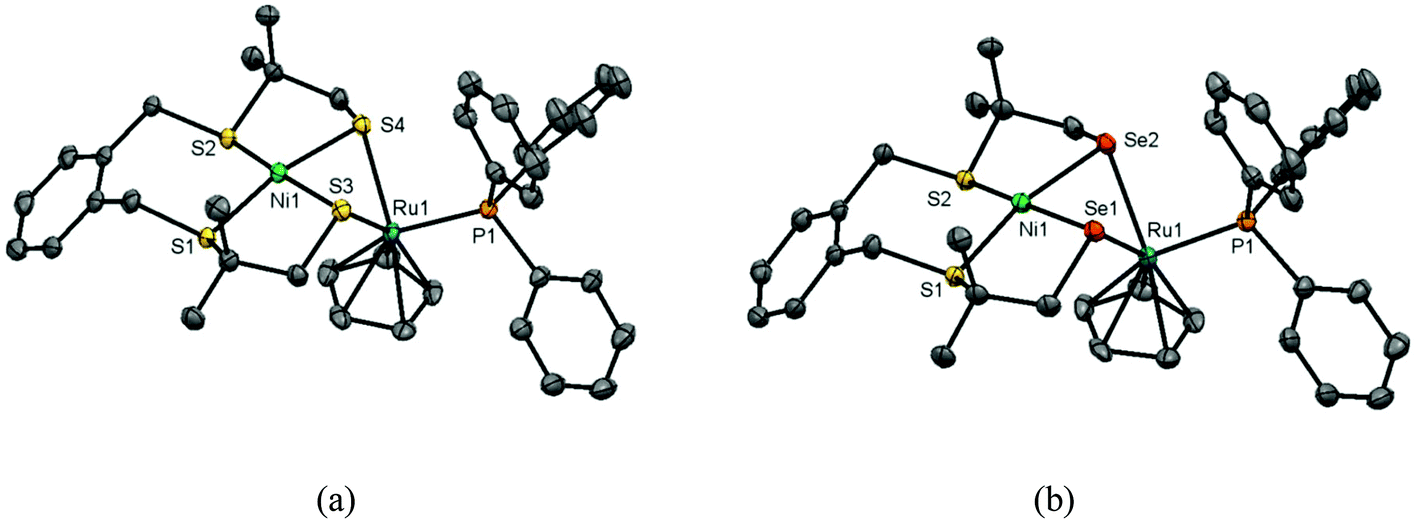
Single crystals of the compounds [Ni(xbSmS)RuCp(PPh3)]PF6 (1) and [Ni(xbSmSe)RuCp(PPh3)]PF6 (2) were obtained by vapor diffusion of pentane into acetone solutions of the complexes; crystallographic data are provided in Table S1.† Projections of the molecular structures of the heterodinuclear complexes are shown in Fig. 1 and selected bond distances and angles are provided in Table 1. The complexes (1) and (2) both crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and are isomorphous. In both structures, the PF6− counter ion, the lattice pentane solvent and the phenyl rings of the triphenylphosphane ligands are disordered over two orientations. Both heterodinuclear [NiRu] complexes contain an Ni(II) center in a square-planar geometry formed by the two thioether and two thiolate or selenolate donor atoms from the tetradentate ligand. Both thiolate or selenolate donors are bridging to a Ru(II) center that is coordinated in a pseudo-octahedral ‘piano stool’ geometry that is completed by the Cp− and the PPh3 ligand. This ‘piano stool’ configuration is most common for cyclopentadienyl complexes with a Ru(II) centre.9,10,16,17 The Ni–Ru distance (2.8435(4) Å) in complex (1) is determined by the sulfur atoms from the thiolate groups, which are involved in the bent Ni(μ-SR)2Ru butterfly core, and is relatively short compared to previously reported [NiRu] complexes which also contain a Cp− ligand, being 3.11, 2.99 and 2.91 Å.10 For complex (2) the Ni–Ru distance (2.9246(5) Å) is slightly longer because of the larger ionic radius of the selenolate donor atom. The hinge angle of the butterfly core, which is defined by the intersection of the least-square planes defined by NiS2/NiSe2 and RuS2/RuSe2, is much sharper (98.80° for (1) and 96.57° for (2)) than those in previously reported [NiRu] compounds (ranging between 108.4–120.9°).9,10
and are isomorphous. In both structures, the PF6− counter ion, the lattice pentane solvent and the phenyl rings of the triphenylphosphane ligands are disordered over two orientations. Both heterodinuclear [NiRu] complexes contain an Ni(II) center in a square-planar geometry formed by the two thioether and two thiolate or selenolate donor atoms from the tetradentate ligand. Both thiolate or selenolate donors are bridging to a Ru(II) center that is coordinated in a pseudo-octahedral ‘piano stool’ geometry that is completed by the Cp− and the PPh3 ligand. This ‘piano stool’ configuration is most common for cyclopentadienyl complexes with a Ru(II) centre.9,10,16,17 The Ni–Ru distance (2.8435(4) Å) in complex (1) is determined by the sulfur atoms from the thiolate groups, which are involved in the bent Ni(μ-SR)2Ru butterfly core, and is relatively short compared to previously reported [NiRu] complexes which also contain a Cp− ligand, being 3.11, 2.99 and 2.91 Å.10 For complex (2) the Ni–Ru distance (2.9246(5) Å) is slightly longer because of the larger ionic radius of the selenolate donor atom. The hinge angle of the butterfly core, which is defined by the intersection of the least-square planes defined by NiS2/NiSe2 and RuS2/RuSe2, is much sharper (98.80° for (1) and 96.57° for (2)) than those in previously reported [NiRu] compounds (ranging between 108.4–120.9°).9,10
 |
| | Fig. 1 Displacement ellipsoids plots (50% probability level) of (a) [Ni(xbSmS)RuCp(PPh3)]PF6 (1) and (b) [Ni(xbSmSe)RuCp(PPh3)]PF6 (2) at 110(2) K. Hydrogen atoms, PF6− anion, lattice solvent molecules, and disorder are omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for the complexes (1) and (2)
| |
(1) |
(2) |
|
Distances (Å)
|
| Ni1–S1 |
2.1847(6) |
2.1898(8) |
| Ni1–S2 |
2.1824(6) |
2.1881(8) |
| Ni1–S3/Se1 |
2.1935(6) |
2.3107(5) |
| Ni1–S4/Se2 |
2.1876(6) |
2.3050(5) |
| Ni1–Ru1 |
2.8435(4) |
2.9246(5) |
| Ru1–Cp(centroid) |
2.191 |
2.189 |
| Ru1–P1 |
2.3180(5) |
2.3174(7) |
| Ru1–S4/Se2 |
2.4256(5) |
2.5271(3) |
| Ru1–S3/Se1 |
2.4275(5) |
2.5298(3) |
|
Angles (°)
|
| P1–Ru–S4/Se2 |
92.362(18) |
91.999(19) |
| P1–Ru–S3/Se1 |
92.674(19) |
92.271(19) |
| S4/Se2–Ru–S3/Se1 |
73.502(17) |
74.449(10) |
| S4/Se2–Ni–S3/Se1 |
83.03(2) |
83.024(17) |
| S1–Ni–S3/Se1 |
90.45(2) |
90.73(2) |
| S2–Ni–S4/Se2 |
90.21(2) |
90.52(2) |
| S2–Ni–S1 |
94.98(2) |
94.24(3) |
| Ni–S3/Se1–Ru |
75.767(18) |
74.188(14) |
| Ni–S4/Se2–Ru |
75.909(18) |
74.334(14) |
The metal–selenolate bond distances in complex (2) are approximately 0.1 Å longer than the metal–thiolate bond lengths in complex (1), similar to the differences observed in the reported [NiFe] complexes also containing [Ni(xbSmS)] and [Ni(xbSmSe)].21 The Ni–thiolate distance in [Ni(xbSmS)RuCp(PPh3)]PF6 is 2.19 Å, which is comparable to the distance of 2.21 Å in the [NiFe] hydrogenase active site.27 The Ni–Se distance in [Ni(xbSmSe)RuCp(PPh3)]PF6 is 2.31 Å, significantly shorter than the 2.46 Å found in the [NiFeSe] hydrogenase active site.28
Electrochemical analyses
The electrochemical properties of the nickel–ruthenium complexes using cyclic voltammetry were investigated in acetonitrile with 0.1 M tetrabutylammonium hexafluoridophosphate as the supporting electrolyte with a scan rate of 200 mV s−1. A glassy carbon electrode was used as a working electrode and Ag/AgCl was used as a reference electrode, but all the potentials are reported vs. the ferrocene/ferrocinium (Fc0/+) couple. The voltammograms of the complexes (1) and (2) are highly similar; both show one irreversible wave at −1.70 V and −1.65 V vs. Fc/Fc+ followed by two small waves at −2.01, −2.25 V and −2.18, −2.40 vs. Fc/Fc+, respectively (Fig. 2). The cyclic voltammograms of the mononuclear nickel complexes show one irreversible wave at −1.96 V and −1.93 V vs. Fc/Fc+ for the compounds [Ni(xbSmS)] and [Ni(xbSmSe)], respectively (Fig. S8 and 9†). The cyclic voltammogram of the reference compound [RuCp(PPh3)(MeCN)2]PF6 shows one irreversible reduction at −2.54 V vs. Fc/Fc+ (Fig. S12†).
 |
| | Fig. 2 Cyclic voltammograms of (1) (a) and (2) (b) (1 mM) in an MeCN solution containing TBAPF6 (0.1 M) as the supporting electrolyte and using a glassy carbon electrode at a scan rate of 200 mV s−1. | |
Electrocatalytic hydrogen evolution in the presence of HOAc
The activity of the compounds in electrocatalytic proton reduction was investigated using cyclic voltammetry with addition of varying amounts of acetic acid to acetonitrile solutions of the NiRu complexes. Both complexes show a catalytic wave at around −2.20 V vs. Fc/Fc+, which shifts to more negative potentials with the addition of higher amounts of acid (Fig. 3). The overpotential for electrocatalytic proton reduction of the complexes (1) and (2) at an acetic acid concentration of 10 mM has been calculated using the half-wave potentials, taking homoconjugation of the acid into account.29 Both complexes display quite similar overpotentials, being 810 mV for complex (1) and 830 mV for complex (2). In order to prove that indeed dihydrogen gas is formed in the electrocatalytic reaction, controlled-potential coulometry (CPC) experiments were carried out on 1.0 mM solutions of the complexes (1) and (2) in acetonitrile (5 ml) in the presence of 7 μl of HOAc (10 equivalents) at −2.35 V vs. Fc/Fc+. The produced dihydrogen gas was quantified volumetrically by GC analysis. The CPC experiments were run for 1 h, while the solution was stirred continuously. Using complex (1) as the electrocatalyst for proton reduction, a total of 92 μl H2was produced for 1 mM complex in 1 h with 74% faradaic yield. For complex (2) a total of 106 μl H2 was produced in 1 h with 73% faradaic yield. In the absence of the catalyst formation of H2 is not observed.
 |
| | Fig. 3 Cyclic voltammograms of (1) (a) and (2) (b) (1 mM) in an MeCN solution of TBAPF6 (0.1 M) using a glassy carbon electrode at a scan rate of 200 mV s−1 in the presence of 0 (black), 10 (red), 20 (orange), 30 (brown), 40 (green), 50 (blue) mM of acetic acid. | |
Discussion
In this paper the compounds [Ni(xbSmS)RuCp(PPh3)]PF6 and [Ni(xbSmSe)RuCp(PPh3)]PF6 are described as potential mimics of the active site of the [NiFe] and [NiFeSe] hydrogenases. Single crystal X-ray crystallography has shown that the two structures are isomorphous and both have some structural similarities with the active site of the [NiFe] and [NiFeSe] hydrogenases, but with a Ru ion rather than an Fe center. Although it was anticipated that the compounds would have different electrochemical properties because of the different physical properties of sulfur and selenium, the electrochemical studies of the two compound showed quite similar results: changing the thiolate to selenolate donor atoms does not result in a significant difference of the redox potentials of the compounds. Comparison of the cyclic voltammograms of the NiRu compounds with those of the mononuclear nickel complexes and the reference compound [RuCp(PPh3)(MeCN)2]PF6 indicates that the metal centers do not dissociate during catalytic turnover. The catalytic proton reduction of the mononuclear nickel complexes occur at more negative potentials in identical conditions, although CPC showed the production of lower amounts of H2 compared to the NiRu compounds, namely 80 μl H2 for [Ni(xbSmS)] and 70 μl H2 for [Ni(xbSmSe)]. These data show that the binding of the ruthenium fragment to the mononuclear nickel compounds results in higher catalytic activities, especially for [Ni(xbSmSe)] for which the amount of produced dihydrogen increased with 30%. Coordination of the cationic ruthenium center to the nickel center results in an overall positively charged compound, which might help to lower the reduction potential of the nickel center thereby facilitating the reduction of protons. The compound [RuCp(PPh3)(MeCN)2]PF6 is also active in proton reduction, but only at a much more negative potential, which also indicates that dissociation of the NiRu compounds in solution does not occur (see Fig. S10, 11 and 13†). The electrocatalytic properties of a number of related [Ni(xbSmS)RuCp(L)]+ complexes based on the compound [Ni(xbSmS)] have been reported.10 The complexes [Ni(xbSmS)RuCp(CO)]PF6 and [Ni(xbSmS)RuCp(dmso)]PF6 were shown to have higher catalytic activity than [Ni(xbSmS)RuCp(PPh3)]PF6 whereas the compound [Ni(xbSmS)RuCp(PCy3)]PF6 has a lower activity.10 From this study it is apparent that the monodentate ligand bound to the ruthenium center has a large influence on the electrocatalytic activity of the dinuclear NiRu compound; it seems that the catalytic activity is lower when the ligand binds more strongly to the ruthenium center. Unfortunately, because of the different reaction conditions used by us the catalytic activity of our NiRu systems cannot be compared with those reported.10 It is difficult to compare the reactivity of different Ni(S4)Ru and Ni(S2Se2)Ru compounds, as the activity of the compounds not only may be influenced by the nature of the chalcogenide bridging donor atoms, but also on the ligand environment of the ruthenium center. The main structural difference between the Ni(S4)Ru and Ni(S2Se2)Ru compounds is in the nickel thiolate or selenolate distances.21–23 From our study it appears that this difference does not have a large effect on the catalytic activity of the compounds. It is difficult to discriminate the different effects that the ligands and the two metal centers have on the catalytic efficiency of the compound, because of the irreversible reduction waves of the two NiRu complexes. The irreversibility of the reduction processes in the NiRu compounds might indicate that the electrocatalysis is due to the formation of a heterogeneous catalyst by the deposition of nickel onto the glassy carbon electrode. However, the electrode was polished in between each single measurement and proton reduction was not observed when the electrode was used without polishing in a new solution in the absence of fresh NiRu catalyst. Although these experiments confirm that our complexes retain their structures during the catalytic reaction, the understanding of the active species is still not complete.
Conclusion
Two NiRu complexes are reported as mimics of the active sites of [NiFe] and [NiFeSe] hydrogenases. Both complexes are structurally highly similar and differ only in the bridging thiolate/selenolate donor atoms. The crystallographic studies show that the compounds in fact are isomorphous, with the only difference being the longer bond distances in the selenolate analogue. Although cyclic voltammetry and GC analysis of electrocatalytic proton reduction show that both complexes catalyze the hydrogen evolution reaction, the results show that changing the thiolate donor to a selenolate does not make a significant difference in either the activity or the overpotential. Further investigations will be done in order to improve catalytic activity and lower the overpotential for the hydrogen evolution reaction.
Experimental
Materials
All experiments were performed using standard Schlenk techniques or in a glovebox under an argon or nitrogen atmosphere unless otherwise noted. Chemicals were purchased from Acros or Aldrich and were used without further purification. Organic solvents were deoxygenated by the freeze–pump–thaw method and were dried over molecular sieves prior to use. The NMR solvent CD2Cl2 for metal complexes was deoxygenated by the freeze–pump–thaw method and was stored over molecular sieves in a glovebox. The complexes [Ni(xbSmS)],12 [Ni(xbSmSe)],21 and [RuCp(PPh3)2Cl]26 were synthesized according to published methods.
Physical measurements
NMR spectra were recorded on a 300 MHz Bruker DPX 300 spectrometer and chemical shifts were referenced against the solvent peak. Mass spectra were obtained with a Finnigan TSQ-quantum instrument using ESI. Elemental analyses were performed by the Microanalytical Laboratory Kolbe in Germany. Electrochemical measurements were performed at room temperature under an argon atmosphere using an Autolab PGstat10 potentiostat controlled by GPES4 software. A three-electrode cell system was used with a glassy carbon working electrode, a platinum counter electrode and an Ag/AgCl reference electrode. All electrochemistry measurements were done in acetonitrile solution with tetrabutylammonium hexafluoridophosphate as the supporting electrolyte; after each run ferrocene was added as an internal reference. All potentials are reported vs. the internal reference system Fc/Fc+, which under these conditions was found at −0.43 V vs. Ag/AgCl in MeCN. Electrocatalysis experiments were carried out by adding different concentrations of acetic acid to the MeCN solution of complexes. Controlled-potential coulometry (CPC) experiments were done with the same three-electrode cell system and electrodes. CPC experiments were recorded with an Autolab PGstat10 potentiostat controlled by GPES4 software. Gas chromatographic analysis was performed on a Shimadzu gas chromatograph GC-2010 at 35 °C fitted with a Supelco Carboxen 1010 molecular sieve column. Helium was used as the carrier gas, and analytes were detected using a thermal conductivity detector operated at 80 mA. The total volume of H2 produced during the reaction was calculated using a calibration line, which was obtained using the external reference method by injection of known amounts of H2 into the GC using a Hamilton gas-tight syringe (see Fig. S7†). Complexes (1) and (2) (1 mmol in 5 ml of acetonitrile) were placed into the three-electrode cell and prior to the each measurement the systems were deaerated by bubbling with helium for 10 min. The system was closed, and the headspace was pumped through the solution for 1 min. Before each GC sampling the headspace pumping was temporarily stopped to allow equilibration of the pressure, then GC measurement was started with a 0.5 mL sample of the headspace injection. The GC valve and the pump (KNF NMS 010L micro diaphragm pump) were enclosed in a helium-purged housing to prevent air leaking into the system.
Single crystal X-ray crystallography
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Version 1.171.36.32 Agilent Technologies, 2013). The same program was used to refine the cell dimensions and for data reduction. The structures were solved with the program SHELXS-2014/7 and were refined on F2 with SHELXL-2014/7.30 Analytical numeric absorption correction using a multifaceted crystal model was applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions using the instructions AFIX 23, AFIX 43 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5Ueq of the attached C atoms. The structures are partly disordered. The three phenyl groups of the triphenylphosphane ligand, the PF6− counterion, and the lattice pentane solvent molecule are found to be disordered over two orientations (all occupancy factors can be retrieved from the .cif file). The two structures are isomorphous. CCDC 1561282 and 1561283† contain the supplementary crystallographic data for [Ni(xbSmS)RuCp(PPh3)](PF6) and [Ni(xbSmSe)RuCp(PPh3)](PF6).
Synthesis of [Ni(xbSmS)RuCp(PPh3)](PF6) (1)
[RuCp(PPh3)2Cl] (179 mg; 0.246 mmol) and [Ni(xbSmS)] (99 mg; 0.246 mmol) were dissolved in DCM (10 mL) and the mixture was stirred for 5 days. The obtained solution was filtered to remove an insoluble precipitate and evaporated until dryness. To the resulting solid 10 ml ethanol was added, the obtained solution was filtered and evaporated under reduced pressure. A solution of NH4PF6 (81.2 mg; 0.498 mmol) in 10 ml acetonitrile was added to the residual solid and the solution was stirred at room temperature for 4 hours. The solvent was evaporated until dryness, the remaining solid was dissolved in dichloromethane (5 ml) and the solution was filtered to remove NH4I. To the filtrate an excess of diethyl ether was added and the mixture was placed in the freezer (−35 °C) overnight. The precipitate was filtered and dried in vacuo to obtain the pure dark purple product in a yield of 49 mg (20%). Single crystals suitable for X-ray structure determination were obtained from vapor diffusion of pentane into acetone solutions of the complex. 1H NMR [300 MHz, CD2Cl2, 298 K] δ 7.45–7.35 (m, 19H, Ph–![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3–6, P(C65)3), 4.46 (s, 5H, η5-C55), 4.19 (d, J = 12.4 Hz, 2H; Ph–Ceqax–S–), 3.66 (d, J = 12.4 Hz, 2H; Ph–Ceqax–S–), 2.14 (d, J = 13.4 Hz, 2H; C(CH3)2–Ceqax–S–), 1.98 (d, J = 13.4 Hz, 2H; C(CH3)2–Ceqax–S–), 1.70 (s, 6H, Meax), 1.61 (s, 6H, Meeq); 31P {1H} NMR [121.5 MHz, CD2Cl2, 298 K] 48.12 (s,
3–6, P(C65)3), 4.46 (s, 5H, η5-C55), 4.19 (d, J = 12.4 Hz, 2H; Ph–Ceqax–S–), 3.66 (d, J = 12.4 Hz, 2H; Ph–Ceqax–S–), 2.14 (d, J = 13.4 Hz, 2H; C(CH3)2–Ceqax–S–), 1.98 (d, J = 13.4 Hz, 2H; C(CH3)2–Ceqax–S–), 1.70 (s, 6H, Meax), 1.61 (s, 6H, Meeq); 31P {1H} NMR [121.5 MHz, CD2Cl2, 298 K] 48.12 (s, ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) Ph3), −145.16 (sept, JPF = 710 Hz; PF6); 13C NMR [75 MHz, CD2Cl2, 298 K] 135, 132, 131, 128, 79, 47, 35, 26, 24 ppm. ESI-MS (CH3OH): 830.8, calcd: 831.0 [M − PF6]+.
Ph3), −145.16 (sept, JPF = 710 Hz; PF6); 13C NMR [75 MHz, CD2Cl2, 298 K] 135, 132, 131, 128, 79, 47, 35, 26, 24 ppm. ESI-MS (CH3OH): 830.8, calcd: 831.0 [M − PF6]+.
Synthesis of [Ni(xbSmSe)RuCp(PPh3)](PF6) (2)
[RuCp(PPh3)2Cl] (179 mg; 0.246 mmol) and [Ni(xbSmSe)] (99 mg; 0.246 mmol) were dissolved in DCM (10 mL) and the mixture was stirred for 5 days. The obtained solution was filtered to remove an insoluble precipitate and evaporated until dryness. To the resulting solid 10 ml ethanol was added, the obtained solution was filtered and evaporated under reduced pressure. A solution of NH4PF6 (81.2 mg; 0.498 mmol) in 10 ml acetonitrile was added to the residual solid and the solution was stirred at room temperature for 4 hours. The solvent was evaporated until dryness, the remaining solid was dissolved in dichloromethane (5 ml) and the solution was filtered to remove NH4I. To the filtrate an excess of diethyl ether was added and the mixture was placed in the freezer (−35 °C) overnight. The precipitate was filtered and dried in vacuo to obtain the pure dark purple product in a yield of 130 mg (29%). Single crystals suitable for X-ray structure determination were obtained from vapor diffusion of pentane into acetone solutions of the complex. 1H NMR [300 MHz, CD2Cl2, 298 K] δ 7.43–7.24 (m, 19H, Ph–3–6, P(C65)3), 4.45 (s, 5H, η5-C55), 4.23 (d, J = 12.6 Hz, 2H; Ph–Ceqax–S–), 3.63 (d, J = 12.6 Hz, 2H; Ph–Ceqax–S–), 2.38 (d, J = 12.0 Hz, 2H; C(CH3)2–Ceqax–Se–), 2.13 (d, J = 12.3 Hz, 2H; C(CH3)2–Ceqax–Se–), 1.75 (s, 6H, Meax), 1.61 (s, 6H, Meeq); 31P {1H} NMR [121.5 MHz, CD2Cl2, 298 K] 46.97 (s, Ph3), −144.08 (sept, JPF = 714 Hz; PF6); 13C NMR [75 MHz, CD2Cl2, 298 K] 135, 132, 130, 128, 78, 35, 27, 25 ppm. ESI-MS (CH3OH): 926.7, calcd: 926.9 [M − PF6]+. Elemental Analysis calcd (%) for C39H44F6NiP2RuS2Se2·0.30C5H12 (1106.57): C 44.86, H 4.50; found: C 44.80, H 4.83.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
Mr J. J. M. van Brussel and Mr W. Jesse are gratefully acknowledged for performing the ESI-MS measurements. We thank Dr D. G. H. Hetterscheid for useful discussions.
References
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577 RSC.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS PubMed.
- H. R. Pershad, J. L. C. Duff, H. A. Heering, E. C. Duin, S. P. J. Albracht and F. A. Armstrong, Biochemistry, 1999, 38, 8992 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 249, 1518 CrossRef CAS.
- K. Weber, I. Heise, T. Weyhermüller and W. Lubitz, Eur. J. Inorg. Chem., 2014, 148 CrossRef CAS.
- S. Kaur-Ghumaan and M. Stein, Dalton Trans., 2014, 43, 9392 RSC.
- S. Canaguier, V. Fourmond, C. U. Perotto, J. Fize, J. Pecaut, M. Fontecave, M. J. Field and V. Artero, Chem. Commun., 2013, 49, 5004 RSC.
- Y. Oudart, V. Artero, J. Pécaut, C. Lebrun and M. Fontecave, Eur. J. Inorg. Chem., 2007, 2613 CrossRef CAS.
- S. Canaguier, L. Vaccaro, V. Artero, R. Ostermann, J. Pecaut, M. J. Field and M. Fontecave, Chem. – Eur. J., 2009, 15, 9350 CrossRef CAS PubMed.
- T. R. Simmons and V. Artero, Angew. Chem., Int. Ed., 2013, 52, 6143 CrossRef CAS PubMed.
- J. A. W. Verhagen, D. D. Ellis, M. Lutz, A. L. Spek and E. Bouwman, Dalton Trans., 2002, 1275 RSC.
- T. Liu, S. Chen, M. J. O'Hagan, M. Rakowski DuBois, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 134, 6257 CrossRef CAS PubMed.
- W. Zhu, A. C. Marr, Q. Wang, F. Neese, D. J. Spencer, A. J. Blake, P. A. Cooke, C. Wilson and M. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18280 CrossRef CAS PubMed.
- S. Canaguier, M. Field, Y. Oudart, J. Pecaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876 RSC.
- Y. Oudart, V. Artero, L. Norel, C. Train, J. Pécaut and M. Fontecave, J. Organomet. Chem., 2009, 694, 2866 CrossRef CAS.
- G. M. Chambers, R. Angamuthu, D. L. Gray and T. B. Rauchfuss, Organometallics, 2013, 32, 6324 CrossRef CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 271, 127 CrossRef.
- K. Weber, O. F. Erdem, E. Bill, T. Weyhermuller and W. Lubitz, Inorg. Chem., 2014, 53, 6329 CrossRef CAS PubMed.
- C. Wombwell and E. Reisner, Dalton Trans., 2014, 43, 4483 RSC.
- C. Wombwell and E. Reisner, Chem. – Eur. J., 2015, 21, 8096 CrossRef CAS PubMed.
- G. Gezer, D. Durán Jiménez, M. A. Siegler and E. Bouwman, Dalton Trans., 2017, 46, 7506 RSC.
- L.-C. Song, Y. Lu, L. Zhu and Q.-L. Li, Organometallics, 2017, 36, 750 CrossRef CAS.
- C. A. Downes, J. W. Yoo, N. M. Orchanian, R. Haiges and S. C. Marinescu, Chem. Commun., 2017, 53, 7306 RSC.
- D. Steinmann, T. Nauser and W. H. Koppenol, J. Org. Chem., 2010, 75, 6696 CrossRef CAS PubMed.
- J. L. Clark and S. B. Duckett, Dalton Trans., 2014, 43, 1162 RSC.
- M. V. Rampersad, S. P. Jeffery, M. L. Golden, J. Lee, J. H. Reibenspies, D. J. Darensbourg and M. Y. Darensbourg, J. Am. Chem. Soc., 2005, 127, 17323 CrossRef CAS PubMed.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549 CrossRef CAS PubMed.
- V. Fourmond, P. A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, C71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1561282 and 1561283. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02631e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a