
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyridylpyrazole N^N ligands combined with sulfonyl-functionalised cyclometalating ligands for blue-emitting iridium(III) complexes and solution-processable PhOLEDs†
Helen
Benjamin
a,
Mark A.
Fox
 a,
Andrei S.
Batsanov
a,
Hameed A.
Al-Attar
b,
Chensen
Li
c,
Zhongjie
Ren
c,
Andrew P.
Monkman
b and
Martin R.
Bryce
*a
a,
Andrei S.
Batsanov
a,
Hameed A.
Al-Attar
b,
Chensen
Li
c,
Zhongjie
Ren
c,
Andrew P.
Monkman
b and
Martin R.
Bryce
*a
aDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
bDepartment of Physics, Durham University, Durham DH1 3LE, UK
cState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China
First published on 4th August 2017
Abstract
A series of blue iridium(III) complexes (12–15) comprising sulfonyl-functionalised phenylpyridyl cyclometalating ligands and pyridylpyrazole N^N ligands are reported, with an X-ray crystal structure obtained for 12. The complexes are highly emissive with photoluminescence quantum yields of 0.52–0.70 in dichloromethane solutions: two of the complexes (12 and 14) show emissions at λPLmax 457 nm which is considerably blue-shifted compared to the archetypal blue emitter FIrpic (λmax 468 nm). The short excited state lifetimes (1.8–3.3 μs) and spectral profiles are consistent with phosphorescence from a mixture of ligand-centred and MLCT excited states. Density functional (DFT) and time dependent DFT (TD-DFT) calculations are in agreement with the electrochemical properties and the blue phosphorescence of the complexes. The additional mesityl substituent on the pyridylpyrazole ligand of 12 and 13 enhances the solubility of the complexes facilitating thin film formation by solution processing. Phosphorescent organic light-emitting diodes (PhOLEDs) have been fabricated using 12 or 13 in a solution-processed single-emitting layer using either poly(vinylcarbazole) (PVK) or 1,3-bis(N-carbazolyl)benzene (mCP) as host. The most blue-shifted electroluminescence (λELmax 460 nm, CIEx,y 0.15, 0.21) is obtained for an OLED containing complex 12 and mCP, with a brightness of 5400 cd m−2 at 10 V which is high for PhOLEDs with similar blue CIE coordinates using a solution-processed emitter layer.
Introduction
The varied applications of luminescent transition metal complexes1 include biological labelling probes, ion sensors, water splitting, solar cells and emitters for phosphorescent organic light-emitting diodes (OLEDs)2–8 and for solid-state lighting.9,10 Cyclometalated iridium(III) complexes are especially prominent in the OLED field due to their ability to harvest both singlet and triplet excitons, which can result in internal quantum efficiencies approaching 100%.11–13 Many complexes incorporate cyclometalated 2-phenylpyridine (ppy) ligands where the highest occupied molecular orbital (HOMO) is localised mostly on the iridium atom and the phenyl ring, and the lowest unoccupied MO (LUMO) is on the pyridyl ring. This spacial separation of HOMO and LUMO means that the emission wavelength of the complexes, which occurs from a mixture of triplet metal-to-ligand charge-transfer (3MLCT) states and π–π* transitions of the ligands,14,15 can be tuned with precision by the presence of electron-withdrawing or electron-donating substituents at specific sites on the ppy framework. Additional attractive features are that the complexes generally emit with high quantum yields and relatively short excited state lifetimes on the microsecond timescale, and they possess high photochemical and chemical stability.11–13Blue emitting iridium complexes are important current targets.16,17 Four main approaches are known to be effective in shifting the emission of iridium complexes towards the blue.18 These are: (1) Lowering the HOMO with respect to the LUMO by introducing electron-withdrawing groups, such as fluorine19 sulfonyl,20 phosphoryl,20 perfluoroalkyl,21 perfluoroalkyl carbonyl,22 and cyano13 substituents at specific positions on the phenyl ring. (2) Raising the LUMO by either adding electron-donating groups such as methoxy to the pyridyl ring,23,24 or replacing the pyridyl ring with another heterocycle with higher LUMO energy.25 (3) Using electron-withdrawing N^N ligands, e.g. pyridyl-azole, to modulate the electron density around the metal centre, thereby affecting the HOMO energy.26–28 (4) Using strong σ-donor ligands, e.g. carbenes, to increase the crystal field splitting, thereby widening the HOMO–LUMO gap and shifting non-emissive d–d* metal-centred (MC) states to higher energies.19
The present study combines a pyridylpyrazole-based N^N ligand with sulfonyl-containing cyclometalating ligands29 to obtain the new heteroleptic complexes 12–15 with emission blue-shifted compared to the archetypal blue emitter, FIrpic,16 and the analogous picolinate-complexes 1 and 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 (Fig. 1). Pyridylpyrazole ligands blue-shift the emission due to their high acidity, reducing the electron density around the Ir centre and hence reducing the HOMO level relative to the LUMO.30 They have also been exploited as the chromophoric ligands in N-heterocyclic carbene complexes31 and in conjunction with other ligands.32–34 The strongly electron-withdrawing CF3 group on the pyrazole ring should further blue shift the emission.35 The p-tolylsulfonyl and methylsulfonyl groups (complexes 1 and 2) both blue shift the emission to the same extent,29 and methoxy substituents can lead to a blue shift depending on their position on the ppy ligand.36,37 Reducing the number of aromatic fluorine substituents, e.g. by selective replacement with methoxy, can maintain the blue colour and reduce chemical degradation of a complex during device operation.38 Complexes 12–15 incorporate a mesityl substituent at C4 of the ppy ligand; it is known that a mesityl group at this position enhances the quantum yields of some complexes by reducing triplet–triplet quenching without affecting blue colour purity.39–42 We also report the new mesityl substituted pyridylpyrazole ligand 9 which achieves the desired aims of increasing the solubility of the complexes 12 and 13, in comparison with 14 and 15, thereby facilitating film formation by solution processes without affecting the emission colour.
29 (Fig. 1). Pyridylpyrazole ligands blue-shift the emission due to their high acidity, reducing the electron density around the Ir centre and hence reducing the HOMO level relative to the LUMO.30 They have also been exploited as the chromophoric ligands in N-heterocyclic carbene complexes31 and in conjunction with other ligands.32–34 The strongly electron-withdrawing CF3 group on the pyrazole ring should further blue shift the emission.35 The p-tolylsulfonyl and methylsulfonyl groups (complexes 1 and 2) both blue shift the emission to the same extent,29 and methoxy substituents can lead to a blue shift depending on their position on the ppy ligand.36,37 Reducing the number of aromatic fluorine substituents, e.g. by selective replacement with methoxy, can maintain the blue colour and reduce chemical degradation of a complex during device operation.38 Complexes 12–15 incorporate a mesityl substituent at C4 of the ppy ligand; it is known that a mesityl group at this position enhances the quantum yields of some complexes by reducing triplet–triplet quenching without affecting blue colour purity.39–42 We also report the new mesityl substituted pyridylpyrazole ligand 9 which achieves the desired aims of increasing the solubility of the complexes 12 and 13, in comparison with 14 and 15, thereby facilitating film formation by solution processes without affecting the emission colour.
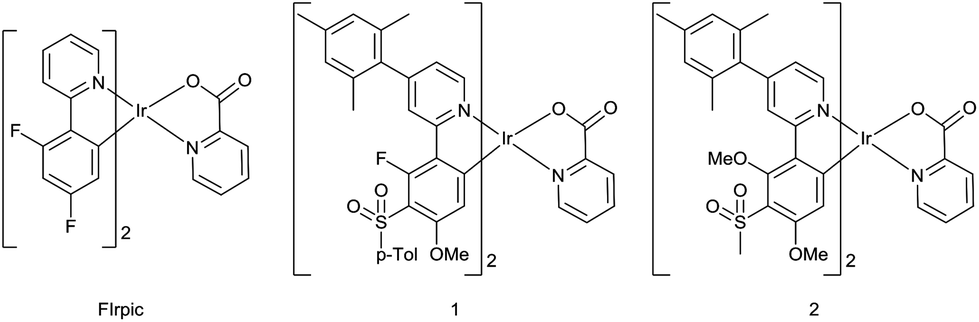 | ||
| Fig. 1 Previously reported picolinate complexes. | ||
Synthesis and characterisation
The route to ligand 9 is shown in Scheme 1. 2-Acetyl-4-chloropyridine 6 was synthesised by reaction between 4-nitro-α-picoline-N-oxide 3 and acetyl chloride to give 443 in 23% yield. An intermediate oxime has been proposed in this step.43 The N-oxide was reduced using PCl3 to produce 543 in 73% yield, followed by reaction with methyl magnesium chloride to give 6 in 92% yield.44 The mesityl group was then introduced via a Suzuki coupling of 6 with 2,4,6-trimethylphenylboronic acid to give 7 in 52% yield. The final steps, described in the literature for other analogues,45 involved a crossed Claisen condensation reaction with ethyl trifluoroacetate to give a presumed diketone intermediate 8, followed by a ring closing condensation reaction with hydrazine hydrate to give 9 in 20% yield.
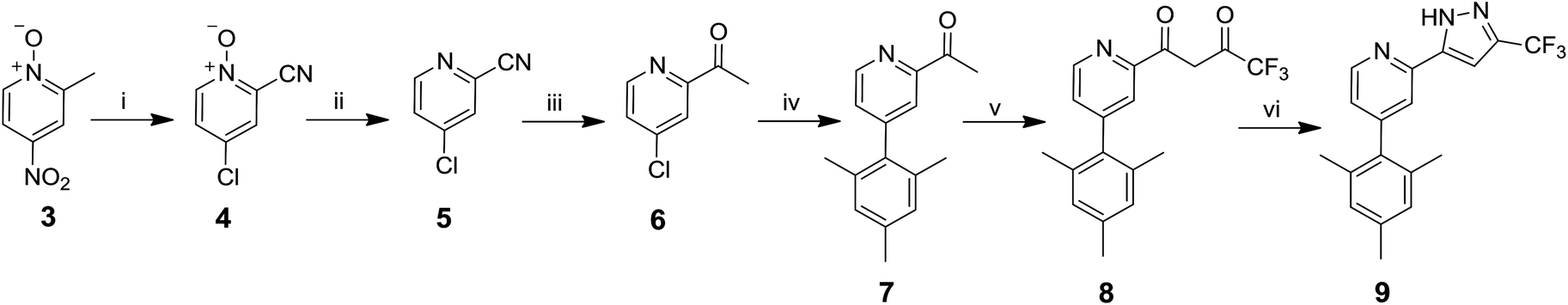 | ||
| Scheme 1 Synthesis of ligand 9. Reagents and conditions: i, CH3COCl, 60 °C; ii, PCl3, CHCl3, reflux; iii, (1) CH3MgCl, ether, 0 °C, (2) dilute HCl; iv, Pd(OAc)2, PPh3, 2,4,6-trimethylphenylboronic acid, Na2CO3, DME/water, reflux; v, KOtBu, F3CCO2Et, THF, reflux; vi, N2H4·H2O, ethanol, reflux. | ||
The desired complexes 12 and 13 were then synthesised under standard conditions;46,47 namely, reaction of the ligand 10 with IrCl3·3H2O and ligand 1129 with [Ir(COD)Cl]2 in 2-ethoxyethanol to give a species presumed to be the bridged μ-dichloro diiridium dimer [Ir(L)2Cl]2. This intermediate species was then reacted in situ with ligand 9 to give the desired complexes 12 and 13 (Scheme 2). The attempted synthesis of 13 using IrCl3·3H2O failed to yield any product and the ligand 11 could not be recovered. Difficulties in the cyclometalation of ligands containing methoxy substituents using IrCl3·3H2O have been reported previously.29,36 Complexes 14 and 15 were synthesised from 10 and 11 by analogous reactions with 3-trifluoromethyl-5-(2-pyridyl)pyrazole ligand 16.30 Thermal gravimetric analysis (TGA) shows that all the complexes possess good thermal stability (Table S5 in ESI†), suggesting the complexes should be stable under device operation.
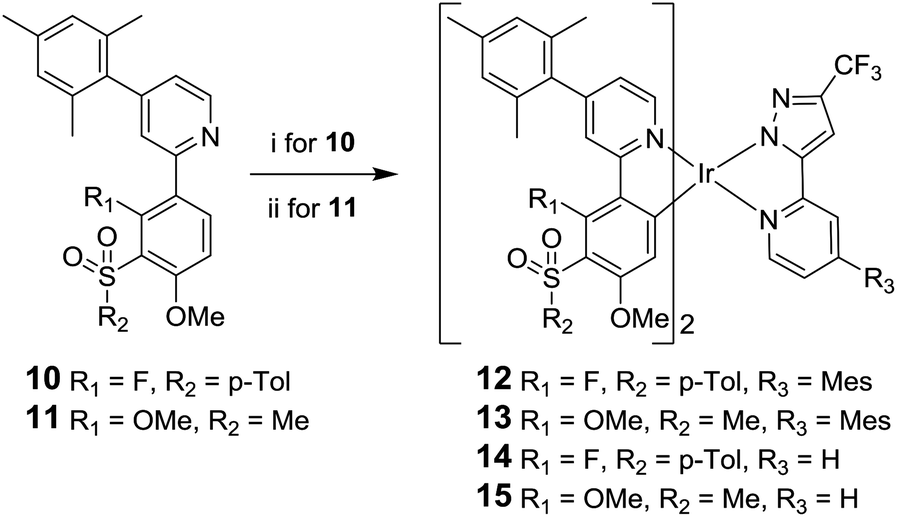 | ||
| Scheme 2 Synthesis of complexes 12–15. Reagents and conditions: i, IrCl3·3H2O, 2-ethoxyethanol, 130 °C, followed by 9 (for 12 and 13) or 3-trifluoromethyl-5-(2-pyridyl)pyrazole 16 (for 14 and 15) DCM/ethanol, 55 °C; ii, [Ir(COD)Cl]2, 2-ethoxyethanol, 130 °C, followed by 9 or 16, DCM/ethanol, 55 °C. | ||
X-Ray crystal structure
The single crystal X-ray structure of the highly-functionalised complex 12 (Fig. 2) shows a distorted octahedral coordination of the Ir atom, similar to that in its analogues.32 The two Ir–C bonds are in cis positions to one another and trans to the Ir–N bonds of the N^N ligand, which are, therefore, elongated compared to the mutually trans bonds Ir–N(4) and Ir–N(5). The mesityl groups are near-perpendicular to the corresponding pyridyl rings. The crystal packing of 12 is dominated by edge-to-face contacts between arene groups, without π–π stacking. | ||
| Fig. 2 X-ray molecular structure of 12. H atoms and the disorder of CF3 group are omitted; thermal ellipsoids are drawn at the 30% probability level. Bond distances (Å): Ir–N(1) 2.102(6), Ir–N(3) 2.142(8), Ir–N(4) 2.040(6), Ir–N(5) 2.057(6), Ir–C(10) 2.006(7) and Ir–C(22) 1.991(9). Interplanar angles (°): i/ii 7, ii/iii 90, iv/v 7, iv/vi 74, vii/viii 4, vii/ix 89. | ||
Photophysical and electrochemical properties
The absorption and emission spectra of complexes 12–15 (Fig. 3) are consistent with pyridylpyrazole subtly lowering the energy of the HOMO. All the complexes show strong absorption bands in the 250–325 nm region, assigned to the π–π* transitions on the ligands.16,18 Absorption bands with lower extinction coefficients in the 350–450 nm range are ascribed to singlet and triplet metal-to-ligand charge-transfer (1MLCT and 3MLCT) states.48,49 Here, we see differences between the picolinate complexes 1 and 2,29 and complexes 12–15; the 1MLCT bands move to higher energies on replacing the picolinate ligands with ligands 9 or 16, suggesting the pyridylpyrazole has indeed decreased the energy of the metal d-orbitals. The emission spectra of the complexes are shown in Fig. 3b. Complexes 12–15 emit in the blue region, and display a similar emission profile to that of FIrpic and the analogous picolinate complexes 1 and 2. A notable feature is that the λmax value for 12 and 13 is blue-shifted by 7 and 8 nm relative to their analogous picolinate complexes 1 and 2, respectively. This represents a blue-shift of 11 nm and 2 nm relative to that of the benchmark sky-blue emitter FIrpic.16 The impressive 11 nm shift for complex 12 is primarily a consequence of the combined benefits of the sulfone substituents and the N^N ligand. The UV-Vis absorption and emission of complexes 14 and 15 are similar to those of complexes 12 and 13, indicating the introduction of the mesityl substituent on the N^N ligand does not impact the emission colour. | ||
| Fig. 3 (a) UV-Vis absorption spectra of the complexes in DCM [<10−5 M]. Inset shows an expansion of the 350–475 nm region. (b) Emission spectra in deaerated DCM, λex = 380 nm. | ||
The photoluminescence quantum yield (PLQY) and lifetime data are listed in Table 1. The PLQYs for complexes 12–15 are comparable to their pic analogues 1 and 2, with lifetimes and spectral profiles consistent with phosphorescence from a mixture of ligand-centred and MLCT excited states.18,49
| Complex | λ absmax (ε)a/nm (×103 M−1 cm−1) |
λ
emmaxb/nm |
PLQY/ΦPLa,c |
τ
Pa,d/μs |
k r/105 s−1 | k nr/105 s−1 |
|---|---|---|---|---|---|---|
| a Data obtained in DCM solution at 20 °C. b Data obtained in degassed DCM solution with λex = 380 nm. c Measured relative to Ir(ppy)3ΦPL = 0.46 in degassed DCM at 20 °C; estimated error ±5%. d Estimated error ±5%. | ||||||
| FIrpic | 277 (50.1), 301 (34.2), 304 (32.6), 337 (13.8, sh), 357 (8.9, sh), 400 (6.2), 454 (0.8) | 468, 496, 531 (sh) | 0.67 | 1.72 | 3.90 | 1.92 |
|
129 |
258 (s sh, 108.5), 271 (128.6), 297 (123.4), 307 (sh, 112.5), 339 (s sh, 29.2), 379 (15.7), 450 (1.0) | 464, 490, 524 (sh) | 0.67 | 1.78 | 3.76 | 1.85 |
|
229 |
275 (104.4), 291 (101.9), 324 (sh, 47.8), 381 (14.5), 405 (sh, 7.9), 456 (0.9) | 474, 497, 536 (sh) | 0.58 | 2.13 | 2.72 | 1.97 |
| 12 | 274 (144.4), 293 (136.7), 370 (19.4), 419 (sh, 2.7), 447 (0.7) | 457, 486, 521 (sh) | 0.62 | 2.30 | 2.70 | 1.65 |
| 13 | 255 (sh, 100.4), 277 (125.8), 292 (133.5), 321 (59.6) (sh), 375 (16.3), 406 (sh, 5.1), 454 (0.8) | 466, 494, 531 (sh) | 0.52 | 3.13 | 1.66 | 1.53 |
| 14 | 274 (171.0), 291 (161), 374 (17.9), 418 (2.1), 450 (0.5) | 457, 486, 522 (sh) | 0.70 | 2.74 | 2.55 | 1.09 |
| 15 | 276 (129.3), 295 (122.75), 325 (50.8), 371 (18.2), 411 (3.2), 454 (0.7) | 467, 494, 531 (sh) | 0.64 | 3.30 | 1.94 | 1.09 |
The electrochemical behaviour of complexes 12–15 was investigated by cyclic voltammetry (CV) in a 0.1 M NnBu4PF6 acetonitrile solution, and compared to FIrpic and picolinate analogues 1 and 2.29 The voltammograms of FIrpic, 12–15 are shown in Fig. 4 and S13,† and the key parameters are listed in Table 2. Whereas the picolinate complexes (FIrpic, 1 and 2) show a single quasi-reversible oxidation wave, complexes 12–15 show two oxidation waves. In the case of 12 and 13 the two waves are irreversible, whereas for 14 and 15 the first oxidation wave is quasi-reversible.
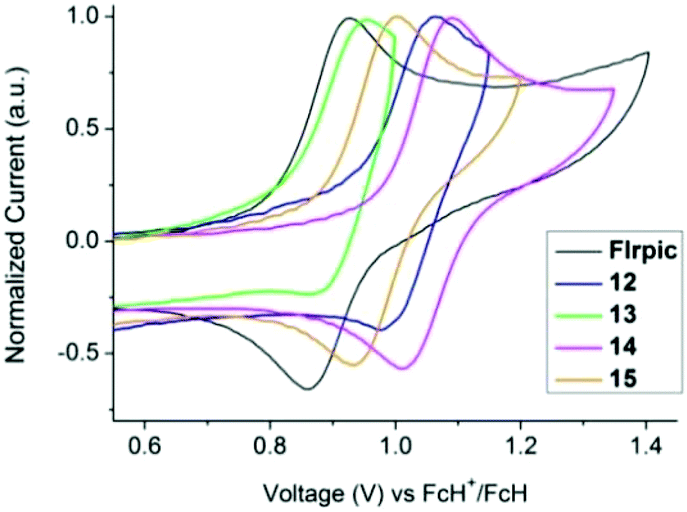 | ||
| Fig. 4 Cyclic voltammograms of the iridium complexes. | ||
| Complex |
E
ox11/2a/V |
E
ox21/2a/V |
Obs E(HOMO)c/V | Complex | Calc. HOMO/eV | Calc. LUMO/eV | Calc. HLGd/eV |
|---|---|---|---|---|---|---|---|
| a Redox data were obtained in 0.1 M NnBu4PF6 acetonitrile solutions and are reported vs. FcH/FcH+.50 b Not reversible. c E(HOMO) = −4.8 − Eox11/2 with respect to the FcH/FcH+ couple at 4.8 V. d HLG = HOMO–LUMO energy gap. | |||||||
| FIrpic29 | 0.89 | — | −5.69 | FIrpic′ | −5.49 | −1.87 | 3.62 |
|
129 |
1.01 | — | −5.81 | 1′ | −5.64 | −1.94 | 3.70 |
|
229 |
0.89 | — | −5.69 | 2′ | −5.53 | −1.89 | 3.64 |
| 12 | 1.02b | 1.28b | −5.82 | 12′ | −5.74 | −1.83 | 3.91 |
| 13 | 0.91b | 1.17b | −5.71 | 13′ | −5.62 | −1.77 | 3.85 |
| 14 | 1.07 | 1.41b | −5.87 | 14′ | −5.77 | −1.89 | 3.88 |
| 15 | 0.97 | 1.28b | −5.77 | 15′ | −5.64 | −1.84 | 3.80 |
As complexes 12–15 contain a pyridylpyrazole-based ligand it is assumed the second oxidation occurs either on this ligand or is due to an interaction of this ligand with the metal. Exchanging the picolinate ancillary ligand for pyridylpyrazole results in an increase in the first oxidation potential by only 10–20 mV (1vs. 12, 2vs. 13), consistent with pyridylpyrazole subtly lowering the energy of the HOMO. The presence of the mesityl group on the N^N ligand (complexes 12 and 13) decreases the oxidation potentials by ca. 50 mV compared to the unsubstituted complexes 14 and 15. No reduction waves were observed within the solvent window to −2.0 V. This is consistent with the calculated large HOMO–LUMO gap (Table 2).
Computations
Electronic structure calculations were carried out on the four new iridium complexes 12–15 to explore the frontier orbitals (Fig. 5 and Tables S1–S4†) in order to support the electrochemical and photophysical observations. The full geometries were optimised at B3LYP/LANL2DZ:3-21G* to compare directly with computed data of FIrpic, 1 and 2 reported earlier.29 The optimised geometries are denoted as 12′–15′ to identify them as computed models and distinguish the predicted data from experimental data. Comparison between optimised (12′) and X-ray determined (12) geometry reveals good agreement with differences in bond lengths below 0.04 Å.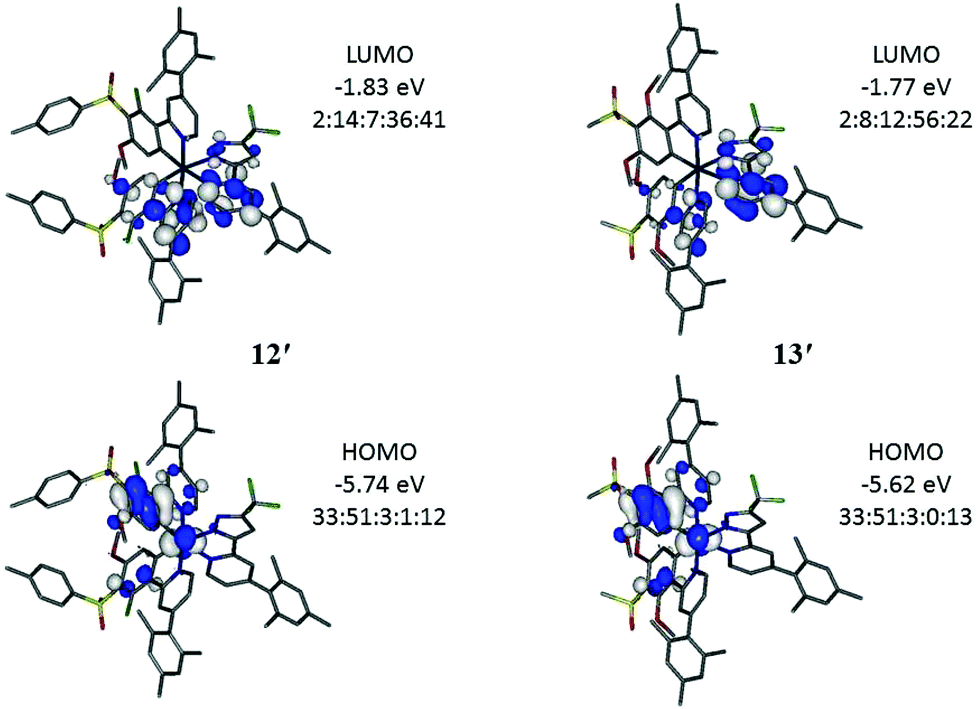 | ||
| Fig. 5 Frontier molecular orbitals for 12′ and 13′. All contours are plotted at ±0.04 (e per bohr3)1/2. Ir:phenyl:pyrazolyl:pyridyl(pypz):pyridyl(ppy) % orbital contribution ratio are listed for each orbital. | ||
Complexes 12′–15′ have HOMOs similar to FIrpic with the HOMO on iridium and phenyl (ppy) (Fig. 5). The lower HOMO energies on the complexes 12′–15′, compared to FIrpic, reflect the electron-withdrawing properties of the sulfone groups. Frontier orbital energies for all complexes are listed in Table 2 for direct comparison with the observed CV data. The trends in observed and computed HOMO energies are in very good agreement. The iridium contributions (32–33%, Table 3) in the HOMOs of 12′–15′ are lower than the corresponding contribution in FIrpic′ (44%) suggesting increased ligand contributions in the MLCT transitions involved in the emissions of 12′–15′ with respect to MLCT transitions for FIrpic′.
| Complex | % Ir HOMO | S0 ← T1a/nm | Complex | Observed λemmaxb/nm |
|---|---|---|---|---|
| a Values from TD-DFT data on S0 optimised geometries with scaling energy factor of 0.945 based on DCM at 298 K. b Observed highest energy band from emission spectra (Table 1). | ||||
| FIrpic′29 | 44 | 471 | FIrpic | 471 |
|
1′29 |
39 | 467 | 1 | 464 |
|
2′29 |
40 | 473 | 2 | 474 |
| 12′ | 33 | 455 | 12 | 457 |
| 13′ | 33 | 459 | 13 | 466 |
| 14′ | 32 | 454 | 14 | 457 |
| 15′ | 32 | 459 | 15 | 467 |
The LUMOs in 12′–15′ have substantial contributions from the pyridyl groups of the ppy and the pyridylpyrazole ligands which raise their orbital energies compared to FIrpic′ where the LUMO is entirely located on the pyridyl group of the ppy ligands. The pyridylpyrazole ligands are, therefore, not ancillary. Consequently, the HOMO–LUMO energy gaps (HLGs) in complexes 12′–15′ are larger than the HLG in FIrpic′ (Table 2) which is consistent with the observed blue-shifted emissions of 12–15 compared to FIrpic. The LUMOs in 13′ and 15′ contain more pyridylpyrazole character than ppy character (Fig. 5) which is explained by the increased number of electron-donating methoxy groups present in the ppy ligands of these complexes.
TD-DFT computations were carried out on the S0 optimised geometries of 12′–15′ to predict emission wavelengths of these complexes. The initial excitation is assumed to give the lowest energy singlet excited state S1 which in the presence of the iridium centre results in intersystem crossing (ISC) to form the triplet excited state T1 and phosphorescence is observed from the S0 ← T1 process. The small Stokes shifts observed for these complexes suggest that the T1 geometry is similar to the corresponding S0 geometry. The reverse process S0 ← T1 is thus considered to have the same nature as the computed S0 → T1 process with the predicted emission wavelength adjusted to take into account the Stokes shift. Table 3 shows there is close agreement between the predicted S0 ← T1 wavelengths and the observed emission wavelengths for all the complexes.
Phosphorescent organic light-emitting devices
PhOLEDs containing complex 12, 13 or 2 as the dopant complex were fabricated to compare directly with our reference devices containing FIrpic and 1. The initial standard device architecture comprised of a simple single-emissive-layer which was a blend of poly(vinylcarbazole) (PVK) as the host material, OXD-7 (an electron-transporting material) and the Ir complex to give the architecture: ITO/PEDOT:PSS (45 nm)/PVK:OXD-7 (30 wt%):Ir complex (15 wt%) (60 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm). The emissive layer was spin-coated from chlorobenzene solution to prevent possible degradation of the complexes which is known to occur during thermal evaporation of complexes which have fluorinated ligands.38,51–54 TPBi was used as an additional electron transporting layer adjacent to the cathode to improve charge balance and to ensure that excitons are confined in the emitter layer.55 The efficiency and luminance data of the devices are summarised in Table 4 and the electroluminescence (EL) spectra are shown in Fig. 6a. The EL and PL spectra are similar for all the complexes due to efficient exciton confinement on the emissive molecules. The λELmax of complex 12 is significantly bluer (by 6 nm) than the corresponding pic complex 1. However, as a trade-off, 12 has lower brightness (panel e), lower EQE (panel c), and lower current and power efficiencies (panels d and f). Based on the electrochemical data, the HOMO of 12 is lower in energy than 1, which could be partially responsible for the reduced performance. Complex 13, however, performed better than 2, with comparable brightness and turn-on voltage (panel e), and with superior EQE, current and power efficiencies. The λELmax of 13 is also blue-shifted relative to 2; however, this is offset by a broader emission resulting in similar CIE coordinates for both complexes. The additional mesityl substituent on the N^N ligand of 12 and 13 enhances the solubility of the complexes facilitating thin film formation by solution processing. | ||
| Fig. 6 Electroluminescent device data for complexes FIrpic, 1, 2, 12 and 13 in PVK host and for 12 in mCP host. | ||
| Complex | λ ELmax/nm | Brightness (max, and at 10 V)/cd m−2 | Turn-on voltagec/V | EQEmax/% | Current efficiency/cd A−1 | Power efficiency/lm W−1 | CIE coordinates/(x,y)d |
|---|---|---|---|---|---|---|---|
| a In PVK host. b In mCP host. c Measured at a brightness of 10 cd m−2. d Measured at 12 V. | |||||||
| FIrpica | 475 | 7340, 2380 | 6.2 | 5.4 | 12.2 | 5.1 | (0.19, 0.38) |
| 1 | 470 | 2072, 175 | 8.5 | 3.5 | 6.1 | 1.9 | (0.16, 0.30) |
| 2 | 478 | 516, 160 | 8.5 | 1.2 | 2.6 | 0.9 | (0.22, 0.40) |
| 12 | 464 | 1066, 55 | 9.5 | 2.3 | 4.1 | 1.2 | (0.17, 0.28) |
| 13 | 473 | 456, 195 | 7.9 | 1.7 | 3.9 | 1.4 | (0.23, 0.40) |
| 12 | 460 | 5428, 5400 | 6.2 | 6.3 | 6.9 | 3.5 | (0.15, 0.21) |
We further explored the bluest complex 12 using mCP as a wide energy gap host which is more appropriate for a deep blue emitter in the architecture: ITO/TAPC (45 nm)/mCP:Ir complex 12 (15 wt%) (45 nm)/Tm3PyBP (5 nm)/TPBi (30 nm)/LiF (1 nm)/Al (100 nm). TAPC served as a hole-transporting layer and Tm3PyBP serves as an additional hole/exciton-blocking layer to optimise the device structure.56 Compared to the 12:PVK device, the 12:mCP device displayed a significantly lower turn-on voltage and further blue shifted electroluminescence to λELmax 460 nm, CIEx,y (0.15, 0.21) with an increased maximum EQE of 6.3% and increased brightness at 10 V of 5400 cd m−2 which is high compared to reported PhOLEDs with similar blue CIE coordinates using a solution-processed emitter layer.57
Conclusions
To summarise, replacing the picolinate ancillary ligand of the heteroleptic iridium complexes 1 and 2 with a pyridylpyrazole-based ligand affords the new complexes 12–15 with blue shifted emission. This is ascribed to a reduction of electron density around the metal centre which results in a lowering of the energy of the HOMO of the complexes, as observed in the solution electrochemical data. (TD-)DFT calculations are in excellent agreement with the observed photophysical and electrochemical properties of the complexes. Blue-emitting PhOLEDs have been fabricated with a solution-processed emissive layer structure. Notably, devices of complex 12 possess λELmax 460 nm and CIEx,y (0.15, 0.21) with brightness at 10 V of 5400 cd m−2. This study demonstrates that the rational design of new complexes with specific functionalities on the cyclometalating and the N^N ligands is an effective route to new efficient blue emitters and PhOLEDs using relatively simple device architectures.Experimental section
Materials, synthesis and characterisation
All commercially available chemicals were used without further purification. Reactions requiring an inert atmosphere were performed under a blanket of argon gas, which was dried over a phosphorus pentoxide column. Anhydrous solvents were dried through an HPLC column on an Innovative Technology Inc. solvent purification system. Column chromatography was performed using 40–60 μm mesh silica gel. Analytical TLC was performed on plates pre-coated with silica gel (Merck, silica gel 60F254) and visualised using UV light (254, 315, 365 nm). NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. Chemical shifts are referenced to tetramethylsilane [TMS, Si(CH3)4] at 0.00 ppm. Melting points were determined in open-ended capillaries using a Stuart Scientific SMP3 melting point apparatus at a ramping rate of 1 °C min−1 and were recorded to the nearest 0.1 °C. ESI and MALDI mass spectra were recorded on a Thermo-Finnigan LTQ FT (7.0 T magnet) spectrometer. ASAP mass spectra were recorded on a Waters Xevo QTOF spectrometer. GCMS spectra were recorded on a Thermo-Finnigan Trace GCMS (EI and CI ion sources). Elemental analyses were obtained on an Exeter Analytical Inc. CE-440 elemental analyzer.Solution electrochemistry and photophysics
Cyclic voltammetry experiments were recorded using a BAS CV50 W electrochemical analyzer fitted with a three-electrode system consisting of a Pt disk (∅ = 1.8 mm) as the working electrode, a Pt wire as an auxiliary electrode and an Ag/AgNO3 (0.1 M [NEt4][ClO4] in CH3CN) system as the reference electrode. Experiments were conducted in dry acetonitrile solution with NnBu4PF6 (0.1 M) as the supporting electrolyte at a scan rate of 100 mV s−1. The reference electrode was assumed to be stable and was referenced externally to ferrocene (Cp2Fe) and decamethylferrocene (Cp*2Fe) which displayed potentials (E1/2) of −0.41 V and +0.10 V, respectively, versus Ag/AgNO3 under these conditions. It was not possible to use Cp2Fe or Cp*2Fe as an internal reference because their addition to the solutions of the complexes resulted in a distortion of the redox waves of the complexes.Solution state photophysical data were obtained using freshly prepared solutions of the complexes in DCM. Emission and lifetime measurements used thoroughly degassed solutions achieved by three freeze–pump–thaw cycles, and quartz cuvettes with a path length of 1 cm. The solutions had absorbance below 0.10 to minimise inner filter effects. UV-vis absorption measurements were recorded using a Unicam UV2-100 spectrometer operated with Unicam Vision (ver. 3.50) software. Baseline correction was achieved by reference to pure solvent in the same cuvette. Absorption measurements were obtained using quartz cuvettes with a path length of 2 cm. Excitation and emission spectra were recorded on a Jobin–Yvon–Horiba SpexFluoromax 3 Spectrometer. Solutions of the complexes in degassed DCM [<10−5 M] were used for decay measurements. Samples were excited with a pulsed nitrogen laser emitting at 337.1 nm. Emission was focused onto a spectrograph and detected on a sensitive gated iCCD camera (Stanford Computer Optics) with sub-nanosecond resolution. Solution PLQYs were recorded in degassed solvent, and determined using the relative method, with Ir(ppy)3 (ΦPL = 0.46 in degassed dichloromethane, determined in-house vs. ΦPL = 0.546 in 0.5 M H2SO4) as the reference. The PLQYs were calculated according to the following equation:

Thermal analyses
Thermogravimetric analysis (TGA) was performed in a nitrogen atmosphere using a PerkinElmer Pyris 1 TGA instrument. The complexes were heated at a rate of 10 °C min−1 from room temperature up to 600 °C.Computational studies
All calculations were carried out with the Gaussian 09 package.58 All optimised S0 geometries for 12′–15′ were carried out using B3LYP59 with the pseudopotential (LANL2DZ)60 for iridium and 3-21G* basis set for all other atoms.61 This model chemistry was selected for direct comparison with computed data on FIrpic′, 1′ and 2′.29 Electronic structure and TD-DFT calculations were also from the optimised geometries at B3LYP/LANL2DZ:3-21G*. The MO diagrams and orbital contributions were generated with the aid of Gabedit62 and GaussSum63 packages, respectively.Phosphorescent organic light-emitting devices (PhOLEDs)
PhOLEDs with poly(vinylcarbazole) (PVK) host were fabricated on indium tin oxide (ITO)-coated glass substrates of thickness 125 nm and possessing a sheet resistance of 15 Ω per square. Poly(3,4-ethylenedioxythiophene) doped with high work function hole injection layer poly(styrenesulfonic acid) (PEDOT:PSS) (HIL1.3) from CLEVIOS was spin-coated at 2500 rpm for 60 s to produce a ∼50 nm thick hole-injecting/transporting layer (HTL). The PEDOT:PSS layer was annealed at ca. 200 °C for 5 min to remove any residual water. A chlorobenzene solution of 25 mg ml−1 of poly(vinylcarbazole) (PVK) (Mw = 90000) was doped with 30% w/w of (1,3-phenylene)bis[5-(4-tert-butylphenyl)-1,3,4-oxadiazole] (OXD-7). Blended devices were made by mixing 15% w/w of the Ir complexes. The prepared mixtures were filtered with a 0.45 μm pore filter and spin-coated at 2500 rpm for 1 min on the top of the PEDOT:PSS layer and baked for 10 min at 120 °C. Each sample was shadow masked to produce four identical devices of area 4 × 5 mm; the samples were then introduced into a nitrogen glove box, where 30 nm of 2,2′,2′′-(1,3,5-benzenetriyl)tris-[1-phenyl-1H-benzimidazole] (TPBi) was evaporated as an electron injection/hole blocking layer at a rate of ∼1 A s−1 under vacuum at a pressure of ca. 1 × 10−6 Torr, followed by 0.8 nm LiF and a 100 nm capping layer of aluminium under the same evaporation conditions. Therefore, the device configuration for all complexes was: ITO/PEDOT:PSS (50 nm)/PVK:OXD-7 (35%):Ir-complex (15%)/TPBi (30 nm)/LiF (0.8 nm)/Al (100 nm). All samples were encapsulated inside a glove box using DELO UV cured epoxy (KATIOBOND) and capped with a 1.2 × 1.2 cm microscope glass slide then exposed to UV light for 3 min. Current–voltage data, device efficiency, brightness and electroluminescence spectra were measured in a calibrated Labsphere LMS-100 integrating sphere. A home written NI LabVIEW programme was used to control an Agilent 6632B DC power supply, and the emission properties of the device were measured using an Ocean Optics USB4000 CCD fibre optic spectrometer. The thicknesses of the various layers in the device were measured with a J A Woolam VASE Ellipsometer using thin films which had been spin-coated on Si/SiO2 substrates under the same conditions as the device films.
Devices with 1,3-bis(N-carbazolyl)benzene (mCP) host were fabricated as follows. TAPC (1,1-bis{4-[N,N-di(p-tolyl)amino]phenyl}cyclohexane), TPBi, and Tm3PyPB (1,3,5-tris(3-pyridyl-3-phenyl)benzene) were obtained from commercial sources. Indium tin oxide (ITO)-coated glass with a sheet resistance of 10 Ω per square was used as the substrate. Before device fabrication, the ITO-coated glass substrate was precleaned and exposed to UV-ozone for 15 min. PEDOT:PSS was then spin-coated onto the clean ITO substrate as a hole-injection layer. Next a mixture of complex 12 and mCP in chlorobenzene was spin-coated (10 mg mL−1; 3000 rpm) to form a ca. 45 nm thick emissive layer and annealed at 80 °C for 30 min to remove the residual solvent. Finally, a 5 nm-thick hole/exciton blocking layer of Tm3PyPB was vacuum deposited followed by a 30 nm-thick electron-transporting layer (ETL) of TPBi, and a cathode composed of a 1 nm-thick layer of LiF and aluminum (100 nm) was sequentially deposited through shadow masking at a pressure of 10−6 Torr. The OLED devices had a pixel sise of 4 mm × 4 mm. All measurements were performed at room temperature under ambient conditions.
Synthesis of ligands and complexes
44 (1.5 g, 9.68 mmol), 2,4,6-trimethylphenylboronic acid (1.75 g, 10.67 mmol) and PPh3 (657 mg, 2.50 mmol) were dissolved in DME (70 mL) and the solution was degassed for 20 min by bubbling with argon. Separately, an aqueous solution of Na2CO3 (4.18 g, 39.43 mmol in 17 mL) was degassed. The solutions were combined and Pd(OAc)2 (130 mg, 0.58 mmol) was added. The mixture was heated to reflux overnight under an atmosphere of argon. The solution was then cooled to RT, and the majority of the solvent was removed in vacuo. DCM and water were added, and the organic layer separated. The aqueous layer was extracted with additional DCM, and the extracts combined and the solvent removed in vacuo. The crude residue was purified by column chromatography (DCM), followed by recrystallisation from hexane to give 1-(4-mesitylpyridin-2-yl)ethanone 7 (1.2 g, 52%); m.pt. 118.9–119.9 °C; δH (400 MHz; CDCl3) 8.74 (1H, dd, J 4.9, 0.8), 7.88 (1H, dd, J 1.7, 0.8), 7.31 (1H, dd, J 4.9, 1.7), 6.95–6.94 (2H, m), 2.79 (3H, s), 2.33 (3H, s), 1.97 (6H, s); δC (101 MHz; CDCl3) 200.07, 153.73, 151.14, 149.18, 138.05, 135.64, 135.08, 128.61, 128.41, 123.06, 26.19, 21.19, 20.72; HRMS (FTMS + ESI): calcd for [C16H17NO + H]+: 240.1388. Found: 240.1388.
:3 EtOAc:DCM v/v) followed by recrystallisation from hexane to give an off-white solid, 4-mesityl-2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 9 (277 mg, 20%); m.pt. 171.1–172.9 °C; δH (400 MHz; CDCl3) 11.95 (1H, br s), 8.70 (1H, dd, J 5.0, 0.9), 7.47 (1H, dd, J 1.5, 0.9), 7.15 (1H, dd, J 5.0, 1.5), 6.96–7.00 (2H, m), 6.91 (1H, s), 2.35 (3H, s), 2.02 (6H, s); δF (376 MHz; CDCl3) −62.34 (3F, s); δC (101 MHz; CDCl3) 151.57, 149.86, 146.91, 144.42 (q, J 38.4), 143.10, 138.08, 135.43, 134.97, 128.52, 125.07, 121.40, 121.14 (q, J 267.9), 101.34, 21.06, 20.57; HRMS (FTMS + ESI): calcd for [C18H16N3F3 + H]+: 332.1375. Found: 332.1379.
29 (169 mg, 0.38 mmol) in 2-ethoxyethanol (5 mL). The solution was heated to 130 °C under an argon atmosphere overnight, during which time a bright yellow precipitate formed, presumed to be the dichloro bridged dimer. The solvent was removed in vacuo and the precipitate was redissolved in DCM/ethanol (20 mL, 3:1 v/v). 4-Mesityl-2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 9 (77 mg, 0.28 mmol) was added and the solution was heated at 60 °C overnight under an argon atmosphere. The solution was cooled and the solvent was removed in vacuo. The residue was purified by column chromatography (EtOAc in DCM, 2.5% increased to 4%) to give a yellow solid, iridium complex 12 (190 mg, 70%); Anal. calc. for C74H65F5IrN5O6S2: C, 60.39; H, 4.45; N, 4.76. Found: C, 60.53; H, 4.71; N, 4.50; δH (400 MHz; CDCl3) 8.12–8.17 (1H, m), 8.03–8.08 (1H, m), 7.89 (2H, d, J 8.2), 7.88 (2H, d, J 8.2), 7.67 (1H, d, J 5.9), 7.64 (1H, d, J 5.9), 7.59 (1H, dd, J 1.7, 0.8), 7.57 (1H, d, J 5.9), 7.27 (4H, t, J 8.7), 7.02–6.89 (8H, s), 6.78 (2H, td, J 6.1, 1.8), 5.79 (1H, s), 5.74 (1H, s), 3.47 (3H, s), 3.44 (3H, s) 2.39 (3H, s), 2.38 (3H, s), 2.34 (3H, s), 2.33 (6H, s), 2.09 (3H, s), 2.01 (3H, s), 1.98 (3H, s), 1.97 (3H, s), 1.86 (6H, s); δF (376 MHz; CDCl3) −59.87 (3F, s), −110.50 (1F, s), −111.36 (1F, s); HRMS (FTMS + ESI): calcd for [C74H65N5F5O6S2191Ir + H]+: 1470.3981. Found: 1470.3995. Single crystals of 12·3CH2Cl2 for X-ray study were grown by layering of hexane over a DCM solution of 12; they instantly decomposed when taken out of the solvent. The X-ray experiment was carried out at T = 120 K on a D8 Venture (Bruker AXS) 3-circle diffractometer equipped with a IμS Mo microsource (Mo-Kα radiation, focussing mirrors, ![[small lambda, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cc.gif) = 0.71073 Å) and a PHOTON 100 CMOS area detector. The structure was solved by direct methods (SHELXS v. 2013/1 program64) and refined by full-matrix least squares using SHELXL v. 2014/7 (ref. 65) and OLEX266 software. Crystal data: C74H67F5IrN5O6S2·3CH2Cl2, M = 1726.40, triclinic, space group P
= 0.71073 Å) and a PHOTON 100 CMOS area detector. The structure was solved by direct methods (SHELXS v. 2013/1 program64) and refined by full-matrix least squares using SHELXL v. 2014/7 (ref. 65) and OLEX266 software. Crystal data: C74H67F5IrN5O6S2·3CH2Cl2, M = 1726.40, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 14.209(1), b = 18.215(1), c = 18.780(1) Å, α = 67.984(2), β = 86.202(2), γ = 69.653(2)°, V = 4213.6(5) Å3, Z = 2, Dc = 1.361 g cm−3, μ = 1.89 mm−1, 57359 reflections with 2θ ≤ 50°, 14940 unique, Rint = 0.048, R = 0.069 [11521 data with I ≥ 2σ(I)], wR(F2) = 0.201 (all data). CCDC 1495066.† The structure contains DCM of crystallisation, of which 1.5 molecules per asymmetric unit were located and the rest (presumably disordered along infinite channels parallel to the x axis) was masked using PLATON SQUEEZE program.67,68 The large atomic displacement parameters, together with practical absence of Bragg diffraction at 2θ > 50°, indicate severe internal strain, probably caused by partial solvent loss.
29 (226 mg, 0.55 mmol) in 2-ethoxyethanol (5 mL). The solution was heated to 130 °C under an argon atmosphere overnight before being cooled to RT. Water was added and a yellow precipitate formed which was filtered, washed with more water and dried. The precipitate, presumed to be the intermediate dichloro bridged dimer, was dissolved in DCM/ethanol (40 mL, 3:1 v/v) and 4-mesityl-2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 9 (90 mg, 0.27 mmol) was added and the solution was heated at 55 °C overnight under an argon atmosphere. The solution was cooled and the solvent was removed in vacuo. The residue was purified by column chromatography (EtOAc in DCM: gradient elution from 0% to 15%) to give a yellow solid, iridium complex 13 (98 mg, 27%); Anal. calc. for C64H63F3IrN5O8S2: C, 57.21; H, 4.73; N, 5.21. Found: C, 57.17; H, 5.07; N, 4.68; δH (400 MHz; CD2Cl2) 8.47 (1H, d, J 1.4), 8.38 (1H, d, J 1.4), 7.80–7.74 (3H, m), 7.65 (1H, dd, J 1.8, 0.8), 6.98 (5H, s), 6.95 (2H, s), 6.91 (1H, dd, J 5.7, 1.8), 6.84 (1H, dd, J 5.9, 1.9), 6.81 (1H, dd, J 5.9, 1.9), 6.03 (1H, s), 6.01 (1H, s), 3.88 (3H, s), 3.85 (3H, s), 3.61 (3H, s), 3.60 (3H, s), 3.22 (3H, s), 3.19 (3H, s), 2.33 (3H, s), 2.32 (3H, s), 2.31 (3H, s), 2.09 (3H, s), 2.06 (3H, s), 2.05 (3H, s), 2.01 (3H, s), 1.88 (3H, s), 1.85 (3H, s); δF (376 MHz; CDCl3) −58.48 (s); HRMS (FTMS + ESI): calcd for [C64H63F3N5O8S2191Ir + H]+: 1342.3754. Found: 1342.3802.
:EtOAc 3:2 v/v) to give an off-white solid, 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (1.31 g, 75%); δH (400 MHz; CDCl3) 11.74 (1H, s), 8.65 (1H, ddd, J 4.9, 1.8, 1.0), 7.82 (1H, td, J 7.8, 1.7), 7.65 (1H, dt, J 7.9, 1.1), 7.33 (1H, ddd, J 7.6, 4.9, 1.1), 6.96 (1H, s); δF (376 MHz; CDCl3) −62.35 (s).
29 (110 mg, 0.23 mmol) and IrCl3·3H2O (37 mg, 0.10 mmol) were combined in 2-ethoxyethanol (5 mL), and the mixture was heated to reflux overnight under a nitrogen atomosphere. The reaction was the cooled to RT, and the solvent removed in vacuo. The residue was redissolved in DCM/EtOH (3:1 v/v, 20 mL) and 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (28 mg, 0.13 mmol) and Na2CO3 (11 mg, 0.10 mmol) were added. The solution was heated to 55 °C for 6 h, before being cooled to RT and the solvent removed in vacuo. The residue was purified by column chromatography (0–3% EtOAc in DCM) to give the product as a yellow solid, iridium complex 14 (109 mg, 77%); Anal. calc. for C65H55F5IrN5O6S2: C, 57.68; H, 4.10; N, 5.17. Found: C, 57.84; H, 4.33; N, 4.92; δH (400 MHz, CDCl3) 8.11 (1H, s), 8.05 (1H, s), 7.95–7.78 (6H, m), 7.70–7.64 (1H, m), 7.56 (1H, d, J 6.0), 7.50–7.43 (2H, m), 7.29 (1H, d, J 0.8), 7.26–7.22 (2H, m), 7.18–7.11 (1H, m), 7.01 (1H, s), 6.93 (4H, d, J 6.4), 6.77 (1H, dd, J 5.9, 1.8), 6.71 (1H, dd, J 6.0, 1.8), 5.72 (1H, s), 5.70 (1H, s), 3.44 (3H, s), 3.41 (3H, s), 2.39 (3H, s), 2.38 (3H, s), 2.33 (6H, s), 1.99 (3H, s), 1.97 (6H, s), 1.93 (3H, s); δF (376 MHz; CDCl3) −59.85 (3F, s), −110.73 (1F, s); −111.48 (1F, s); HRMS (FTMS + ESI): calcd for [C65H55N5O6F5S2191Ir + H]+: 1352.3198. Found: 1352.3193.
29 (198 mg, 0.48 mmol) and [Ir(COD)Cl]2 (79 mg, 0.12 mmol) were combined in 2-ethoxyethanol (5 mL), and the mixture was heated to reflux overnight under a nitrogen atmosphere. The reaction was the cooled to RT, and the solvent removed in vacuo. The residue was redissolved in DCM/EtOH (3:1 v/v, 20 mL) and 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (63 mg, 0.30 mmol) and Na2CO3 (50 mg, 0.47 mmol) were added. The solution was heated to 55 °C for 5 h, before being cooled to RT and the solvent removed in vacuo. The residue was purified by column chromatography (0–10% EtOAc in DCM) to give the product as a yellow solid, iridium complex 15 (28 mg, 56%); Anal. calc. for C55H53F3IrN5O8S2: C, 53.91; H, 4.36; N, 5.72. Found: C, 53.48; H, 4.43; N, 5.55; δH (400 MHz, CDCl3) 8.46 (1H, d, J 1.8), 8.40 (1H, d, J 1.8), 7.88 (2H, m), 7.82 (1H, d, J 5.9), 7.75 (1H, d, J 5.6), 7.52 (1H, d, J 5.9), 7.16 (1H, td, J 6.0, 2.6), 7.04 (1H, s), 6.98 (2H, s), 6.96 (2H, s), 6.85 (1H, dd, J 5.9, 1.9), 6.72 (1H, dd, J 5.9, 1.9), 5.96 (1H, s), 5.89 (1H, s), 3.91 (3H, s), 3.91 (3H, s), 3.60 (3H, s), 3.58 (3H, s), 3.30 (3H, s), 3.27 (3H, s), 2.35 (6H, s), 2.03 (6H, s), 2.01 (3H, s), 1.97 (3H, s); δF (376 MHz; CDCl3) −60.00 (s); HRMS (FTMS + ESI): calcd for [C55H53N5O8F3S2191Ir + H]+: 1224.2972. Found: 1224.2987.
(no. 2), a = 14.209(1), b = 18.215(1), c = 18.780(1) Å, α = 67.984(2), β = 86.202(2), γ = 69.653(2)°, V = 4213.6(5) Å3, Z = 2, Dc = 1.361 g cm−3, μ = 1.89 mm−1, 57359 reflections with 2θ ≤ 50°, 14940 unique, Rint = 0.048, R = 0.069 [11521 data with I ≥ 2σ(I)], wR(F2) = 0.201 (all data). CCDC 1495066.† The structure contains DCM of crystallisation, of which 1.5 molecules per asymmetric unit were located and the rest (presumably disordered along infinite channels parallel to the x axis) was masked using PLATON SQUEEZE program.67,68 The large atomic displacement parameters, together with practical absence of Bragg diffraction at 2θ > 50°, indicate severe internal strain, probably caused by partial solvent loss.
29 (226 mg, 0.55 mmol) in 2-ethoxyethanol (5 mL). The solution was heated to 130 °C under an argon atmosphere overnight before being cooled to RT. Water was added and a yellow precipitate formed which was filtered, washed with more water and dried. The precipitate, presumed to be the intermediate dichloro bridged dimer, was dissolved in DCM/ethanol (40 mL, 3:1 v/v) and 4-mesityl-2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 9 (90 mg, 0.27 mmol) was added and the solution was heated at 55 °C overnight under an argon atmosphere. The solution was cooled and the solvent was removed in vacuo. The residue was purified by column chromatography (EtOAc in DCM: gradient elution from 0% to 15%) to give a yellow solid, iridium complex 13 (98 mg, 27%); Anal. calc. for C64H63F3IrN5O8S2: C, 57.21; H, 4.73; N, 5.21. Found: C, 57.17; H, 5.07; N, 4.68; δH (400 MHz; CD2Cl2) 8.47 (1H, d, J 1.4), 8.38 (1H, d, J 1.4), 7.80–7.74 (3H, m), 7.65 (1H, dd, J 1.8, 0.8), 6.98 (5H, s), 6.95 (2H, s), 6.91 (1H, dd, J 5.7, 1.8), 6.84 (1H, dd, J 5.9, 1.9), 6.81 (1H, dd, J 5.9, 1.9), 6.03 (1H, s), 6.01 (1H, s), 3.88 (3H, s), 3.85 (3H, s), 3.61 (3H, s), 3.60 (3H, s), 3.22 (3H, s), 3.19 (3H, s), 2.33 (3H, s), 2.32 (3H, s), 2.31 (3H, s), 2.09 (3H, s), 2.06 (3H, s), 2.05 (3H, s), 2.01 (3H, s), 1.88 (3H, s), 1.85 (3H, s); δF (376 MHz; CDCl3) −58.48 (s); HRMS (FTMS + ESI): calcd for [C64H63F3N5O8S2191Ir + H]+: 1342.3754. Found: 1342.3802.
:EtOAc 3:2 v/v) to give an off-white solid, 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (1.31 g, 75%); δH (400 MHz; CDCl3) 11.74 (1H, s), 8.65 (1H, ddd, J 4.9, 1.8, 1.0), 7.82 (1H, td, J 7.8, 1.7), 7.65 (1H, dt, J 7.9, 1.1), 7.33 (1H, ddd, J 7.6, 4.9, 1.1), 6.96 (1H, s); δF (376 MHz; CDCl3) −62.35 (s).
29 (110 mg, 0.23 mmol) and IrCl3·3H2O (37 mg, 0.10 mmol) were combined in 2-ethoxyethanol (5 mL), and the mixture was heated to reflux overnight under a nitrogen atomosphere. The reaction was the cooled to RT, and the solvent removed in vacuo. The residue was redissolved in DCM/EtOH (3:1 v/v, 20 mL) and 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (28 mg, 0.13 mmol) and Na2CO3 (11 mg, 0.10 mmol) were added. The solution was heated to 55 °C for 6 h, before being cooled to RT and the solvent removed in vacuo. The residue was purified by column chromatography (0–3% EtOAc in DCM) to give the product as a yellow solid, iridium complex 14 (109 mg, 77%); Anal. calc. for C65H55F5IrN5O6S2: C, 57.68; H, 4.10; N, 5.17. Found: C, 57.84; H, 4.33; N, 4.92; δH (400 MHz, CDCl3) 8.11 (1H, s), 8.05 (1H, s), 7.95–7.78 (6H, m), 7.70–7.64 (1H, m), 7.56 (1H, d, J 6.0), 7.50–7.43 (2H, m), 7.29 (1H, d, J 0.8), 7.26–7.22 (2H, m), 7.18–7.11 (1H, m), 7.01 (1H, s), 6.93 (4H, d, J 6.4), 6.77 (1H, dd, J 5.9, 1.8), 6.71 (1H, dd, J 6.0, 1.8), 5.72 (1H, s), 5.70 (1H, s), 3.44 (3H, s), 3.41 (3H, s), 2.39 (3H, s), 2.38 (3H, s), 2.33 (6H, s), 1.99 (3H, s), 1.97 (6H, s), 1.93 (3H, s); δF (376 MHz; CDCl3) −59.85 (3F, s), −110.73 (1F, s); −111.48 (1F, s); HRMS (FTMS + ESI): calcd for [C65H55N5O6F5S2191Ir + H]+: 1352.3198. Found: 1352.3193.
29 (198 mg, 0.48 mmol) and [Ir(COD)Cl]2 (79 mg, 0.12 mmol) were combined in 2-ethoxyethanol (5 mL), and the mixture was heated to reflux overnight under a nitrogen atmosphere. The reaction was the cooled to RT, and the solvent removed in vacuo. The residue was redissolved in DCM/EtOH (3:1 v/v, 20 mL) and 2-(3-(trifluoromethyl)-1H-pyrazol-5-yl)pyridine 16 (63 mg, 0.30 mmol) and Na2CO3 (50 mg, 0.47 mmol) were added. The solution was heated to 55 °C for 5 h, before being cooled to RT and the solvent removed in vacuo. The residue was purified by column chromatography (0–10% EtOAc in DCM) to give the product as a yellow solid, iridium complex 15 (28 mg, 56%); Anal. calc. for C55H53F3IrN5O8S2: C, 53.91; H, 4.36; N, 5.72. Found: C, 53.48; H, 4.43; N, 5.55; δH (400 MHz, CDCl3) 8.46 (1H, d, J 1.8), 8.40 (1H, d, J 1.8), 7.88 (2H, m), 7.82 (1H, d, J 5.9), 7.75 (1H, d, J 5.6), 7.52 (1H, d, J 5.9), 7.16 (1H, td, J 6.0, 2.6), 7.04 (1H, s), 6.98 (2H, s), 6.96 (2H, s), 6.85 (1H, dd, J 5.9, 1.9), 6.72 (1H, dd, J 5.9, 1.9), 5.96 (1H, s), 5.89 (1H, s), 3.91 (3H, s), 3.91 (3H, s), 3.60 (3H, s), 3.58 (3H, s), 3.30 (3H, s), 3.27 (3H, s), 2.35 (6H, s), 2.03 (6H, s), 2.01 (3H, s), 1.97 (3H, s); δF (376 MHz; CDCl3) −60.00 (s); HRMS (FTMS + ESI): calcd for [C55H53N5O8F3S2191Ir + H]+: 1224.2972. Found: 1224.2987.
Acknowledgements
We thank EPSRC grant EL/L02621X/1 for funding the work in Durham.References
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Darmstadt, 2008 Search PubMed.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970 CrossRef CAS PubMed.
- K. K.-W. Lo, W.-K. Hui, C.-K. Chung, K. H.-K. Tsang, D. C.-M. Ng, N. Zhu and K.-K. Cheung, Coord. Chem. Rev., 2005, 249, 1434 CrossRef CAS.
- K. Müllen and U. Scherf, Organic Light Emitting Devices: Synthesis, Properties and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed.
- R. Mertens, The OLED Handbook. A Guide to OLED Technology, Industry and Market, 2014: http://www.oled-info.com/handbook Search PubMed.
- Z.-R. Li and H. Meng, Organic Light-Emitting Materials and Devices, CRC Press, Boca Raton, Florida, 2006 Search PubMed.
- M. Du, Y. Feng, D. Zhu, T. Peng, Y. Liu, Y. Wang and M. R. Bryce, Adv. Mater., 2016, 28, 5963 CrossRef CAS PubMed.
- L. Ying, C.-L. Ho, H. Wu, Y. Cao and W.-Y. Wong, Adv. Mater., 2014, 26, 2459 CrossRef CAS PubMed.
- E. Holder, B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109 CrossRef CAS.
- P. T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380 CrossRef CAS PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484 RSC.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267 RSC.
- S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef CAS.
- E. Baranoff and B. F. Curchod, Dalton Trans., 2015, 44, 8318 RSC.
- S. H. Kim, J. Jang, S. J. Lee and J. Y. Lee, Thin Solid Films, 2008, 517, 722 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992 CrossRef CAS PubMed.
- C. Fan, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2012, 24, 4581 CrossRef CAS.
- J. B. Kim, S. H. Han, K. Yang, S. K. Kwon, J. J. Kim and Y. H. Kim, Chem. Commun., 2015, 51, 58 RSC.
- S. Lee, S. O. Kim, H. Shin, H. J. Yun, K. Yang, S. K. Kwon, J. J. Kim and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14321 CrossRef CAS PubMed.
- L.-L. Wu, C.-H. Yang, I. W. Sun, S.-Y. Chu, P.-C. Kao and H.-H. Huang, Organometallics, 2007, 26, 2017 CrossRef CAS.
- H. Oh, K.-M. Park, H. Hwang, S. Oh, J. H. Lee, J.-S. Lu, S. Wang and Y. Kang, Organometallics, 2013, 32, 6427 CrossRef CAS.
- E. Orselli, G. S. Kottas, A. E. Konradsson, P. Coppo, R. Frohlich, L. de Cola, A. van Dijken, M. Buchel and H. Borner, Inorg. Chem., 2007, 46, 11082 CrossRef CAS PubMed.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713 CrossRef CAS PubMed.
- J. Li, P. I. Djurovich, B. D. Alleyne, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, Polyhedron, 2004, 23, 419 CrossRef CAS.
- H.-J. Seo, K.-M. Yoo, M. Song, J. S. Park, S.-H. Jin, Y. I. Kim and J.-J. Kim, Org. Electron., 2010, 11, 564 CrossRef CAS.
- H. Benjamin, Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, Inorg. Chem., 2016, 55, 8612 CrossRef CAS PubMed.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684 CrossRef CAS.
- C. H. Hsieh, F. I. Wu, C. H. Fan, M. J. Huang, K. Y. Lu, P. Y. Chou, Y. H. Yang, S. H. Wu, I. C. Chen, S. H. Chou, K. T. Wong and C. H. Cheng, Chem. – Eur. J., 2011, 17, 9180 CrossRef CAS PubMed.
- H. Fu, Y.-M. Cheng, P.-T. Chou and Y. Chi, Mater. Today, 2011, 14, 472 CrossRef CAS.
- L. Shi, B. Hong, W. Guan, Z. Wu and Z. Su, J. Phys. Chem. A, 2010, 114, 6559 CrossRef CAS PubMed.
- J. M. Fernandez-Hernandez, J. I. Beltran, V. Lemaur, M. D. Galvez-Lopez, C. H. Chien, F. Polo, E. Orselli, R. Frohlich, J. Cornil and L. De Cola, Inorg. Chem., 2013, 52, 1812 CrossRef CAS PubMed.
- C.-H. Chang, C.-C. Chen, C.-C. Wu, C.-H. Yang and Y. Chi, Org. Electron., 2009, 10, 1364 CrossRef CAS.
- J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazerruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667 RSC.
- V. N. Kozhevnikov, K. Dahms and M. R. Bryce, J. Org. Chem., 2011, 76, 5143 CrossRef CAS PubMed.
- S. Schmidbauer, A. Hohenleutner and B. Konig, Adv. Mater., 2013, 25, 2114 CrossRef CAS PubMed.
- V. N. Kozhevnikov, Y. Zheng, M. Clough, H. A. Al-Attar, G. C. Griffiths, K. Abdullah, S. Raisys, V. Jankus, M. R. Bryce and A. P. Monkman, Chem. Mater., 2013, 25, 2352 CrossRef CAS.
- A. F. Henwood, A. K. Bansal, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2016, 4, 3726 RSC.
- (a) D. Rota Martir, A. K. Bansal, V. Di Mascio, D. B. Cordes, A. F. Henwood, A. M. Z. Slawin, P. C. J. Kamer, L. Martinez-Sarti, A. Pertegas, H. J. Bolink, I. D. W. Samuel and E. Zysman-Colman, Inorg. Chem. Front., 2016, 3, 218 RSC; (b) D. Rota Martir, G. J. Hedley, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, T. Kosikova, D. Philip, D. M. Dawson, S. E. Ashbrook, I. D. W. Samuel and E. Zysman-Colman, Dalton Trans., 2016, 45, 17195 RSC; (c) D. Rota Martir, C. Momblona, A. Pertegás, D. B. Cordes, A. M. Z. Slawin, H. J. Bolink and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2016, 8, 33907 CrossRef PubMed.
- J. Li, Q. Zhang, H. He, L. Wang and J. Zhang, Dalton Trans., 2015, 44, 8577 RSC.
- E. Bisagni, M. Rautureau and C. Huel, Heterocycles, 1989, 29, 1815 CrossRef CAS.
- E. Busto, V. Gotor-Fernández and V. Gotor, Tetrahedron: Asymmetry, 2006, 17, 1007 CrossRef CAS.
- C.-C. Cheng, W.-S. Yu, P.-T. Chou, S.-M. Peng, G.-H. Lee, P.-C. Wu, Y.-H. Song and Y. Chi, Chem. Commun., 2003, 2628 RSC.
- C.-H. Lin, Y.-C. Chiu, Y. Chi, Y.-T. Tao, L.-S. Liao, M.-R. Tseng and G.-H. Lee, Organometallics, 2012, 31, 4349 CrossRef CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082 CrossRef CAS.
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- D. Tordera, J. J. Serrano-Pérez, A. Pertegás, E. Ortí, H. J. Bolink, E. Baranoff, M. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 3391 CrossRef CAS.
- V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, J. Fluorine Chem., 2009, 130, 640 CrossRef CAS.
- V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. Loebl, V. Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, Cent. Eur. J. Chem., 2009, 7, 836 Search PubMed.
- R. Seifert, I. Rabelo de Moraes, S. Scholz, M. C. Gather, B. Lüssem and K. Leo, Org. Electron., 2013, 14, 115 CrossRef CAS.
- J.-H. Jou, W.-B. Wang, S.-M. Shen, S. Kumar, I. M. Lai, J.-J. Shyue, S. Lengvinaite, R. Zostautiene, J. V. Grazulevicius, S. Grigalevicius, S.-Z. Chen and C.-C. Wu, J. Mater. Chem., 2011, 21, 9546 RSC.
- S.-J. Su, Y. Takahashi, T. Chiba, T. Takeda and J. Kido, Adv. Funct. Mater., 2009, 19, 1260 CrossRef CAS.
- D. Sun, H. Zhou, H. Li, X. Sun, Y. Zheng, Z. Ren, D. Ma, M. R. Bryce and S. Yan, J. Mater. Chem. C, 2014, 2, 8277 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- (a) T. H. Dunning Jr. and P. J. Hay, Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976, vol. 3 Search PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (c) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- (a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS; (b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef CAS.
- A. R. Allouche, J. Comput. Chem., 2011, 32, 174 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59 CrossRef CAS PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric data; NMR spectra; additional computational and CV data. CCDC 1495066. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02289a |
| This journal is © The Royal Society of Chemistry 2017 |