
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical and chemical space partitioning in new intermetallic borides MNi21B20 (M = In, Sn)†‡
Frank R.
Wagner
a,
Qiang
Zheng§
 *a,
Roman
Gumeniuk
ab,
David
Bende
a,
Yurii
Prots
a,
Matej
Bobnar
a,
Dong-Li
Hu
c,
Ulrich
Burkhardt
a,
Yuri
Grin
a and
Andreas
Leithe-Jasper
a
*a,
Roman
Gumeniuk
ab,
David
Bende
a,
Yurii
Prots
a,
Matej
Bobnar
a,
Dong-Li
Hu
c,
Ulrich
Burkhardt
a,
Yuri
Grin
a and
Andreas
Leithe-Jasper
a
aMax-Planck-Institut für Chemische Physik fester Stoffe, Nöthnitzer Str. 40, 01187 Dresden, Germany. E-mail: zheng@cpfs.mpg.de; qiangzhengsic@gmail.com
bInstitut für Experimentelle Physik, TU Bergakademie Freiberg, Leipziger Str. 23, 09596 Freiberg, Germany
cMaterials Genome Institute and School of Materials Science and Engineering, Shanghai University, Shanghai 200444, China
First published on 26th September 2017
Abstract
The compounds MNi21B20 (M = In, Sn) have been synthesized and their cubic crystal structure determined (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, lattice parameters a = 7.1730(1) Å and a = 7.1834(1) Å, respectively). The structure can be described as a hierarchical partitioning of space based on a reo-e net formed by Ni3 species with large cubical, cuboctahedral and rhombicuboctahedral voids being filled according to [Ni1@Ni38], [M@Ni312], and [Ni26@B20@Ni324], respectively. The [Ni6@B20] motif inside the rhombicuboctahedral voids features an empty [Ni6] octahedron surrounded by a [B20] cage recently described in E2Ni21B20 (E = Zn, Ga). Position-space bonding analysis using ELI-D and QTAIM space partitioning as well as 2- and 3-center delocalization indices gives strong support to an alternative chemical description of space partitioning based on face-condensed [B@Ni6] trigonal prisms as basic building blocks. The shortest B–B contacts display locally nested 3-center B–B–Ni bonding inside each trigonal prism. This clearly rules out the notion of [Ni6@B20] clusters and leads to the arrangement of 20 face-condensed [B@Ni23Ni33] trigonal prisms resulting in a triple-shell like situation Ni26@B20@Ni324(reo-e), where the shells display comparable intra- and inter-shell bonding. Both compounds are Pauli paramagnets displaying metallic conductivity.
m, lattice parameters a = 7.1730(1) Å and a = 7.1834(1) Å, respectively). The structure can be described as a hierarchical partitioning of space based on a reo-e net formed by Ni3 species with large cubical, cuboctahedral and rhombicuboctahedral voids being filled according to [Ni1@Ni38], [M@Ni312], and [Ni26@B20@Ni324], respectively. The [Ni6@B20] motif inside the rhombicuboctahedral voids features an empty [Ni6] octahedron surrounded by a [B20] cage recently described in E2Ni21B20 (E = Zn, Ga). Position-space bonding analysis using ELI-D and QTAIM space partitioning as well as 2- and 3-center delocalization indices gives strong support to an alternative chemical description of space partitioning based on face-condensed [B@Ni6] trigonal prisms as basic building blocks. The shortest B–B contacts display locally nested 3-center B–B–Ni bonding inside each trigonal prism. This clearly rules out the notion of [Ni6@B20] clusters and leads to the arrangement of 20 face-condensed [B@Ni23Ni33] trigonal prisms resulting in a triple-shell like situation Ni26@B20@Ni324(reo-e), where the shells display comparable intra- and inter-shell bonding. Both compounds are Pauli paramagnets displaying metallic conductivity.
Introduction
Metal-borides feature a perplexing variety of crystal structures. Numerous and systematic studies suggest that saturation of the valence requirements of the electron-deficient boron constituent is the actual driving force of this complexity.1–3 In borides, the complexity of boron-based structural units often depends on metal-to-boron ratios, resulting in formation of one-, two-, and three-dimensional arrangements of covalently bonded boron atoms.1,3–10 Boron-rich materials mostly exhibit a three-dimensional boron partial structure formed by interlinked boron clusters.3,11,12 Two-dimensional boron networks in turn can be often observed at intermediate metal-to-boron ratios between 3/2 and 2,3,13,14 whereas, with decreasing boron content, separated chains or rings and, further, isolated boron atoms prevail.15–19Therefore, studying the phase diagram at intermediate compositions enables one to follow the structural evolution of and bonding interactions between boron aggregates and metal framework observed in complex intermetallic compounds. The recently discovered compounds MNi9B8 (M = Al, Ga)20 represent this structural complexity in an instructive way. There, networks composed of puckered [B16] rings embed arrays of interlinked [M@Ni12] clusters. This extremely unusual crystal structure prompted us to search for isotypic compounds comprising In or Sn as minority elements. However, neither Sn nor In was found to center [Ni12] clusters embedded into 2D boron networks, instead, new compounds MNi21B20 (M = In, Sn) formed. They feature M-filled [Ni12] cuboctahedra and [B20] cages embedded in a Ni atom matrix in such a way that [Ni6@B20] motifs can be recognized. Arrays formed of [Ni6@B20] have been recently observed in the crystal structures of Ga2Ni21B20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and Zn2Ni21B20.22
21 and Zn2Ni21B20.22
In this study, we report on the synthesis, crystal structure, chemical bonding and physical properties of MNi21B20 (M = In, Sn) and place these compounds into context with structurally related metal-rich borides Zn2Ni21B20 and Cr23C6 derivatives.
Experimental section
Sample preparation
Powders of tin (Chempur, <100 μm, 99.9 mass%), indium (Alfa Aesar, −325 mesh, 99.99 mass%), nickel (Chempur, −100 mesh, 99.99 mass%) and crystalline boron (Chempur, <100 μm, 99.9 mass%) were used to prepare the SnNi21B20 and InNi21B20 samples via two-step solid state reactions. Mixtures of nickel and boron with a ratio nearly 1:1 were pressed into pellets, placed into Al2O3 crucibles, sealed in tantalum tubes, and finally enclosed in quartz ampoules. The ampoules were heated to 910 °C, and kept for 7 days. Subsequently, the as-sintered pellets were ground to fine powders (<20 μm), mixed with tin or indium powders with Sn (or In):Ni ratio 1:21. These mixtures were pressed into pellets, heated to 980 °C (for SnNi21B20) or 950 °C (for InNi21B20), and kept for 7 days. Sample handling operations were carried out in argon-filled glove boxes (MBraun, p(O2/H2O) ≤ 1 ppm). All resulting samples are stable in air for long time.
X-ray diffraction
Powder X-ray diffraction (XRD) patterns were recorded on a Huber G670 imaging plate Guinier camera using a curved germanium (111) monochromator and Cu Kα1 radiation (λ = 1.540598 Å). Phase analysis and indexing were done using WinXPow program package.23 Lattice parameters were refined by least-squares fitting with LaB6 [a = 4.15689(8) Å] as internal standard within the WinCSD program package.24Single crystal XRD data for SnNi21B20 were collected on a Rigaku AFC 7 diffraction system equipped with a Saturn 724+ CCD using Mo Kα radiation (λ = 0.71073 Å). A multi-scan procedure was used for absorption corrections.
Since for both compounds, SnNi21B20 and InNi21B20, no single crystals of sufficient quality could be obtained, fine powders of both samples with particle size <20 μm were filled and sealed in quartz capillaries (ϕ = 0.5 mm) for high-resolution synchrotron powder XRD experiments. The data were collected at the high-resolution beamline ID22 of the European Synchrotron Radiation Facility (ESRF) in Grenoble, equipped with a multianalyzer stage with nine detectors and Si (111) monochromator (λ = 0.40073 Å, scan step of 0.002°, 1° ≤ 2θ ≤ 40°). Low temperature data were also collected at 100 K and 80 K for SnNi21B20 and InNi21B20, respectively.
For the crystal structure solutions and refinements a direct phase determination method and a full-pattern fitting (powder data) or full-matrix least-square (single-crystal data) procedures were used within WinCSD program package, respectively.24
Metallography
Small pieces were embedded in a conductive resin and then submitted to multistep grinding and polishing processes with final polishing using 0.25 μm diamond powder. The microstructure was investigated on a light-optical microscope (Axioplan2, Zeiss) as well as a field emission scanning electron microscope (JSM-7800F, JEOL). The composition of the observed phase was analyzed by energy dispersive X-ray spectroscopy (EDXS, Quantax 400 EDSX system, Silicon drift detector, Bruker) and wavelength dispersive X-ray spectroscopy (WDXS, SX100, Cameca) using B Kα, Ni Kα, Sn Lα, and In Lα signals and Ni3B, Sn and InP as standards. The Sn:Ni and In:Ni atomic ratios of the target phases from EDXS were measured to be 1.00(2):21.07(2), and 1.00(2):21.12(2), respectively. Their compositions from WDXS were measured to be Sn3.3(1)Ni51.9(9)B44.8(9), In2.6(1)Ni52.6(9)B44.8(9), respectively. All values are very close to the ones obtained from crystal structure determination.
Furthermore, the 2-center delocalization indices (DIs) between QTAIM atoms, a position-space variant of an effective covalent bond order, were calculated according to ref. 37 and 38. For the computationally demanding calculation of the delocalization indices a reduced k-point mesh of size 23 had to be used. Extension of this approach to multi-center bonding indices is well known.39 Along this line, 3-center bonding indices were calculated and analyzed with the program DISij40 using the previously proposed bond delocalization ratios G41 as an additional classification tool. Values of G(A,B) for bond A–B are obtained from the ratio between 3-center delocalization and 2-center localization of the bond charge A–B. A value of G(A, B) = 0 indicates a perfect 2-center localization of the bond between atoms A and B, while G(A, B) ≥ 1 indicates the absence of 2-center bonding, where the value of the 2c-DI in the range 0 ≪ δ(A, B) < 1 then only represents a down-sampled value of all the higher-center bond indices. As a rough guide, values 0 ≤ G(A, B) < 0.5 indicate a dominating 2-center bonding scenario, 0.5 < G(A, B) ≤ 1 a dominating three-center bonding scenario. Exemplarily, bond G values of 0.2 and 0.3 are obtained for the diamond structures of C and Ge, respectively, contrasting with G(B, B′) = 1 for the endohedral bonds in closo-B6H62− and CaB6.38
Results and discussion
Crystal structure determination
All reflections in the powder synchrotron X-ray diffraction patterns of SnNi21B20 and InNi21B20 samples at 293 K (Fig. 1a and b) were indexed in simple cubic unit cells with lattice parameters a = 7.1834(1) Å and a = 7.1730(1) Å, respectively. Details on crystal data collection are listed in Table 1. Analysis of the extinction conditions indicated possible space groups (SG) P23, Pm, P432, P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, and Pmm. The SG with highest symmetry Pmm was chosen to determine the crystal structures. The direct phase determination method was used to acquire positions of heavy Sn and Ni or In and Ni atoms, while positions of B were found from difference maps of electron density. Low residuals (RI = 0.053, RP = 0.088 and RI = 0.062, RP = 0.099 for SnNi21B20 and InNi21B20, respectively) indicate the reliability of the obtained structures.
3m, and Pmm. The SG with highest symmetry Pmm was chosen to determine the crystal structures. The direct phase determination method was used to acquire positions of heavy Sn and Ni or In and Ni atoms, while positions of B were found from difference maps of electron density. Low residuals (RI = 0.053, RP = 0.088 and RI = 0.062, RP = 0.099 for SnNi21B20 and InNi21B20, respectively) indicate the reliability of the obtained structures.
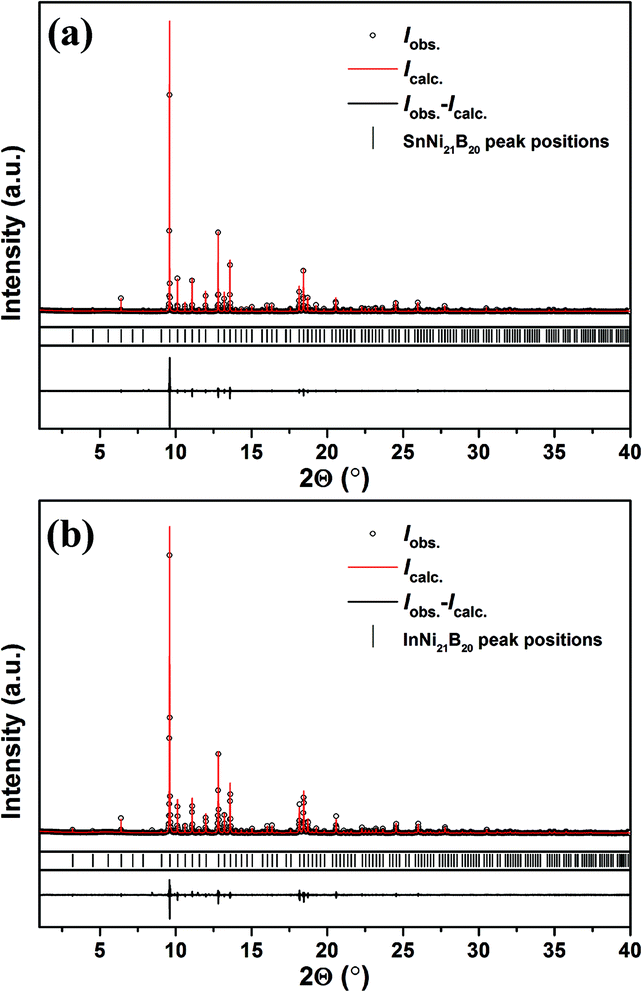 | ||
| Fig. 1 Synchrotron powder X-ray diffraction patterns (black dots) of SnNi21B20 (a) and InNi21B20 (b) at 293 K with the calculated profiles (red lines) after full-profile Rietveld refinements. | ||
| Composition | SnNi21B20 | InNi21B20 |
|---|---|---|
| Space group |
Pmm |
|
| a (Å) | 7.1834(1) | 7.1730(1) |
| V (Å3) | 370.67(2) | 369.06(2) |
| Calculated density/(g cm−3) | 7.023 | 7.036 |
| Z | 1 | |
| λ (Å) | 0.40073 | |
| 2θ range (°) | 1 to 40 | |
| T/K | 293 | |
| μ/mm−1 | 8.571 | 8.399 |
| Reflns in measured range | 243 | 240 |
| Refined parameters | 15 | |
| Refinement method | Full-profile Rietveld | |
| R I; Rp | 0.053; 0.088 | 0.062; 0.099 |
In both crystal structures, [Ni6] octahedra formed by Ni2 atoms are empty, and distances from their centers to Ni2 are 1.922 Å and 1.931 Å in SnNi21B20 and InNi21B20, respectively. Since B–Ni distances may be similarly short (cf. 1.967 Å in Ni3B42) occupation of the octahedra by B is possible. In order to study such a scenario and reduce the thermal vibrations of the B atoms, low temperature synchrotron powder XRD data were collected at 100 K and 80 K, respectively, for SnNi21B20 and InNi21B20. Details on diffraction data collection are listed in Table S2 in the ESI.‡ However, when additional B atoms were placed at this position, the displacement parameters of B atoms became quite large, indicating that this position should remain unoccupied. This was undoubtedly confirmed by the refinement of the occupation parameter for this position. Final atomic coordinates and anisotropic atomic displacement parameters for the two compounds at 293 K are listed in Table 2, and interatomic distances are listed in Table S1 in the ESI.‡ Corresponding atomic coordinates and anisotropic atomic displacement parameters at 100 K and 80 K are listed in Table S3 in the ESI.‡
| Atom | Site | x | y | z | B iso/Beq | B 11 | B 22 | B 33 | B 12 | B 13 | B 23 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SnNi21B20 | |||||||||||
| Sn | 1a | 0 | 0 | 0 | 1.67(2) | 1.67(4) | B 11 | B 11 | 0 | 0 | 0 |
| Ni1 | 3d | 0 | 0 | 1/2 | 1.17(4) | 0.93(5) | B 11 | 1.65(8) | 0 | 0 | 0 |
| Ni2 | 6f | 1/2 | 0.2324(2) | 1/2 | 1.10(3) | 0.93(3) | 1.44(6) | B 11 | 0 | 0 | 0 |
| Ni3 | 12i | 0.28891(7) | x | 0 | 1.23(2) | 1.36(2) | B 11 | 0.98(5) | 0.30(3) | 0 | 0 |
| B1 | 8g | 0.2911(6) | x | x | 1.0(2) | ||||||
| B2 | 12j | 1/2 | 0.2007(6) | y | 1.00(9) | ||||||
| InNi21B20 | |||||||||||
| In | 1a | 0 | 0 | 0 | 1.40(2) | 1.40(3) | B 11 | B 11 | 0 | 0 | 0 |
| Ni1 | 3d | 0 | 0 | 1/2 | 0.97(3) | 0.87(4) | B 11 | 1.16(7) | 0 | 0 | 0 |
| Ni2 | 6f | 1/2 | 0.2308(2) | 1/2 | 0.91(2) | 0.84(3) | 1.05(5) | B 11 | 0 | 0 | 0 |
| Ni3 | 12i | 0.28764(6) | x | 0 | 1.01(2) | 1.19(2) | B 11 | 0.64(4) | 0.22(3) | 0 | 0 |
| B1 | 8g | 0.2921(6) | x | x | 1.1(2) | ||||||
| B2 | 12j | 1/2 | 0.1992(5) | y | 0.76(7) | ||||||
Despite many efforts, a very small single crystal of moderate quality could be found only for SnNi21B20 for single crystal XRD experiment, while no single crystals could be obtained for InNi21B20. Details on XRD data collection for SnNi21B20 are listed in Table S4 in the ESI.‡ The refined structural model is in good agreement with that obtained from the synchrotron powder XRD data. Final atomic coordinates and anisotropic atomic displacement parameters are listed in Table S5 in the ESI.‡ The single crystal XRD data also unambiguously confirm that the centers of the [Ni6] octahedra remain empty.
The SnNi21B20 sample was also investigated by (S)TEM. The electron diffraction patterns along relevant zone axes are shown in Fig. 2a. All patterns can be well indexed in a single cubic lattice with the lattice parameters obtained from powder XRD diffraction. In agreement with the results from the powder XRD and single crystal diffraction data, no extinction conditions were observed. The atomic arrangement in the MNi21B20 structure determined from synchrotron diffraction (as shown in Fig. 3–5) is confirmed by HAADF images. HAADF images for SnNi21B20 along [001] and [011] zone axes are shown in the left and right of Fig. 2b, respectively, which is consistent with the simulated images using the structure model solved from synchrotron diffraction data. Moreover, the HAADF images along these two directions also reveal no superstructure formations and defects in SnNi21B20.
 | ||
| Fig. 2 (a) Electron diffraction patterns for SnNi21B20; (b) HAADF images of SnNi21B20 along [001] (left) and [011] (right); the two insets marked with arrows are the simulated images. | ||
 | ||
| Fig. 3 (a) Nickel (small spheres) and tin (large spheres) substructures in high-pressure cubic modification of Ni3Sn, (b) reo-e framework formed by Ni3 (small white spheres) in MNi21B20 highlighting Sn-centered cuboctahedra (Sn: large white spheres), distorted cubes centered by Ni1 (small turquoise spheres), and the rhombicuboctahedron (outlined by blue lines) with empty octahedron formed by Ni2 (small red spheres) inside. | ||
 | ||
| Fig. 4 Embedding of the [B20] cluster (outlined by bold black lines connecting B atoms visualized as black spheres) in the metal polyhedral framework of (a) MNi21B20 (M = In, Sn) and (b) M2Ni21B20 (M = Ga, Zn). | ||
 | ||
| Fig. 5 (a) The Ni324 rhombicuboctadron (blue lines) embedding the [Ni26@B20] structural unit: [Ni26@B20@Ni324]; Ni2 – red spheres, Ni3 – grey spheres, B – black spheres. (b) Exhaustion of the rhombicuboctahedron by triangular prims [B2@Ni22Ni34] which are additionally capped by Ni1 atoms (white spheres). (c) The prisms [B1@Ni23Ni33] generate the faces of the empty (□@Ni26) octahedron at the core of the crystal structure. | ||
m. For the ideal case of x = (2 + √2)−1 ≈ 0.2929 the resulting network represents one of 28 possible uniform partitions of 3-dimensional space, which is based on purely mathematical work dating back to Andreini in 190743 and subsequently completed and extended.44–46 The network corresponds to number 9 of Grünbaum's original enumeration45 and has been named “reo-e” in the Reticular Chemistry Structure Resource database.47,48
The actual positions of Ni3 atoms with x = 0.28764(6) and 0.28891(7) for M = In, Sn, respectively, slightly deviate from the ideal value, but still lead to a Ni3 network exclusively consisting of interconnected cubes, cuboctahedra and rhombicuboctadra with certain deviations from their ideal Platonic or Archimedean shapes. These polyhedra represent the voids in the Ni3 network, that are found to be filled in a specific way in MNi21B20 (M = In, Sn). The distorted cubic voids are filled by Ni1 atoms according to Ni1@Ni38(reo-e). The [Ni1@Ni38] polyhedra interconnect filled [M@Ni312] cuboctahedra. The recently discovered mineral Nisnite49 (Ni3Sn) adopts a Cu3Au-type of structure, which is constructed from the same [Sn@Ni12] motif (Fig. 3a). This structural arrangement is known as the high pressure modification of Ni3Sn, which adopts the hexagonal Mg3Sn structure at ambient pressure,50 and [M@Ni12] cuboctahedra are also commonly observed in Cr23C6-type borides.51,52 Moreover, in comparison to the In-containing compound, the Ni3–Ni3 distances in the cuboctahedron in the Sn-containing compound are longer, resulting in a spacious Sn-containing cage, consistent with an enlarged Biso value of Sn (see Tables 2 and 3). The large rhombicuboctahedral voids are found to be filled by a [B20] cage of 8 three-bonded B1 and 12 two-bonded B2 boron atoms surrounding a [Ni26] octahedron, which results in a triple-shell-like arrangement Ni16@B20@Ni324(reo-e). The same [Ni6B20] structural motif has been identified only recently in the isotypic structures of Zn2Ni21B2022 and Ga2Ni21B20.21 Body-centered tetragonal Ga2Ni21B20 derives from cubic MNi21B20 by a simple shift of the layers formed by the aforementioned rhombicuboctahedra (Fig. 4). However, while [Ni6@B20@Ni24] rhombicuboctahedra and [Ni@Ni8] cubes are retained, [M@Ni12] cuboctahedra do not appear in Ga2Ni21B20. Instead, a bicapped square antiprism [Ga@Ni10] is formed for the coordination of the Ga atoms.
| Polyhedra/or clusters | SnNi21B20 | InNi21B20 | |
|---|---|---|---|
| [M@Ni312] cuboctahedron | M–Ni3 | 2.9350(4) | 2.9179(4) |
| Ni3–Ni3 | 2.9350(4) | 2.9179(4) | |
| [Ni26@B110B210@Ni324] | B1–B2 | 1.760(6) | 1.764(5) |
| Ni2–Ni2 | 2.719(2) | 2.731(2) | |
| B1–Ni2 | 2.164(4) | 2.154(5) | |
| B2–Ni2 | 2.162(5) | 2.170(4) | |
| Ni3–Ni3 | 2.9350(5) | 2.9179(4) | |
| Ni3–Ni3 | 3.0327(1) | 3.0465(1) | |
| [B1@Ni23Ni33]B23 | Ni2–Ni3 | 2.7176(9) | 2.7169(9) |
| [B2@Ni22Ni34]B12Ni1 | B1–Ni3 | 2.091(4) | 2.096(4) |
| B2–Ni3 | 2.186(3) | 2.183(3) | |
| [Ni1@Ni38] cube | Ni3–Ni3 | 2.9350(7) | 2.9179(6) |
| Ni3–Ni3 | 3.0327(7) | 3.0465(6) | |
| Ni1–Ni3 | 2.5703(3) | 2.5646(3) | |
| Ni1–B2 | 2.039(4) | 2.021(4) |
A complementary view of the [Ni6B20] motif is obtained focusing on the boron structural chemistry in intermetallic borides with roughly equiatomic boron contents, where capped trigonal prismatic coordination of the boron atoms is frequently observed.3,5,53 Indeed, the two boron species are found in tricapped trigonal prismatic coordination according to [B1@Ni23Ni33B23] and [B2@Ni22Ni34B12Ni1], where the specific arrangement of the face-condensation at the B1–B2 contacts yields the [Ni16@B20@Ni324] motif described above (Fig. 5). This view automatically implies, that a separate [Ni6B20] unit does not occur, instead, the [Ni6B20] motif has to be extended by the outer Ni3 atoms forming the rhombicuboctahedral Ni324reo-e cage, which is supported by comparison of the relevant interatomic distances (Table 3 and ESI Table S1‡). Comparing B–Ni distances to the inner Ni26 octahedron, d(B1–Ni2) = d(B2–Ni2) = 2.16 Å, with those to the outer Ni324 cage atoms, d(B1–Ni3) = 2.09 Å, d(B2–Ni3) = 2.19 Å, reveals that they are rather similar. This supports the trigonal prismatic coordination assignment. Moreover, distances d(Ni2–Ni2) = 2.72 Å inside the Ni26 octahedron are comparable to distances between the inner Ni2 and the outer Ni3 shell, with d(Ni2–Ni3) = 2.72 Å. They are even slightly shorter than the distances d(Ni3–Ni3) = 2.94 Å within the Ni3 framework but longer than the shortest Ni–Ni distances d(Ni1–Ni3) = 2.57 Å. This clearly shows that the interconnecting Ni matrix is not just formed from Ni3 atoms, but includes all Ni atoms of the structure. The most conspicuous feature concerns the distance d(B1–B2) = 1.77 Å, which is rather long for a 2c–2e B–B bond and more comparable to those in deltahedral boron clusters with 3-center bonding although the homoatomic coordination number is only 2 or 3 for B2 and B1, respectively. However, elongated B–B distances are a common feature in intermetallic borides even if they do not display deltahedral boron clusters. This aspect is investigated in detail by analysis of atomic interactions.
Electronic structure and chemical bonding
The optimized structure displays only small deviations from the experimental one, which points to a faithful representation of the electronic structure by the chosen DFT/PBE method. The calculated density of states DOS (ELK code, Fig. 6) displays a small pseudo gap close to the Fermi energy EF (set to 0 eV), where DOS(EF) = 22 states per eV per cell. Decomposition of the total DOS in terms of atomic muffin-tin contributions (pDOS) shows the dominating contributions of Ni 3d states in the region around EF. Calculation of the center of gravity for each component pDOS yields an energetic sequence value of −6.8 eV and −6.5 eV for B1 and B2, respectively, which correlates with the corresponding number of homoatomic neighbors of each B species, namely 3 and 2. Similar to previous results20 this is considered a hint on the homoatomic interaction being energetically dominating one for the B atoms. Application of this procedure to the Ni atoms’ pDOS yields center of gravity values of −2.3 eV, −2.5 eV, and −2.3 eV for Ni1, Ni2, Ni3, respectively. Given the optimized average distances Ni–B 2.03 Å, 2.16 Å, 2.15 Å (respective experimental values are 2.04 Å, 2.16 Å, and 2.15 Å) for each Ni species, respectively, a correlation of these with the Ni orbitals’ energetic center of gravity cannot be found, most probably because of the different numbers of boron neighbors (4, 8, 6) in each case. | ||
| Fig. 6 Electronic density of states (DOS, muffin-tin part) of SnNi21B20. | ||
Position-space chemical bonding analysis yields QTAIM effective charges (Sn0.51+)(Ni10.04−)(Ni20.28+)(Ni30.23+)(B10.31−) (B20.19−) which is consistent with the sequence of Allred-Rochow electronegativities 2.0 (B) > 1.8 (Ni) > 1.7 (Sn), although the differences are very small. The boron species display rather small negative effective charges, which look too small to represent a Zintl anion with clear homoatomic 2-center bonds. This issue is clarified in the following analysis employing the electron localizability indicator on the one hand and two- and three-center delocalization indices between QTAIM atoms on the other hand.
The ELI-D distribution (Fig. 7b) yields only three types of local maxima (attractors) in the valence region. For each B1–B2 contact (d(B1–B2)exp. = 1.77 Å) an attractor lies very close to the internuclear line (light brown localization domains in Fig. 7b). The associated ELI-D basin for the B1–B2 bond attractor displays a population of 3.00 electrons. ELI-D/QTAIM basin intersection reveals that 1.08 and 0.97 e belong to QTAIM atoms B1 and B2, respectively, and the remaining 0.96 e are contributed by 4 neighboring nickel QTAIM atoms 2Ni2 + 2Ni3 (Fig. 7c) spanning the common rectangular face between both [B@Ni6] trigonal prisms. With this bond basin's effective atomicity of 6 (2B + 4Ni) the situation can be interpreted as a B2Ni4 multicenter bonding scenario. Even deeper insight will be gained below from the analysis of 3-center DIs and bond G values. Each Ni2 atom being located in the center of the [B8] ring takes part in 8 such bonds, each Ni3 atom in 4 such bonds.
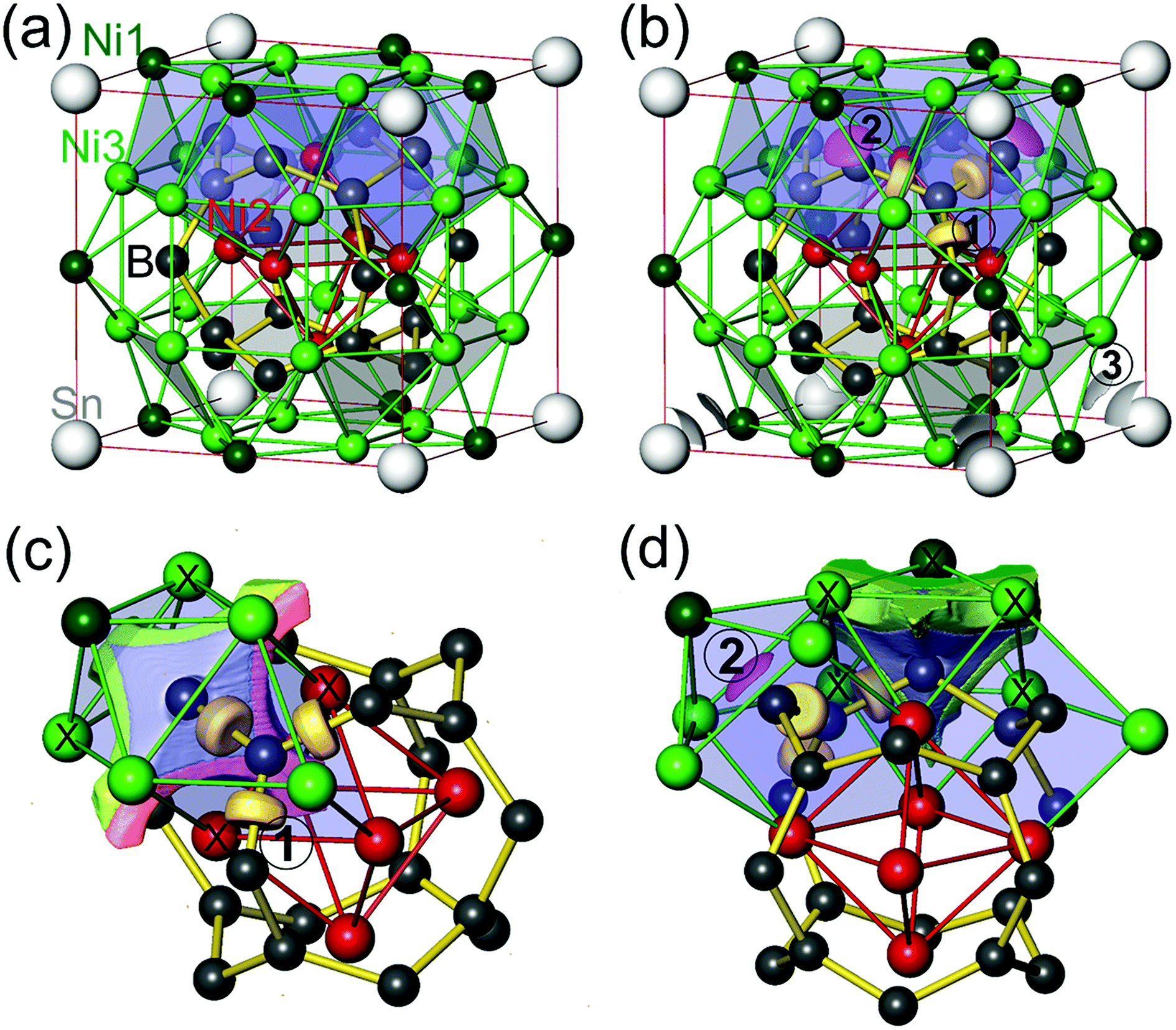 | ||
| Fig. 7 Crystal structure and ELI-D chemical bonding signatures in SnNi21B20: (a) relevant structural features; atomic positions are shown by spheres for Sn (white), Ni1 (dark green), Ni2 (red), Ni3 (green) and B (dark grey); bonds B1–B2 are indicated by yellow lines; trigonal prisms B@Ni6 by blueish translucent faces; inner Ni26 octahedron by red edges, selected trigonal faces of Sn@Ni312 cuboctahedra related to Sn–Ni3 bonding in translucent grey; (b) ELI-D localization domains for bonds B1–B2 (type 1, light brown, YD = 1.30), Ni1–B2 (type 2, magenta, YD = 1.23), and Sn–Ni3 (type 3, grey, YD = 0.942); (c) ELI-D/QTAIM basin intersection for ELI-D bond basin B1–B2 (type 1), intersected atoms of common prism face 2Ni2 + 2Ni3 are marked by “X”, spatial regions of bond basin belonging to boron, Ni2, Ni3 are colored in grey, light red, and light green, respectively; (d) ELI-D/QTAIM basin intersection of bond basin Ni1–B2 (type 2); intersecting Ni3 + 4Ni1 atoms marked with “X”, spatial regions parts attributed to QTAIM B, Ni1, and Ni3 atoms, are colored in light grey, green and light green, respectively. | ||
The second type of ELI-D attractor is found on the internuclear line B2–Ni1 with d(B2–Ni1)exp. = 2.16 Å (Fig. 7b, magenta-colored localization domains). The associated ELI-D basin displays a population of 2.50 electrons, which can be decomposed into 1.17 e from B2, 1.32 e from 1 Ni1 (0.59 e) and 4 × 0.18 e from 4 Ni3 atoms. With an effective atomicity of 6 (1 B + 5 Ni) this basin describes a BNi5 multicenter bonding scenario (Fig. 7d). Each Nil atom takes only part in 4 B2–Ni1 bonds, while each Ni3 atom takes part in 4 B2–Ni1 bonds and 4 B1–B2 bonds.
The third type of ELI-D attractor arises from Sn–Ni3 bonding (d(Sn–Ni3)exp. = 2.94 Å). The endohedral Sn atoms display a nearly spherical ELI-D distribution with a comparably low value of the structuring index32 of ε ≈ 0.01 for this basin set, which characterizes weak bonding but with a large number of 12 partners. The 24 attractors occur in triangles pointing to the corresponding 3 Ni3 atoms of the 8 trigonal faces of the [Ni312] cuboctahedron (grey localization domains in Fig. 7b containing 3 attractors). Each of the associated 24 basins is populated by 0.24 electrons. ELI-D/QTAIM intersection reveals them to represent effectively (1 Sn + 1 Ni3)-diatomic polar bonds, with 0.14 e belonging to QTAIM Sn and 0.10 e to Ni3 atom. Each vertex Ni atom is connected to two such basins. All the basins around a Sn atom were merged into a superbasin containing 5.79 electrons, with 3.49 electrons contributed by the QTAIM Sn atom and 2.30 electrons coming from the neighboring 12 Ni3 atoms, i.e. 0.19 e from each.
The bonding situations B–B, B–Ni, and Sn–Ni described above are comparable to the ones discussed recently for GaNi9B8 with condensed endohedral clusters [Ga@Ni12] nested with a polyanionic B network to yield multicenter B–B–Ni bonding.20 As an extension to this previous study, the high local symmetry and the smaller unit cell size of SnNi20B21 permits a more detailed investigation of chemical bonding employing a complementary method. Analysis of 2c- and 3-center delocalization indices (DIs) between QTAIM atoms (QTAIM/DI analysis) complemented by bond G values yields direct information of the deviation of the bonding from the classical 2-center case.
The highest 2c-delocalization indices are found between boron atoms, where δ(B1, B2) = 0.53 with bond delocalization ratio G(B1, B2) = 0.84. The high G value indicates a high degree of 3c-bonding character, which is consistent with the clearly fractional effective covalent bond order of the B–B bond and the elongated bond distance d(B–B)exp. = 1.77 Å (opt. 1.76 Å) observed also for the deltahedral bonds in hexaborides.41 Decomposition of G(B1, B2) into a sum of all three-center contributions reveals that four similar 3c-DIs of type δ(B1, B2, Ni) play the dominant role. The four Ni atoms involved are just those 2Ni2 and 2Ni3 atoms already identified above with the ELID/QTAIM basin intersection method. They form the surrounding rectangular face of the condensed [B@Ni6] trigonal prisms (Fig. 5b and c). With their contributions to G(B1, B2) summing up to 71% of the total value they represent the main delocalization channel of the B1–B2 bond. Thus, the B1–B2 contact represents four nested 3-center bonds B–B–Ni. Boron species B1 features three, species B2 two of such contacts.
Ni–B bonding is characterized by nearest-neighbor DIs ranging from δ(Ni1, B) = 0.53 for the shortest (opt. 2.03 Å, exp. 2.04 Å) to δ(Ni2, B) = 0.31 for the longest distance (opt. 2.18 Å, exp. 2.16 Å). The corresponding G(Ni, B) values are found to increase with decreasing 2c-DIs from 0.42 to 0.64 as expected. This marks a gradual transition from a mainly two-center Ni–B bonding regime towards a three-center bonding Ni–B–B one, indicated by the dominating 3-center contributions δ(Ni, B1, B2) to G(Ni, B).
DIs(Ni, Ni) take values between δ(Ni1, Ni3) = 0.25 (opt. 2.58 Å, exp. 2.57 Å) and δ(Ni3, Ni3) = 0.11 (opt. 2.98 Å, exp. 2.94 Å) with remarkably clear 2c-character shown by G(Ni, Ni) ≈ 0.2. These values are roughly similar to the spin-averaged values obtained for fcc-Ni.54
The view of a metallic Ni matrix nested with B20 units to form multicenter B–B–Ni bonds is the resulting picture for the MNi20B21 compounds (M = Sn, In). The condensed trigonal prisms [B1@Ni23Ni33]B23 and [B2@Ni22Ni34]B12Ni1 with their three capping atoms are the real building blocks of this motif and a consideration of only [Ni26@B20] would be incomplete.
According to the ELI-D/QTAIM intersection method, the endohedral Sn atoms form polar bonds Sn–Ni without evidence for dominating multicenter bonding. This is corroborated by the DI analysis, which yields δ(Sn, Ni) = 0.24 with G(Sn, Ni) = 0.41 being still in the 2-center regime. The sum over all 12 Sn–Ni DIs reveals that 2.9 bonds are effectively formed in total between Sn and the Ni cage atoms.
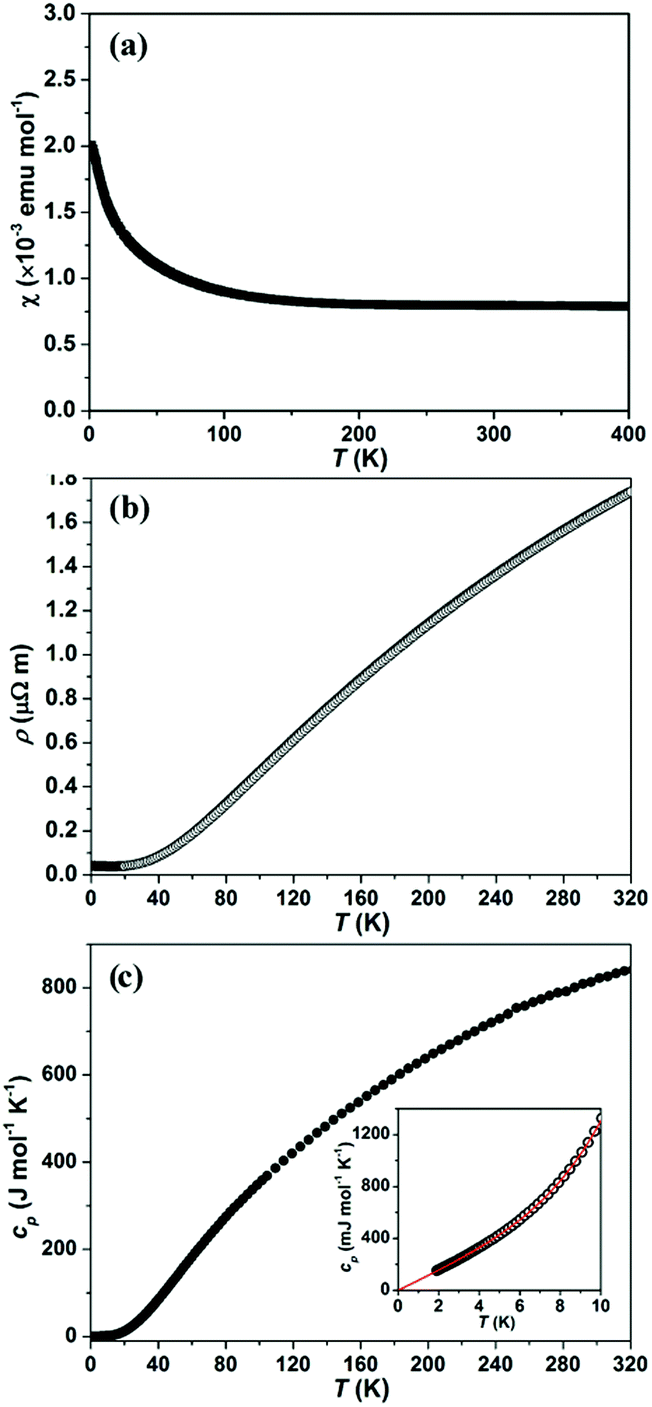 | ||
| Fig. 8 Physical properties of SnNi21B20: (a) temperature-dependent magnetic susceptibility χ(T) in the external magnetic field of μ0H = 7.0 T; (b) temperature dependence of electrical resistivity ρ(T); (c) temperature dependence of heat capacity cp(T), the inset shows the fitting by cp(T) = γT + βT3 + δT5 for 3.0 K < T < 10 K. | ||
The electrical resistivity as a function of temperatureρ(T) of SnNi21B20 and InNi21B20 (Fig. 8b and Fig. S1b in the ESI‡) shows typical metallic characteristics in accordance with the electronic structure calculations (residual resistivity ρ0 and the room temperature resistivity ρ(300 K) as well as calculated RRR values are summarized in Table S6‡.
Temperature-dependent heat capacity cp(T) for SnNi21B20 and InNi21B20 are shown in Fig. 8c and Fig. S1c in the ESI,‡ respectively. Low-T cp(T) (the insets to Fig. 8c and Fig. S1c in the ESI‡) can be fitted by the model cp(T) = γT + βT3 + δT5, where γ is the Sommerfeld coefficient of the electronic heat capacity, βT3 + δT5 are the first terms of the harmonic lattice approximation for the phonon contribution. From the fits (3.0 K < T < 10 K for SnNi21B20 and 2.0 K < T < 10 K for InNi21B20, respectively) the parameters, γ, β and δ as well as the corresponding initial Debye temperatures ΘD(0) have been evaluated and their values are compiled in Table S6.‡
Conclusions
New borides MNi21B20 (M = In, Sn) were synthesized and their crystal structures were solved from synchrotron powder X-ray diffraction data. The cubic crystal structure can be related to a uniform 3D tiling of space by cuboctahedra, cubes and rhombicuboctahedra, i.e. a filled reo-e net of Ni3 atoms, with unusual [Ni6@B20] units filling the [Ni24] rhombicuboctahedra leading to [Ni6@B20@Ni24] triple shell clusters, and M atoms centering the cuboctahedra [M@Ni12]. The triple-shell-like aggregation is the result of the condensation pattern of the two types of boron centered tri-capped trigonal prisms [B@Ni6X3], where the capping X3 features either B3 or B2Ni, leading to 3 and 2-bonded B species, respectively. Position-space chemical bonding analysis reveals these prisms to be the chemically relevant building blocks of the structures of MNi21B20 (M = In, Sn), a consideration of only the [Ni6@B20] part would neglect half of the relevant 3-center B–B–Ni bonds detected by the bonding analysis. Due to their spatial arrangement the [B1@Ni6B23] prisms generate the faces of the empty [Ni6] octahedron at the core, while the [B2@Ni6B12Ni] prims fill the space in-between them hosting the capping B2 atoms of the [B1@Ni6B23] prisms. This leads to the [B20] units and completes the [Ni24] rhombicuboctahedra on the outside of the clusters. The [M@Ni12] atoms display polar covalent bonds M–Ni, with a small effective bond order leading in sum to a significant number of 2.9 covalent bonds formed between Sn and the Ni cage.The electronic structure reveals them to be metals, which was corroborated by physical properties measurements indicating metallic conductivity as well as Pauli paramagnetism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr S. Hückmann and Dr H. Borrmann for performing powder X-ray diffraction measurements, Ms P. Scheppan, Ms M. Eckert and Ms. S. Kostmann for the metallographic analysis, Mr R. Koban for assistance with physical property measurements, and Dr W. Schnelle for valuable discussions. We also acknowledge the ORNL's Center for Nanophase Materials Sciences (CNMS), which is sponsored by an Office of Science User Facility, the U.S. Department of Energy (DOE). Qiang Zheng would like to thank Miaofang Chi for her help. The authors thank Dr C. Curfs from ID22 (former beamline ID31) ESRF, Grenoble for assistance with synchrotron data collection. Open Access funding provided by the Max Planck Society.References
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640–8668 CrossRef CAS PubMed.
- T. Mori, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner, J.-C. Bünzli and V. Pecharsky, North-Holland, Amsterdam, 2008, vol. 38, pp. 105–173 Search PubMed.
- P. Rogl, in Inorganic Reactions and Methods, ed. A. P. Hagen, Wiley, 1991, DOI:10.1002/9780470145289.ch20.
- T. Mori, in Handbook on the Physics and Chemistry of Rare-Earths, ed. K. A. Gschneidner, J. C. Bunzli and V. K. Pecharsky, Elsevier, Amsterdam, 2007, vol. 38, pp. 105–173 Search PubMed.
- Y. B. Kuz'ma, Crystallochemistry of Borides, Lviv University Publishers, Lviv, 1983 Search PubMed.
- H. Nowotny and P. Rogl, in Boron and Refractory Borides, ed. V. Matkovich, Springer, New York, 1977, ch. 23, pp. 413–438, DOI:10.1007/978-3-642-66620-9_23.
- Y. B. Kuz'ma and N. F. Chaban, Binary and Ternary Systems Containing Boron, Metallurgiya, Moscow, 1990 Search PubMed.
- T. Fehlner, J.-F. Halet and J.-Y. Saillard, Molecular clusters: a bridge to solid-state chemistry, Cambridge University Press, 2007 Search PubMed.
- T. Mori, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2012 Search PubMed.
- G. Akopov, M. T. Yeung and R. B. Kaner, Adv. Mater., 2017, 29, 1604506 CrossRef PubMed.
- L. Pauling and S. Weinbaum, Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem., 1934, 87, 181–182 CAS.
- P. Blum and F. Bertaut, Acta Crystallogr., 1954, 7, 81–86 CrossRef CAS.
- Y. B. Kuz'ma, Kristallografiya, 1970, 15, 372–374 Search PubMed.
- P. Rogl and H. Nowotny, Monatsh. Chem., 1974, 105, 1082–1098 CrossRef CAS.
- A. Westgren, Nature, 1933, 132, 480 CrossRef CAS.
- Q. Zheng, M. Kohout, R. Gumeniuk, N. Abramchuk, H. Borrmann, Y. Prots, U. Burkhardt, W. Schnelle, L. Akselrud, H. Gu, A. Leithe-Jasper and Y. Grin, Inorg. Chem., 2012, 51, 7472–7483 CrossRef CAS PubMed.
- C. Goerens and B. P. T. Fokwa, J. Solid State Chem., 2012, 192, 113–119 CrossRef CAS.
- B. P. T. Fokwa, J. von Appen and R. Dronskowski, Chem. Commun., 2006, 4419–4421, 10.1039/b608903h.
- Q. Zheng, G. Roman, R. Helge, W. Schnelle, Y. Prots, U. Burkhardt, Y. Grin and A. Leithe-Jasper, J. Phys.: Condens. Matter, 2015, 27, 415701 CrossRef PubMed.
- Q. Zheng, F. R. Wagner, A. Ormeci, Y. Prots, U. Burkhardt, M. Schmidt, W. Schnelle, Y. Grin and A. Leithe-Jasper, Chem. – Eur. J., 2015, 21, 16532–16540 CrossRef CAS PubMed.
- Q. Zheng, R. Gumeniuk, W. Schnelle, Y. Prots, U. Burkhardt and A. Leithe-Jasper, Solid State Sci., 2016, 55, 93–97 CrossRef CAS.
- Z. P. Malik, O. Sologub, A. Grytsiv, G. Giester and P. F. Rogl, Inorg. Chem., 2011, 50, 7669–7675 CrossRef CAS PubMed.
- WinXPow, STOE and Cie GmbH, Darmstadt, 2003 Search PubMed.
- L. Akselrud and Y. Grin, J. Appl. Crystallogr., 2014, 47, 803–805 CrossRef CAS.
- C. Koch, PhD, Arizona State University, 2002.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Program ELK, version 2.2.10, http://www.sourceforge.net Search PubMed.
- M. Kohout, Program DGrid, version 4.7, Radebeul, Germany, 2015 Search PubMed.
- R. F. W. Bader, Atoms in Molecules, Oxford University Press, Oxford, 1990 Search PubMed.
- M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef CAS.
- F. R. Wagner, V. Bezugly, M. Kohout and Y. Grin, Chem. – Eur. J., 2007, 13, 5724–5741 CrossRef CAS PubMed.
- A. Martin-Pendas, M. Kohout, M. Blanco and E. Francisco, in Modern Charge-Density Analysis, ed. C. Gatti and P. Macchi, Springer, Heidelberg, 2012, ch. 9, pp. 303–358, DOI:10.1007/978-90-481-3836-4_9.
- S. Raub and G. Jansen, Theor. Chem. Acc., 2001, 106, 223–232 CrossRef CAS.
- D. Bende, F. R. Wagner and Y. Grin, Inorg. Chem., 2015, 54, 3970–3978 CrossRef CAS PubMed.
- D. Bende, Y. Grin and F. R. Wagner, in Heusler Alloys - Properties, Growth, Applications, ed. C. Felser and A. Hirohata, Springer International Publishing, 2016, pp. 133–156 Search PubMed.
- J. G. Angyan, M. Loos and I. Mayer, J. Phys. Chem., 1994, 98, 5244–5248 CrossRef CAS.
- A. I. Baranov and M. Kohout, J. Comput. Chem., 2011, 32, 2064–2076 CrossRef CAS PubMed.
- R. Bochicchio, R. Ponec, A. Torre and L. Lain, Theor. Chem. Acc., 2001, 105, 292–298 CrossRef CAS.
- F. R. Wagner, Program DISij, version 7.2.7, Max-Planck-Institut für Chemische Physik fester Stoffe, Dresden, Germany, 2017 Search PubMed.
- C. Börrnert, Y. Grin and F. R. Wagner, Z. Anorg. Allg. Chem., 2013, 639, 2013–2024 CrossRef.
- G. F. Kayser and F. X. Kayser, J. Alloys Compd., 1996, 233, 74–79 CrossRef CAS.
- A. Andreini, Mem. Societa Italiana delle Scienze, 1907, 3, 76–129 Search PubMed.
- R. Williams, The Geometrical Foundation of Natural Structure: A Source Book of Design, Dover Publications, 1st edn, 1979 Search PubMed.
- B. Grünbaum, Geombinatorics, 1994, 4, 49–56 Search PubMed.
- M. Deza and M. Shtogrin, Eur. J. Combinatorics, 2000, 21, 807–814 CrossRef.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- http://rcsr.anu.edu.au .
- R. Rowe, J. D. Grice, G. Poirier, C. J. Stanley and L. Horváth, Can. Mineral., 2011, 49, 651–656 CrossRef CAS.
- J. F. Cannon, Effects of high pressure on the structures of AB3 – type layered compounds, Materials Research Society symposium proceedings, 1984, vol. 22, pp. 113–116 Search PubMed.
- D. Kotzott, M. Ade and H. Hillebrecht, J. Solid State Chem., 2009, 182, 538–546 CrossRef CAS.
- D. Kotzott, M. Ade and H. Hillebrecht, J. Solid State Chem., 2010, 183, 2281–2289 CrossRef CAS.
- T. Lundström, Pure Appl. Chem., 1985, 57, 1383–1390 CrossRef.
- F. R. Wagner , unpublished work.
- Z. Malik, A. Grytsiv, H. Michor, G. Rogl, S. Puchegger, H. Müller, M. Kriegisch, E. Bauer, C. Eisenmenger-Sittner and P. Rogl, J. Alloys Compd., 2013, 550, 302–307 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Dr. Franz Weitzer on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1560948–1560950, 1561246 and 1561247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02501g |
| § Present address: Materials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. |
| This journal is © The Royal Society of Chemistry 2017 |