
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of unexpected silicon- and disiloxane-bridged multiferrocenyl derivatives bearing Si–O–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CH2 and Si–(CH2)2C(CH3)3 substituents via cleavage of tetrahydrofuran and trapping of its ring fragments†
CH2 and Si–(CH2)2C(CH3)3 substituents via cleavage of tetrahydrofuran and trapping of its ring fragments†
Sonia
Bruña
 ab,
Ana Mª
González-Vadillo
a,
Marta
Ferrández
a,
Josefina
Perles
c,
M. Merced
Montero-Campillo
d,
Otilia
Mó
bd and
Isabel
Cuadrado
*ab
ab,
Ana Mª
González-Vadillo
a,
Marta
Ferrández
a,
Josefina
Perles
c,
M. Merced
Montero-Campillo
d,
Otilia
Mó
bd and
Isabel
Cuadrado
*ab
aDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain. E-mail: isabel.cuadrado@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Spain
cLaboratorio de Difracción de Rayos X de Monocristal, Servicio Interdepartamental de Investigación (SIDI), Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain
dDepartamento de Química, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain
First published on 24th July 2017
Abstract
The formation of a family of silicon- and siloxane-bridged multiferrocenyl derivatives carrying different functional groups attached to silicon, including Fc2(CH3)3C(CH2)2SiCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (5), Fc2(CH2CH–O)SiCHCH2 (6), Fc2(OH)SiCHCH2 (7), Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8) and Fc2(CH2CH–O)Si–O–SiFc3 (9) is described. Silyl vinyl ether molecules 6, 8 and 9 and the heteroleptic vinylsilane 5 resulted from the competing metathesis reaction of lithioferrocene (FcLi), CH2CH–OLi or (CH3)3C(CH2)2Li with the corresponding multifunctional chlorosilane, Cl3SiCHCH2 or Cl3Si–O–SiCl3. The last two organolithium species have been likely formed in situ by fragmentation of the tetrahydrofuran solvent. Diferrocenylvinyloxyvinylsilane 6 is noteworthy since it represents a rare example of a redox-active silyl mononomer in which two different CC polymerisable groups are directly connected to silicon. The molecular structures of the silicon-containing multiferrocenyl species 5, 6, 8 and 9 have been investigated by single-crystal X-ray diffraction studies, demonstrating the capture and storage processes of two ring fragments resulting from the cleavage of cyclic THF in redox-active and stable crystalline organometallic compounds. From electrochemical studies we found that by changing the anion of the supporting electrolyte from [PF6]− to [B(C6F5)4]−, the redox behaviour of tetrametallic disiloxane 8 can be switched from a poorly resolved multistep redox process to four consecutive well-separated one-electron oxidations, corresponding to the sequential oxidation of the four ferrocenyl moieties.
CH2 (5), Fc2(CH2CH–O)SiCHCH2 (6), Fc2(OH)SiCHCH2 (7), Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8) and Fc2(CH2CH–O)Si–O–SiFc3 (9) is described. Silyl vinyl ether molecules 6, 8 and 9 and the heteroleptic vinylsilane 5 resulted from the competing metathesis reaction of lithioferrocene (FcLi), CH2CH–OLi or (CH3)3C(CH2)2Li with the corresponding multifunctional chlorosilane, Cl3SiCHCH2 or Cl3Si–O–SiCl3. The last two organolithium species have been likely formed in situ by fragmentation of the tetrahydrofuran solvent. Diferrocenylvinyloxyvinylsilane 6 is noteworthy since it represents a rare example of a redox-active silyl mononomer in which two different CC polymerisable groups are directly connected to silicon. The molecular structures of the silicon-containing multiferrocenyl species 5, 6, 8 and 9 have been investigated by single-crystal X-ray diffraction studies, demonstrating the capture and storage processes of two ring fragments resulting from the cleavage of cyclic THF in redox-active and stable crystalline organometallic compounds. From electrochemical studies we found that by changing the anion of the supporting electrolyte from [PF6]− to [B(C6F5)4]−, the redox behaviour of tetrametallic disiloxane 8 can be switched from a poorly resolved multistep redox process to four consecutive well-separated one-electron oxidations, corresponding to the sequential oxidation of the four ferrocenyl moieties.
Introduction
Reactions with organolithium compounds are of fundamental and practical interest, particularly because of their valuable role in organic and organometallic chemistry.1 The deprotonation reactions of organolithiums are often carried out in the presence of ethereal solvents, such as diethyl ether and tetrahydrofuran (THF), to enhance the reactivity of organometallic reagents. However, it is well-known that these polar organometallic reagents commonly decompose ethereal solvents.1–3 In particular, THF, the most widely employed cyclic ether, is a strongly activating solvent, highly susceptible to undergo deprotonation by alkyllithium bases such as t-BuLi, i-PrLi and s-BuLi.1–4Metal-induced cleavage reactions of ethers are rather complicated and, depending on the organometallic reagent and conditions, can take place in many different forms. Scheme 1 summarises an overview of the main described degradation processes of THF. Route A represents the principal pathway for decomposition of THF by strong organolithium bases RLi (R = Me, t-Bu, n-Bu) and involves the deprotonation of the α carbon, adjacent to oxygen, to generate a C–Li bond. Subsequently, the resultant 2-furyl anion undergoes a facile reverse [3 + 2] rearrangement to afford ethylene gas and the lithium enolate of acetaldehyde (CH2CH–OLi).3 In rare cases, for example using a t-BuLi/HMPA mixture (HMPA = hexamethylphosphoric triamide ((Me2N)3PO)) (Route B) ring opening occurs but a THF ring retains the five atoms forming lithium but-3-en-1-oxide.2 Using novel and more sophisticated synergic mixed-metal strategies, Mulvey and co-workers have observed remarkable unprecedented degradation pathways of THF. Thus, in the presence of a moderately reactive bimetallic Na/Zn base (TMEDA)Na(TMP)(CH2SiMe3)Zn(CH2SiMe3) (TMEDA = N,N,N′,N′-tetramethylethylenediamine; TMP = 2,2,6,6-tetramethylpiperidide) (Route C), α-metallation of THF occurs but the α deprotonated anion remains intact without any opening of the heterocyclic OC4 ring.5 In marked contrast to this, the more reactive heterometallic Na/Mg [(TMEDA)Na(TMP)(CH2SiMe3)Mg(TMP)] or Na/Mn [(TMEDA)Na(TMP)(CH2SiMe3)Mn(TMP)] bases promote a noteworthy cleavage of six bonds in the five-membered THF ring (Route D).6 The two anionic fragments of the THF deconstruction shown in Route D have been trapped in separated complexes, which have been crystallographically characterised.6
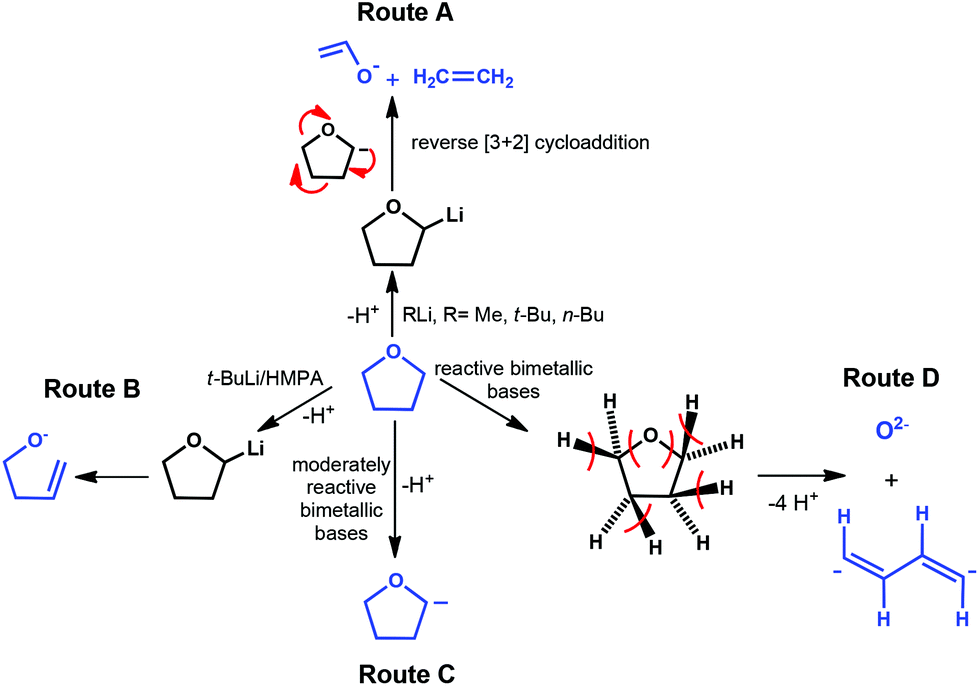 | ||
| Scheme 1 Summary of the main known fragmentation reactions that the THF ring can experiment in the presence of different organometallic bases. | ||
In a related context, deprotonative metallation has proved to be of particular importance in the chemistry of ferrocene since this synthetic transformation has been frequently applied for the derivatisation of the ferrocene backbone. The reagents classically used for this purpose are organolithiums such as n-BuLi and t-BuLi.7 Accordingly, both 1-lithioferrocene (FcLi)8 and dimetallated 1,1′-dilithioferrocene Fe(η5-C5H4Li)2, (fcLi2)9 have been prepared and often used as valuable intermediates in the synthesis of new ferrocene derivatives as they readily undergo transmetallation or nucleophilic substitution reactions. In addition, recent work by Mulvey and co-workers has shown that direct multimetallation of ferrocene with mixed bimetallic synergic bases is also possible.10 One of the most noteworthy examples of this chemistry is the 1,1′,3,3′-tetramagnesiation of ferrocene using the sodium tris(amido)magnesiate base NaMg(NiPr2)3.10b,c In addition to promoting unique polymetallation of ferrocene, some of these bimetallic reagents are particularly useful for the functionalisation of substituted ferrocenes bearing sensitive organic functional groups such as nitriles, esters or carboxylic acids.11–13
Over the years, our research interest concerns the chemistry of silicon-containing multiferrocenyl compounds.14,15 As contributions to this field, we have recently synthesised and studied Si-vinyl and Si–H functionalised triferrocenyl molecules Fc3SiCHCH2 (1)16 and Fc3SiH (2)17 (Fc = (η5-C5H4)Fe(η5-C5H5)). Triferrocenylvinylsilane 1 was prepared from 1-lithioferrocene, in turn generated by the transmetallation of 1-(tri-n-butylstannyl)ferrocene (FcSnBu3) with n-BuLi, followed by quenching with the electrophile trichlorovinylsilane. In contrast, the synthesis of homoleptic hydrosilane 2 was effected in a one-pot procedure, in which 1-lithioferrocene was directly generated in situ, by the reaction of ferrocene and t-BuLi, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF and n-hexane.17 Over the course of this reaction we found that, in addition to the desired Si–H-functionalised triferrocenylsilane 2, diferrocenyl(3,3-dimethylbutyl)silane Fc2(CH3)3C(CH2)2SiH (3) was unexpectedly generated. The formation of heteroleptic silicon hydride 3 is proposed to occur via the competitive metathesis reaction of Cl3SiH with organolithium bases FcLi and (CH3)3C(CH2)2Li, which, in turn, has been formed in situ via the carbolithiation of ethene, generated by degradation of THF.
:1 mixture of THF and n-hexane.17 Over the course of this reaction we found that, in addition to the desired Si–H-functionalised triferrocenylsilane 2, diferrocenyl(3,3-dimethylbutyl)silane Fc2(CH3)3C(CH2)2SiH (3) was unexpectedly generated. The formation of heteroleptic silicon hydride 3 is proposed to occur via the competitive metathesis reaction of Cl3SiH with organolithium bases FcLi and (CH3)3C(CH2)2Li, which, in turn, has been formed in situ via the carbolithiation of ethene, generated by degradation of THF.
 CH2 with 1-lithioferrocene, directly generated from ferrocene and t-BuLi in THF, and furthermore to test the reactivity of a more complex chlorosilane, Cl3Si–O–SiCl3. In addition, to improve the yield of triferrocenylvinylsilyl 1, the ultimate focus of this work was to obtain new functionalised ferrocenes by the capturing of molecular fragments generated from the fragmentation of cyclic THF.
CH2 with 1-lithioferrocene, directly generated from ferrocene and t-BuLi in THF, and furthermore to test the reactivity of a more complex chlorosilane, Cl3Si–O–SiCl3. In addition, to improve the yield of triferrocenylvinylsilyl 1, the ultimate focus of this work was to obtain new functionalised ferrocenes by the capturing of molecular fragments generated from the fragmentation of cyclic THF.
Herein we report on the formation of structurally new multiferrocenyl silanes and disiloxanes, namely Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6), Fc2(OH)SiCHCH2 (7), Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8) and Fc2(CH2CH–O)Si–O–SiFc3 (9). These vinylsilyl compounds have been serendipitously generated from the reaction of ferrocene, t-BuLi and the corresponding multifunctional chlorosilane in the presence of a cyclic THF solvent. The formation of 6, 8 and 9, bearing the reactive silyl vinyl ether group (–Si–O–CHCH2), is noteworthy since it not only provides new experimental proof about the fragmentation mechanism of cyclic THF, but also points to a new potentially useful synthetic way to functionalised ferrocenes. It is interesting to note that, among vinylsilanes, those bearing oxygen substituents at silicon are particularly useful, as these reagents consistently provide superior reactivity compared to their alkylsilyl counterparts.18
Results and discussion
Reaction of monolithioferrocene with Cl3SiCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH2: formation of Fc3SiCHCH2 (1), Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6) and Fc2(OH)SiCHCH2 (7)
CH2: formation of Fc3SiCHCH2 (1), Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6) and Fc2(OH)SiCHCH2 (7)
Two different synthetic strategies were explored for the formation of triferrocenylvinylsilane (1). Both routes, A and B, are shown in Scheme 2 and involve the lithiation of ferrocene followed by the low temperature salt metathesis reaction of 1-lithioferrocene (FcLi) and the chlorosilane Cl3SiCHCH2. Within these approaches, the use of pure FcLi is a key factor, in order to exclude the formation of dimetallated ferrocene, Fe(η5-C5H4Li)2, (fcLi2), which could produce undesirable disubstituted- and oligo-ferrocene products. While the dilithiation of ferrocene with n-butyllithium in the presence of the chelating diamine TMEDA to afford fcLi2 is well-established,9 in marked contrast, the selective formation of monolithioferrocene is a very challenging synthetic task in organometallic chemistry. In our long experience, the yields of this reaction were quite variable. A convenient procedure for FcLi was reported by Guillaneux and Kagan,8a and involved the use of 1-(tri-n-butylstannyl)ferrocene as an excellent precursor of 1-lithioferrocene, leading, through reaction with suitable electrophiles, to monosubstituted ferrocenes in high yields.
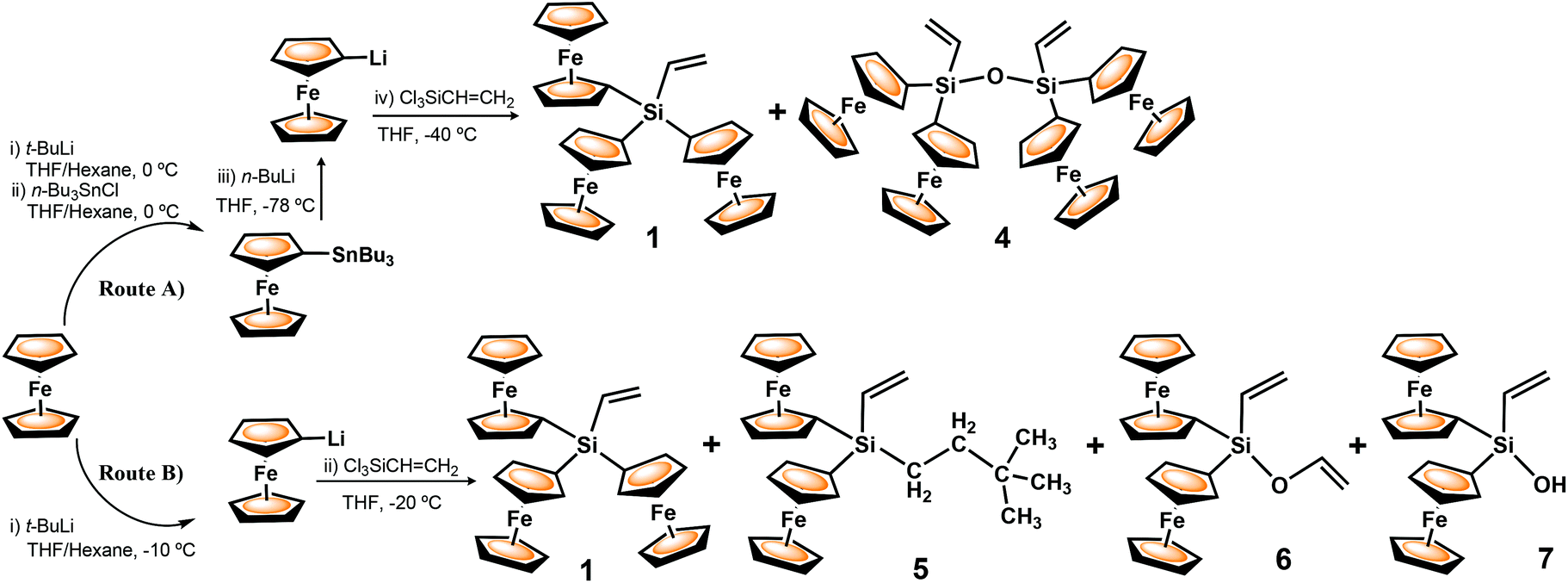 | ||
| Scheme 2 Alternative routes for the reaction of 1-lithioferrocene and trichlorovinylsilane. | ||
Accordingly, in our first attempt to prepare 1, with three ferrocenyl moieties attached to the vinylsilane functionality, we used a two-step synthetic procedure starting from FcSnBu3 as the monolithioferrocene precursor (Scheme 2, Route A).16 Subsequently, the transmetallation of FcSnBu3 was achieved by treatment with n-BuLi at −78 °C, followed by quenching with Cl3SiCHCH2. After removing the insoluble LiCl and subsequent purification via column chromatography, the target Si-vinyl-terminated 1 was isolated in high purity and reasonable yield (42%). During the purification of the reaction mixture, tetraferrocenyl disiloxane 4 was also obtained. Most likely, this initially unexpected tetrametallic compound 4 was formed under the used reaction conditions, as a result of the condensation reaction of a silanol-containing diferrocenyl molecule Fc2(OH)SiCHCH2, in turn generated accidentally, either by partial hydrolysis of the starting trichlorovinylsilane or by hydrolysis of chlorodiferrocenylvinylsilane Fc2(Cl)SiCHCH2, as it is shown in Scheme S1 (see the ESI†).16 Triferrocenyl compound 1 has proven to be a particularly reactive vinylsilane and has been successfully incorporated, via hydrosilylation chemistry, around the surface of polyhedral octasilsesquioxane cages (POSS) and linear and cyclic siloxane scaffolds.14c
With the aim to improve the yield of vinylsilane 1, to reduce the number of reaction steps, and mainly, to check whether new functionalised ferrocenes were formed, in an alternative approach 1-lithioferrocene was directly generated in situ in a single synthetic step, from the reaction between ferrocene and t-BuLi in a 1:1 mixture of THF and n-hexane at −10 °C (Scheme 2, Route B). Subsequently, without isolation of the pyrophoric solid FcLi, a THF solution of trichlorovinylsilane was added dropwise to the reaction mixture cooled at −20 °C.
After the separation of the solid LiCl and appropriate workup of the crude product, the 1H NMR spectrum of the reaction mixture (see the ESI, Fig. S1†) exhibited the signals of targeted compound 1 and the apparition of new cyclopentadienyl resonances due to unknown monosubstituted ferrocenyl species. Thus, two complex multiplets centred at δ 1.00–1.50 ppm characteristic of linked CH2 groups, and a very intense singlet at δ 0.95 ppm, clearly attributable to the protons of decoupled methyl units were observed. In addition, informative resonances in the vinyl region also appeared. These key NMR spectroscopic data motivated us to isolate the unknown compounds and properly investigate their origins and molecular structures.
The purification of the crude reaction product by careful column chromatography on silica gel allowed us to separate a first band containing unreacted ferrocene, followed by other orange bands. The first one, eluted with n-hexane/CH2Cl2 (10:1), contained a yellow-orange crystalline compound which was isolated in 6% yield, in high purity, and unequivocally identified as diferrocenyl(3,3-dimethylbutyl)vinylsilane (5). This heteroleptic silane supports the resonances observed at δ 1.00–1.50 and 0.95 ppm in the 1H NMR spectrum of the reaction mixture (see the ESI, Fig. S1†). The second band, eluted with n-hexane/CH2Cl2 (10:2) was found to contain a mixture of the expected compound 1 and a new mysterious vinylsilane 6. Unfortunately, both compounds have very similar physical properties and have been therefore particularly difficult to separate. Luckily after repeated column chromatographies and several recrystallization processes using different mixtures of n-hexane/CH2Cl2, we achieved the separation and isolation of pure samples of the targeted vinylsilane 1 (in 65% yield) and the new ferrocenyl species 6 (in 9% yield). On the basis of multinuclear NMR spectroscopy, mass spectrometry and X-ray studies (see below) the unknown compound was unequivocally identified as Fc2(CH2CH–O)SiCHCH2 (6), an interesting molecule bearing two ferrocenyl moieties and two types of CC double bonds (one directly connected to silicon, and the other one to oxygen), thus explaining the striking 1H NMR resonances observed in the vinyl region in the spectrum of the reaction mixture.
The origin of the initially unexpected vinylsilanes Fc2(CH3)3C(CH2)2SiCHCH2 (5) and Fc2(CH2CH–O)SiCHCH2 (6) is certainly noteworthy and deserves an explanation. Their formation can be rationalised by the sequence of processes outlined in Scheme 3, and presumably involves the THF cleavage. As already mentioned in the introduction (Scheme 1), the main pathway for the THF degradation by RLi reagents first comprises the deprotonation at the carbon α to oxygen. Then, the resultant 2-furyl anion undergoes a reverse [3 + 2] cycloaddition to generate ethylene and the lithium enolate of acetaldehyde. Therefore, probably, compound 6 carrying both a vinyloxy and a vinyl group has been formed under the used reaction conditions, as a result of the competing salt-metathesis reaction of the organolithium reagents FcLi and CH2CH–OLi with Cl3SiCHCH2. Meanwhile, diferrocenyl compound 5 is likely formed as a result of the competing metathesis reaction of FcLi and (CH3)3C(CH2)2Li with Cl3SiCHCH2. As observed during the formation of heteroleptic hydrosilane 3,17 the only plausible source of the lithiated reagent (CH3)3C(CH2)2Li is the intermolecular carbolithiation of ethylene arising from the decomposition of THF (Scheme 3).1–3
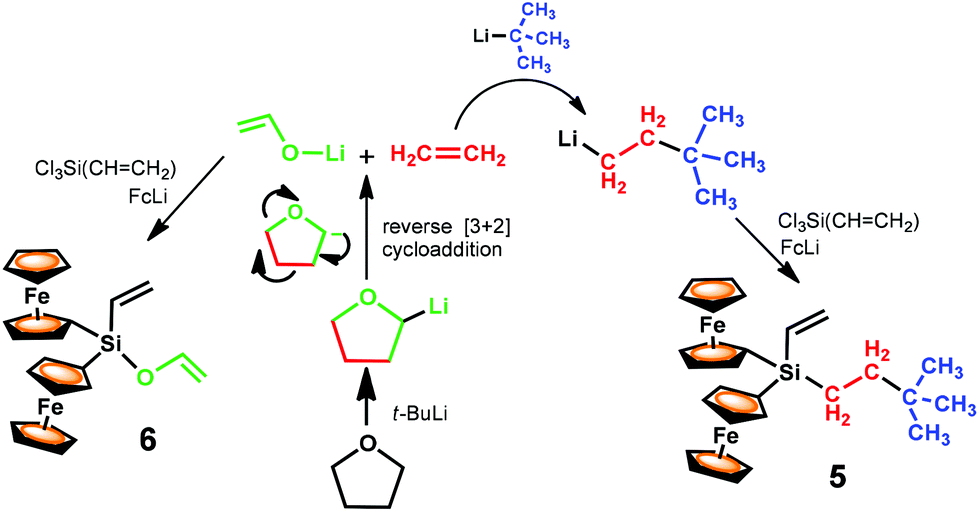 | ||
| Scheme 3 Pathway for the degradation of THF and reactions that presumably occur in the system THF/t-BuLi/FcLi/Cl3SiCHCH2. | ||
When comparing the two synthetic procedures used to prepare 1 it can be concluded that Route B resulted more efficient and less complicated than the approach using FcSnBu3 as a starting material and that, in addition, newly interesting products, 5 and 6, have been formed. The reaction of FcLi with Cl3SiCHCH2 following Route B (Scheme 2) was repeatedly performed and in all the cases compounds 1, 5 and 6 were obtained. In addition, in some reactions, diferrocenylvinylsilanol (7) was also isolated. The formation of compound 7 provides significant evidence to affirm that disiloxane 4, obtained by Route A, was generated as a result of the condensation reaction of silanol-containing intermediates, as we previously proposed (see the ESI, Scheme S1†).16 Compound 7 was purified by column chromatography and was isolated as an air stable, yellow-orangish crystalline solid in 5% yield.
It must be emphasised that compounds 6 and 7 carry two different reactive and polymerisable groups, allowing them to participate in different chemical transformations. In addition, the polar vinyl ether reactive group present in 6 is an attractive functionalisation due to its versatile and rich reactivity.18
Once separated, the new multiferrocenyl vinylsilanes 5–7 were thoroughly characterised by elemental analysis, multinuclear NMR spectroscopy, IR and MALDI-TOF mass spectrometry. In addition to the ferrocenyl resonances, the 1H NMR spectra of 5–7 show three double doublets in the expected integrated ratios, which are consistent with the vinyl AMX system. Furthermore, the 1H NMR spectrum of 5 shows the peculiar pattern for the –(CH2)2C(CH3)3 group, a singlet at δ 0.94 ppm corresponding to the nine protons of the three decoupled methyl groups, and two complex multiplets centred at δ 1.02 and 1.44 ppm characteristic of the –CH2–CH2– unit. The –O–CHCH2 group of 6 appears as a double doublet at δ 6.59 and two doublets, one centred at δ 4.58 while the other one cannot be observed because it is overlapped with the C5H5 resonance at δ 4.16 ppm. This last signal could be identified performing a {1H–13C} HMQC experiment (see the ESI, Fig. S9†), as the carbon resonance of the –O–CHCH2 unit correlates, not only with the multiplet at δ 4.58 ppm, but also with the other that is under the C5H5 resonance at δ 4.16 ppm. Meanwhile, the spectrum of compound 7 exhibits a singlet at δ 2.15 ppm, distinctive of the OH group.
Besides the characteristic carbon signals of the ferrocenyl units and of the vinyl groups (at 133 and 137 ppm), the 13C NMR spectrum of 5 shows the resonances of the 3,3-dimethylbutyl group. Thus, the CH2 carbons appear at δ 9.0 and 38.3 ppm, the CH3 at δ 28.9 ppm, and the ipso-carbon at δ 31.3 ppm. In the spectrum of compound 6 two resonances of the –O–CHCH2 group appear at δ 94.2 and 146.3 ppm. The 29Si NMR spectrum displays a single resonance at δ −17.0 ppm (for 1), δ −12.1 ppm (for 5), δ −7.8 ppm (for 6), and at δ −8.9 ppm (for 7). Therefore, the replacement of a ferrocenyl moiety by a 3,3-dimethylbutyl shifts the silicon signals downfield. These resonances are even more deshielded in the spectra of compounds 6 and 7, due to the electronic influence of the –OCHCH2 and –OH groups.
Reaction of monolithioferrocene with hexachlorodisiloxane: formation of Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8), Fc2(CH2CH–O)Si–O–SiFc3 (9) and Fc3Si–O–SiFc3 (10)
Encouraged by the results described above and with the idea to increase the members of the multiferrocenyl family supported by Si–O–Si bridges, our efforts were next directed toward the synthesis of a disiloxane maximally functionalised with as many ferrocenyl units as chemically possible. Thus, we sought to prepare the hexaferrocenyldisiloxane molecule 10 shown in Scheme 4. To this end, monolithioferrocene was generated by treating ferrocene with t-BuLi, in a 1:1 mixture of THF and n-hexane at −10 °C. Subsequently, FcLi was reacted with hexachlorodisiloxane (Cl3Si–O–SiCl3) at −20 °C and the reaction mixture was purified by column chromatography. To our surprise, the tetrametallic compound Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8) was isolated as the major reaction product (29% yield). Compound 8 is a disiloxane functionalised with four ferrocenyl units and two vinyloxy reactive groups, which was isolated as an orange, crystalline and air stable solid. It is interesting to observe that, in addition to the peak of 8 at m/z 898.1, the MALDI-TOF mass spectrum of the reaction mixture (see Fig. 1) exhibits two peaks at m/z 1040.0 and 1182.0, thus confirming the formation of the targeted hexaferrocenyldisiloxane 10 and of an asymmetric disiloxane molecule, Fc2(CH2CH–O)Si–O–SiFc3 (9), also shown in Scheme 4.
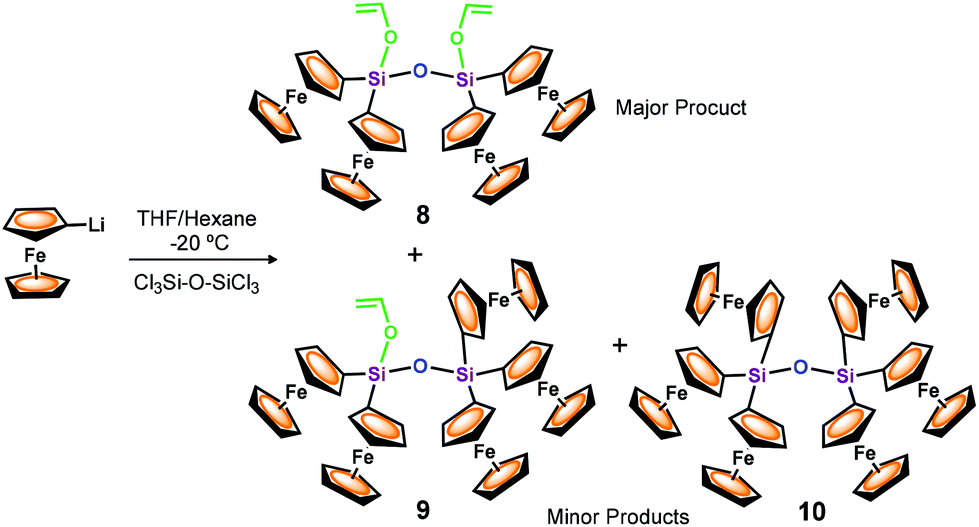 | ||
| Scheme 4 Synthesis of polyferrocenes 8–10 with disiloxane bridges. | ||
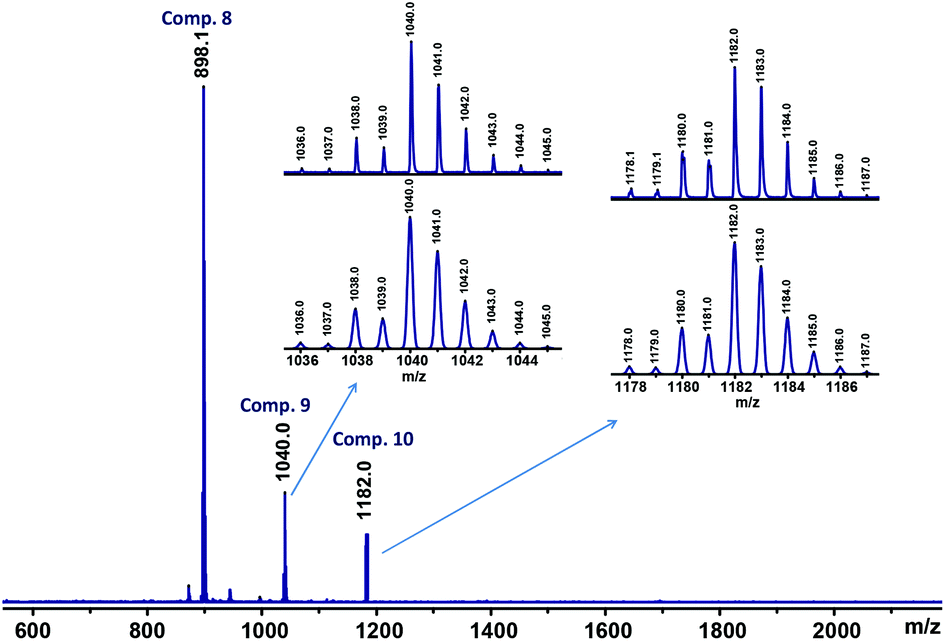 | ||
| Fig. 1 MALDI-TOF mass spectrum of the reaction mixture of disiloxanes 8–10. The insets show the experimental (top) and calculated (bottom) isotopic patterns for compounds 9 and 10. | ||
Unfortunately, significant amounts of pure samples of multiferrocenyl disiloxanes 9 and 10 could not be isolated, so far, due to their similar solubility properties. However, their existence is fully supported by the MALDI-TOF mass spectrometric study and, in the case of disiloxane 9 by X-ray diffraction analysis (see below).
On the basis of the formation of silylated acetaldehyde species 6, it seems reasonable to assume that disiloxanes 8 and 9 were formed as a result of the competing metathesis reactions of FcLi and CH2CH–OLi to react with the hexachlorodisiloxane, according to Scheme 5.
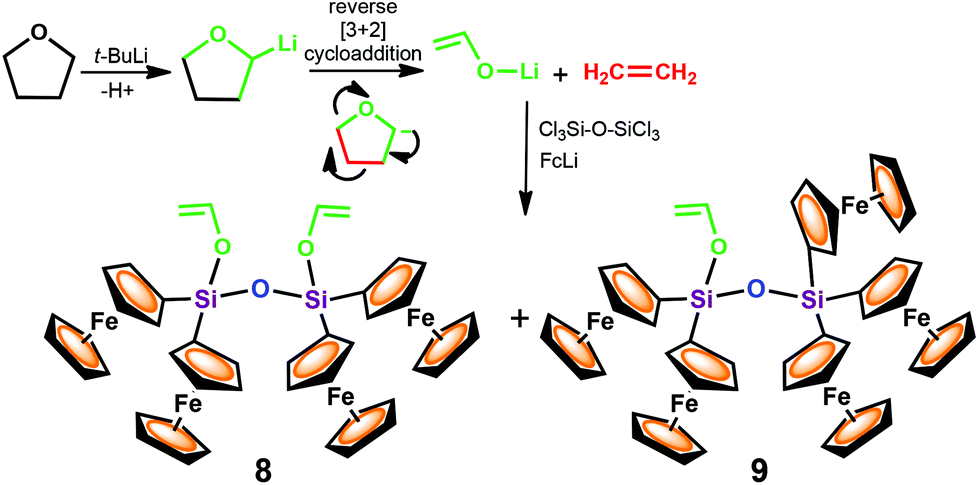 | ||
| Scheme 5 Reactions that presumably occur in the system THF/t-BuLi/FcLi/Cl3Si–O–SiCl3. | ||
As can be observed, the reaction between FcLi and Cl3Si–O–SiCl3 fails to give the desired hexaferrocenyl 10 in preparative amounts, which might be due to the steric repulsion of the six ferrocenyl moieties in close contact across the siloxane bond. Therefore, it is very likely that the difficulty entailed in effectively bonding six ferrocenyl units to the Si–O–Si bridge facilitates, as an alternative reaction route, the incorporation of the less bulky vinyloxy group.
We have to note that neither disiloxane 8 nor vinylsilanes 5 and 6 are accessible by conventional organosilicon chemistry methods, illustrating again the synthetic potential of THF-cleavage reactions.
Multinuclear NMR (1H, 13C and 29Si) analyses of the new disiloxane 8 were consistent with the proposed structure. Thus, the protons of the –O–CHCH2 groups appear in the 1H NMR spectrum at 6.82 ppm (double doublet) and at δ 4.68 and 4.26 ppm (doublets). The 29Si NMR of 8 presents a single resonance at δ −27.7 ppm, in good agreement with values found for other ferrocenyl disiloxanes.14
Crystal structures of compounds 5–9
The ultimate confirmation that the molecular fragments generated from the cleavage of the THF ring have been certainly captured in crystalline redox-active ferrocenyl compounds was achieved by single-crystal X-ray diffraction analysis of silanes 5 and 6 and disiloxanes 8 and 9.Diffraction-grade single crystals were obtained by crystallisation at 4 °C in n-hexane/CH2Cl2 (10:2) for 5, at room temperature in CH2Cl2 for 6 and 7 and at 4 °C in n-hexane/CH2Cl2 (10:3) for 8 and 9. The resulting structures are shown in Fig. 2 and 3, and complete structural information is collected in the ESI.† The three bimetallic silanes 5–7 and the pentametallic disiloxane 9 crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one (6, 7 and 9) or two (5) molecules per asymmetric unit, while tetrametallic disiloxane 8 belongs to the monoclinic P21/n space group. The two crystallographic independent molecules in the asymmetric unit cell of 5 (5A and 5B) are shown in Fig. 2 (top); they differ only slightly in their bond lengths and angles.
with one (6, 7 and 9) or two (5) molecules per asymmetric unit, while tetrametallic disiloxane 8 belongs to the monoclinic P21/n space group. The two crystallographic independent molecules in the asymmetric unit cell of 5 (5A and 5B) are shown in Fig. 2 (top); they differ only slightly in their bond lengths and angles.
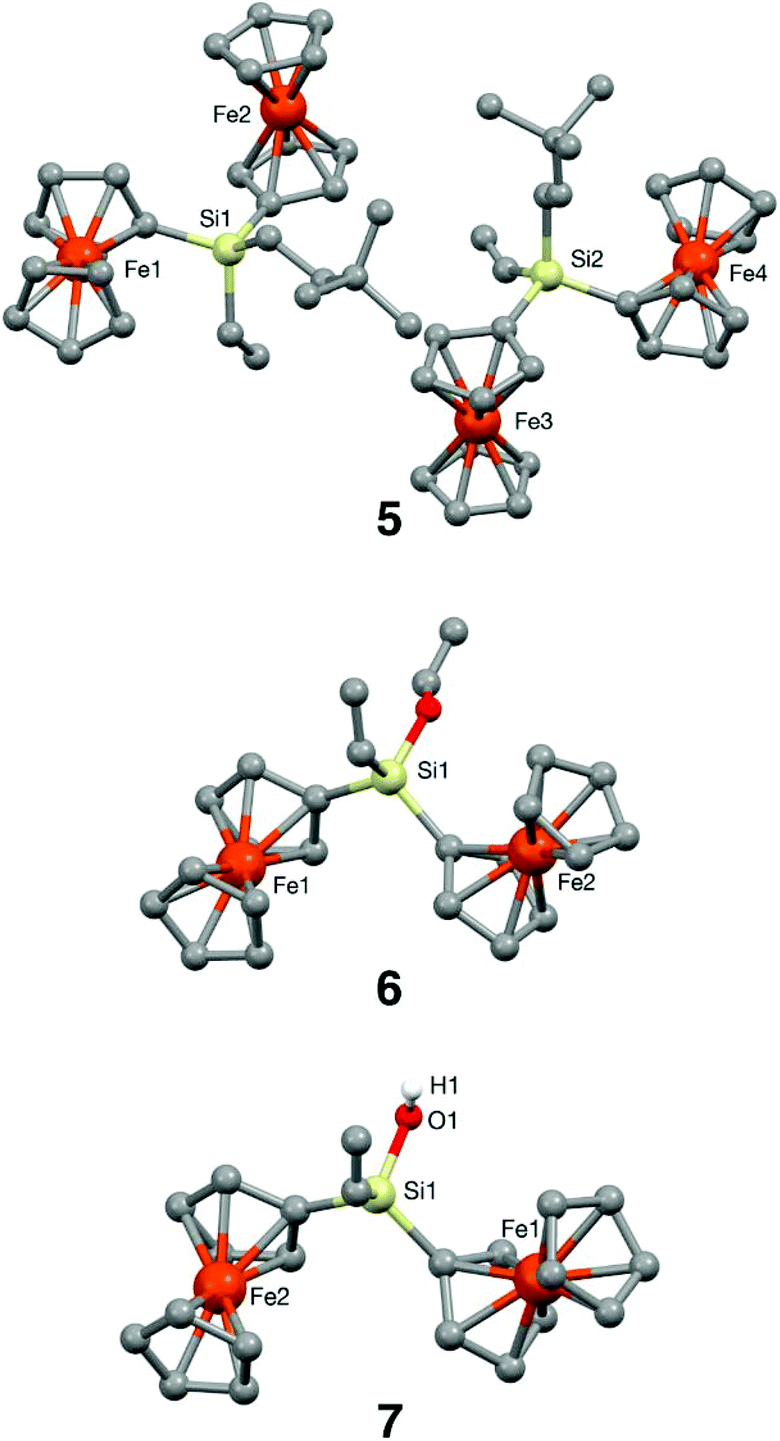 | ||
| Fig. 2 Molecular structures of silanes 5–7 with selected atoms labelled. Hydrogen atoms (except the hydroxylic one) have been removed for clarity. | ||
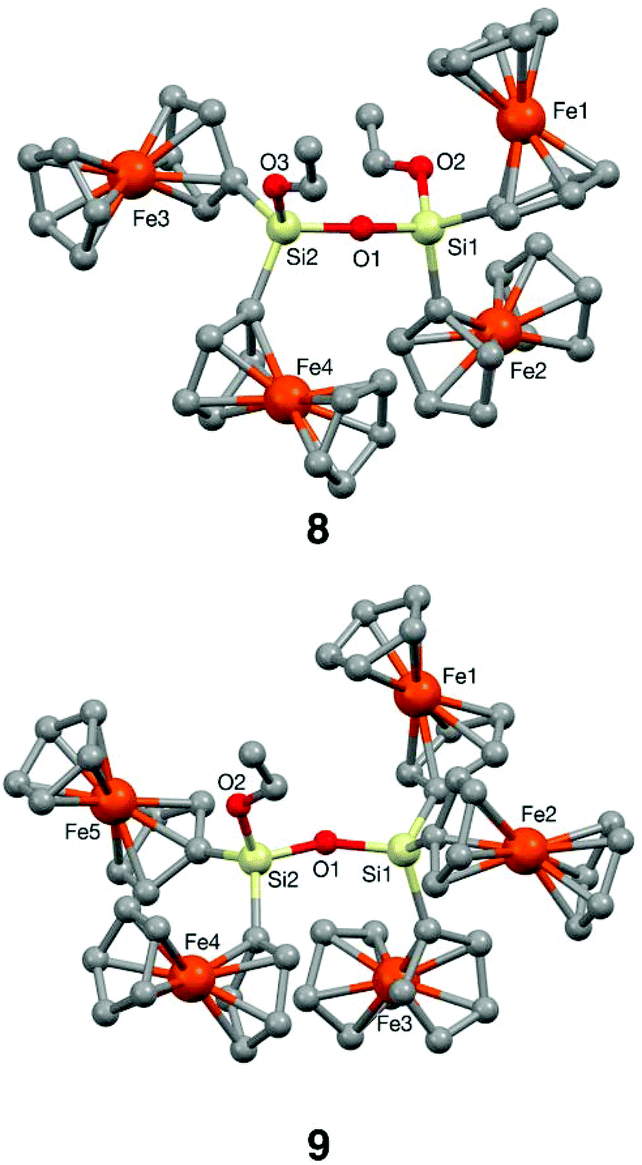 | ||
| Fig. 3 Molecular structures of disiloxanes 8 and 9 with selected atoms labelled. Hydrogen atoms have been removed for clarity. | ||
In both molecules, the two ferrocenyl groups attached to the silicon atoms are disposed in a nearly perpendicular arrangement. The cyclopentadienyl rings in 5 are parallel (maximum deviation 2.22° for the Cps attached to Fe2) and essentially eclipsed in three of the ferrocenyl moieties, while in the fourth one (the one comprising Fe3) they are completely staggered. The intramolecular distances between Fe atoms are 6.0556(8) Å (Fe1–Fe2) and 6.1853(8) Å (Fe3–Fe4). The supramolecular arrangement is achieved by van der Waals forces, giving rise to four-molecule rings.
In the bifunctional compound 6 (Fig. 2, middle), the cyclopentadienyl rings are also essentially parallel (maximum deviation 2.81° in the ferrocene containing Fe1) and eclipsed. Iron atoms are separated by 6.0857(6) Å and no relevant supramolecular interactions have been found.
In diferrocenylsilanol 7 (Fig. 2, bottom) the disposition of the Fc units is very similar to the one observed in 6, with the cyclopentadienyl rings parallel (maximum deviation 2.36° in the Fc with Fe2) and eclipsed, presenting an intramolecular distance between Fe atoms of 6.1174(6) Å. However, in this structure hydrogen bonds between the pairs of molecules are responsible for the formation of dimeric units (see the ESI, Fig. S31† and Table S12†). This is in agreement with the IR data, where a ν(OH) band at 3413 cm−1 was observed, which corresponds to Si–OH groups linked by hydrogen bonds. The fact that the two diferrocenylsilanols 6 and 7 show an almost identical molecular conformation and very similar packing patterns (see the ESI, Fig. S32†) regardless of the difference in supramolecular interactions, proves the important role that the shape and size of these molecules play in their arrangement in the crystal state. In this case, the smaller substituent in 7 together with the existence of hydrogen bonds, lead to a closer packing of the molecules involved in the supramolecular interactions, which in turn yields a denser crystal (calculated density values are 1.473 for 6 and 1.526 Mg cm−3 for 7).
In disiloxanes 8 and 9 (Fig. 3) the ferrocenyl units are placed as distant as possible from each other in order to avoid steric congestion around the silicon atoms, and the Si–O–CHCH2 groups are arranged in zig-zag chains. These two disiloxanes show a bent arrangement of the disiloxane linkage, with Si–O–Si angles of 160.0(1)° for 8 and 152.9(2)° for 9. The Fe atoms of the ferrocenyl substituents attached to the same silicon centre are separated by 5.9037(5) and 6.1052(5) Å (8) and by 5.444(1), 6.501(1) and 6.077(1) Å (9). Meanwhile, the iron centres of the ferrocenyl units attached to different silicon atoms (see the ESI, Table S11†) are separated by larger distances, ranging from 6.3853(7) to 8.4029(6) Å (for 8) and from 6.501(1) to 9.327(1) Å (for 9).
In all of the crystal structures 5–9, the silicon atoms are nearly tetrahedral with C–Si–C (for 5–9), C–Si–O (for 6–9) and O–Si–O (for 8 and 9) bond angles close to 109° (see the ESI, Fig. Table S13†) and similar to the ones obtained for 1.9
Electrochemistry and electronic structure calculations
The redox properties of 5–8, featuring ferrocenyl moieties linked by a silicon atom and/or a disiloxane bridge, are also of considerable interest as these molecules display multielectron redox chemistry.19 The electrochemical behaviour was investigated by cyclic voltammetry (CV) and square wave voltammetry (SWV), using CH2Cl2 as a non-nucleophilic solvent, and tetra-n-butylammonium hexafluorophosphate ([n-Bu4N][PF6]) or tetra-n-butylammonium tetrakis(pentafluorophenyl)borate ([n-Bu4N][B(C6F5)4]) as supporting electrolytes containing anions of different coordinating ability. Geiger and co-workers have performed remarkable studies on the perfluoroarylborate anion [B(C6F5)4]− and have ascribed the improvements observed in the oxidative electrochemical processes of organometallic compounds to its intrinsically low ion-pairing strengths and nucleophilicities, and to the increased solubility of its salts in lower polarity solvents as compared to classical electrolytes such as [PF6]−, [ClO4]− and [BF4]−.20Diferrocenyl silanes 5–7 constitute the simplest molecules described here and they are structurally related to the homoleptic vinylsilane 1 and heteroleptic diferrocenyl-vinylsilane Fc2(CH3)SiCHCH2 previously investigated by us.14d,16 They are constituted by a vinyl reactive group and two identical ferrocenyl units, linked together by a silicon bridge, and they vary in the nature of the non-electroactive –O–CHCH2, –(CH2)2C(CH3)3 or –OH substituent (R in Fig. 4 top). As expected, this leads to qualitatively similar voltammetric responses which differ only slightly in the half-wave potentials E1/2, reflecting the different electronic influences of the fourth substituent (R) at silicon. As representative examples, Fig. 4(A–D) present the CV and SWV responses obtained for 5. In CH2Cl2 using either the traditional [PF6]− or the non-coordinating [B(C6F5)4]− electrolyte anion, the electrochemical oxidation of 5 takes place in two well-resolved voltammetric waves, which correspond to the sequential oxidation of the two ferrocenyl moieties, giving the ferrocenium species 5+ and 52+. Experimentally measured potential values are 1E1/2 = 0.420 and 2E1/2 = 0.620 V (ΔE1/2 = 2E1/2 − 1E1/2 = 200 mV) (in [n-Bu4N][PF6]) and 1E1/2 = 0.422 and 2E1/2 = 0.737 V (ΔE1/2 = 315 mV) (in [n-Bu4N][B(C6F5)4]). Similarly, the CVs and SWVs of 6 and 7 also showed two successive clearly defined ferrocene-based oxidation waves separated by ΔE1/2 = 180 and 170 mV (in [n-Bu4N][PF6]) and by ΔE1/2 = 280 and 264 mV (in [n-Bu4N][B(C6F5)4]). These findings suggest the existence of appreciable iron–iron interactions between the two neighboring ferrocenyl units linked to the silicon atom. The structurally related linear and cyclic oligo- and polyferrocenylsilanes, having similar silicon-bridged ferrocenyl moieties, have been extensively studied by Manners21 and Pannell22 and exhibit electronic communication between adjacent iron centres.23
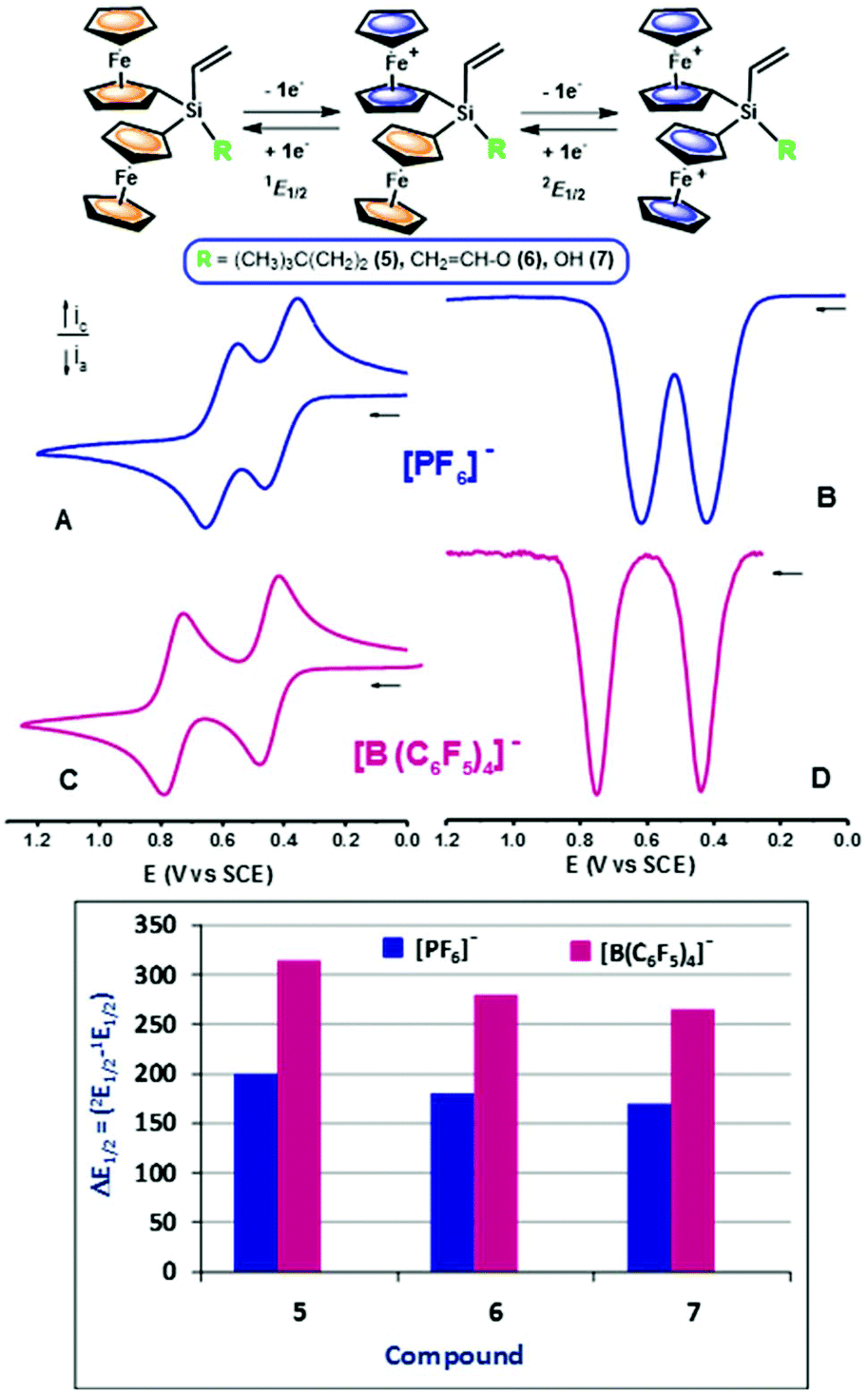 | ||
| Fig. 4 Top: Redox processes experimented by diferrocenylvinylsilanes 5–7. Voltammetric responses, on a Pt electrode, of 5: (A) and (B) recorded in CH2Cl2 containing 0.1 M [n-Bu4N][PF6]; (C) and (D) recorded in CH2Cl2 containing 0.1 M [n-Bu4N][B(C6F5)4]. CVs are at a scan rate of 0.1 V s−1. Bottom: ΔE1/2 values for the two successive oxidations of 5–7 measured in CH2Cl2 in the presence of the weakly coordinating [B(C6F5)4]− (red) or the traditional [PF6]− (blue) supporting electrolyte anions. | ||
For redox-active multimetallic compounds, the magnitude of the separation between the half-wave potentials (ΔE1/2) of two redox sites has usually been taken as a measure of electronic interactions between metal centres. However, one should be extremely careful when using electrochemical data since voltammetric separations are influenced by the effects of the solvent/supporting electrolyte media, which can modify the electrostatic interactions in polycationic species.
The graphical comparison of ΔE1/2 values shown in the bottom of Fig. 4 is also representative of the changes in the redox splitting between the first and second redox events observed for diferrocenylsilanes 5–7 when changing the anion of the supporting electrolyte from nucleophilic [PF6]− to weakly coordinating fluoroarylborate anion. The fact that in the three compounds the low ion-pairing supporting electrolyte [n-Bu4N][B(C6F5)4] increases the separation of the two one-electron oxidations suggests that the through-space interaction is also significant. In addition, this figure shows that the incorporation of –(CH2)2C(CH3)3 substituent in the bridging silicon of diferrocenyl 5 results in an enlarged peak separation, which can be attributed to more effective intermetallic communication between the two ferrocenyl units.
The determination of redox potentials for the successive oxidations of 5–7 allowed us to estimate the comproportionation constant, Kc, relative to the equilibrium [FeII–FeII] + [FeIII–FeIII] ↔ [FeII–FeIII].24 The wave splitting (ΔE1/2) between the first and second oxidations measured in the weakly coordinating supporting electrolyte [n-Bu4N][B(C6F5)4] and the Kc are both indicative of the thermodynamic stability of the mixed valence state of these molecules relative to other redox systems.25 The resulting values of Kc are 211.15 × 103 (for 5), 54.07 × 103 (for 6) and 29.01 × 103 (for 7), calculated from ΔE1/2 electrochemical values, indicating that intermetallic interaction exists in the partially oxidised multiferrocenyl compounds 5–7.19b,26
Many times, a strong electronic communication between active redox centres is allowed by the presence of unsaturated or aromatic units in the bridge. This is nicely reflected in the shape of the HOMO, which involves the bridge, as a sign of delocalisation of the charge. However, neither 6–727 nor 5 (see Fig. 5A) present a HOMO orbital involving the silicon bridges, something not surprising in class II mixed-valence compounds. When dealing with these systems in a vacuum, i.e., looking strictly at the chemical structure, weak non-covalent interactions in the van der Waals range are already observed between the substituents in the silicon bridge and the ferrocenyl units, as evidenced by the NCI (Non Covalent Interaction) analysis represented by green isosurfaces in Fig. 5B. Undoubtedly, the solvent/electrolyte medium plays a very important role on modulating and reinforcing these interactions between centres, as previously observed in the voltammetric measurements. Upon oxidation, the corresponding SOMO orbital occupies the next ferrocenyl unit to be oxidised (Fig. 5A). We obtain a vertical ionisation potential value of 6.08 eV for 5, in agreement with previous values for compounds 6 and 7 (6.16 and 6.08 eV).27 The calculated UV-Vis spectrum of 5 in CH2Cl2 along with the different orbital contributions to each transition is provided in Fig. S35 and Table S14 (see the ESI†).
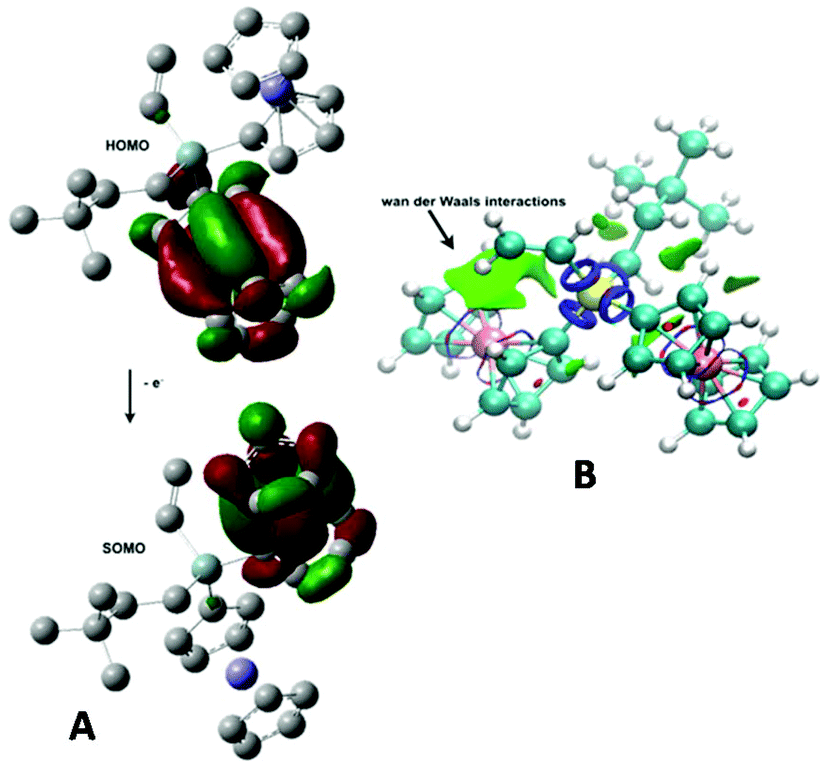 | ||
| Fig. 5 (A): HOMO and SOMO orbitals of compound 5 at the BPW91/6-31+G(d) level of theory. (B): NCI analysis carried out by means of the NCIPLOT program. Blue and red colors denote strong attractive and repulsive interactions, respectively, whereas green color indicates interactions in the van der Waals range. | ||
In addition, electronic structure calculations show that compound 5 presents a different variation of the partial charge of both Fe atoms (0.119 and 0.190 a.u.) after the first oxidation, in agreement with the electrochemical results. This compound exhibits slightly larger iron charge variations on their response to oxidation with respect to values reported for compounds 6 and 7 at the same level of theory.27 Accordingly, spin densities on iron atoms in the radical species present values of 0.699 and 0.398 a.u.
We have also explored the electrochemical oxidation of 8 since the four ferrocenyl redox moieties linked by the Si–O–Si bridge provide an excellent opportunity to study multi-step electron-transfer processes. As shown in Fig. 6, the CV of 8 in CH2Cl2/0.1 M [n-Bu4N][PF6] is considerably more complex than that of the diferrocenyl compounds 5–7, as it exhibits a sequence of several overlapped, and poorly resolved, waves (from about +0.45 to +0.85 V vs. SCE), thereby preventing a detailed analysis of the successive individual oxidations. The more anodic wave is not a diffusion-controlled process and a sharp cathodic stripping wave is observed on the return sweep. This indicates that for disiloxane 8, a change in solubility accompanies the change in the oxidation state, very likely due to the rapid precipitation of the hexafluorophosphate salt [84+][PF6]4 on the electrode surface. On the reverse scan, this tetraoxidised cation is redissolved as it is reduced. Such electrochemical phenomena are well known for multi-ferrocenyl compounds,28 where solubility problems and follow-up reactions result in deviations from ideality when using the traditional nucleophilic electrolyte anion [PF6]− in low-polarity solvents.
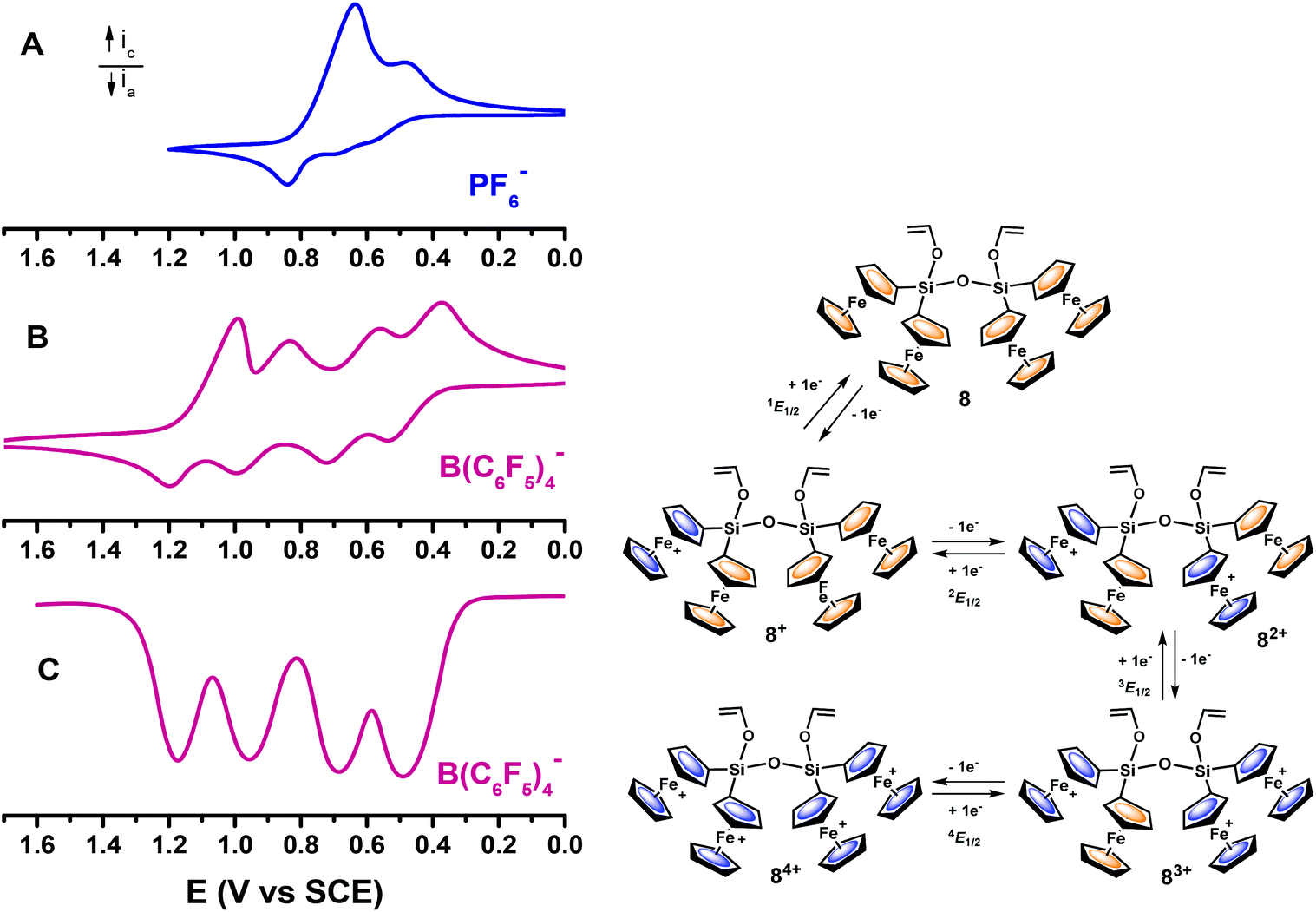 | ||
| Fig. 6 Cyclic and SWV responses, on a platinum electrode of tetrametallic 8, recorded in CH2Cl2 containing 0.1 M [n-Bu4N][PF6] (A) or 0.1 M [n-Bu4N][B(C6F5)4] (B and C) as supporting electrolytes. CVs at a scan rate of 0.1 V s−1. | ||
By using [n-Bu4N][B(C6F5)4] as an electrolyte, we hoped to accurately investigate the multistep electron-transfer processes of tetrametallic 8, because the weakly coordinating [B(C6F5)4]− anion is extremely effective in solubilising positively charged species produced in anodic processes.
Thus, Fig. 6 compares the CV of 8 in CH2Cl2/[NBu4][PF6] with those of the same molecule in CH2Cl2 with 0.1 M [n-Bu4N][B(C6F5)4], and shows the striking improvement observed for the anodic reaction of this tetrametallic disiloxane. With [B(C6F5)4]− the adsorption effects on the working electrode were minimised, which indicates an increase in solubility of the tetracationic species 84+ in this solvent/electrolyte medium. In addition, in agreement with the results reported by Geiger and coworkers, as compared to the small inorganic [PF6]− anion, the [B(C6F5)4]− electrolyte anion has a lower coordinating power and restrains ion pairing, allowing the development of interactions between the four metallocene units in 8, which are electrostatic in nature, that is, mostly through-space interactions. This results in the observation of four well-resolved one-electron oxidation waves, at different potentials, for each of the four ferrocenyl moieties. Likewise, SWV of 8 measured in CH2Cl2/[n-Bu4N][B(C6F5)4] (Fig. 6C) also shows four well-separated waves. The height peak of the two more anodic waves is somewhat smaller than that of the first two waves. This effect has been previously observed in ferrocenyl29 and fulvalenediyl multimetallic compounds30 and can be taken as a qualitative indication that the last one-electron transfers are slightly slower than that of the first two oxidations.31
The mechanism for the electrochemical oxidation of this tetrametallic disiloxane is shown in Fig. 6 and involves a first oxidation (at 1E1/2 = +0.484 V vs. SCE) corresponding to the generation of monocationic species 8+. At a higher potential (2E1/2 = +0.696 V) a second electron is removed from a ferrocenyl moiety attached to the neighbouring silicon atom, at the other end of the Si–O–Si bridge, yielding the dicationic species 82+. The potential separation between the second and third redox processes, ΔE1/2 = 3E1/2 − 2E1/2 = 256 mV, is larger than 2E1/2 − 1E1/2 = 212 mV and 4E1/2 − 3E1/2 = 228 mV, implying that the third oxidation (at 3E1/2 = +0.952 V) occurs at one of the two remaining ferrocenyl moieties, the adjacent to the last oxidised ferrocenyl subunit.14e,16 The final oxidation of the last neutral ferrocenyl centre is the most difficult one and takes place at more anodic potential (4E1/2 = +1.180 V), giving the tetracationic species 84+. The considerable spread of the four oxidations in disiloxane 8 suggests appreciable intermetallic interactions between the Si- and Si–O–Si-bridged ferrocenyl moieties.14e,16 The observed shifts of the oxidation potential values for the 8/8+/82+/83+/84+ couples (see Fig. 6) when going from [PF6]− to [B(C6F5)4]− media are consistent with a considerable decrease in the ion pairing of the FeIII centres with the weakly coordinating fluoroarylborate anion as the ferrocenyl moieties are progressively oxidised.
Experimental section
General considerations
:5. Then, 0.5–1 μL of the mixture was deposited on the target plate using the dried droplet method. The positive ion and the reflectron mode were used for these analyses.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2: synthesis of Fc3Si–CHCH2 (1), Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6) and Fc2(OH)SiCHCH2 (7).
15 g of sublimed ferrocene (80.6 mmol) was dissolved in a mixture of 100 mL of dry THF and 100 mL of dry n-hexane, under argon at room temperature, and then cooled to −10 °C. To this stirred system, a 1.7 M solution of t-BuLi in n-pentane (72.0 mL, 121.0 mmol) was added dropwise. The mixture was stirred for another 30 min and cooled to −20 °C before a solution of Cl3SiCHCH2 (3.2 mL, 24.2 mmol) in 15 mL of dry THF was added dropwise. The solution was allowed to warm to room temperature and was stirred overnight. The mixture was filtered, treated with n-hexane, and then filtered once again to remove the LiCl byproduct. Solvent removal yielded a red-orange oily product, which was purified by column chromatography on silica gel (6 cm × 10 cm) using n-hexane as an eluent. A first band containing unreacted ferrocene was eluted, and subsequently, a second orange band was obtained with n-hexane/CH2Cl2 (10:1). Solvent removal afforded the reaction side product 5, which was obtained as an analytically pure, air-stable, yellow-orangish crystalline solid. Subsequently, on eluting with n-hexane/CH2Cl2 (10:2) a third major band was collected. Solvent removal afforded a mixture of two compounds (1 and 6). This mixture was subjected to a second column chromatography on silica gel (6 cm × 16 cm). A first band was eluted with n-hexane/CH2Cl2 (10:2) and solvent removal afforded compound 1 as an analytically pure, air-stable, orange crystalline solid. The other fractions of this column were again mixtures of compounds 1 and 6. After several columns and recrystallisations in n-hexane/CH2Cl2, compound 6 was finally isolated as an analytically pure, air-stable, orange crystalline solid. In some of the repeated reactions, a final fraction was obtained from the first column chromatography, eluted with CH2Cl2. After the appropriate solvent removal process, compound 7 was obtained as a pure, air-stable, light orange crystalline solid.
CH2: synthesis of Fc3Si–CHCH2 (1), Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6) and Fc2(OH)SiCHCH2 (7).
15 g of sublimed ferrocene (80.6 mmol) was dissolved in a mixture of 100 mL of dry THF and 100 mL of dry n-hexane, under argon at room temperature, and then cooled to −10 °C. To this stirred system, a 1.7 M solution of t-BuLi in n-pentane (72.0 mL, 121.0 mmol) was added dropwise. The mixture was stirred for another 30 min and cooled to −20 °C before a solution of Cl3SiCHCH2 (3.2 mL, 24.2 mmol) in 15 mL of dry THF was added dropwise. The solution was allowed to warm to room temperature and was stirred overnight. The mixture was filtered, treated with n-hexane, and then filtered once again to remove the LiCl byproduct. Solvent removal yielded a red-orange oily product, which was purified by column chromatography on silica gel (6 cm × 10 cm) using n-hexane as an eluent. A first band containing unreacted ferrocene was eluted, and subsequently, a second orange band was obtained with n-hexane/CH2Cl2 (10:1). Solvent removal afforded the reaction side product 5, which was obtained as an analytically pure, air-stable, yellow-orangish crystalline solid. Subsequently, on eluting with n-hexane/CH2Cl2 (10:2) a third major band was collected. Solvent removal afforded a mixture of two compounds (1 and 6). This mixture was subjected to a second column chromatography on silica gel (6 cm × 16 cm). A first band was eluted with n-hexane/CH2Cl2 (10:2) and solvent removal afforded compound 1 as an analytically pure, air-stable, orange crystalline solid. The other fractions of this column were again mixtures of compounds 1 and 6. After several columns and recrystallisations in n-hexane/CH2Cl2, compound 6 was finally isolated as an analytically pure, air-stable, orange crystalline solid. In some of the repeated reactions, a final fraction was obtained from the first column chromatography, eluted with CH2Cl2. After the appropriate solvent removal process, compound 7 was obtained as a pure, air-stable, light orange crystalline solid.
Data for 1. Yield: 9.59 g (65%). 1H NMR (CDCl3, 300 MHz, ppm): δ 4.05 (s, 15H, C5H5), 4.25, 4.39 (m, 12H, C5H4), 6.08 (dd, 3J = 20.2 Hz, 2J = 4.0 Hz, 1H, CH
CHtransHcis), 6.25 (dd, 3J = 14.7 Hz, 2J = 4.0 Hz, 1H, CHCHtransHcis), 6.69 (dd, 3J = 20.2 Hz, 3J = 14.7 Hz, 1H, CHCH2). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 68.7 (C5H5), 70.8, 74.0 (C5H4), 132.2 (CHCH2), 136.4 (CHCH2). 29Si {1H} NMR (CDCl3, 59 MHz, ppm): δ −17.0 (SiFc). IR (KBr, cm−1): ν(CC) 1163, ν(Si–C) 820. MS (MALDI-TOF): m/z 610.1 [M+]. Anal. calcd for C32H30Fe3Si: C, 62.95; H, 4.96. Found: C, 62.68; H, 4.98.
Data for 5. Yield: 0.74 g (6%). 1H NMR (CDCl3, 300 MHz, ppm): δ 0.94 (s, 9H, CH3), 1.02, 1.44 (m, 4H –CH2–), 4.10 (s, 10H, C5H5), 4.15, 4.37 (m, 8H, C5H4), 5.83 (dd, 3J = 20.3 Hz, 2J = 4.0 Hz, 1H, CH
CHtransHcis), 6.13 (dd, 3J = 14.7 Hz, 2J = 4.0 Hz, 1H, CHCHtransHcis), 6.46 (dd, 3J = 20.3 Hz, 3J = 14.7 Hz, 1H, CHCH2). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 9.0 (Si–CH2), 28.9 (CH3), 31.3 (C–CH3), 38.3 (CH2–C), 68.4 (C5H5), 68.5 (ipso-Fc), 70.76, 70.78, 73.7, 73.8 (C5H4), 133.1 (CHCH2), 136.9 (CHCH2). 29Si {1H} NMR (CDCl3, 59 MHz, ppm): δ −12.1 (SiFc). IR (KBr, cm−1): ν(C–C) 2950, ν(CC) 1162, ν(Si–C) 819. MS (MALDI-TOF): m/z 510.2 [M+]. Anal. calcd for C28H34Fe2Si: C, 65.87; H, 6.71. Found: C, 65.59; H, 6.85.
Data for 6. Yield: 1.02 g (9%). 1H NMR (CDCl3, 300 MHz, ppm): δ 4.16 (s, 10H, C5H5), 4.16 (d, 3J = 5.7 Hz, 1H, O–CH
CHtransHcis), 4.25, 4.29, 4.42 (m, 8H, C5H4), 4.58 (d, 3J = 13.5 Hz, 1H, O–CHCHtrans Hcis), 6.11 (dd, 3J = 20.0 Hz, 2J = 4.2 Hz, 1H, CHCHtransHcis), 6.28 (dd, 3J = 14.9 Hz, 2J = 4.2 Hz, 1H, CHCHtransHcis), 6.46 (dd, 3J = 20.0 Hz, 3J = 14.9 Hz, 1H, CHCH2), 6.59 (dd, 3J = 13.5 Hz, 3J = 5.7 Hz, 1H, O–CHCH2). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 68.5 (ipso-Fc), 68.7 (C5H5), 71.3, 71.4, 73.7, 73.8 (C5H4), 94.2 (O–CHCH2), 134.3 (CHCH2), 135.6 (CHCH2), 146.3 (O–CHCH2). 29Si {1H} NMR (CDCl3, 59 MHz, ppm): δ −7.8 (SiFc). IR (KBr, cm−1): ν(CC) 1633, 1167, ν(Si–C) 824. MS (MALDI-TOF): m/z 468.1 [M+]. Anal. calcd for C24H24Fe2SiO: C, 61.53; H, 5.17. Found: C, 61.36; H, 5.05.
Data for 7. Yield: 0.53 g (5%). 1H NMR (CDCl3, 300 MHz, ppm): δ 2.15 (s, 1H, OH), 4.17 (s, 10H, C5H5), 4.26, 4.29, 4.42 (m, 8H, C5H4), 6.11 (dd, 3J = 20.2 Hz, 2J = 4.1 Hz, 1H, CH
CHtransHcis), 6.23 (dd, 3J = 15.0 Hz, 2J = 4.1 Hz, 1H, CHCHtransHcis), 6.51 (dd, 3J = 20.2 Hz, 3J = 15.0 Hz, 1H, CHCH2). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 67.8 (ipso-Fc), 68.5 (C5H5), 71.2, 71.3, 73.4, 73.5 (C5H4), 134.0 (CHCH2), 136.3 (CHCH2). 29Si {1H} NMR (CDCl3, 59 MHz, ppm): δ −8.9 (SiFc). IR (KBr, cm−1): ν(O–H) 3604, 3413, ν(CC) 1166, ν(Si–C) 822. MS (MALDI-TOF): m/z 442.1 [M+]. Anal. calcd for C22H22Fe2SiO: C, 59.73; H, 5.02. Found: C, 59.60; H, 5.15.
CH–O)Si–O–Si(O–CHCH2)Fc2 (8), Fc2(CH2CH–O)Si–O–SiFc3 (9) and Fc3Si–O–SiFc3 (10).
Following the same procedure as the one described for the previous reaction, sublimed ferrocene (4.5 g, 24.2 mmol) was dissolved in 30 mL of dry THF and 30 mL of dry n-hexane and treated with a solution of Cl3Si–O–SiCl3 (0.69 mL, 3.6 mmol) in 7 mL of THF. The red-orange oily product obtained after appropriate treatment was purified by column chromatography on silica gel (3 cm × 11 cm). Unreacted ferrocene was firstly eluted with n-hexane and, subsequently, a second orange band was obtained with n-hexane/CH2Cl2 (10:3). Solvent removal afforded disiloxane 8 as an analytically pure, air-stable, yellow-orangish crystalline solid. Subsequently, on eluting with different n-hexane/CH2Cl2 mixtures, diverse bands were collected. After MALDI-TOF analysis, these fractions were found to correspond to a mixture of compounds 9 and 10, impossible to isolate in quantitative yields.
Data for 8. Yield: 0.94 g (29%). 1H NMR (CDCl3, 300 MHz, ppm): δ 4.16 (s, 20H, C5H5), 4.26 (d, 3J = 5.8 Hz, 1H, O–CH
CHtransHcis), 4.29, 4.36, 4.40 (m, 16H, C5H4), 4.68 (d, 3J = 13.5 Hz, 1H, O–CHCHtrans Hcis), 6.82 (dd, 3J = 13.5 Hz, 3J = 5.8 Hz, 1H, O–CHCH2). 13C{1H} NMR (CDCl3, 75 MHz, ppm): δ 65.8 (ipso-Fc), 68.8 (C5H5), 71.1, 71.2, 73.6, 73.8 (C5H4), 94.6 (O–CHCH2), 145.6 (O–CHCH2). 29Si{1H} NMR (CDCl3, 59 MHz, ppm): δ −27.7 (SiFc). IR (KBr, cm−1): ν(CC) 1636, 1171, ν(Si–O–Si) 1075, ν(Si–C) 815. MS (MALDI-TOF): m/z 898.1 [M+]. Anal. calcd for C44H42Fe4Si2O3: C, 58.80; H, 4.71. Found: C, 58.95; H, 4.63.
Conclusions
In this work, we have successfully isolated and characterised several silicon- and siloxane-bridged multiferrocenyl compounds bearing different reactive groups in the solid and solution states. We propose that species Fc2(CH3)3C(CH2)2SiCHCH2 (5), Fc2(CH2CH–O)SiCHCH2 (6), Fc2(CH2CH–O)Si–O–Si(O–CHCH2)Fc2 (8) and Fc2(CH2CH–O)Si–O–SiFc3 (9) have resulted from the competing salt-metathesis reactions of the corresponding chlorosilanes (Cl3SiCHCH2 or Cl3Si–O–SiCl3) with lithioferrocene and the organolithium reagents CH2CH–OLi or (CH3)3C(CH2)2Li which, in turn, have been generated in situ by degradation of THF. Multiferrocenyl species 6, 8 and 9 are noteworthy since, for the first time, a CH2CH–O− fragment resulting from the cleavage of the THF ring has been entrapped and stored in very stable and redox-active compounds, which have been crystallographically characterised. To the best of our knowledge, these compounds are the first redox-active molecules containing a reactive silyl vinyl ether group. In addition, biferrocenyl 6, carrying both a vinyl and a vinyloxy group, is also of interest since it represents the first, and so far the only example of an electroactive silyl mononomer with two different reactive polymerisable groups.
Electrochemical studies reveal that, in both electrolyte systems containing either the traditional [PF6]− or the weakly coordinating [B(C6F5)4]− anion, the silicon-bridged diferrocenylvinyl compounds 5–7 undergo two well-separated reversible one-electron oxidations, indicating intermetallic interactions between the ferrocenyl moieties. The introduction of the 3,3-dimethylbutyl group in the silicon bridge of 5 seems to increase the metal–metal interaction with respect to that of the structurally related diferrocenyl systems 6 and 7. In addition, tetraferrocenyl disiloxane 8 displays an interesting redox behaviour that can be switched from a poorly unresolved multistep redox process, to four consecutive, clearly defined, one-electron oxidations by changing the anion of the supporting electrolyte from [PF6]− to the weak nucleophilic and ion-pairing [B(C6F5)4]− anion. Future studies will focus on studying the reactivity of 6, 7 and 8 as redox-active bifunctional silyl monomers.
Acknowledgements
The authors are grateful to the Spanish Ministerio de Economía y Competitividad (MINECO) (projects CTQ2012-30728 and CTQ2015-63997-C2-1-P) for the generous support of this work. M. M. M.-C. and O. M. thank the STSM COST Action CM1204 and the Project FOTOCARBON-CM S2013/MIT-2841 of the Comunidad Autónoma de Madrid. Computational time at Centro de Computación Científica (CCC) of Universidad Autónoma de Madrid is acknowledged.References
- (a) M. Schlosser, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, John Wiley & Sons Ltd, Chichester, 2nd edn, 2002, ch. 1, pp. 1–352 Search PubMed; (b) J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, Elsevier, Oxford, 2002, ch. 7, pp. 273–280 Search PubMed.
- J. Clayden and S. A. Yasin, New J. Chem., 2002, 26, 191–192 RSC.
- R. B. Bates, L. M. Kroposki and D. E. Potter, J. Org. Chem., 1972, 37, 560–562 CrossRef CAS.
- P. Stanetty and M. D. J. Mihovilovic, Org. Chem., 1997, 62, 1514–1515 CrossRef CAS.
- A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706–708 CrossRef CAS PubMed.
- R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588–591 CrossRef CAS PubMed.
- Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons Ltd, West Sussex, England, 2008 Search PubMed.
- (a) D. Guillaneux and H. B. Kagan, J. Org. Chem., 1995, 60, 2502–2505 CrossRef CAS; (b) B. Bildstein, M. Malaun, H. Kopacka, K. Wurst, M. Mitterböck, K.-H. Ongania, G. Opromolla and P. Zanello, Organometallics, 1999, 18, 4325–4336 CrossRef CAS; (c) R. Sander and U. T. Mueller-Westherhoff, J. Organomet. Chem., 1996, 512, 219–224 CrossRef.
- (a) M. D. Rausch and D. J. Ciappenelli, J. Organomet. Chem., 1967, 10, 127–136 CrossRef CAS; (b) J. J. Bishop, A. Davison, M. L. Katcher, D. W. Lichtenberg, R. E. Merrill and J. C. Smart, J. Organomet. Chem., 1971, 27, 241–249 CrossRef CAS; (c) R. F. Kovar, M. D. Rausch and H. Rosenberg, Organomet. Chem. Synth., 1970/1971, 1, 173 Search PubMed; (d) D. A. Rider, K. A. Cavicchi, N. K. Power-Billard, T. P. Russell and I. Manners, Macromolecules, 2005, 38, 6931–6938 CrossRef CAS.
- (a) K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Chem. Commun., 2001, 1678–1679 RSC; (b) W. Clegg, K. W. Henderson, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and D. M. Tooke, Angew. Chem., Int. Ed., 2001, 40, 3902–3905 CrossRef CAS; (c) P. C. Andrikopoulos, D. R. Armstrong, W. Clegg, C. J. Gilfillan, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, J. A. Parkinson and D. M. Tooke, J. Am. Chem. Soc., 2004, 126, 11612–11620 CrossRef CAS PubMed; (d) W. Clegg, E. Crosbie, S. H. Dale-Black, E. Hevia, G. W. Honeyman, A. R. Kennedy, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Organometallics, 2015, 34, 2580–2589 CrossRef CAS.
- E. Hevia, A. R. Kennedy and M. D. McCall, Dalton Trans., 2012, 41, 98–103 RSC.
- G. Dayaker, A. Sreeshailam, F. Chevallier, T. Roisnel, P. R. Krishna and F. Mongin, Chem. Commun., 2010, 46, 2862–2864 RSC.
- A. H. Stoll, P. Mayer and P. Knochel, Organometallics, 2007, 26, 6694–6697 CrossRef CAS.
- (a) S. Bruña, A. M. González-Vadillo, D. Nieto, C. J. Pastor and I. Cuadrado, Macromolecules, 2012, 45, 781–779 CrossRef; (b) M. Zamora, S. Bruña, B. Alonso and I. Cuadrado, Macromolecules, 2011, 44, 7994–8007 CrossRef CAS; (c) S. Bruña, S. D. Nieto, A. M. González-Vadillo, J. Perles and I. Cuadrado, Organometallics, 2012, 31, 3248–3258 CrossRef; (d) I. Cuadrado, C. M. Casado, B. Alonso, M. Morán, J. Losada and V. Belsky, J. Am. Chem. Soc., 1997, 119, 7613–7614 CrossRef CAS; (e) S. Bruña, A. F. Garrido-Castro, J. Perles, M. M. Montero-Campillo, O. Mó, A. E. Kaifer and I. Cuadrado, Organometallics, 2016, 35, 3507–3519 CrossRef; (f) I. Cuadrado, in Silicon-Containing Dendritic Polymers, ed. P. R. Dvornic and M. J. Owen, Springer, Germany, 2009, vol. 2, ch. 8, pp. 141–196 Search PubMed.
- For interesting examples of multiferrocenyl compounds see for instance: (a) L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC; (b) J.-K. Ou-Yang, L.-J. Chen, L. Xu, C.-H. Wang and H.-B. Yang, Chim. Chem. Lett., 2013, 14, 471–474 CrossRef; (c) Q.-J. Li, G.-Z. Zhao, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2012, 31, 7241–7247 CrossRef CAS; (d) G.-Z. Zhao, Q.-J. Li, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang and H.-B. Yang, Organometallics, 2011, 30, 5141–5146 CrossRef CAS; (e) M. S. Inkpen, S. Scheerer, M. Linseis, A. J. P. White, R. F. Winter, T. Albrecht and N. J. Long, Nat. Chem., 2016, 8, 825–830 CrossRef CAS PubMed; (f) A. Hildebrandt and H. Lang, Organometallics, 2013, 32, 5640–5653 CrossRef CAS; (g) U. Pfaff, G. Filipczyk, A. Hildebrandt, M. Korb and H. Lang, Dalton Trans., 2014, 43, 16310–16321 RSC; (h) Y. V. Zatsikha, C. D. Holstrom, K. Chanawanno, A. J. Osinski, C. J. Ziegler and V. N. Nemykin, Inorg. Chem., 2017, 56, 991–1000 CrossRef CAS PubMed.
- S. Bruña, A. M. González-Vadillo, D. Nieto, C. J. Pastor and I. Cuadrado, Organometallics, 2010, 29, 2796–2807 CrossRef.
- S. Bruña, J. Perles, D. Nieto, A. M. González-Vadillo and I. Cuadrado, J. Organomet. Chem., 2014, 751, 769–780 CrossRef.
- See for example: (a) S. E. S. Martin and D. A. Watson, J. Am. Chem. Soc., 2013, 135, 13330–13333 CrossRef CAS PubMed; (b) T. Moyori, T. Hayashi and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3516–3522 CrossRef CAS; (c) C. Chen, S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2008, 130, 12892–12893 CrossRef CAS PubMed; (d) S. Luo and R. F. Jordan, J. Am. Chem. Soc., 2006, 128, 12072–12073 CrossRef CAS PubMed.
- (a) S. Santi, A. Bisello, R. Cardena and A. Donoli, Dalton Trans., 2015, 44, 5234–5257 RSC; (b) Inorganic Electrochemistry, ed. P. Zanello, F. Fabrizi de Biani and C. Nervi, Royal Society of Chemistry, Cambridge, 2nd edn, 2012 Search PubMed; (c) S. Barlow and D. O'Hare, Chem. Rev., 1997, 97, 637–670 CrossRef CAS PubMed; (d) D. Astruc, Electron Transfer and Radical Processes in Transition-Metal Chemistry, VCH, New York, 1995 Search PubMed.
- (a) W. E. Geiger and F. Barriére, Acc. Chem. Res., 2010, 43, 1030–1039 CrossRef CAS PubMed; (b) F. Barriére and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980–3989 CrossRef PubMed; (c) A. Nadafy, T. T. Chin and W. E. Geiger, Organometallics, 2006, 25, 1654–1663 CrossRef; (d) F. Barrière, N. Camire, W. E. Geiger, U. T. Mueller-Westerhoff and R. Sanders, J. Am. Chem. Soc., 2002, 124, 7262–7263 CrossRef; (e) R. J. LeSuer and W. E. Geiger, Angew. Chem., Int. Ed., 2000, 39, 248–250 CrossRef CAS.
- (a) D. A. Foucher, C. H. Honeyman, J. M. Nelson, B. Z. Tang and I. Manners, Angew. Chem., Int. Ed. Engl., 1993, 32, 1709–1711 CrossRef; (b) R. Rulkens, A. J. Lough, I. Manners, S. R. Lovelace, C. Grant and W. E. Geiger, J. Am. Chem. Soc., 1996, 118, 12683–12695 CrossRef CAS; (c) D. E. Herbert, J. B. Gilroy, W. Y. Chan, L. Chabanne, A. Staubitz, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2009, 131, 14958–14968 CrossRef CAS PubMed.
- (a) V. V. Dement'ev, F. Cervantes-Lee, L. Parkanyi, H. Sharma, K. H. Pannell, M. T. Nguyen and A. F. Diaz, Organometallics, 1993, 12, 1983–1987 CrossRef; (b) M. T. Nguyen, A. F. Diaz, V. V. Dementiev and K. H. Pannell, Chem. Mater., 1993, 5, 1389–1394 CrossRef CAS; (c) K. H. Pannell, V. V. Dementiev, H. Li, F. Cervantes-Lee, M. T. Nguyen and A. F. Diaz, Organometallics, 1994, 13, 3644–3650 CrossRef CAS.
- When [n-Bu4N][PF6] is used as the supporting electrolyte in the low polarity dichloromethane solvent, the strong ion pairing between the positively charged electrogenerated ferrocenium species and the [PF6]− anion of the electrolyte, leave little or any electrostatic effect. See: A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef CAS PubMed.
- The value of Kc can be determined using the equation Kc = exp[FΔE1/2/RT]. See: D. E. Richardson and H. Taube, Inorg. Chem., 1981, 20, 1278–1285 CrossRef CAS.
- Both the E1/2 and the splitting ΔE1/2 values are strongly dependent on the solvent and supporting electrolyte anion employed. See ref. 19 and references therein: (a) R. F. Winter, Organometallics, 2014, 33, 4517–4536 CrossRef CAS; (b) D. M. D'Alessandro and F. R. Keene, Dalton Trans., 2004, 3950–3954 RSC.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS.
- M. M. Montero-Campillo, S. Bruña, I. Cuadrado and O. Mó, Comput. Theor. Chem., 2015, 1053, 281–288 CrossRef CAS.
- This stripping-type behaviour was observed for the first time in poly(vinylferrocene). See for example: (a) J. B. Flanagan, S. Margel, A. J. Bard and F. C. Anson, J. Am. Chem. Soc., 1978, 100, 4248–4253 CrossRef CAS; (b) A. Merz and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 3222–3223 CrossRef CAS.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Rüffer, T. Hahn, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761–6771 CrossRef CAS.
- A. Nadafy and W. E. Geiger, Organometallics, 2008, 27, 5624–5631 CrossRef.
- J. Osteryoung and J. J. O'Dea, in Electroanalytical Chemistry, ed. A. J. Bard, Marcel Dekker, Inc., New York, 1986, vol. 14 Search PubMed.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS Version 2.03, Program for Empirical 951 Absorption Correction, University of Göttingen, Germany, 1997–2001 Search PubMed.
- SAINT+NT Version 6.04, SAX Area-Detector Integration Program, Bruker Analytical X-ray Instruments, Madison, WI, 1997–2001 Search PubMed.
- Bruker AXS SHELXTL Version 6.10, Structure Determination Package, Bruker Analytical X-ray Instruments, Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure 960 Refinement, Germany, 1997 Search PubMed.
- M. J. Frisch, et al., Gaussian09, Revision D.01, Gaussian, Inc., Wallingford, 2009 Search PubMed.
- (a) J. P. Perdew, in Electronic structure of solids, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991 Search PubMed; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon, Oxford, 1990 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures referenced in the text; spectroscopic, theoretical, X-ray crystallographic and CV and SWV data. CCDC 1556608–1556612. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt02286g |
| This journal is © The Royal Society of Chemistry 2017 |