
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and investigation of neptunium zirconium phosphate, a member of the NZP family: crystal structure, thermal behaviour and Mössbauer spectroscopy studies
D. M.
Bykov
 *ab,
R. J. M.
Konings
b,
C.
Apostolidis
b,
A.
Hen
b,
E.
Colineau
b,
T.
Wiss
b and
P.
Raison
b
*ab,
R. J. M.
Konings
b,
C.
Apostolidis
b,
A.
Hen
b,
E.
Colineau
b,
T.
Wiss
b and
P.
Raison
b
aDelft University of Technology, Faculty of Applied Sciences, Department of Radiation Science and Technology, Mekelweg 15, 2629 JB Delft, The Netherlands. E-mail: d.bykov@tudelft.nl
bEuropean Commission, Joint Research Center (JRC) – Karlsruhe, P.O. Box 2340, D-76125 Karlsruhe, Germany
First published on 23rd August 2017
Abstract
A new double neptunium zirconium phosphate of the type MxZr2(PO4)3 (M = Np), crystallizing in the structure type NaZr2(PO4)3 (NZP, NASICON), was synthesized by solid state reactions at high temperatures and characterized by X-ray diffraction, infrared spectroscopy and Mössbauer spectroscopy. The Rietveld refinement of the XRD pattern together with the analysis of the IR spectra of the sample confirmed the space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, the same as that for the lanthanide analogues Ln0.33Zr2(PO4)3. However, Mössbauer studies revealed the presence of neptunium in the two oxidation states +3 and +4, indicating a two-phase NZP system with different crystallographic environments of the neptunium atoms. The thermal behaviour of the sample was followed up to 1400 °C by thermogravimetric analysis.
c, the same as that for the lanthanide analogues Ln0.33Zr2(PO4)3. However, Mössbauer studies revealed the presence of neptunium in the two oxidation states +3 and +4, indicating a two-phase NZP system with different crystallographic environments of the neptunium atoms. The thermal behaviour of the sample was followed up to 1400 °C by thermogravimetric analysis.
Introduction
Anhydrous phosphates with the crystal structure of NaZr2(PO4)3 (NZP, NASICON) constitute a family of compounds with a number of useful properties: high thermal1 and chemical stability,2,3 resistance to radiation,4,5 low coefficients of thermal expansion,6 luminescence properties7–10 and ionic conductivity.11–13A special feature peculiar to this family of compounds is the ability to incorporate actinides and other heavy metals, which makes them potentially suitable ceramic hosts for the storage and disposal of radioactive waste. This property is attributed to the high isomorphic capacity of the NZP structure, which facilitates the incorporation of a variety of elements of different ionic radii, valence states, structures of electronic shells (s-, p-, d- and f-elements) and electronegativities.1 Taking into consideration the complexity of nuclear waste streams, this property is an asset, since it potentially allows incorporation of different waste components into a single-phase ceramic form.
The framework structure of the parent compound NaZr2(PO4)3 with the space group Rc and the crystal chemical formula (M1)[6](M2)[8]3[L2[6](P[4]O4)3] consists of units of two ZrO6 octahedra linked to three PO4 tetrahedra, which are aligned along the crystallographic c-direction of the unit cell, forming endless chains.14 Zirconium cations in the octahedral coordination (positions of the framework or L positions) can be substituted preferentially by cations of higher ionic charge and smaller radii in the range of 0.53–0.9 Å.15 Interstitial M1 positions, occupied by sodium, are located between the structure units of two ZrO6 octahedra and three PO4 tetrahedra. These positions are suitable for preferential substitution by larger cations of lower charge with radii in the range of 0.71–1.65 Å.15 In sodium zirconium phosphate the interstitial M2 positions remain vacant but if the concentration of monovalent cations needed for the compensation of framework charges increases (for example, as a result of substitution of zirconium by trivalent cations), these sites will become occupied as well. However, such examples of filling the M2 positions are almost completely limited to the monovalent cations Li+, Na+ and K+.
Isomorphic substitutions of cations in the interstitial and framework positions of the prototype compound NaZr2(PO4)3 can be accompanied by the reduction of unit cell symmetry without major distortions of the framework structure units. Consequently, the NZP class of materials includes several space groups of related symmetry: Rc, R, R3, R32, Pc, P.1,16,17 In a number of cases monoclinic distortion of the lattice can lead to the appearance of a superstructure with space groups C2/c or Cc.1
The inclusion of tri- and tetravalent cations of lanthanides and some of the actinides into the structure of NZP phosphates was reported in a number of papers. These elements can be incorporated into octahedral zirconium positions (L positions) or into interstitial M1 positions of the structure. Several studies have described the existence of limited solid solutions of the following types at room temperature: KZr2−xUx(PO4)3 (0 ≤ x ≤ 0.2),18 NaZr2−xNpx(PO4)3 (0 ≤ x ≤ 0.6), NaZr2−xPux(PO4)3 (0 ≤ x ≤ 0.2),19,20 KZr2−xPux(PO4)3 (0 ≤ x ≤ 0.35, x = 2).21 The actinide end members of these series, the phosphates AM2(PO4)3 (M = U, A = K, Rb and M = Np, Pu, A = Na, K, Rb), were shown to have polymorphic high-temperature NZP modifications (prepared at 1000 °C or above), while crystallizing in other structure types at lower temperatures.22–24 Solid solutions with trivalent rare earth elements at the L positions of the NZP structure with general formula Na1+xRxZr2−x(PO4)3 (R = In, Yb, Er, Y, Dy, Tb, Gd, Sm, 0 ≤ x ≤ 1) are also known.11,25 It was shown that the limits of isomorphic substitutions of the REE ions in the framework positions strongly depended on the ionic radii of trivalent cations and rapidly decreased with increasing the latter. Thus, the maximum value of x for Sm3+, the largest of the studied cations in the paper by Miyajima et al.11 (r(Sm3+) = 0.958 Å (CN = 6)), was close to 0.2.
In the above examples the 4f- and 5f-elements occupy smaller zirconium positions of the structure. At the same time, the bigger M1 positions are expected to be more suitable for the larger cations of lanthanides and actinides in oxidation states +3 and +4. Such examples are known: phosphates of trivalent lanthanides Ln0.33Zr2(PO4)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,16 and actinides Am0.33Zr2(PO4)3,26 and of tetravalent actinides Th0.25Zr2(PO4)3,27,28 U0.25Zr2(PO4)327 and Pu0.25Zr2(PO4)3.26 The isoformula phosphates with d-elements Zr0.25Zr2(PO4)3 and Hf0.25Hf2(PO4)429–31 were also described. However, in the case of actinide compounds detailed crystal structure characterization is often lacking. Such studies are needed to understand the structural chemistry of actinides in this class of materials, proposed as alternative waste forms.
7,16 and actinides Am0.33Zr2(PO4)3,26 and of tetravalent actinides Th0.25Zr2(PO4)3,27,28 U0.25Zr2(PO4)327 and Pu0.25Zr2(PO4)3.26 The isoformula phosphates with d-elements Zr0.25Zr2(PO4)3 and Hf0.25Hf2(PO4)429–31 were also described. However, in the case of actinide compounds detailed crystal structure characterization is often lacking. Such studies are needed to understand the structural chemistry of actinides in this class of materials, proposed as alternative waste forms.
Neptunium is one of the long-lived components of nuclear waste and contributes substantially to the long term radiotoxicity of waste. The chemistry of the light actinides, to which neptunium belongs, is complex since these elements exhibit a transitional behaviour between the 5d- and 4f-metals. This is due to the proximity of the energies of 7s-, 6d- and 5f-shells which can easily contribute to the chemical bonding depending on the experimental conditions. This results in the possibility of existence of multiple oxidation states for these elements, sometimes simultaneously. Therefore, the question of oxidation state is important when characterizing compounds containing light actinides. With regard to the simple neptunium cations, both tri- and tetravalent oxidation states are expected to have reasonable stability, which is indicated by the slightly positive value of the reduction potential for Np4+/Np3+ pair (Eo = 0.15 V).32
The analysis of the literature data given above suggests several possibilities for the incorporation of tri- and tetravalent neptunium cations into the NZP structure: interstitial sodium positions M1 can accommodate both Np4+ and Np3+ cations to form theoretical compositions Np0.25Zr2(PO4)3 and Np0.33Zr2(PO4)3 with maximum occupancies of 0.25 and 0.33, respectively, while framework zirconium positions would accommodate Np4+ and possibly Np3+ ions with the formation of solid solutions NaZr2−xNpIVx(PO4)3 and Na1+xNpIIIxZr2−x(PO4)3. In the latter case the maximum value of x is not known but is expected to be less than 0.2, the limit established in ref. 11 and 25 for Sm (r(Sm3+) = 0.958 Å, r(Np3+) = 1.01 Å).
In this paper we report the synthesis of a new double neptunium zirconium phosphate of the type NpxZr2(PO4)3, belonging to the NZP family, and the study of its crystal structure, thermal stability and the oxidation state of neptunium.
Experimental
Synthesis
All operations with isotope 237Np were carried out in nitrogen-filled gloveboxes, designed for working with radioactive substances, especially α-emitters. The theoretical composition Np0.25Zr2(PO4)3 was targeted, since previously published research indicated the stabilization of the tetravalent state of neptunium in the NZP phosphates. Therefore, neptunium dioxide NpO2 was taken for the synthesis. Several methods were used for the preparation of the neptunium zirconium phosphate.In the first method, we used solutions of zirconium oxychloride (ZrOCl2) (1.385 M), phosphoric acid (H3PO4) (1.483 M) and a powder of neptunium dioxide (NpO2). Stoichiometric amounts of these materials were mixed together in a glass beaker and carefully dried on a sand bath (T ≈ 50–60 °C) to prevent possible change of the oxidation state of neptunium. The thus obtained precursor powder of light-yellow colour was then ground in an agate mortar with a small amount of isopropyl alcohol. Heat treatments were performed in a furnace under a pure argon atmosphere. After the first heat treatment at 600 °C for 24 h, the powder turned in colour to dark-grey, almost black. It was ground again before the next heating at 800C for 24 h. The sample became black-green after this temperature. This step was followed by XRD analysis. The scheme of the synthesis is summarized in Fig. 1.
 | ||
| Fig. 1 The scheme of synthesis of neptunium zirconium phosphate with the NZP structure. | ||
The second method of synthesis was via solid state reagents. Stoichiometric amounts of the NpO2 powder, ZrOCl2·8H2O and (NH4)2HPO4 in solid forms were placed in an agate-lined stainless steel jar with agate grinding bodies and ground for 23 hours using a vibrational mill RETSCH MM301 at a frequency of 20 s−1. The precursor (yellow colour) was then split into two parts, which were heated under different atmospheres: argon or air (oxygen). The temperature regimes applied for the heat treatments were in both cases 600 °C, 72 h and 800 °C, 24 h. After the first heat treatment at 600 °C the sample synthesized in argon was grey in colour, which remained as such after further heating at 800 °C. In contrast, the sample prepared in air almost did not change its colour (light-yellow) after both heating steps.
It should be noted that both of these routes represent schemes for the synthesis of starting precursor powders needed for obtaining a final ceramic nuclear waste form. The fabrication of such waste forms would require additional steps to produce monolithic materials having acceptable durability. These methods include hot uniaxial pressing (HUP), hot isostatic pressing (HIP), pressing followed by sintering, etc.33
X-ray diffraction analysis
XRD patterns at room temperature were measured using a Seifert 3000 diffractometer, CuKα1,2 radiation, a scintillation detector, Bragg–Brentano geometry, and θ–θ configuration. The diffraction pattern for the Rietveld analysis was collected with a Bruker D8 diffractometer operated in a Bragg–Brentano configuration with a curved Ge (111) monochromator having a ceramic Cu tube and a Vantec position sensitive detector. The measurements were performed at 40 kV and 40 mA. In both machines the goniometers were mounted directly inside the gloveboxes.The XRD samples were prepared by depositing an analysed powder mixed with an epoxy glue on the surface of a specially designed plastic sample holder. This technique allowed avoiding spreading of contamination and simplified further the handling of waste.
Phase analysis was performed using the databases ICSD and PDF4 and the crystallographic software “Match!” (version 1.10). Rietveld analysis was carried out using the FullProf software package.34 Structure visualization was realized with the computer program VESTA.35
SEM analysis
The SEM-EDX analysis was performed on a FEI (Philips) XL 40 instrument with 3 nm resolution using an accelerating voltage of 25 kV. The samples were measured without coating. The detection and quantitative calculations were based on the measurements of the intensities of Mα-lines of neptunium, Lα-lines of zirconium and Kα-lines of phosphorus. Oxygen was not analysed. The EDX spectra were quantified by a standardless method using the PAP correction.36DTA/TG analysis
Differential thermal analysis (DTA) and thermogravimetry (TG) were carried out using Netzsch STA 409 C/CD apparatus. Samples were placed in alundum crucibles and heated in the temperature range from room temperature to 1400 °C. The mass of the analysed powders did not exceed 40 mg. Measurements were performed under a pure nitrogen atmosphere at a flow rate of 70 ml min−1. The heating and cooling rates were 7 K min−1.Infrared spectroscopy
The IR spectrum of the sample at room temperature was obtained using a PerkinElmer 2000 Fourier transform infrared (FT-IR) spectrophotometer with a resolution of 1 cm−1. The powder was diluted in KBr and pressed into pellets for the measurements.Mössbauer spectroscopy
The 237Np Mössbauer spectrum was recorded in a transmission geometry, using the sinusoidal drive motion of a ∼100 mCi 241Am metal source. The maximum velocity scale of the spectrometer (125 mm s−1) was determined with respect to the NpAl2 calibration standard (Bhf = 330 T at 4.2 K). Both the source and the absorber were kept at a constant temperature of 4.2 K using a liquid helium cryostat. The absorber was prepared by encapsulating ∼150 mg of a fine powdered sample of neptunium zirconium phosphate in three concentric aluminium containers.Results and discussion
XRD characterization
Both synthetic routes resulted in the formation of products crystallizing in the structure type NaZr2(PO4)3 but only the sample prepared by method 1 (Fig. 1) gave a powder with enough crystallinity to perform the Rietveld analysis after 800 °C (Fig. 2a). | ||
| Fig. 2 Normalized X-ray powder diffraction patterns of neptunium zirconium phosphate obtained by (a) method 1 at 800 °C; (b) method 2 in argon at 800 °C (only reflections of NpO2 are present); (c) method 2 in argon at 874 °C, 25 h (NZP phase starts to form but NpO2 is still the main phase); (d) method 2 in argon at 900 °C (the main phases are NZP, NpO2 and ZrP2O7). | ||
Heating of the precursor powder prepared by method 2 under an Ar or O2 atmosphere gave powders of grey and light-yellow colours, respectively. Despite the difference in colour, XRD measurements performed after successive heat treatments up to 800 °C both gave practically the same patterns corresponding to NpO2 (Fig. 2b), except that the sample obtained in oxygen also showed small reflections belonging to other phases. Both samples were further heated at 900 °C for 25 h (Ar) and 15 h (O2). Only treatment in argon resulted in the formation of a powder with the NZP structure (Fig. 2d). At the same time, the diffraction peaks were broad and of rather low intensity, which indicated the low crystallinity of the material. In addition, the sample also contained significant amounts of ZrP2O7 and NpO2 admixtures. Heating of the powder, prepared at 600 °C in argon, at 874 °C gave an intermediate XRD pattern (Fig. 2c), which means that NpO2 powder started to react around this temperature.
Comparison of the results of the two methods indicates that the history of the reactants (synthesis route, annealing temperature etc.) needs to be taken into account during the solid state synthesis procedure. Since method 1 resulted in the most crystalline material, only this sample was used for further characterization.
Infrared (IR) spectroscopy
Factor group analysis of vibrations of the orthophosphate ion in compounds with the space group Rc was carried out by Kurazhkovskaya et al.27 For this space group one can expect one ν1 band, two ν2 bands, and five bands of each of the ν3 and ν4 vibrations. Here the symbols ν1, ν2, ν3 and ν4 denote symmetric stretching, symmetric bending, asymmetric stretching and asymmetric bending P–O vibrations, respectively. In the spectra of double zirconium phosphates with alkali elements five bands of asymmetric stretching vibrations overlap, and in the spectra of NaZr2(PO4)3 they appear as one broad band with a maximum at about 1045 cm−1 and a weak high-frequency band (1200 cm−1) due to the contribution of the electron density of Zr to the P–O bond.27 However, with decreasing symmetry (from space group Rc to space groups R and Pc) these bands split and the number of possible bands increases.27 For the phosphates crystallizing in NZP type, this symmetry change corresponds to the compositional change from AM2(PO4)3 (sp. gr. Rc) to B0.5Zr2(PO4)3 (sp. gr. R) and to R0.33Zr2(PO4)3 (sp. gr. Pc) (A, B, and R stand for mono-, di- and trivalent cations). The reduction of symmetry in phosphates Ln0.33Zr2(PO4)3 with the appearance of additional lines in the IR spectra was explained by the ordering of Ln atoms at the M1 positions of the structure.16 It should be also noted that the IR spectra of phosphates M0.25Zr2(PO4)3 (M = Th, U), which were described in the same paper of Kurazhkovskaya et al.,27 resembled much more the spectrum of the parent compound NaZr2(PO4)3, which led to the conclusion that the actinide atoms were distributed over M1 sites statistically and the symmetry Rc remained.
The IR spectrum of the Np phase, synthesized in the present work using method 1, is shown in Fig. 3. It is identical to the spectra of the rare earth phases with the space group Rc.27,37 The IR bands observed in the region from 500 to 640 cm−1 are assigned to the asymmetrical bending ν4 vibrations of the PO4 units, bands from 920 to 980 cm−1 – to symmetrical stretching ν1 vibrations, and from about 1000 to 1280 cm−1 – to asymmetrical stretching ν3 vibrations. As in the case of lanthanide zirconium phosphates, the position of the weak high-frequency band of asymmetric vibrations at 1280 cm−1, which is generally not characteristic of phosphates, is explained by the contribution of a part of the electronic density of small and highly charged polarizing zirconium cations to the P–O bond.27,38 The spectrum is also characterized by the presence of molecular water – bands at 3435 and 1631 cm−1 – which could be a result of absorption of some moisture by the sample during storage, or could be introduced with potassium bromide used for the preparation of pellets for the IR measurements. The band at 2662 cm−1 is assigned to the vibrations of OH-groups in the acidic POH-ions, which is possibly due to the fact that phosphoric acid (H3PO4) was used for the synthesis.
 | ||
| Fig. 3 Infrared spectrum of the studied neptunium zirconium phosphate with NZP structure. | ||
The analogy of the IR spectrum of the neptunium phase, obtained in the present study, with the spectra of double phosphates Ln0.33Zr2(PO4)3 indicates that the neptunium compound possesses the same symmetry as the lanthanide zirconium phosphates, and regarding the oxidation state points to the conclusion that neptunium is present in the trivalent state.
In order to obtain information on the valence state of neptunium in the sample, we performed Mössbauer spectroscopy, a method which provides direct indication of the oxidation state.
Mössbauer spectroscopy
For the investigation, we used a sample prepared by method 1 at 800 °C (see the Experimental section). The Mössbauer spectrum of the neptunium sample is shown in Fig. 4. Taking into account the IR data (a possible trivalent oxidation state of neptunium), the measured spectrum was fitted with three sets of hyperfine parameters, one for the main hexagonal phase, containing neptunium in the oxidation state +3, the second set for the hexagonal phase in which neptunium has the oxidation state +4, and the last set for a cubic NpO2 (which was also identified from the X-ray diffraction pattern). The values of isomer shifts and parameters of quadrupole interaction for the spectra of the main phase, the secondary phase and NpO2 are shown in Table 1. Estimation of the relative concentrations from the fitted data yields roughly (in mol% Np) 77% for the main hexagonal phase, 14% for the secondary hexagonal phase and 9% for the cubic NpO2. It should be noted that these percentages are based on the area integration of the different sub-spectra and might vary, especially due to the relatively low measured signal, high background and the fact that there is an excess of material that is not visible (no Np in it) and might be caused by a non-homogeneous mixture of the different phases within the sample. | ||
| Fig. 4 Mössbauer spectrum of the double neptunium zirconium phosphate. Dots represent the experimental data, red line – the fitted curve, blue line – the contribution from the main NZP phase with Np3+ (∼77%), green line – the NpO2 impurity (∼9%), purple line – the secondary phase containing Np4+ (∼14%). | ||
| ISO, mm s−1 | QUA, mm s−1 | Relative intensity | |
|---|---|---|---|
| Main phase | 51.04(5) | 66.1(2) | 77% |
| Secondary phase | 2.7(3) | 39.7(1.4) | 14% |
| NpO2 phase | 7.75(0) | 5.1(0) | 9% |
Since both Np0.25Zr2(PO4)3 and Np0.33Zr2(PO4)3 can exist in theory and would differ only in their space groups (Rc vs. Pc, based on the analogy found in the literature), we can assign the hexagonal phases with tetra- and trivalent neptunium, observed in the Mössbauer measurements, to these two theoretical formulas.
Furthermore, taking into account that the targeted stoichiometry was Np0.25Zr2(PO4)3, the obtained molar percentages would correspond to (wt%) ∼62% of Np0.33Zr2(PO4)3, ∼1% of NpO2 and ∼15% of Np0.25Zr2(PO4)3. To compensate for the difference in stoichiometry one also needs to consider the presence of ∼22% of an impurity phase, which does not contain any neptunium and binds the excess of zirconium and phosphorus. This impurity might be, for instance, a compound with a bulk chemical composition, Zr2P2O9, although only small traces of crystalline ZrP2O7 and ZrO2 could be identified from the XRD pattern. At the same time, the presence of an amorphous component could also be seen at the diffractograms, appearing as an increased background with a complex shape, especially at lower angles. The exact composition of all impurity phases in this case is difficult to identify. Important is, however, that the XRD data do not contradict the conclusion of the presence of ∼22% of Np-free phase, drawn from the results of Mössbauer spectroscopy.
Rietveld analysis
Phase analysis of the sample revealed the presence of four crystalline phases: a major component with the structure of NZP and admixtures of ZrP2O7, NpO2 and ZrO2. The presence of several additional reflections, also observed for the double lanthanide zirconium phosphates,16 but not for the sodium zirconium phosphate, clearly indicates the reduction of symmetry compared to the prototype compound NaZr2(PO4)3 (from the space group Rc to the space group Pc). Taking this into account and also the fact that neptunium was found to be mainly trivalent, we used the structure of Eu0.33Zr2(PO4)316 as the starting model for crystal structure refinement, suggesting the chemical formula Np0.33Zr2(PO4)3 for the studied phosphate.
The model was refined by the Rietveld method using the FullProf Suite Program (2.05). Additional phases were also introduced during the refinement procedure, but only their profiles were refined using the LeBail algorithm. The background was modelled by a set of manually selected consecutive points with adjustable intensities. Soft distance constraints were applied to P–O distances of the main phase in order to keep them within reasonable values found for phosphate minerals.39 The progress of the crystallographic refinement was monitored using the values of reliability factors (R-factors). The refinement rounds continued until R-factors reached a stable minimum value with no further improvements. At the last stages, values of atomic displacement parameters were refined for neptunium, zirconium, phosphorus and oxygen atoms. Finally, the occupancy of the neptunium position was refined. Releasing this parameter during the refinement improved the fitting and was accompanied by a slight decrease of R-factors. The refinement converged at RB = 2.5, Rf = 2.06 for the main phase, Rp = 2.31, Rwp = 3.17 (not corrected for background) and χ2 = 3.51. Values of the unit cell parameters and details of the pattern fitting can be found in Table 2. The refined structural parameters are given in Table 3. Observed and calculated XRD patterns and the structure fragment are shown in Fig. 5 and 6, respectively.
 | ||
| Fig. 5 Observed and calculated powder X-ray diffraction patterns of the double neptunium zirconium phosphate, sp. gr. Pc (no. 165). The difference pattern is shown at the bottom of the picture. Vertical bars indicate the calculated positions of the Bragg reflections of the main phase and the impurities of cubic NpO2 (sp. gr. Fmm), ZrP2O7 (sp. gr. Pa) and ZrO2 (sp. gr. Fmm). | ||
 | ||
| Fig. 6 Structure fragment of neptunium zirconium phosphate. | ||
| R f = 2.06%, Rp = 2.31%, Rexp = 1.69%, χ2 = 3.51. | |
|---|---|
| Chemical formula sum | Np0.29Zr2(PO4)3 |
| Structure model | Eu0.33Zr2(PO4)3 |
| Crystal system | Hexagonal |
| Space group |
Pc (165) |
| a, Å | 8.755(1) |
| b, Å | 8.755(1) |
| c, Å | 23.101(3) |
| V, Å3 | 1533.6(1) |
| Z | 6 |
| 2θ-range (°) | 5–150 |
| Number of reflections | 1088 |
| General number of points | 16915 |
| Number of fitted parameters | 121 |
| R B, % | 2.31 |
| R wp, % | 3.17 |
| Atom | Site | x | y | z | U, Å2 | Occ. |
|---|---|---|---|---|---|---|
| Np1 | 2b | 0 | 0 | 0 | 0.0501 | 0.143 |
| Zr1 | 4c | 0 | 0 | 0.15136(5) | 0.0150 | 0.333 |
| Zr2 | 4d | 0.6667 | 0.3333 | 0.47299(7) | 0.0086 | 0.333 |
| Zr3 | 4d | 0.3333 | 0.6667 | 0.81197(7) | 0.0180 | 0.333 |
| P1 | 6f | 0.2849(2) | 0 | 0.25 | 0.0248 | 0.5 |
| P2 | 12g | 0.9466(1) | 0.3262(2) | 0.58042(5) | 0.0219 | 1.0 |
| O1 | 12g | 0.1582(6) | −0.0427(9) | 0.1987(2) | 0.0417 | 1.0 |
| O2 | 12g | 0.8243(8) | 0.292(1) | 0.5278(2) | 0.0417 | 1.0 |
| O3 | 12g | 0.5370(7) | 0.698(1) | 0.8638(1) | 0.0417 | 1.0 |
| O4 | 12g | 0.2012(6) | 0.1608(7) | 0.0946(2) | 0.0417 | 1.0 |
| O5 | 12g | 0.8760(5) | 0.5002(6) | 0.4285(3) | 0.0417 | 1.0 |
| O6 | 12g | 0.5304(4) | 0.8352(5) | 0.7522(2) | 0.0417 | 1.0 |
The lower value of the occupancy factor for neptunium in comparison with the one expected on the basis of the theoretical composition Np0.33Zr2(PO4)3 (0.143 vs. 0.1667, Table 3) corresponds to the empirical formula Np0.29Zr2(PO4)3. This is in good agreement with the results of Mössbauer spectroscopy for the presence of two phases containing neptunium in the oxidation state +3 (77 mol%) and +4 (14 mol%), giving the same gross formula Np0.29Zr2(PO4)3. In this case, the X-ray diffraction provides an averaged pattern for the mixture of two NZP phases with different oxidation states of neptunium. Introducing the model of the Np0.25Zr2(PO4)3 phase into the refinement, alongside Np0.33Zr2(PO4)3, did not improve the fit and the R-factors. This is not surprising, since the anticipated symmetry and diffraction pattern would be very similar to that of Np0.33Zr2(PO4)3, leading to overlapping of the diffraction patterns.
The relatively high value of χ-criteria can be explained by the presence of an amorphous component, which does not give reflections at the XRD pattern (in agreement with the Mössbauer data) and could also be due to imperfection of the structure model (the presence of Np0.25Zr2(PO4)3 is not taken into account during the refinement).
SEM/EDX-analysis
The sample is characterized by a uniform microstructure (Fig. 7). The powder consists of agglomerations of particles of bigger and smaller sizes, some of them reaching a few tenths of microns. The EDX analysis showed that the sample contains regions, the chemical composition of which oscillates around the expected Np:Zr ratio in the targeted material. At the same time, some minor regions were found that were either enriched or depleted in neptunium (Fig. 7). This is also in agreement with the presence of NpO2 and Zr2P2O9 phases, discussed earlier in the section on Mössbauer spectroscopy. EDX analyses of randomly scanned areas, excluding Np-depleted and Np-enriched regions, give the average Np:Zr ratio of 1:8.9 corresponding to the mean chemical composition Np0.23Zr2(PO4)3 (NZP-region in Fig. 7). Among these areas there were also regions corresponding to an average formula Np0.28Zr2(PO4)3, which is closer to the value found by the Rietveld analysis. However, the difference is likely to be attributed to other factors during sample preparation and measurement, rather than the actual difference in chemical composition. These factors are, for example, inhomogeneity of the sample, unpolished surface, porosity of the powder and others.
 | ||
| Fig. 7 Microstructure of the Np-sample obtained by SEM (secondary electrons). EDX analysis shows three phases: (1) NZP, the composition of which oscillates around the expected Np:Zr ratio in the targeted material; (2) Np-R, a neptunium-rich region; and (3) Np-D – a neptunium depleted region. | ||
Thermal behaviour
Fig. 8 shows the TG/DTA curves of the studied neptunium zirconium phosphate recorded from room temperature up to 1400 °C. The appearance of two small exothermal peaks on the DTA curve at ∼970 °C and 1080 °C, shown by arrows in Fig. 8, is accompanied by a small decrease of mass at the TG curve and is attributed to the decomposition of the sample, in agreement with Alami Talbi et al.7 and Bykov et al.40 | ||
| Fig. 8 TG/DTA curves of the studied neptunium zirconium phosphate. Arrows indicate peaks attributed to the decomposition of the sample. | ||
In addition to the TG/DTA analysis, the sample synthesized at 800 °C in argon was further heated at 856 °C (20 h), 900 °C (25 h) and 1000 °C (2 h) in argon and at 805 °C (15 h) and 900 °C (20 h) in a pure oxygen atmosphere in order to study its thermal stability. In both cases, increasing the temperature leads to the increase of the content of the secondary phase of zirconium pyrophosphate, ZrP2O7. This process occurs faster when oxygen is used as an atmosphere. In this case already after 900 °C zirconium pyrophosphate becomes the major phase, whereas in the case of argon most of the powder after 900 °C still consists of the NZP phase and only after 1000 °C does zirconium pyrophosphate start to dominate. Unidentified smaller peaks were also observed at the XRD patterns obtained after treatment of the powder at 900 °C and 1000 °C in argon and after 805 °C and 900 °C in oxygen.
Thus, both data for thermal analysis and X-ray diffraction lead to the conclusion that the studied neptunium zirconium phosphate starts to decompose at temperatures of around 850 °C. This value is close to the decomposition temperature of the phosphates of zirconium with lanthanides Ln0.33Zr2(PO4)3.7,40–42
The diffractograms of all samples heated in argon contained reflections consistent with the space group Pc. This allowed us to perform the LeBail fit of the diffraction data and to calculate the unit cell parameters (Table 4). It can be seen that there is a gradual decrease of the parameters a, c and of the volume V with the increase of the temperature of the heat treatment, leading to the contraction of the unit cell. At the same time, the relative change of the cell parameters is small and does not exceed 0.7% for both a and c.
c) heated in argon and cooled to room temperature
| Temperature, °C | a, Å | c, Å | V, Å3 |
|---|---|---|---|
| 800 | 8.756(1) | 23.100(3) | 1534 |
| 856 | 8.750(3) | 23.072(9) | 1530 |
| 900 | 8.735(4) | 23.029(2) | 1522 |
| 1000 | 8.695(1) | 22.952(5) | 1503 |
The phenomenon of contraction upon heating for the NZP structure is rare but known. For example, the temperature behaviour of the erbium-zirconium phosphate Er0.33Zr2(PO4)3 was investigated by Orlova et al.43 It was found that despite expansion and contraction along different crystallographic directions upon heating, contraction of the structure as a whole takes place. In this case, the contribution of the axial negative and relatively large in absolute value coefficients αa and αb, compared to the positive coefficient αc, is responsible for the negative average value of αav.
With regard to the studied compound, two possibilities should be considered as an explanation:
1. Temperature hysteresis, as observed for the phosphate Zr0.25Zr2(PO4)3.31 The unit cell parameters of the sample at room temperature obtained after the heating cycle from 25 to 575 °C were somewhat smaller compared to the initial values (less than 0.35%). Moreover, for this compound a second-order phase transition upon heating was found, which was accompanied by the contraction of the structure along all crystallographic directions.
2. There is a possibility that an oxidation process with the formation of the oxidation state +4 of neptunium occurs during heating. Since the ionic radius of Np4+ is smaller than Np3+ in hexahedral coordination (0.87 Å vs. 1.01 Å, respectively), partial oxidation of Np3+ to Np4+ would also lead to the contraction of the crystal lattice.
From the data that we have obtained it is not possible to conclude which option would be the most likely.
In the case of samples heated above 800 °C in oxygen the indexing of the diffraction patterns was more difficult due to the increasing complexity of the diffractograms with increasing temperature. Therefore, the unit cell parameters were not calculated. However, after removing from consideration the peaks belonging to the ZrP2O7 phase, the remaining reflections might indicate the appearance of a superstructure (805 °C) and partial decomposition of the sample (900 °C).
Concluding remarks
Based on the analysis of the literature data carried out by Bykov,15 the phosphates MxZr2(PO4)3 (M – mono-, di-, tri- and tetravalent cations) crystallize in two structure types: NaZr2(PO4)3 for the cations with radii from 0.71 to 1.67 Å and the structure of scandium tungstate Sc2(WO4)3 (SW) for the cations with radii 0.49–0.66 Å. Both structures are composed of the same basic framework units [Zr2(PO4)3]−, but differ in the arrangement of these units and the coordination number of an interstitial cation (CN = 4 for SW).44 The radii of both Np3+ and Np4+ in octahedral coordination fall within the first range (1.01 and 0.87 Å, respectively).The existence of a solid solution NaZr2−xNpx(PO4)3 (0 ≤ x ≤ 0.6), similar to solid solutions Na1+xRxZr2−x(PO4)3 (R = In, Yb, Er, Y, Dy, Tb, Gd, Sm, 0 ≤ x ≤ 1),25 suggests that both tri- and tetravalent neptunium cations can substitute zirconium in the ZrO6 framework. However, the analogy of the XRD and IR spectra obtained in the present work with those for the Ln0.33Zr2(PO4)3 phosphates, as well as the relatively low synthesis temperature, leads to the conclusion that the Np atoms occupy the interstitial M1 sites. At the same time, the results of Mössbauer spectroscopy show the presence of a mixture of two Np-containing hexagonal phases and an NpO2 admixture. Since no other crystalline phases containing neptunium were revealed from the XRD pattern during Rietveld analysis (including the fact that NpO2 was identified from the pattern), and also because it is unlikely that Np is present in an amorphous phase, it is assumed that the secondary Np-containing phase belongs to the same structure type, where Np4+ occupies similar but not the same crystallographic positions. It can also be concluded that in the absence of a charge compensator of lower valence it is harder for bigger neptunium cations to compete with the smaller zirconium ones for the occupation of framework positions. This conclusion is further strengthened by the fact that the unit cell parameters of the new neptunium phase correlate well with the data for the lanthanide phosphates,16 as shown in Fig. 9. The mean neptunium radius shown on this picture is calculated based on the atomic percentages of Np3+ and Np4+ found in the present work.
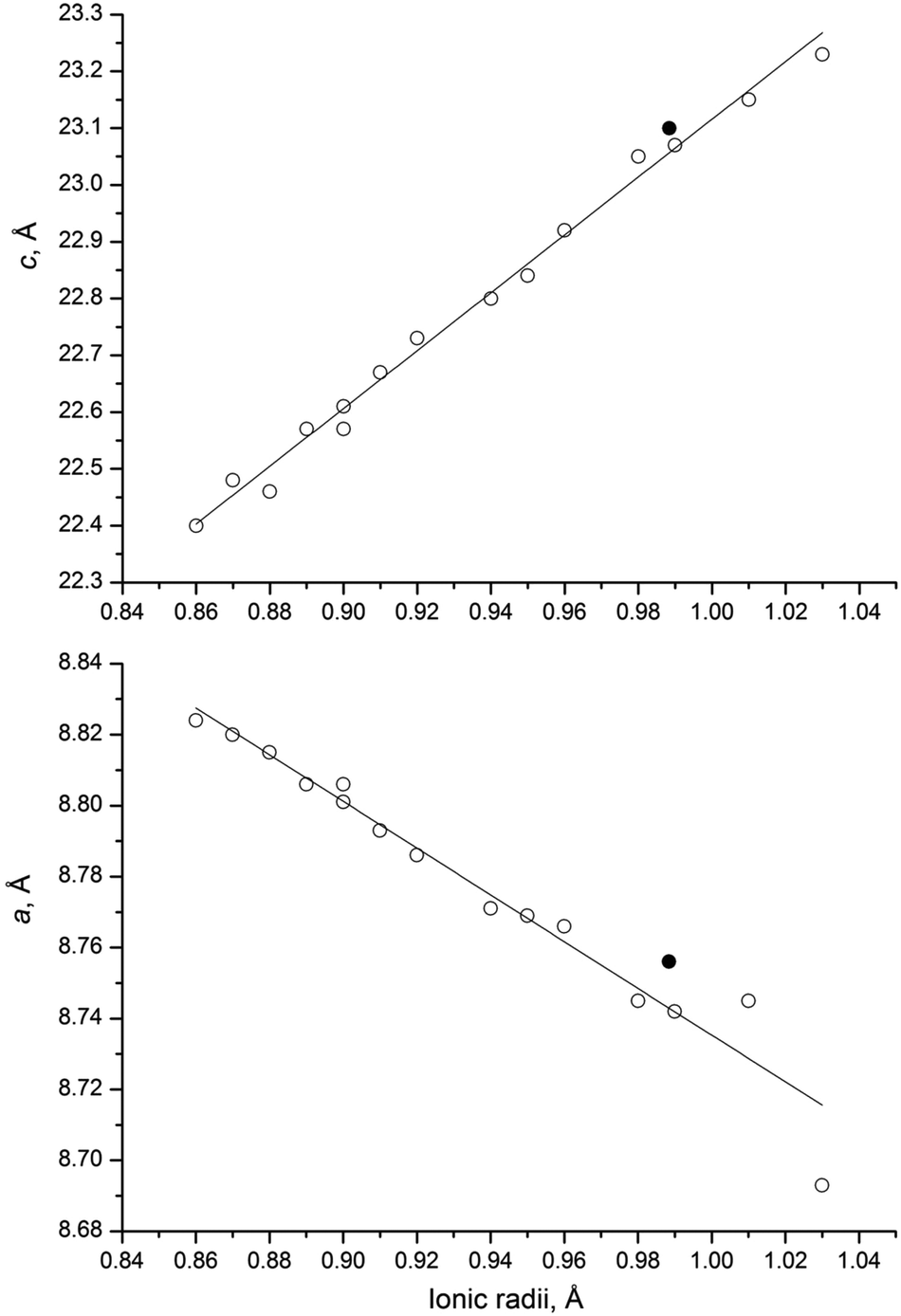 | ||
| Fig. 9 Comparison of the unit cell parameters a and c of neptunium zirconium phosphate (●) with the trend reported in the paper by Bykov et al.16 for Ln0.33Zr2(PO4)3 (○): Ln = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Y, Er, Tm, Yb and Lu. The ionic radius of neptunium shown in this graph corresponds to the average value between Np3+ and Np4+ (CN = 6) based on the molar percentages found by Mössbauer spectroscopy. | ||
Designing crystalline waste forms generally requires detailed crystal chemical knowledge of the site preferences of the waste ions and adjustment of the overall stoichiometry when producing the final product. The results of the present paper illustrate crystal chemical flexibility of the NZP phases, which to some extent allows for variations in stoichiometry, and the ability to incorporate elements with complex redox behaviour, such as the actinide elements. The additional formation of the ZrP2O7 phase, observed as an impurity in this study, can be an advantage when it comes to waste applications. This phase does not contain actinides and can play the role of a chemical buffer during the synthesis of a waste material, so that variations of chemical composition would cause a change in the ratios of the phases present and not the nature of these phases.
In conclusion, the new phosphate with an empirical formula Np0.29Zr2(PO4)3 complements the series of NZP compounds with actinide and lanthanide atoms in the interstitials positions of the structure. The results of the present study show that the possibility of coexistence of several oxidation states in compounds of neighbouring elements U and Pu needs to be taken into account.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank V. S. Kurazhkovskaya for the discussion on the IR spectra. D.B. acknowledges the European Commission and the Deutscher Akademischer Austauschdienst, DAAD, for the fellowship fundings.References
- A. I. Orlova, Radiochemistry, 2002, 44, 423 CrossRef CAS.
- A. I. Orlova, V. N. Zyryanov, A. R. Kotel'nikov, V. T. Demarin and E. V. Rakitina, Radiochemistry, 1993, 35, 717 Search PubMed.
- A. I. Orlova, V. N. Zyryanov, O. V. Egor'kova and V. T. Demarin, Radiochemistry, 1996, 38, 20 Search PubMed.
- A. I. Kryukova, I. A. Kulikov, G. Yu. Artem'eva, O. V. Pechenevskaya and V. A. Alferov, Radiokhimiya, 1992, 34, 82 CAS.
- A. I. Orlova, Yu. F. Volkov, R. F. Melkaya, L. Yu. Masterova, I. A. Kulikov and V. A. Alferov, Radiochemistry, 1994, 36, 322 Search PubMed.
- T. Oota and I. Yamai, J. Am. Ceram. Soc., 1986, 69, 1 CrossRef CAS.
- M. Alami Talbi, R. Brochu, C. Parent, L. Rabardel and G. Le Flem, J. Solid State Chem., 1994, 110, 350 CrossRef.
- M. Hirayama, N. Sonoyama, A. Yamada and R. Kanno, J. Solid State Chem., 2009, 182, 730 CrossRef CAS.
- B. Glorieux, V. Jubera, A. I. Orlova, A. E. Kanunov, A. Garcia, C. Pallier and T. A. Oleneva, Inorg. Mater., 2013, 49, 82 CrossRef CAS.
- A. I. Orlova, A. E. Kanunov, E. N. Gorshkova, A. N. Shushunov, S. N. Pleskova, E. R. Mikheeva, D. O. Savinykh and E. S. Leonov, Inorg. Mater., 2013, 49, 89 CrossRef CAS.
- Y. Miyajima, Y. Saito, M. Matsuoka and Y. Yamamoto, Solid State Ionics, 1996, 84, 61 CrossRef CAS.
- K. Nomura, S. Ikeda, K. Ito and H. Einaga, Bull. Chem. Soc. Jpn., 1992, 65, 3221 CrossRef CAS.
- D. M. Bykov, G. Sh. Shekhtman, A. I. Orlova, V. S. Kurazhkovskaya, E. Yu. Borovikova and V. Yu. Volgutov, Solid State Ionics, 2011, 182, 47 CrossRef CAS.
- L.-O. Hagman and P. Kierkegaard, Acta Chem. Scand., 1968, 22, 1822 CrossRef CAS.
- D. M. Bykov, PhD thesis, Nizhny Novgorod State University, 2006.
- D. M. Bykov, E. R. Gobechiya, Yu. K. Kabalov, A. I. Orlova and S. V. Tomilin, J. Solid State Chem., 2006, 179, 3101 CrossRef CAS.
- M. Barre, M. P. Crosnier-Lopez, F. Le Berre, J. Emery, E. Suard and J.-L. Fourquet, Chem. Mater., 2005, 17, 6605 CrossRef CAS.
- H. T. Hawkins, D. R. Spearing, D. K. Veirs, J. A. Danis, D. M. Smith, C. D. Tait, W. H. Runde, M. N. Spilde and B. E. Scheetz, Chem. Mater., 1999, 11, 2851 CrossRef CAS.
- A. I. Orlova, in Structural Chemistry of Inorganic Actinide Compounds, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, Amsterdam, 2007, ch. 8, pp. 315–339 Search PubMed.
- A. I. Orlova, A. A. Charlamova and Yu. F. Volkov, in Review of Excess Weapons Plutonium Disposition LLNL Contract Work in Russia, ed. L. J. Jardine and G. B. Borisov, Lawrence Livermore National Laboratory, Livermore, 2002, pp. 407–418 Search PubMed.
- H. T. Hawkins, D. R. Spearing, D. M. Smith, F. G. Hampel, D. K. Veirs and B. E. Scheetz, in AIP Conf. Proc, 2000, vol. 532, p. 168 Search PubMed.
- Yu. F. Volkov, R. F. Melkaya, V. I. Spiryakov and G. A. Timofeev, Radiokhimiya, 1994, 36, 205 CAS.
- Yu. F. Volkov, S. V. Tomilin, A. I. Orlova, A. A. Lizin, V. I. Spiryakov and A. N. Lukinykh, Radiochemistry, 2003, 45, 319 CrossRef CAS.
- A. A. Burnaeva, Yu. F. Volkov, A. I. Kryukova, I. A. Korshunov and O. V. Skiba, Radiokhimiya, 1992, 34, 12 Search PubMed.
- Y. Miyajima, T. Miyoshi, J. Tamaki, M. Matsuoka, Y. Yamamoto, C. Masquelier, M. Tabuchi, Y. Saito and H. Kageyama, Solid State Ionics, 1999, 124, 201 CrossRef CAS.
- D. M. Bykov, A. I. Orlova, S. V. Tomilin, A. A. Lizin and A. N. Lukinykh, Radiochemistry, 2006, 48, 234 CrossRef CAS.
- V. S. Kurazhkovskaya, D. M. Bykov and A. I. Orlova, J. Struct. Chem., 2004, 45, 966 CrossRef CAS.
- A. I. Orlova, V. Yu. Volgutov, G. R. Castro, S. García-Granda, S. A. Khainakov and J. R. García, Inorg. Chem., 2009, 48, 9046 CrossRef CAS PubMed.
- J. Alamo and R. Roy, J. Am. Ceram. Soc., 1984, 67, C-80 CrossRef CAS.
- A. E. Mel'nikov and I. A. Bondar’, Russ. J. Inorg. Chem., 1989, 34, 2122 Search PubMed.
- A. I. Orlova, S. G. Samoilov, G. N. Kazantsev, V. Yu. Volgutov, D. M. Bykov, A. V. Golubev and E. Yu. Borovikova, Crystallogr. Rep., 2009, 54, 431 CrossRef CAS.
- S. Cotton, in Lanthanide and Actinide Chemistry, Wiley, Chichester, West Sussex, UK, 2006 Search PubMed.
- B. E. Burakov, M. I. Ojovan and W. E. Lee, Crystalline Materials for Actinide Immobilisation, Imperial College Press, London, 2010 Search PubMed.
- J. Rodríguez-Carvajal, Commission on Powder Diffraction (IUCr), Newsletter, 2001, 26, 12 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653 CrossRef CAS.
- J. L. Pouchou and F. Pichoir, in Electron Probe Quantitation, ed. K. F. J. Heinrich and D. E. Newbury, Plenum Press, New York, 1991, pp. 31–76 Search PubMed.
- V. S. Kurazhkovskaya, D. M. Bykov, E. Yu. Borovikova, N. Yu. Boldyrev, L. Mikhalitsyn and A. I. Orlova, Vib. Spectrosc., 2010, 52, 137 CrossRef CAS.
- P. Tarte, A. Rulmont and C. Merckaert-Ansay, Spectrochim. Acta, Part A, 1986, 42, 1009 CrossRef.
- D. M. C. Huminicki and F. C. Hawthorne, Rev. Mineral. Geochem., 2002, 48, 123 CrossRef CAS.
- D. M. Bykov, R. J. M. Konings and A. I. Orlova, J. Alloys Compd., 2007, 439, 376 CrossRef CAS.
- S. Tamura, N. Imanaka and G. Adachi, Solid State Ionics, 2002, 154–155, 767 CrossRef CAS.
- J. M. Heintz, L. Rabardel, M. Al Qaraoui, M. Alami Talbi, R. Brochu and G. Le Flem, J. Alloys Compd., 1997, 250, 515 CrossRef CAS.
- A. I. Orlova, S. G. Samoilov, G. N. Kazantsev, V. Yu. Volgutov and A. Yu. Kazakova, Radiochemistry, 2010, 52, 666 CrossRef CAS.
- Y. Piffard, A. Verbaere and M. Kinoshita, J. Solid State Chem., 1987, 71, 121 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |