
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chalcogen bonding in synthesis, catalysis and design of materials
Kamran T.
Mahmudov
 *abc,
Maximilian N.
Kopylovich
a,
M. Fátima C.
Guedes da Silva
a and
Armando J. L.
Pombeiro
*a
*abc,
Maximilian N.
Kopylovich
a,
M. Fátima C.
Guedes da Silva
a and
Armando J. L.
Pombeiro
*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: kamran_chem@mail.ru; kamran_chem@yahoo.com; pombeiro@tecnico.ulisboa.pt
bDepartment of Chemistry, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan
cOrganic Chemistry Department, RUDN University, 6 Miklukho-Maklaya str., Moscow 117198, Russian Federation
First published on 22nd June 2017
Abstract
Chalcogen bonding is a type of noncovalent interaction in which a covalently bonded chalcogen atom (O, S, Se or Te) acts as an electrophilic species towards a nucleophilic (negative) region(s) in another or in the same molecule. In general, this interaction is strengthened by the presence of an electron-withdrawing group on the electron-acceptor chalcogen atom and upon moving down in the periodic table of elements, from O to Te. Following a short discussion of the phenomenon of chalcogen bonding, this Perspective presents some demonstrative experimental observations in which this bonding is crucial for synthetic transformations, crystal engineering, catalysis and design of materials as synthons/tectons.
 Kamran T. Mahmudov | Kamran T. Mahmudov was born in Tovuz, Azerbaijan, and received his B.Sc. (2001), M.Sc. (2003), Ph.D. (2007) and Habilitation (2013) degrees at the Baku State University, where he worked as an Assistant Professor (2006–2008). Since 2009, he has been conducting postdoctoral research under the supervision of Prof. A.J.L. Pombeiro at the University of Lisbon. Dr. Mahmudov has authored or coauthored about 92 papers in ISI journals, 4 reviews, 5 book chapters, and he is the co-editor of the book “Noncovalent interactions in synthesis and design of new compounds” (Wiley 2016). His main research interests are related to noncovalent interaction assisted synthesis and catalysis. He is married to Farida Abdiyeva (journalist). |
 Maximilian N. Kopylovich | Maximilian N. Kopylovich graduated in Chemical Engineering (1993) from the Byelorussian State Technological University and completed his PhD degree in Chemistry (1998) at the same institution. Then he worked as a post-doctoral fellow (2000–2007), auxiliary (2008–2013) and senior (2014-now) researcher at the Centro de Química Estrutural, Instituto Superior Técnico (IST), Universidade de Lisboa. The scientific interests of MK lie mainly within synthetic coordination chemistry and catalysis. His current research interests are related to the metal-mediated (template) and noncovalent interaction assisted syntheses of new compounds and supramolecular assemblies. |
 M. Fátima C. Guedes da Silva | M. Fátima C. Guedes da Silva is Associate Professor, at the Instituto Superior Técnico, Lisbon, Portugal. She is the coordinator of the research group of Coordination Chemistry and Catalysis of the Centro de Química Estrutural, member of the Coordination Commission of this Centre and of the Directive Board of the Catalysis and Sustainability (CATSUS) PhD program. Her research activity follows a general streamline that usually starts with the design and synthesis of novel coordination compounds, then evolving to their eventual application by exploring structure–properties (including reactivity)–function (application) relationships. Her main research interests include: structural determination, by X-ray diffraction analysis, of metal complexes and organic compounds, metal polynuclear assemblies and supramolecular structures; activation, by transition metal centres, of small molecules with biological, pharmacological, environmental or industrial significance; metal mediated synthesis and catalysis; molecular electrochemistry and electrocatalysis; and mechanistic investigation of fast reactions mainly by the digital simulation of cyclic voltammetry. She has co-authored over 250 research publications, obtained 12 patents, and presented 15 invited lectures at international conferences. |
 Armando J. L. Pombeiro | Armando J. L. Pombeiro is Full Professor at the Instituto Superior Técnico, Universidade de Lisboa, President of the Centro de Química Estrutural and Coordinator of its Synthesis and Catalysis thematic line, Director of the Catalysis and Sustainability (CATSUS) PhD program, Full Member of the Academy of Sciences of Lisbon (in charge of various directive and representative positions) and former President of the Portuguese Electrochemical Society. His research group investigates the activation of small molecules with industrial, environmental or biological significance, including metal-mediated synthesis and catalysis (e.g., functionalization of alkanes under mild conditions), crystal engineering of coordination compounds, design and self-assembly of polynuclear and supramolecular structures, molecular electrochemistry and theoretical studies. He was the Chairman of the 25th ICOMC and member of organizing/scientific committees of 40 international conferences or schools. He has authored one book (plus four as editor), (co-)authored over 600 research publications, obtained 33 patents, and presented 100 invited lectures at international conferences. His work has received ca. 16 |
1. Introduction
Noncovalent interactions were firstly taken into consideration by van der Waals in 1873, helping to revise the equation of state for real gases.1 In comparison to covalent bonds, intra- and intermolecular noncovalent interactions are in general weak and exhibit much lower energy and directionality, as reflected by the term “noncovalent”. Nevertheless, in many cases these interactions can collectively play a dominant role in the synthesis, catalysis and design of materials.Currently, based on the nature of the particular elements or synthons involved in the interactions, noncovalent bonds are classified into hydrogen, agnostic, anagostic, π–π stacking, n–π*, π-cation, π-anion and others. These usually lead to the formation of molecular associates or clusters with directionalities and distances specific for a particular type of interactions.2–30 Recently, due to the application of modern techniques, such as NMR or X-ray diffraction analysis, new types of noncovalent interactions, such as chalcogen, pnicogen, tetrel and icosagen bonds, have been identified. Although they are usually considered as weak interactions, their strength ranges from a few to several hundreds of kJ mol−1 depending on their type.
In view of the amphoteric (electrophilic and nucleophilic) behaviour of chalcogens, the chalcogen-donor interactions have long been known as “secondary bonding interactions”,31–34 however being recognized as a particular chalcogen (Ch) bonding in recent years. Its general structure (together with the halogen bond, for comparison) is depicted in Scheme 1, in which Ch is an electrophilic chalcogen atom (O, S, Se or Te) and D is a donor centre (electron donor atom, anion, π-system, radical, etc.). Surprisingly, the strength of chalcogen bonding is sometimes comparable with that of hydrogen bonds and other types of noncovalent interactions.35
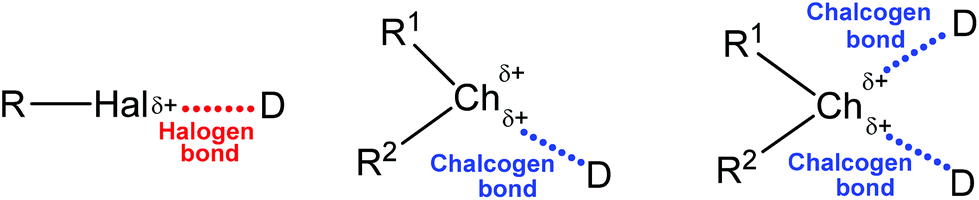 | ||
| Scheme 1 Schematic representation of halogen and chalcogen bonds (R1 is a stronger electron-withdrawing group than R2). | ||
Generally the formation of a chalcogen bond is explained in terms of the electrostatic potential present on the outermost portion of the chalcogen's surface, opposite to the R–Ch bond(s) (Scheme 1).36,37 This region of positive electrostatic potential is called the “σ-hole” and refers to the electron deficient outer lobe of a p orbital involved in a covalent bond, especially when one of the atoms is highly electronegative.36,37 In the cases when a region of positive electrostatic potential is perpendicular to a portion of a molecular framework, it is named the “π-hole”.26,38 Thus, due to the unequal occupation of the valence orbitals, the electrostatic potential around the chalcogen atom is anisotropic. The size of the σ-hole increases from the lighter to the heavier chalcogen atoms as polarizability increases and electronegativity decreases (O < S < Se < Te), and the remainder of the molecule becomes more electron-withdrawing. In the multivalent chalcogen atoms the respective σ-holes are localized on the axes of the covalent bonds (e.g. opposite to R1 and R2, Scheme 1),39,40 while the R–Ch covalent bonds elongate upon the formation of the chalcogen bond.
Since the mode of association is intimately related to the steric and electronic nature of the R substituent, modification of the R group can be used to direct the chalcogen bond assisted synthesis, catalysis and design of materials. Thus, a divalent chalcogen may be involved in multiple chalcogen bonding, as in 1,4-bis(methyltellanyl)buta-1,3-diyne, where each tellurium atom behaves as a two-electron donor and two-electron acceptor centre (Scheme 2).41 Each Te atom, placed at the edge of a stack, is in close contact with four Te atoms of neighbouring molecules. As a result, each Te forms zigzag like contacts (3.74 and 3.82 Å) to two neighbouring atoms (Scheme 2), with less than the van der Waals distances (Te⋯Te 4.12 Å).42
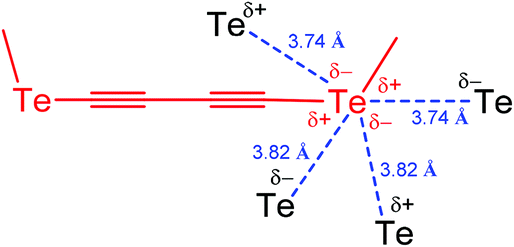 | ||
| Scheme 2 Multiple chalcogen bonding in 1,4-bis(methyltellanyl)buta-1,3-diyne (one fragment is shown).41 | ||
Due to the presence of the lone pairs of the chalcogen atom, a σ-hole is usually surrounded by a region of negative molecular electrostatic potential, and this determines a high directionality of the chalcogen bonds. This linear geometry of the R–Ch⋯D fragment provides a maximal electrostatic attraction between a σ-hole and an area of negative molecular electrostatic potential around lone pairs of the heteroatom or π-system and a minimal repulsion between areas of negative molecular electrostatic potential. For instance, the highly directional interactions between sulphur atoms and aromatic rings in the side chains of amino acids are crucial for the formation, stability and unique properties of α-helices in proteins.43,44
Another interesting property of chalcogen bonds is their tunability, i.e. when their strength and geometric properties can be relatively easy modified or “tuned”.45–47 It should be noted that the recognized chalcogen bonds mainly involve divalent chalcogens, although they are commonly engaged in other valencies,48 namely with four or six separate covalent bonds. However, a few recent works address tetravalent chalcogens.49,50
Similar to the hydrogen bond, the chalcogen bonds are classified into several fundamental types: negative charge assisted chalcogen bond (Ch⋯D−), positive charge assisted chalcogen bond (Ch+⋯D), conventional (or “neutral”) chalcogen bond (Ch⋯D) and resonance assisted chalcogen bond (RACB).40,51–56 Similar to the hydrogen and halogen bonds,29,57–59 chalcogen interactions assisted by a charge or a conjugated π-system (in the case of RACB) can be characterized by the enhanced strength of the chalcogen bond in comparison with the conventional chalcogen bonding.40,51–56
There are several specific features of chalcogen bonds;3,5,6,8,24,60,61 from those we can list the following ones. (i) Directionality: Due to the localization of a region of positive electrostatic potential on the outermost, opposite to R, part of the chalcogen atom, the R–Ch fragment elongates and “linearly” approaches D, with the typical R–Ch⋯D bond angle close to 180°. The stronger the chalcogen bond is, the shorter is the Ch⋯D distance; (ii) Tunability: The strength of the chalcogen R–Ch⋯D bonding can be tuned by the variation of R or D and by the replacement of the Ch atom by another one (O, S, Se or Te); (iii) Hydrophobicity: The chalcogen-bond donors are hydrophobic in comparison to conventional hydrogen bonding. In fact, positive electrostatic potential(s) on the outermost surface of the chalcogen atom is (are) hidden by the next covalent bond(s). In other words, the attached groups on the chalcogen atom prevent the solubility of organochalcogens in aqueous or polar solvents;7 (iv) Donor atom size: Due to the short van der Waals radii of hydrogen atoms (1.20 Å) compared to the chalcogen atoms (1.52, 1.80, 1.90, and 2.06 Å for O, S, Se, and Te, respectively),42 the hydrogen bond is less sensitive to steric hindrance than the chalcogen bond, and this causes differences in some physical and chemical properties; (v) Multiplicity: Unlike the hydrogen atom, the chalcogen atom shows variable valences, e.g., di-, tetra- and hexa-valence, which provide multiple chalcogen bonding. This allows the creation of new types of materials with unique properties, and may be used in synthetic transformations, catalysis, crystal engineering, etc.
As a result of the mentioned distinctive features, application of the noncovalent interactions became a growing area of modern chemistry, while chalcogen bonding started to be used as a driving force toward desired selectivity in synthesis. For example, the reaction of NaSPh with syn or anti hydroxy epoxide in THF afforded a mixture of the syn–syn and syn–anti lactones I and II in a ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 ratio, while other two possible isomeric lactones (anti–syn or anti–anti) were not observed (Scheme 3).62 The crystal structures of syn–syn lactones revealed two attractive 1,4-intramolecular interactions (chalcogen bonds) of divalent sulfur with both hydroxyl and carbonyl oxygens. These stabilizing noncovalent S⋯O interactions were considered to be a driving force for this reaction and an important factor in favouring the observed stereochemical outcome.62
:70 ratio, while other two possible isomeric lactones (anti–syn or anti–anti) were not observed (Scheme 3).62 The crystal structures of syn–syn lactones revealed two attractive 1,4-intramolecular interactions (chalcogen bonds) of divalent sulfur with both hydroxyl and carbonyl oxygens. These stabilizing noncovalent S⋯O interactions were considered to be a driving force for this reaction and an important factor in favouring the observed stereochemical outcome.62
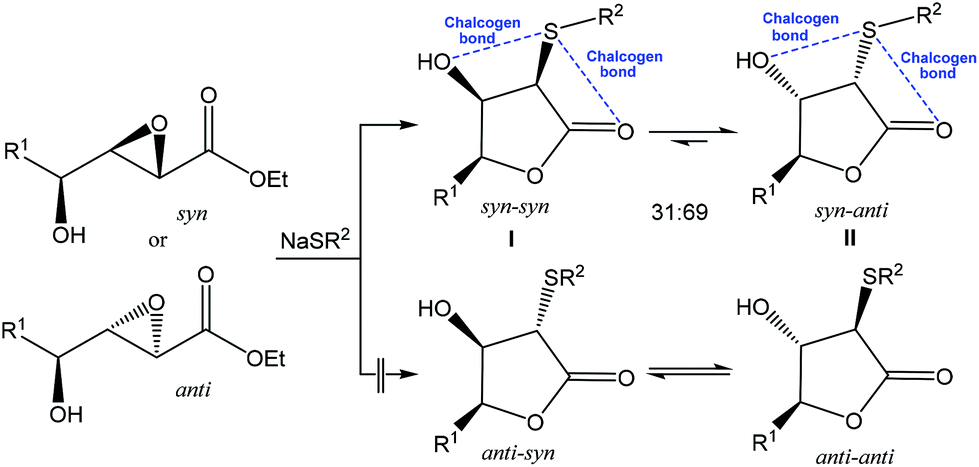 | ||
| Scheme 3 Chalcogen bond assisted stereoselective synthesis of lactones.62 | ||
Furthermore, various modifications of chalcogenated ligands have been performed to drive molecular self-assembly in the synthesis of coordination compounds and to modulate their properties.29,63–68 Thus, the reaction of cis-[PdCl2(CNXyl)2] (Xyl = 2,6-Me2C6H3) with heterocyclic 1,3-thiazol-2-amines or 1,3,4-thiadiazol-2-amines yields a mixture of kinetically (I with S⋯Cl chalcogen bond) and thermodynamically (II with S⋯N chalcogen bond) controlled regioisomers of binuclear diaminocarbene complexes at room temperature and on heating, respectively (Scheme 4).67 The ratio of isomers (I and II) depends on the solvent polarity and temperature (Scheme 4). According to DFT calculations, the chalcogen bond energy of the thermodynamic isomers II (4.6–5.3 kcal mol−1) is higher than that of the kinetic ones I (2.8–3.0 kcal mol−1) (Scheme 4).
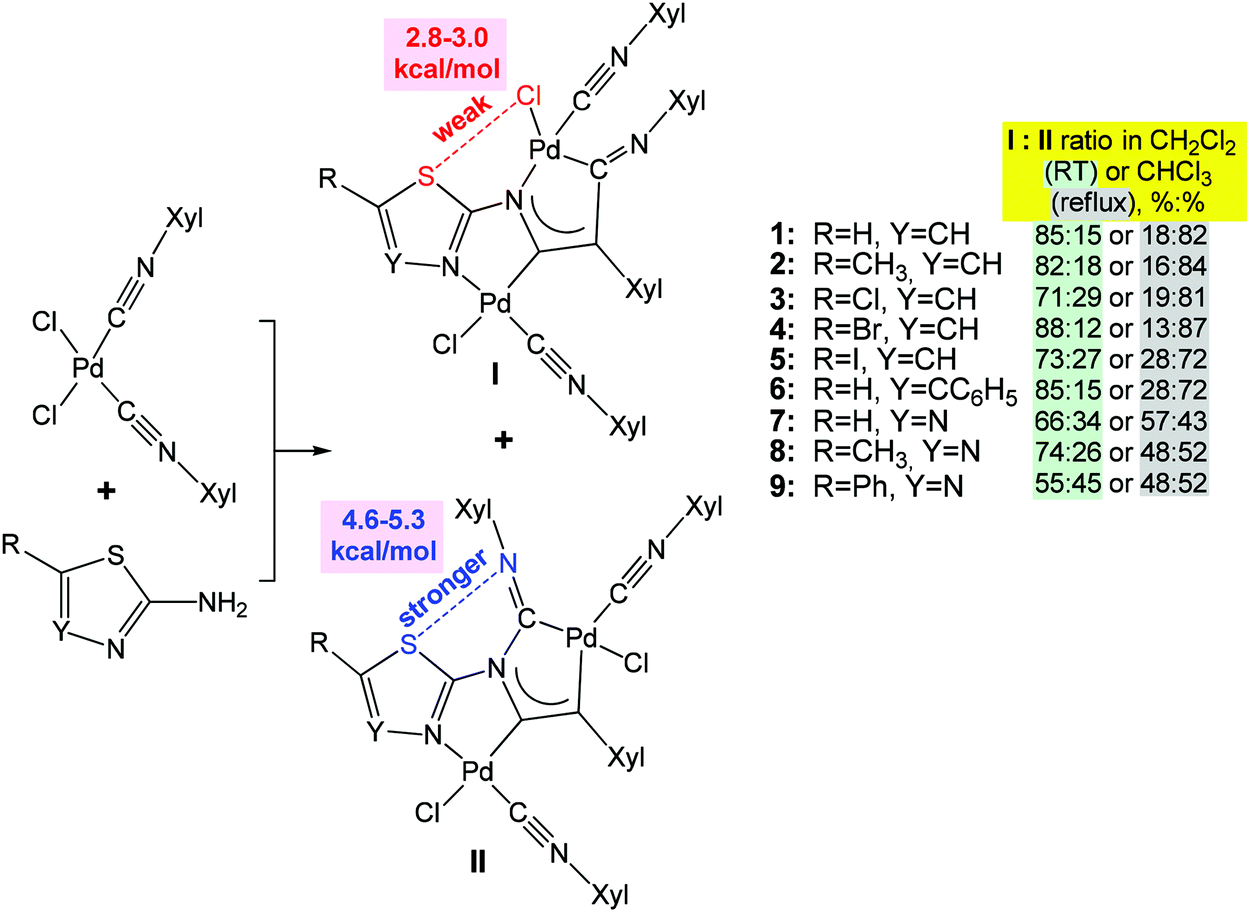 | ||
| Scheme 4 Difference in energy between S⋯Cl and S⋯N types of chalcogen bonds drives regioisomerization of binuclear (diaminocarbene)PdII complexes.67 | ||
As hydrogen and halogen noncovalent interactions, the chalcogen bond can also be used in catalysis providing an additional stabilization of intermediate(s).65,69–72 For example, the chalcogen bond donor catalyst (2S,3R)-3-isopropyl-2-phenyl-3,4-dihydro-2H-benzo[4,5]thiazolo[3,2-a]pyrimidine (Scheme 5, black) promotes the asymmetric annulation of a range of nucleophiles such as 1,3-diketones, β-ketoesters and azaarylketones to (E,E)-α,β-unsaturated anhydrides, yielding either functionalized esters (upon ring opening), dihydropyranones or dihydropyridones with high enantioselectivity (up to 97% ee) and yields (up to 86%) via a chalcogen bond assisted intermediate.72 The interaction of the catalyst with an (E,E)-α,β-unsaturated anhydride leads to an intermediate I, with the carbonyl oxygen adopting a syn-conformation with respect to the isothiourea sulfur atom due to a stabilizing chalcogen bonding in the formed 5-membered ring (Route I in Scheme 5). Michael addition of diketone enolate to the Re face of I gives intermediate III, which can also be obtained by a [3,3]-rearrangement of II. A subsequent proton transfer/lactonisation process yields an enantioenriched dihydropyranone IV, which, upon ring opening with MeOH, produces the ester product V in high (up to 97%) ee (Routes II–V in Scheme 5).
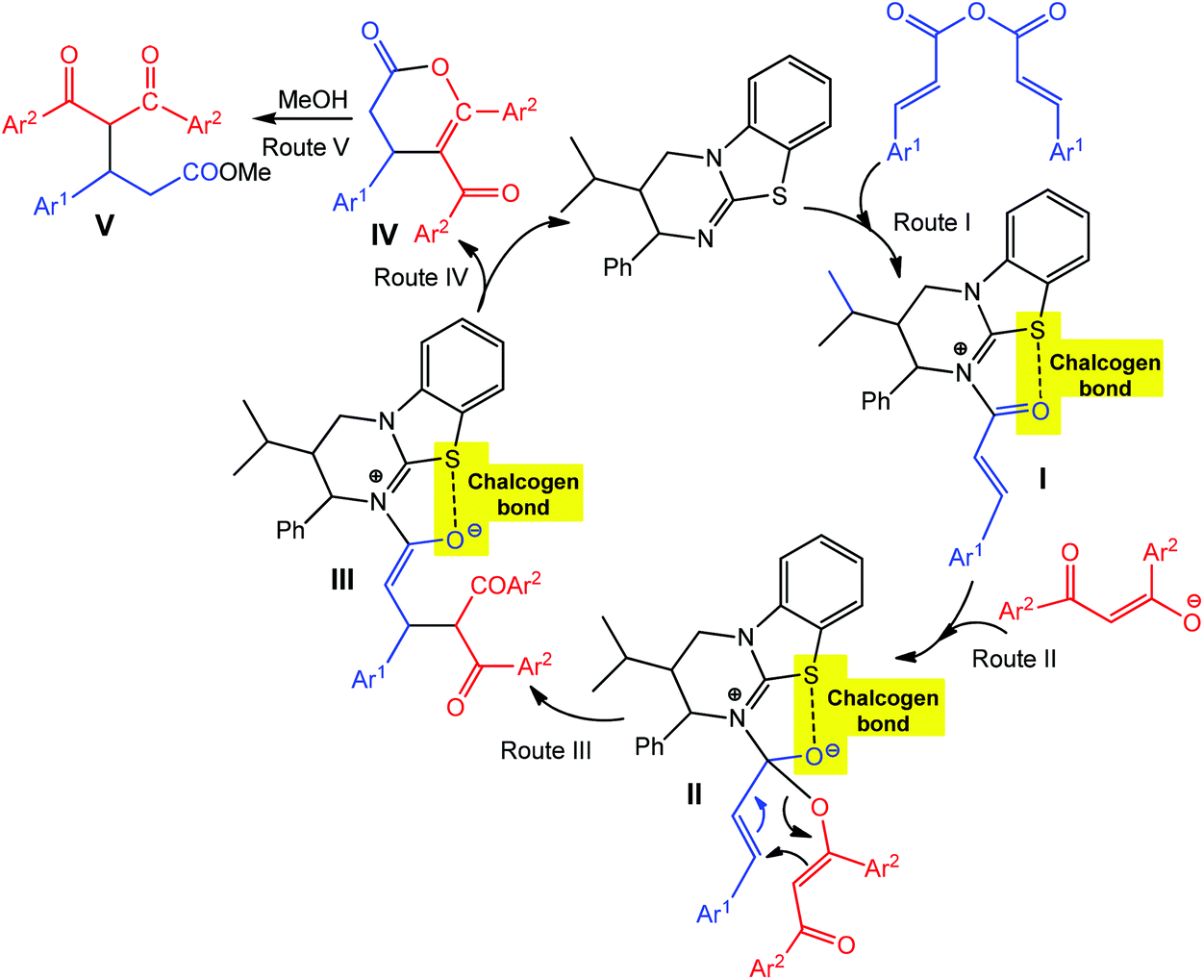 | ||
| Scheme 5 Proposed mechanisms of asymmetric dihydropyranone formation via chalcogen bond involved intermediates.72 | ||
Chalcogen bonding can play a crucial role not only in synthesis and catalysis, but also in the design of materials. For instance, chalcogen S⋯S bonding has been used to construct supramolecular nanotubes which are able to host other molecules (Scheme 6).41 In the case of 1,4,9,12,17,20-hexathiacyclotetracosa-2,10,18-triyne, the pre-made rings are stacked together by chalcogen bonding (Scheme 6).41 The diameter of the thus-built tubes is approximately 6 Å, the size appropriate to constrain n-hexane. The channel size is controllable by a number of –CH2– or –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– units (Scheme 6), as well as by replacement of S by Se or Te atoms. Accordingly, those materials can host various guests such as chlorobenzene, p-xylene, nitrobenzene, etc.41
C– units (Scheme 6), as well as by replacement of S by Se or Te atoms. Accordingly, those materials can host various guests such as chlorobenzene, p-xylene, nitrobenzene, etc.41
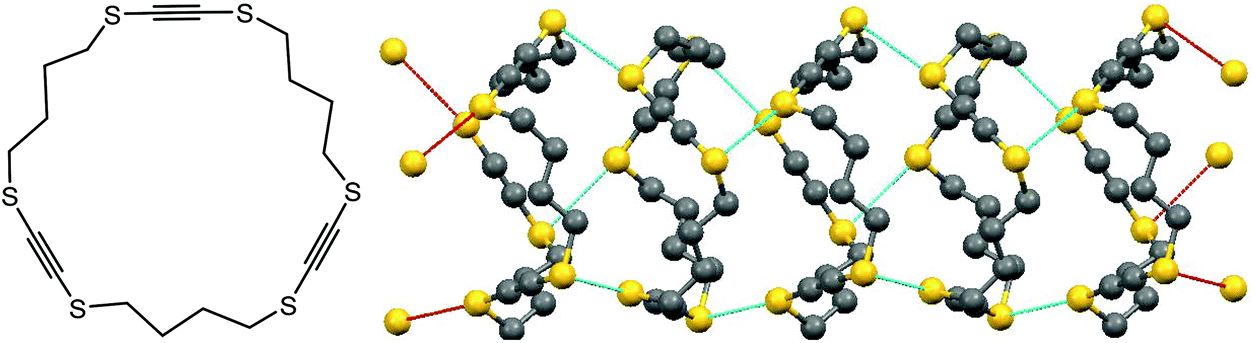 | ||
| Scheme 6 Supramolecular nanotubes constructed from 1,4,9,12,17,20-hexathiacyclotetracosa-2,10,18-triyne.41 | ||
Apart from the above representative cases, new prospective approaches to the application of chalcogen bonding appear every day, and below we highlight some of them. In some cases the crucial role of chalcogen bonds was not recognized by authors, but we spotlight them based on the reported interatomic contacts (which should be less than the sum of the van der Waals radii) and the specific bond directionality.
2. Synthesis
The inter- or intramolecular chalcogen bonding plays a significant role in many cases of chemical synthesis, drug design, enzyme mimetics, vapor deposition, etc.73–78 The intramolecular chalcogen bonding is classified into 1-3, 1-4, 1-5 and 1-6 types (Scheme 7) with the corresponding type of the quasi-ring involving Ch⋯D synthons (D = O, S, Se, Te, N, P, halogen, anion, π-system, radical, etc.).77,78 As expected, in most cases the Ch⋯D distance decreases from 1-3 to 1-6 rings, in parallel with the increase of thermodynamic stability of the corresponding chalcogen bond. Moreover, the steric energy of a molecule, which accounts for its geometry at the energy minimum, is expressed in terms of a number of contributions including chalcogen bonds.78,79 In this section, the chalcogen bond assisted synthesis of organochalcogens and coordination compounds will be demonstrated by several examples where the Ch⋯D interactions play a role as a driving force and/or selectivity synthon in the course of the reaction.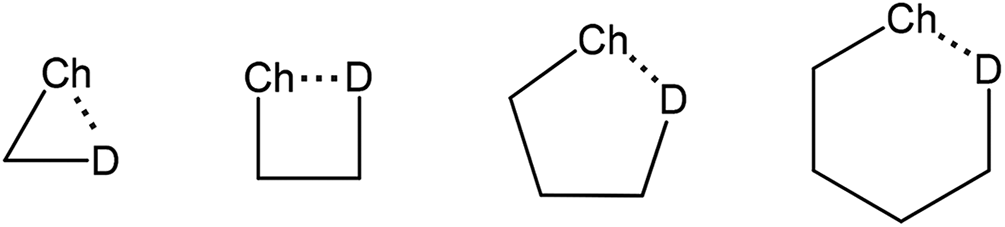 | ||
| Scheme 7 Quasi-ring synthons in organochalcogens.78,79 | ||
2.1. Organic synthesis
Functionalized chalcophenes are key structural units in conducting polymers, microelectronic actuators, chemical sensors, electrochromic devices, organic light emitting diodes, nonlinear optics, bioactive pharmacophores, natural products, and the inter- and intramolecular chalcogen bonding can contribute to their synthetic transformations. For example, the ring-closure addition–elimination reaction of 2,3-dimethoxybuta-1,3-diene with TeCl4 in the presence of CH3SiSiCH3 and CH3COONa produces 3,4-methoxytellurophene stabilized by strong intermolecular chalcogen bonds (Scheme 8).80 The formation of the Te⋯Te bond assisted 6-membered ring in 3,4-methoxytellurophene was accounted for by the metallic character of tellurium, what can be used to create new organic electronic materials.80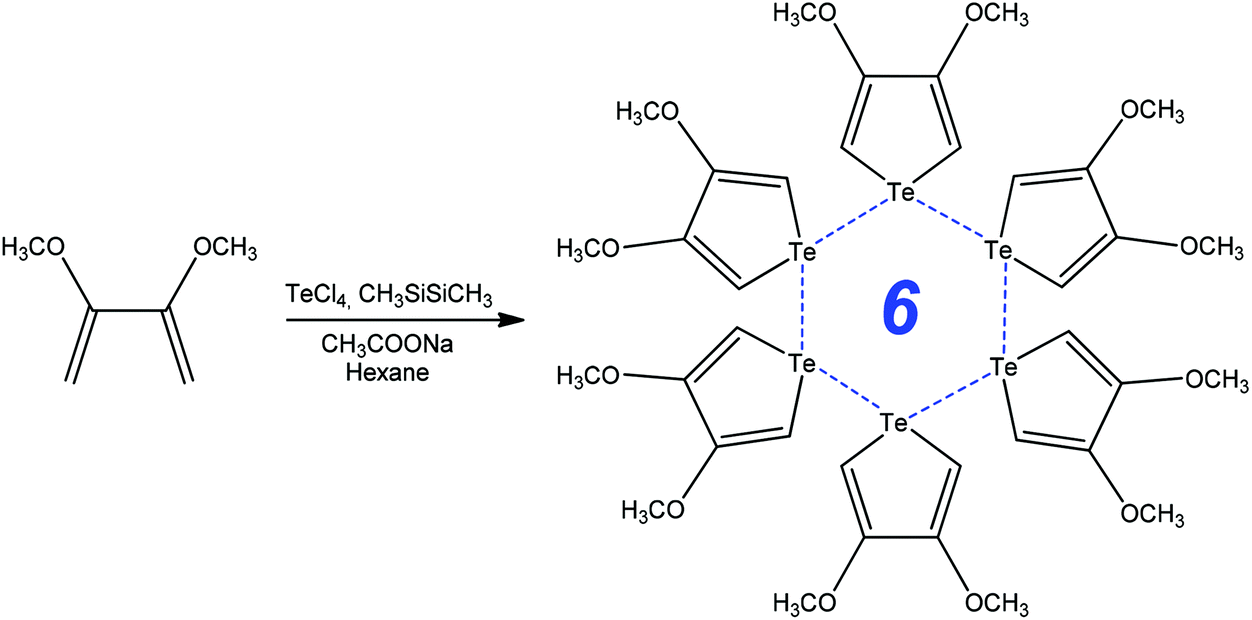 | ||
| Scheme 8 Synthesis of 3,4-dimethoxytellurophene.80 | ||
Another interesting motif that relies upon a 1,5 N⋯Se interaction was encountered in the single-crystal X-ray structure of bis{[2-(N,N-dimethylamino)methyl]phenyl}tetraselenide, obtained by the treatment of N,N-dimethylbenzylamine with n-butyllithium and elemental selenium, followed by oxidation under aqueous conditions (Scheme 9).81 The formation of the two five-membered cycles with the assistance of intramolecular chalcogen bonding results in the production of the corresponding organotetraselenide compound (Scheme 9).
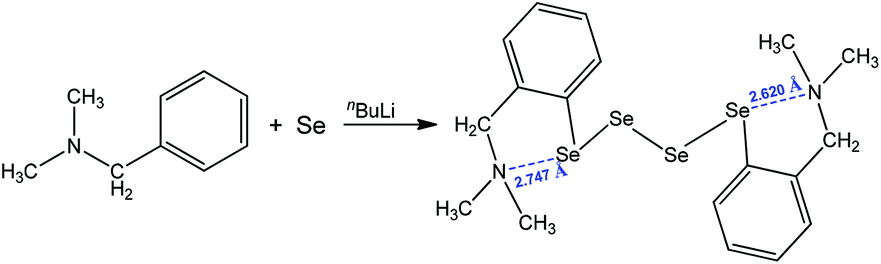 | ||
| Scheme 9 Synthesis of bis{[2-(N,N-dimethylamino)methyl]phenyl}tetraselenide.81 | ||
A 1,4 N⋯S chalcogen bonding appears to influence the biological activity and conformation of heterobiaryls and can also drive the reaction towards the target product.78,82,83 For example, the Suzuki–Miyaura coupling of 6-chloro-2-(4-fluorophenyl)imidazo[1,2-b]pyridazine with thiophen-2-ylboronic acid leads to 2-(4-fluorophenyl)-6-(thiophen-2-yl)imidazo[1,2-b]pyridazine (Scheme 10).84
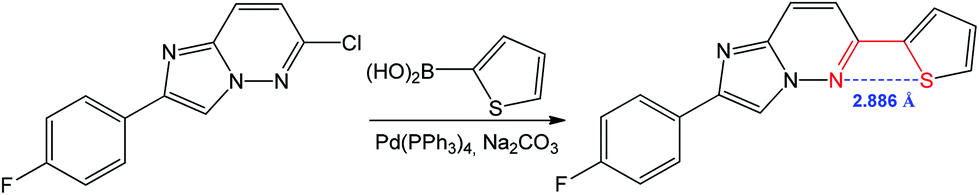 | ||
| Scheme 10 Synthesis of 2-(4-fluorophenyl)-6-(thiophen-2-yl)imidazo[1,2-b]pyridazine.84 | ||
Other 1,4-(S⋯O) and 1,4-(Se⋯Cl) chalcogen bonding synthons were found in N,N-diisopropyl-4-methoxy-3-(methylthio)picolinamide and 3-(benzylselanyl)-4-chloro-N,N-diiso-propylpicolinamide, respectively (Scheme 11).85 The formation of a chalcogen bond assists the C–H bond activation in 4-substituted-N,N-diisopropylpicolinamide, while no interaction is observed between S (or Se) and the carbonyl oxygen as the oxygen atom points away from the S (or Se) centre.
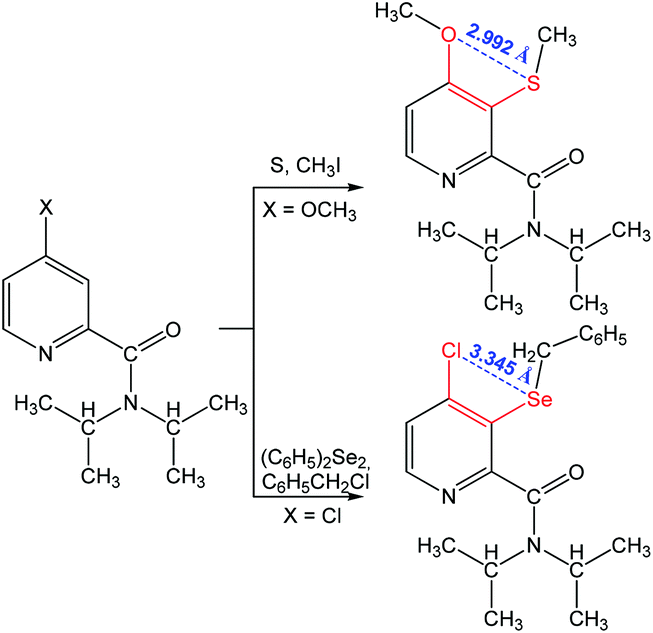 | ||
| Scheme 11 Synthesis of chalcogen (S and Se) derivatives of 4-chloro- and 4-methoxy-N,N-diisopropylpyridine-2-carboxamide.85 | ||
Chalcogen bonding assisted 1,3-dipolar cycloaddition of N3,4-bis(4-bromophenyl)-1,2,4-thiadiazolidine-3,5-diimine with N-(4-bromophenyl)cyanamide produces (E)-1,2-bis(4-bromo-phenyl)-1-(3-((4-bromophenyl)amino)-1,2,4-thiadiazol-5-yl)guanidine, which contains a 1,5-(S⋯N) synthon with a distance of 2.538 Å (Scheme 12), suggesting that there is a strong noncovalent interaction between the electron-donating guanidine N atom and the acceptor S atom of the thiadiazole ring.86
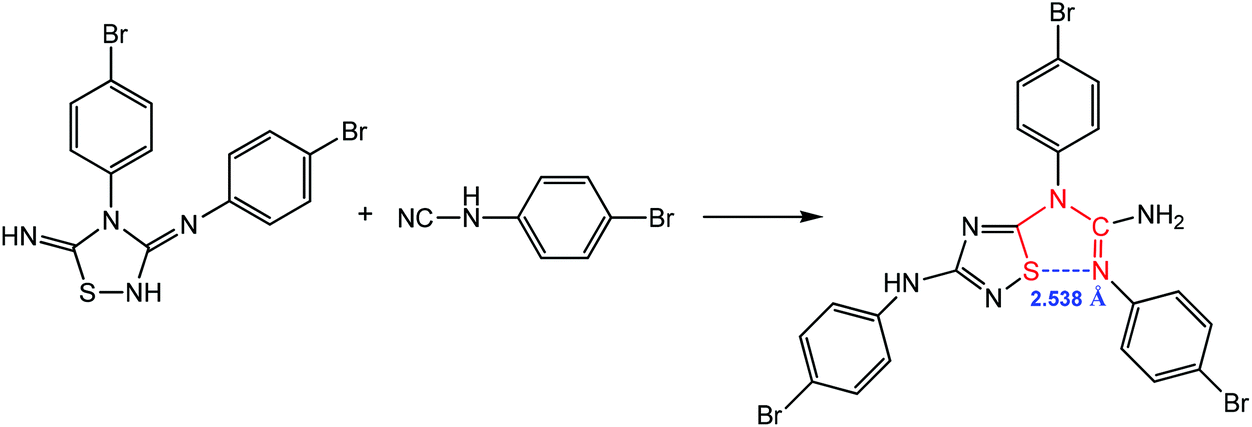 | ||
| Scheme 12 Synthesis of (E)-1,2-bis(4-bromophenyl)-1-(3-((4-bromophenyl)amino)-1,2,4-thiadiazol-5-yl)guanidine.86 | ||
Chalcogen bonding between oxygen atoms O⋯O has been observed in a large number of organic compounds, contributing to their synthesis.79,87,88 From those, one can consider a reaction of (2-methoxyphenyl)-bis(methylperoxy)-λ3-iodane with dimethyl malonate to give dimethyl 2-((2-methoxyphenyl)-λ3-iodanylidene)malonate stabilized by chalcogen bonding with a short O⋯O distance (Scheme 13).87
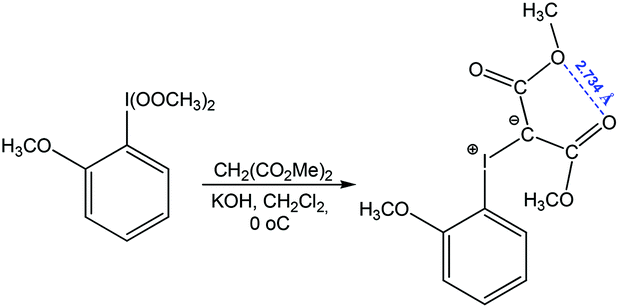 | ||
| Scheme 13 Synthesis of dimethyl 2-((2-methoxyphenyl)-λ3-iodanylidene)malonate.87 | ||
Apart from the stabilization of a particular product, chalcogen bonding can increase the stability of organic radicals.89–92 Thus, the reduction of 1,2,3,5-dithiadiazolyl salt [NC–(CF2)4–CNSNS]SbF6 with ferrocene (FeCp2) led to the 1,2,3,5-dithiadiazolyl radical NC–(CF2)4–CNSSN˙ which is stabilized by multiple chalcogen bonding (Scheme 14).91 In fact, this compound is dimerized in the trans-cofacial fashion with intra-dimeric chalcogen bonding S⋯N, and the dimeric units are also linked by S⋯Ncyano interactions forming an infinite chain. Both S atoms in the dithiadiazolyl ring behave as a two-centred chalcogen bond donor towards the N˙ radical and the nitrogen atom of the cyano group (Scheme 14). When the temperature is increased from 305 to 335 K, NC–(CF2)4–CNSSN˙ shows magnetic bistability with colour change from dark green (dimeric solid) to dark brown (paramagnetic liquid).
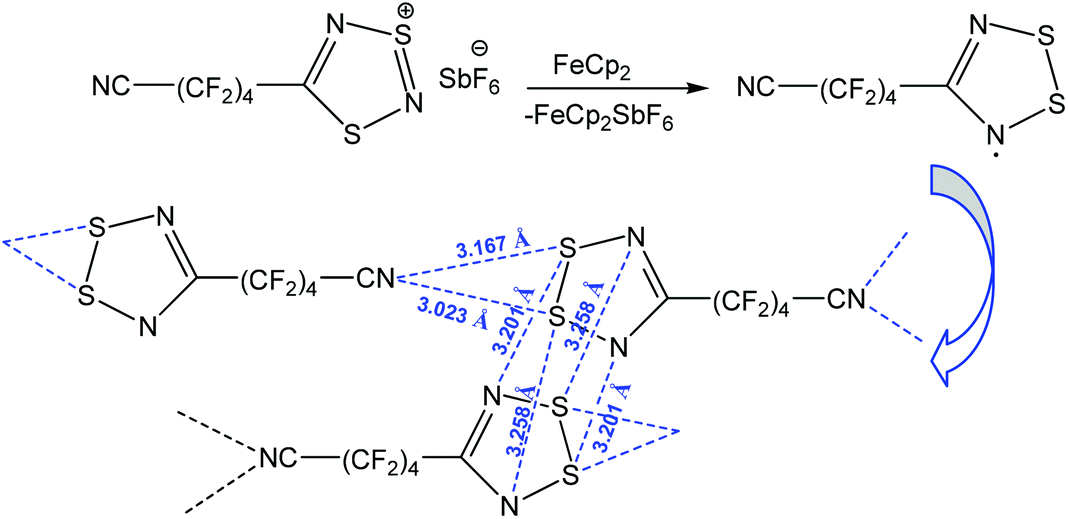 | ||
| Scheme 14 Stabilization of the 1,2,3,5-dithiadiazolyl radical NC–(CF2)4–CNSSN˙.91 | ||
Another example is related to ortho-chalcogen substituted phenoxyl radicals stabilized by strong chalcogen bonding (Scheme 15).92 A homolytic abstraction of the hydrogen atom from the –OH group of 4-methoxy-6-methylbenzo[b]thiophen-7-ol by a peroxyl radical yields the chalcogen bond protected 4-methoxy-6-methylbenzo[b]thio-phenoxyl-7 radical (Scheme 15).92 From this example, one can conclude that the antioxidant properties of phenoxyl radicals can be controlled with the assistance of chalcogen bonding.
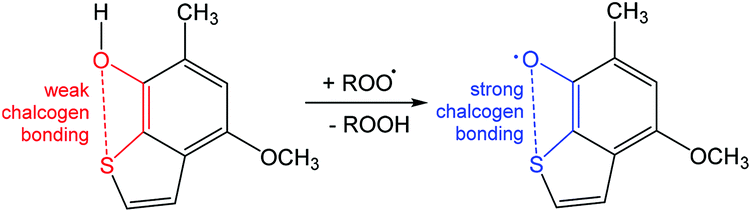 | ||
| Scheme 15 Stabilization of the 1,2,3,5-dithiadiazolyl radical.92 | ||
2.2. Inorganic synthesis
Exploiting the analogy with hydrogen and halogen bonds, the chalcogen bond is comparatively a new type of weak noncovalent interaction, which can be used by organic tectons to control the secondary coordination sphere of metal ions.29,63–68 As for other types of noncovalent interactions, chalcogen bonding can be intra- and intermolecular, in coordination compounds.29,63–68 In intramolecular chalcogen-bonded coordination compounds, the coordination entities can be employed with two different roles: as donors and as acceptors.29 Thus, metal coordination may increase the electrophilicity of the chalcogen atom of a ligand which can act as the chalcogen bond donor towards other coordination entities or species.29 Chalcogen bond containing coordination entities can also act as the chalcogen bond acceptor nodes through their ligands.29,63–68 The potential of chalcogen bonding in the synthesis of coordination compounds has recently been pointed out by us (we refer to the readers),29 but, in this subsection, we shall discuss more recent examples on this topic. An interesting case has already been presented above, in the Introduction section, concerning the regioisomerization of binuclear (diaminocarbene)PdII complexes (Scheme 4).67 Other examples of inorganic synthesis assisted by chalcogen bonding are as follows.Chalcogen bonding may be involved in self-assembly of a variety of supramolecular aggregates that include cyclic tetramers and hexamers, which can act as receptors for host species such as tetrahydrofurane, Pd2+, etc.66 For instance, the auto-association of [Pd(NCCH3)4](BF4)2 with the negative charge assisted chalcogen bond supported tetramer of iso-tellurazole N-oxides in a CH2Cl2/acetonitrile mixture affords a macrocyclic Pd complex (Scheme 16).66 In this compound the metal centre displays a square planar coordination geometry with Pd–Te distances of 2.5804(4) Å, while the Te⋯O− chalcogen bonding is 2.174 Å, being significantly shorter than the sum of the van der Waals radii of the interacting atoms, of 3.58 Å (Te + O = 2.06 + 1.52 = 3.58 Å). Thus, each of this strong intramolecular negative charge assisted chalcogen bonding closes a five-membered metallacycle leading to the Pd2+ hosted macrocycle, which can contribute thermodynamically to the complexation reaction.
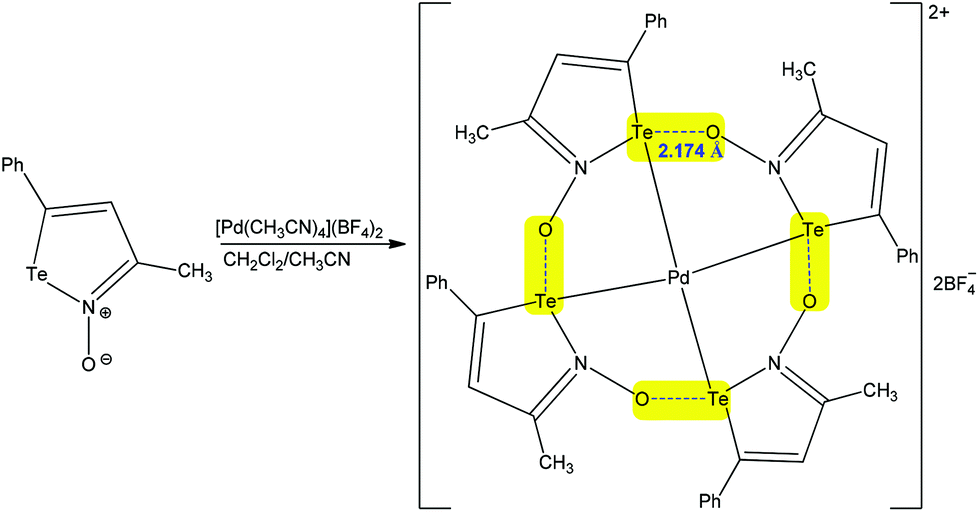 | ||
| Scheme 16 Synthesis of a macrocyclic Pd complex with intramolecular chalcogen bonds.66 | ||
Multiple chalcogen bonding participates in the interaction of [PdCl2(NCPh)2] with the reaction product of SeCl2 with tBuNH2 leading to a dinuclear palladium(II) adduct (Scheme 17).68 This product consists of two different palladium(II) complexes in the same crystalline lattice, linked together by multiple Se⋯Cl chalcogen bonding. The coordination environment of both palladium centres in this chalcogen bond supported adduct is square-planar. The Cl–Se–N and Cl–Se–Cl angles in the range of 145.62–166.69° are in agreement with chalcogen bond formation with a moderate directionality.
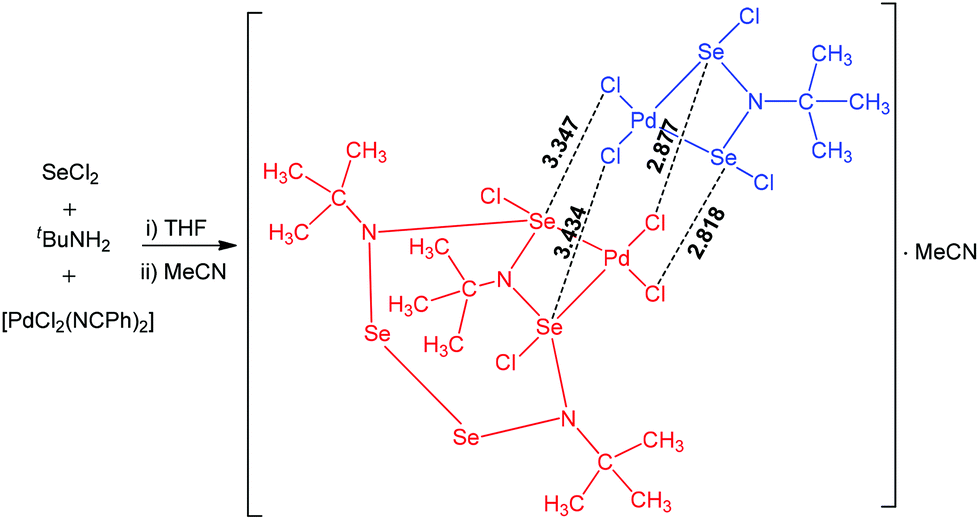 | ||
| Scheme 17 Multiple chalcogen bonding in the synthesis of a dinuclear palladium(II) adduct.68 | ||
3. Catalysis
Noncovalent interactions play an important role in many catalytic cycles, e.g., offering additional stabilization to particular intermediate(s) and thus providing high selectivity in the case of asymmetric catalysis.65,69–72,93–95 The stabilization of an intermediate depends on the types of noncovalent interactions and reaction conditions. Although individually these interactions are relatively weak, their orchestrated action can provide a powerful tool for reaching high yields and controlling selectivity.The role of noncovalent interactions, such as π-stacking interactions,70 hydrogen69 and halogen24 bonding, in catalysis has been well recognized. Taking into account the similarities between those weak forces and chalcogen bonding, the latter should also be explored as an important tool in catalysis. Moreover, the higher directionality of chalcogen bonds may allow achieving a higher selectivity. In particular, interesting catalytic results can be expected if catalysis is performed in nonpolar media when the hydrophobic character of the chalcogen bond plays an additional role.71 Although data on the use of chalcogen bond donor molecules in catalysis still remain rather scarce,71,72,93–95 we found some interesting examples, and one of them has been discussed in the Introduction section (Scheme 5),72 while others follow below.
Thus, the chalcogen bond accelerates the hydrogenation of quinolones and imine when dithieno[3,2-b;2′,3′-d]thiophenes or their diimides are used as chalcogen bond donor catalysts (Scheme 18).71 Cooperation of two chalcogen bonds in a specific binding motif (as “tongs”) facilitates the expected substrate–catalyst interactions. According to the proposed mechanism, an intermediate is stabilized by chalcogen bonds leading to a 5-membered ring, which initiates the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in quinolines and imines (Scheme 18).
N bond in quinolines and imines (Scheme 18).
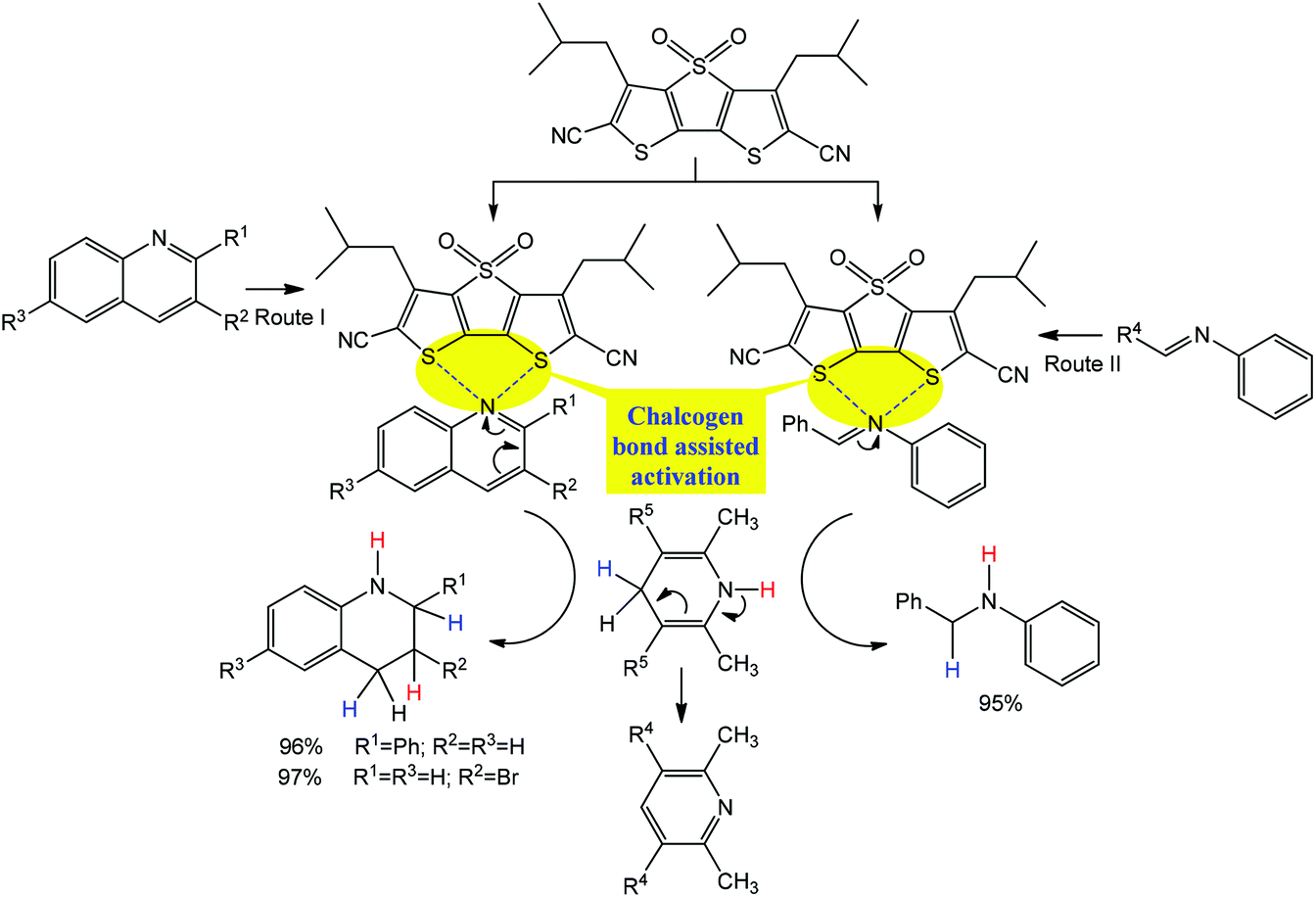 | ||
| Scheme 18 Chalcogen bond assisted hydrogenation of quinolines (Route I) and imines (Route II).71 | ||
Similar to the above example of Scheme 5, a chiral isothiourea catalyst, (R)- or (S)-2-phenyl-2,3-dihydrobenzo[d]imidazo[2,1-b]thiazole, is effectively converted into a chalcogen bond supported α,β-unsaturated acylammonium intermediate by its reaction with an α,β-unsaturated anhydride, which undergoes two sequential chemoselective nucleophilic attacks by 2-aminothiophenols producing 1,5-benzothiazepines I and a negligible amount (<1%) of by-products II and III (Scheme 19a).93
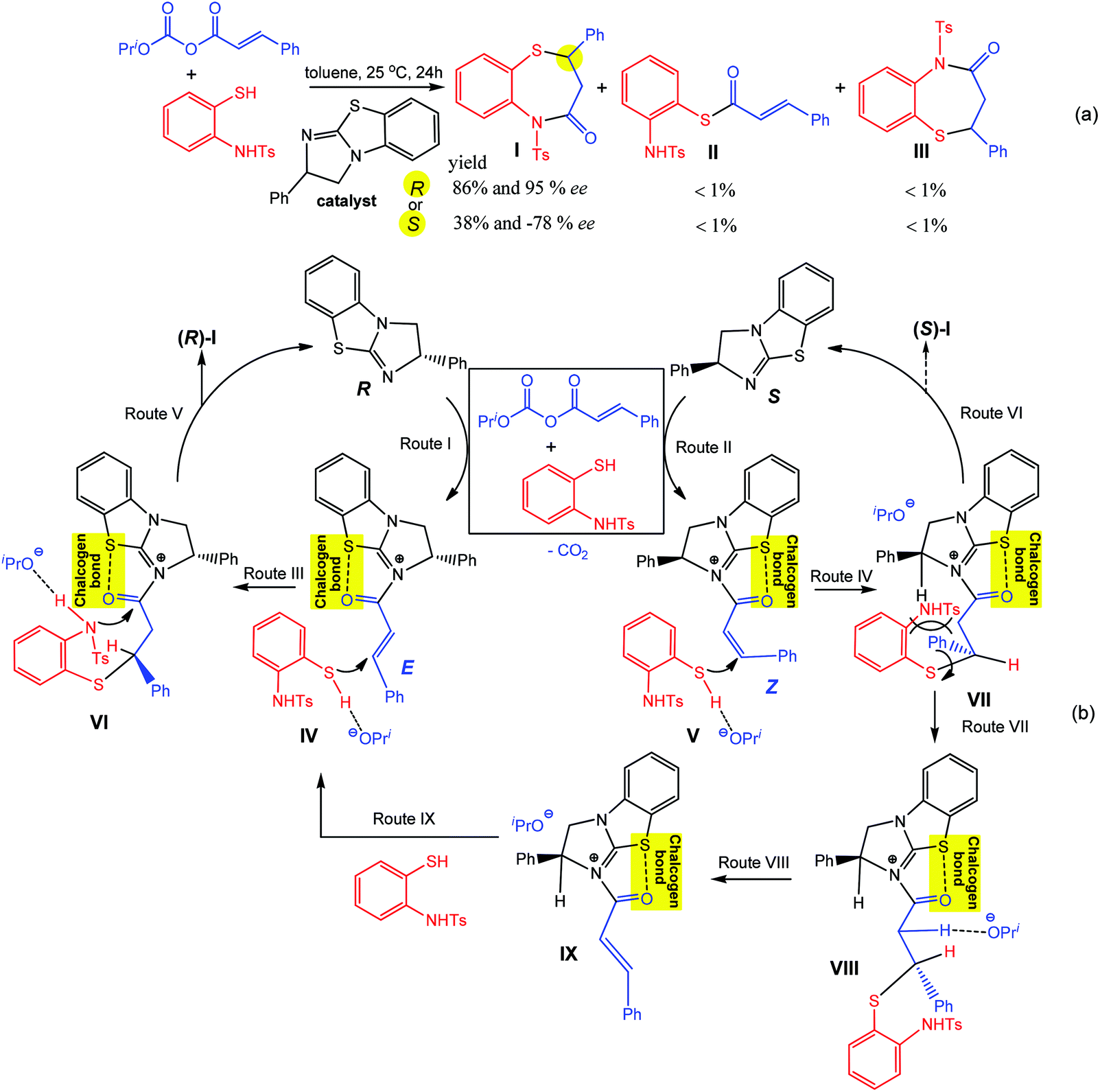 | ||
| Scheme 19 Synthesis of 1,5-benzothiazepines (a) and proposed mechanism (b).93 | ||
According to the proposed mechanism (Scheme 19b), the reaction of (R)- or (S)-2-phenyl-2,3-dihydrobenzo [d]imidazo[2,1-b]thiazole with a mixture of E- or Z-isomers of α,β-unsaturated anhydrides and 2-aminothiophenols goes through (i) the formation of acylammonium intermediates IV and V, stabilized by intramolecular S⋯O chalcogen bonding, with the liberation of iPrO− and CO2 where the former behaves as a hydrogen bond acceptor towards the S–H moiety of a 2-aminothiophenol (Routes I and II in Scheme 19); (ii) a nucleophilic attack of a sulfur atom of 2-aminothiophenol to an olefin carbon atom leading to the intermediates VI and VII (Routes III and IV in Scheme 19), followed by cyclization with the formation of I (in comparison to Route V, Route VI may be inhibited by a favourable conformational change driven by the release of repulsion energy); (iii) due to unstable conformation, an intermediate VII is converted into VIII (Route VII), bearing a more stable conformation, and 2-aminothiophenol eliminates to generate the (E)-intermediate IX (Route VIII), which again affords R-1,5-benzothiazepine via intermediates IV and VI (Routes IX, III and V). In all the proposed intermediates, the chalcogen bond increases the electrophilic character of the olefinic carbon atom towards nucleophilic attack by the sulfur atom of 2-aminothiophenol, the enantioselectivity of this reaction being directed by the difference of cyclization rates between the intermediates VI and VII.
As has been mentioned above and illustrated by the above examples, the directionality and versatility of chalcogen bonding offers several possibilities for the creation and stabilization of different types of intermediates in catalytic cycles. The cooperation of chalcogen bonding with other type(s) of noncovalent interactions can further facilitate a desired reaction, while a particular orientation could enhance the selectivity. For example, cooperating chalcogen and halogen bonds are observed in the peri-substituted naphthalene (Scheme 20a) catalyzed regioselective deiodination of thyroxine (T4) to 3,5,3′-triiodothyronine (T3) and 3,3′,5′-triiodothyronine (rT3) by outer- and inner-ring deiodination pathways, respectively (Scheme 20b).94,96 Although the formation of any I⋯Se halogen bond elongates (weakens) the C–I bond, a regioselective and effective bond activation begins only when the two chalcogen atoms are in the peri position in the catalysts (Scheme 20a). Thus they interact with each other leading to chalcogen bonding that facilitates the transfer of a higher electron density to the C–I σ* orbitals through halogen bonds (Scheme 20c). As a selenol provides stronger chalcogen bonding than a thiol, replacement by selenium of either one or both sulfur atoms of the peri-naphthalene-dithiol considerably increases the yield (75-fold more) of the deiodination.
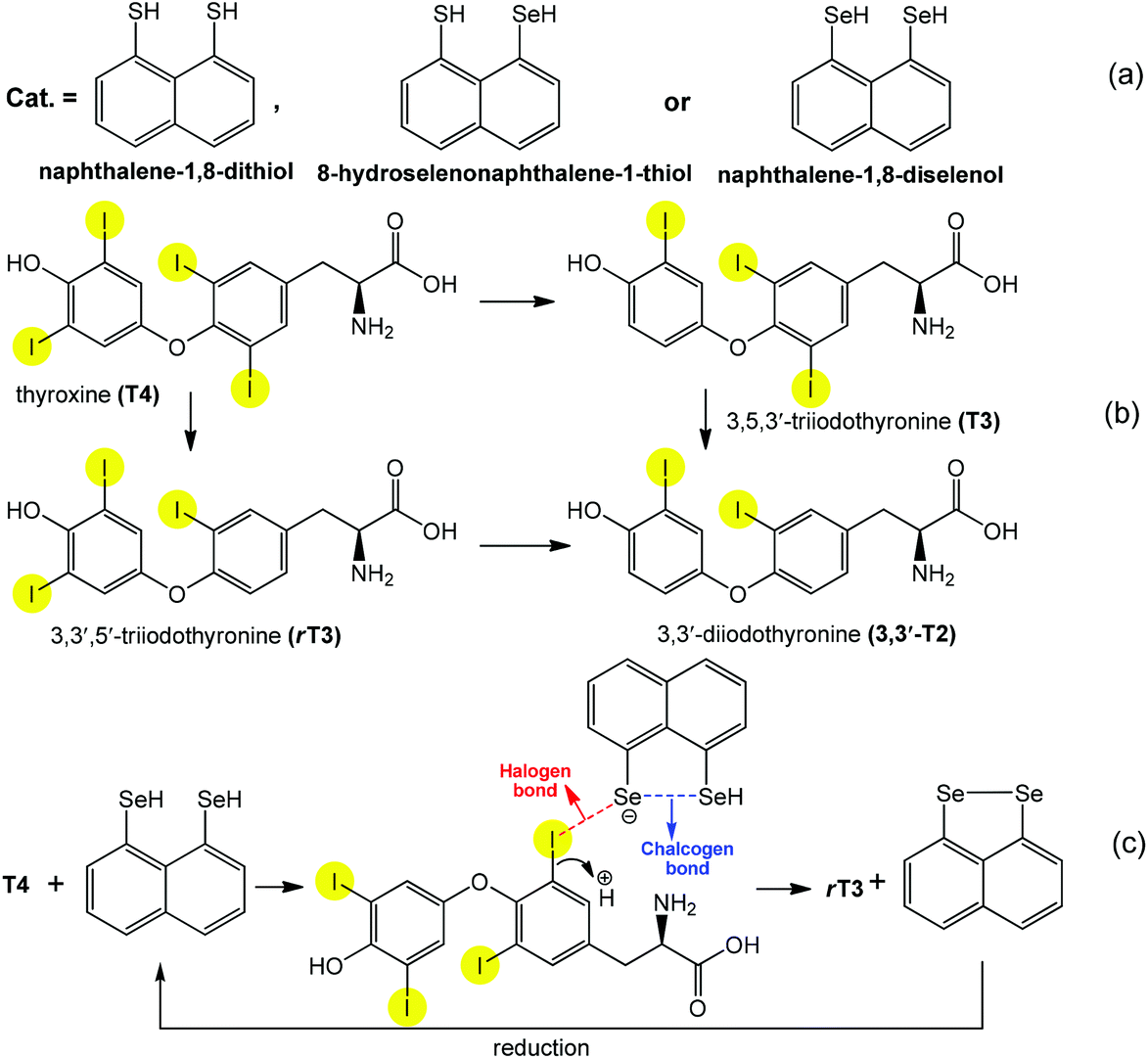 | ||
| Scheme 20 Synthetic transformations of thyroxines (a) and peri-substituted naphthalenes (b). Proposed actions of halogen and chalcogen bonds in the activation of the C–I bond (c).94 | ||
4. Design of materials
Materials containing organochalcogens or coordination compounds with chalcogen bond(s) have been extensively investigated due to their unique properties, such as electric and thermal conductivities, molecular self-assemblies, nanotube formation, ion exchange capacity, molecular recognition, non-linear optics, photoelectrochemical behaviour, drug delivery, etc.78,97–119 All these properties are dependent on the nature of the constituent elements, directionality and tunability of chalcogen bonding, as well as the corresponding size of the quasi-ring involving the Ch⋯D synthon and planarity of this ring. All the Ch⋯D containing quasi-rings are almost planar and can be involved in other types of noncovalent interactions leading to better materials with enhanced device performance.97,98 Accordingly, chalcogen bonding in organic/inorganic materials can not only provide a fundamental understanding of structure–property relationships120 but also has great potential for many technological applications.27,41,117–124 In this respect, switching to heavier chalcogen atoms with a larger polarizability, such as selenium or tellurium, is of particular interest since the larger atomic size may strengthen the inter- or intramolecular chalcogen bond interactions in solution and in the solid state.125 For instance, many polymers reported to date are based on thiophene or its fused ring analogues, while a conjugated polymer with selenophene may exhibit a better performance in organic field-effect transistors, retaining the identical polymer backbone.126–129The variation of substituent(s) and chalcogen bond acceptor side(s) in the monomeric unit can also significantly affect the charge transport properties of these materials. Thus, the use of selenophene spacers in the polymer poly(2,7-carbozole-alt-diselenienylbenzothiadiazole) (Scheme 21a) led to increased crystallinity and higher power conversion efficiency (4.1% vs. 3.6%) in comparison to the analogous thiophene device due to Se⋯Se chalcogen bonding.130 Moreover, the replacement of sulfur or selenium with tellurium in conjugated polymers leads to the narrower HOMO–LUMO gap, red-shifted optical absorption, higher polarizability and dielectric constant.97,131
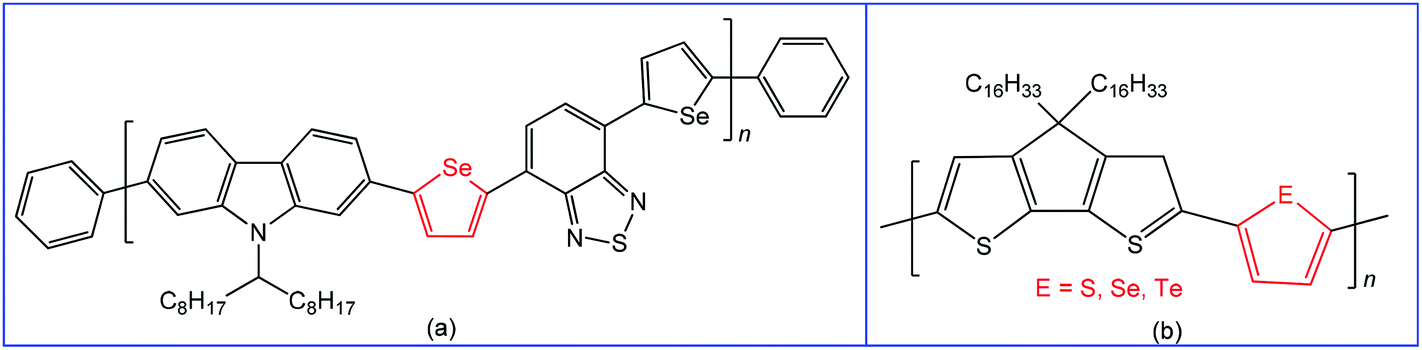 | ||
| Scheme 21 Poly(2,7-carbozole-alt-diselenienylbenzothiadiazole) (a);130 cyclopentadithiophene polymer with different comonomers (thiophene, selenophene and tellurophene) (b).132 | ||
The variation of comonomers (thiophene, selenophene and tellurophene) in cyclopentadithiophene polymer (Scheme 21b)132 offers a red-shifted absorption with narrowing the band gap. At the same time, the polymer tends to aggregate in solution due to the strong Te⋯Te chalcogen bonding. The intensity of the absorption peak depends on the strength of the chalcogen–chalcogen interactions, i.e., a polymer with a tellurophene comonomer demonstrates the most intense aggregation-induced absorption.132
As can be seen from the above examples, chalcogen bonding can control the arrangement of molecules in the solid state, and this is very important for the successful design of materials with desired magnetic, conducting, non-linear optic, etc. properties. Although considerable progress towards the application of hydrogen and halogen bonding for the design of new materials has been achieved, only limited examples are available regarding the related role of chalcogen bonding.62,67,113–116 Hence, in this section we highlight the role of chalcogen bonding in crystal engineering and for molecular recognition.
4.1. Crystal engineering
Along with the well-explored hydrogen- and halogen-bonded motifs, in recent years chalcogen bonding has also attracted significant interest in crystal engineering, drug design,78 supramolecular synthons, etc., as in most cases it combines two essential properties: strength and directionality.51,90,101,102,108,109,133–171 The manipulation of dimensionality can be achieved by modifying the type and/or strength of chalcogen bonds present in the crystal.A variety of chalcogen bond involved synthons with electron donor or acceptor character were employed for controlling the assembly of molecular building blocks into designed supramolecular architectures. Both types of synthons are of great importance owing to the fact that by changing the molecular geometries of organochalcogens one can manipulate the structure of crystals. Hence, an understanding of the nature of the chalcogen bonding towards its control of the molecular arrangement in the solid state can be a fundamental issue for obtaining the desired organic semiconducting or switching materials.
One of the examples demonstrating these possibilities relates to (E/Z)-2,2′-bibenzo[d][1,3]oxaselenolylidene where the inter- and intra-molecular chalcogen bonds contribute to the E ↔ Z switching in solution and to the isolation of E- or Z-isomers in the solid state (Scheme 22).140 The E-isomer is stabilized by multiple inter- and intramolecular noncovalent interactions, such as chalcogen (Se⋯O in a 1,4-membered quasi-ring) and hydrogen (C–H⋯Se) bonds, and two types of π–π interactions (πphenyl–πoxaselenole and Se–πSe⋯Oring).137 Not only intramolecular homonuclear chalcogen bonds, but also intermolecular short Se(2)⋯Se(2) (3.449 Å, Coxaselenole–Se(2)⋯Se(2) 167.81°) interactions have an impact on the stabilization of the Z-isomer in the solid state. Thus, the chalcogen bond involved synthon can play a donor or an acceptor role in the construction of supramolecular architectures.
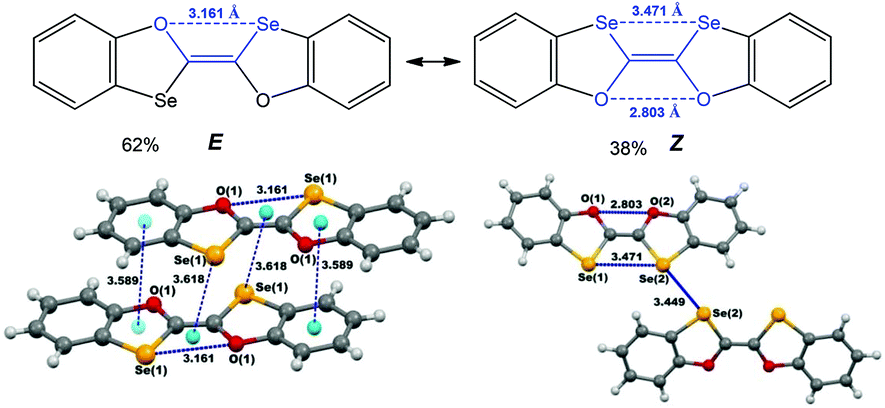 | ||
| Scheme 22 Chalcogen bonding in (E/Z)-2,2′-bibenzo[d][1,3]oxaselenolylidene.140 | ||
Other types of chalcogen bonding have also been identified and applied in crystal engineering. For example, a positive charge assisted chalcogen bonding synthon is found in a series of peri-substituted chalconium acenaphthene salts I–VI (Scheme 23).154 The same equatorial–axial arrangement is observed in I–III, with the methyl group positioning along the acenaphthene plane with axial occupation of a phenyl group. The planar location of the methyl group with the acenaphthene moiety provides the “correct” geometry for the existence of a positively charged assisted chalcogen bonding with moderate directionality, 166–176° (Scheme 23). Upon switching from S to Te, the Ch+⋯Br distances increase in the following order: 3.181(S) < 3.195(Se) < 3.285(Te) Å (Scheme 23), which are 13–16% shorter than the sum of van der Waals radii for the two interacting atoms. A similar tendency occurs in the homonuclear positively charge assisted chalcogen bonding synthons Ch+⋯Ch IV–VI: 3.134(S) < 3.247(Se) < 3.445(Te) Å (16–19% shorter than the sum of van der Waals radii) and Ch+⋯Ch–CH3 angles approaching 180° (170–177°) (Scheme 23), which may promote the delocalization of a chalcogen lone pair in a 5-membered quasi-ring. In each case the two phenyl rings are located axially to the acenaphthene plane but with either a trans (IV) or a cis (V and VI) orientation (Scheme 23).
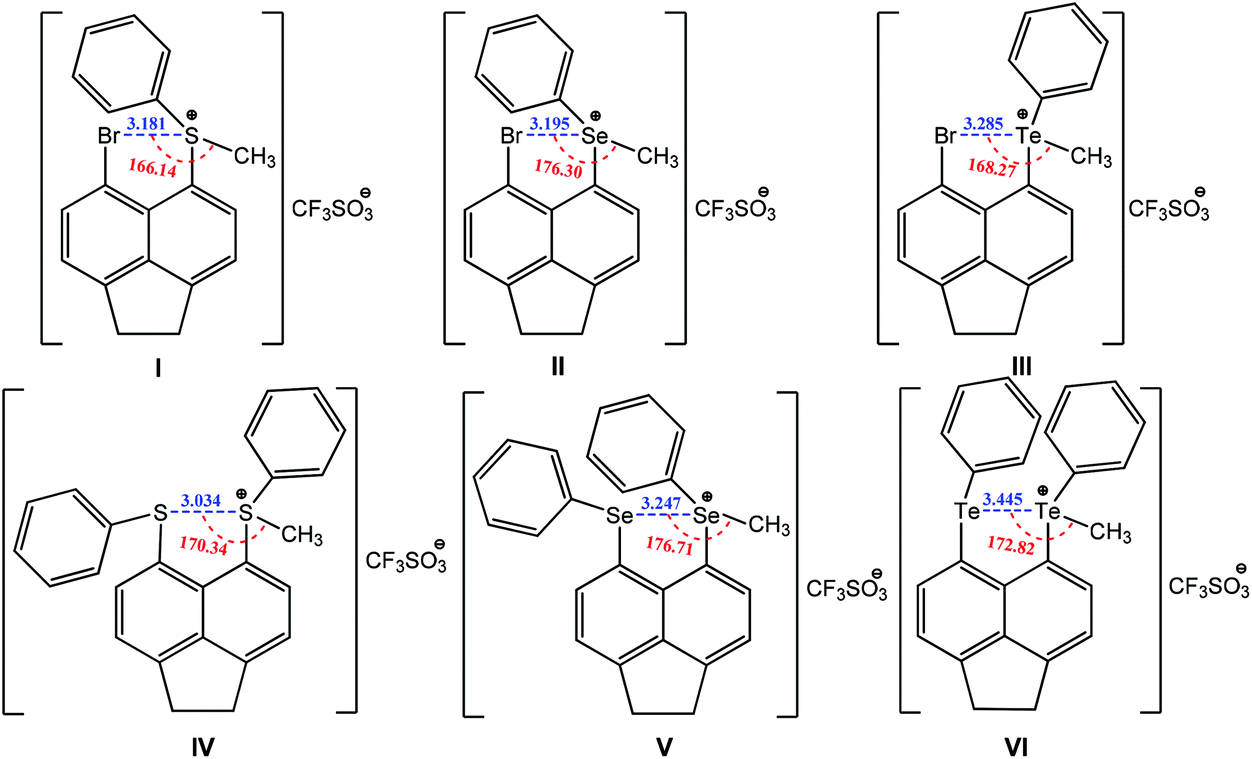 | ||
| Scheme 23 Positive charge assisted chalcogen bonding in peri-substituted chalconium acenaphthene salts.154 | ||
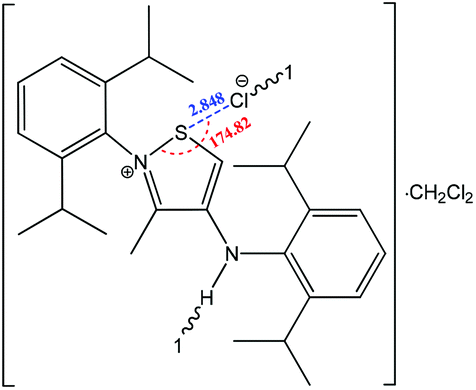
The strength of the chalcogen⋯nucleophile interactions is related to the degree of charge transfer between the chalcogen bond donor and the acceptor, illustrating the importance of electrostatic interactions in the formation of a negative charge assisted chalcogen bond. Thus, a strong and directional chalcogen bonding is expected when an anion is used as a chalcogen bond acceptor. Furthermore, the negative charge assisted chalcogen bond may cooperate with other types of noncovalent interactions, such as hydrogen bonding or ionic interactions, as in 2-(2,6-diisopropylphenyl)-4-((2,6-diisopropylphenyl)amino)-3-methyliso-thiazol-2-ium chloride (Scheme 24), where cooperation of a negative charge assisted chalcogen bonding with ionic interactions and hydrogen bonding was found.148 In this compound, the S⋯Cl− distance (2.848 Å) is significantly lower than the sum of van der Waals radii (S + Cl = 3.55 Å), and the N+–S⋯Cl− angle approaches 180° (174.82°). In the intermolecular charge assisted hydrogen bonding system, the N–H⋯Cl− angle and the N⋯Cl− distance are 146.05° and 3.267 Å, respectively, whereas the intramolecular distance between the Cl− anion and the N+ of the thiazol ring is 4.553 Å, which falls in the recognized range of ionic interaction distances.172 Thus, cooperation of all the above mentioned noncovalent interactions contributes to the stabilization of this organic salt in the solid state.
 | ||
| Scheme 24 Negative charge assisted chalcogen bonding in 2-(2,6-diisopropylphenyl)-4-((2,6-diisopropylphenyl)amino)-3-methylisothiazol-2-ium chloride.148 | ||
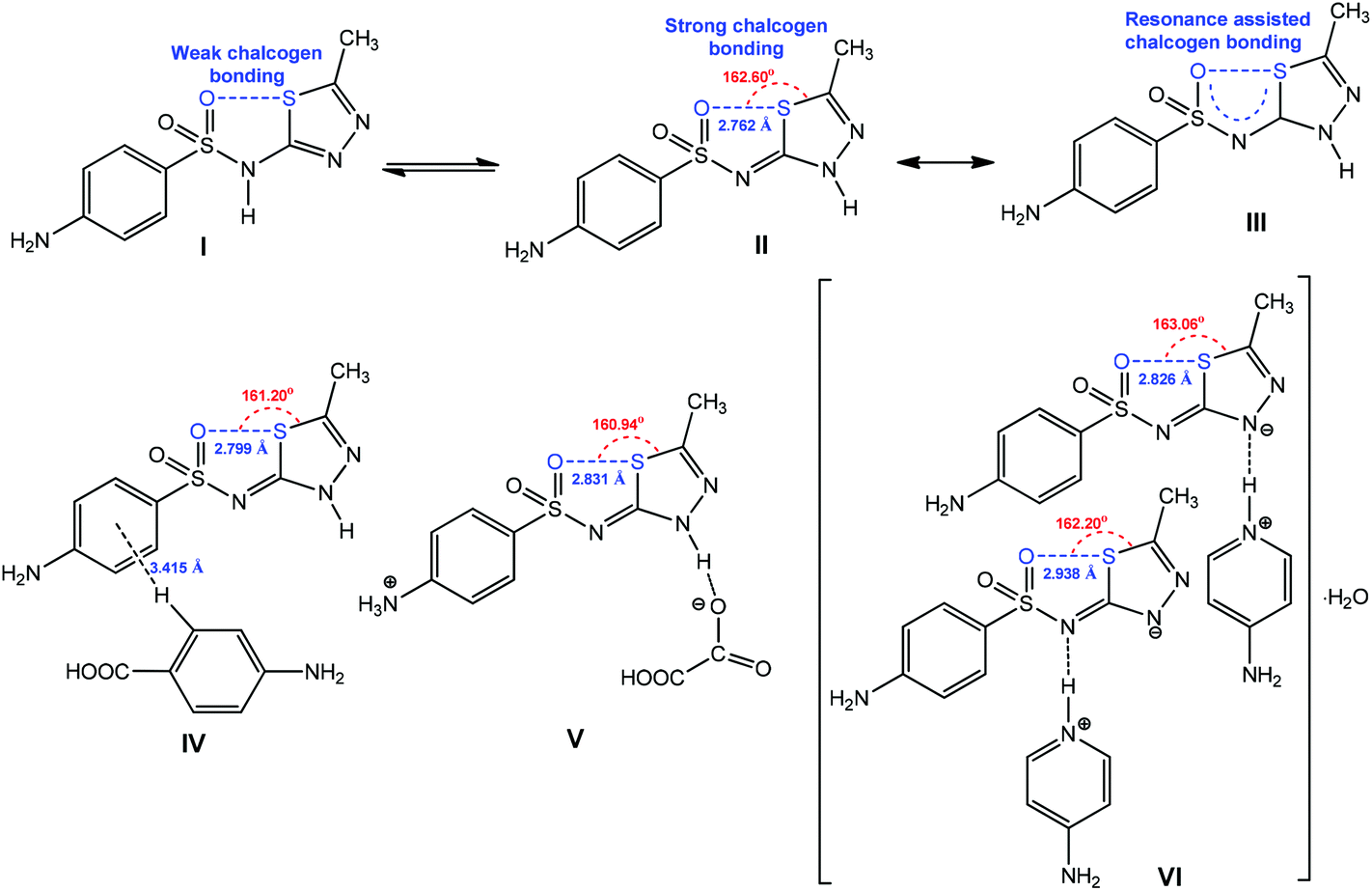
The chalcogen bond contributes to the stabilization of one tautomeric form of the pharmaceutical ingredient sulfamethizole (Scheme 25).159 Due to the π-conjugation in the 5-membered quasi-ring with the formation of RACB, the tautomer III is stable in the solid state (Scheme 25).159 The strengthening and directionality of RACB in III can be manipulated by cocrystalization (IV), protonation (V) or deprotonation (VI) (Scheme 24). In all cases, the O⋯S distance increases (2.799–2.938 Å) in comparison to the starting material II (2.762 Å), whereas the O⋯S–Cthizole angle can decrease (in IV–VI) or increase (in VI) depending on the exhibited noncovalent interactions. Apparently, the participation of the RACB moiety of VI as a hydrogen bond acceptor synthon towards protonated 4-aminopyridine does not affect the O⋯S–Cthizole angle (Scheme 25).
 | ||
| Scheme 25 RACB in molecular (IV), cationic (V) and anionic (VI) forms of sulfamethizole.159 | ||
4.2. Molecular recognition and anion transport
It is well established that noncovalent interactions are responsible for the detection of many organic, inorganic and bio-relevant molecules. In order to develop an artificial receptor (carrier or ionophore) selective towards a specific analyte, the receptor should contain a variety of synthons, organized to complement the size and shape of the analyte. Similar to the halogen bonding,21,173–177 the relatively strong and directional chalcogen bond can also be used for selective recognition; however, quantitative data regarding the chalcogen bonding in solution are sparse.112–115,178–183Due to the hydrophobic character of the chalcogen bond, a desired (high) analytical signal can be expected when the molecular recognition is carried out in apolar media. Moreover, there are three possible ways for the application of chalcogen bonding in molecular recognition: (i) the receptor may have a chalcogen bonding system as a synthon, and it can interact with the analyte, or (ii) the analyte may have a chalcogen bonding system as a synthon, and it can interact with the receptor; or (iii) a chalcogen bond synthon can be formed during the analytic reaction (detection process).
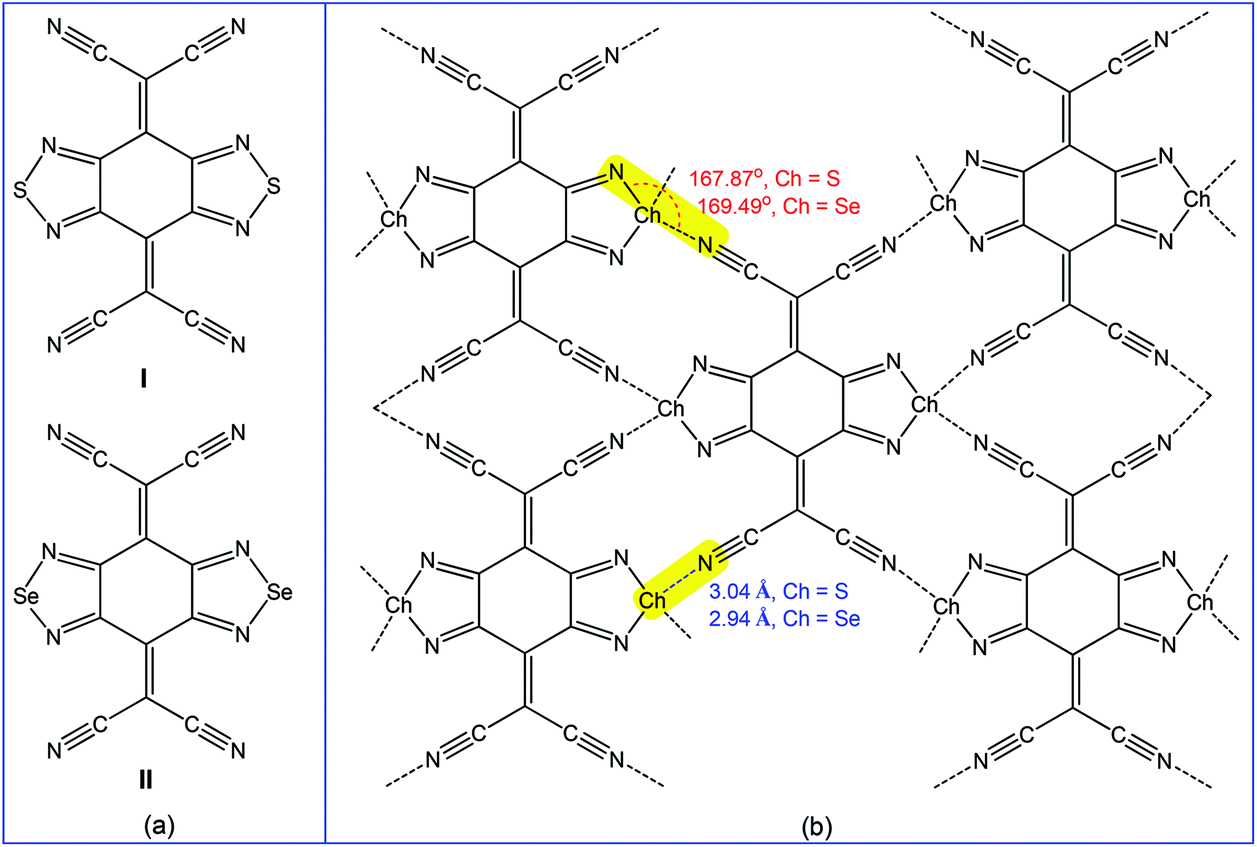
Accordingly, the multiple chalcogen bond containing 2,2′-(benzo[1,2-c:4,5-c′]bis([1,2,5]chalcadiazole)-4,8-diylidene)dimalononitrile receptors (I and II) (Scheme 26a) have been employed in the selective separation of p-xylene or 2,6-dimethylnaphthalene from the corresponding isomeric mixtures.179 Both receptors are planar molecules and chalcogen atoms act as bifurcated chalcogen bond donor centres towards the cyano group nitrogens of neighbouring molecules with moderate directionality (167.87° N–S⋯Ncyano and 169.49° N–Se⋯Ncyano angles). Short chalcogen bonds connect the molecule with four neighbours (Scheme 26b), thus leading to nearly coplanar “sheetlike” networks, which form infinite layers with face-to-face overlapping between heterocycles.179 Thus, in order to maintain the chalcogen bond assisted supramolecular architecture (Scheme 26b), these receptors selectively capture the symmetric para-disubstituted benzene rather than the less favoured ortho- or meta-disubstituted benzenes.179 In fact, the packing diagram of I·p-xylene (analytical form) shows the existence of a similar “sheetlike” network with short S⋯Ncyano contacts (3.22 and 3.24 Å), followed by decreasing the directionality of chalcogen bonding (137.24 and 142.82° N–S⋯Ncyano angles).179
 | ||
| Scheme 26 Chalcogen bonding in 2,2′-(benzo[1,2-c:4,5-c′]bis([1,2,5]chalcadiazole)-4,8-diylidene) dimalononitrile receptors.179 | ||
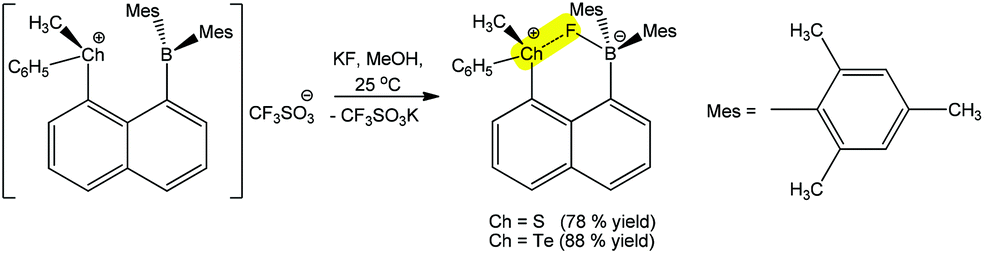
A chalcogen bond assisted synthon can also be formed during the detection process. For instance, the reaction of (R)-(8-(dimesitylboryl)naphthalen-1-yl)(methyl)(phenyl)chalconium trifluoromethane-sulfonate with a fluoride anion (analyte) leads to a positive charge assisted chalcogen bond containing (R)-(8-(dimesitylboryl)naphthalen-1-yl)(methyl)(phenyl)chalconium fluoride (Scheme 27).181 The replacement of S by Te in the receptor increases its fluoride affinity because the larger size of Te allows for a reduction of intraligand repulsions, which generates a more favourable chalcogen bonding with the F− analyte. In fact, the Te+⋯F (2.505 Å) distance is shorter than the S+⋯F (2.550 Å), whereas the latter is more directional (CPh–S+⋯F 176.61°) in comparison to the former one (CPh–Te+⋯F 174.02°).
 | ||
| Scheme 27 Positive charge assisted chalcogen bonding in the recognition of F− by (R)-(8-(dimesitylboryl)naphthalen-1-yl)(methyl)(phenyl)chalconium trifluoromethanesulfonates.181 | ||
The electron-deficient perfluoroaryl-substituted tellurophenes (I–IV) act as anion receptors through negative charge assisted chalcogen bonding in THF (Scheme 28).113 For all the anions, the bidentate chalcogen bonding in IV–X− leads to a significant increase of the association constant (Ka) in comparison to the monodentate ones III–X− (Scheme 28), in view, in the former case, of the effect of an ethynylene linker of chalcogen-based heterocycles. In fact, both associates III–X− and IV–X− were optimized using DFT calculations, the minimum-energy geometry being stabilized by chalcogen bonding interactions having the Te⋯Cl− distances of 3.07 and 3.23 Å and the 168 and 170° C–Te⋯Cl− angles, respectively (Scheme 28).
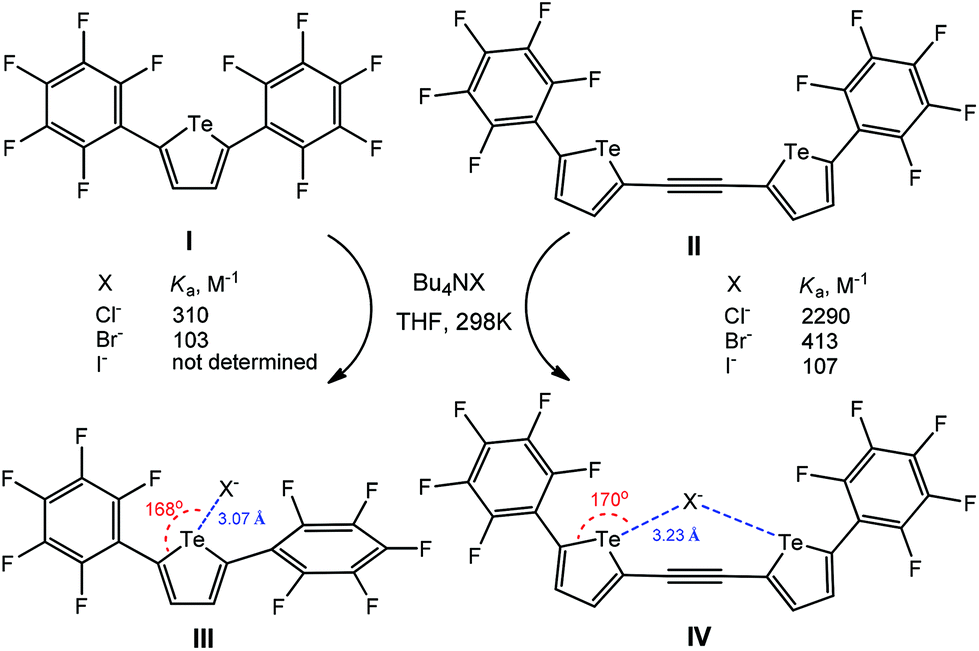 | ||
| Scheme 28 Negative charge assisted chalcogen bonding in the recognition of X− by perfluoroaryl-substituted tellurophenes.113 | ||
The transmembrane transport of anions is another important direction due to various analytical and medical applications, and noncovalent interactions have successfully been employed in this field.184–188 Thus, an application of chalcogen bonding in anion transport with electron-deficient dithieno[3,2-b:2′,3′-d]thiophenes (Scheme 29) has been reported.114 The calculated anion binding energy in 1:1 complexes increases with the enhancement of the electrophilic character of a sulfur atom in a neighbouring ring, as observed by the oxidation of the bridging S atom in I to SO (II) and S(O)2 (III) (Scheme 29). Thus, the electrophilic character of the chalcogen bond donor centre(s) can be regulated by the addition of an electron-withdrawing group(s) to the anionophore moiety.
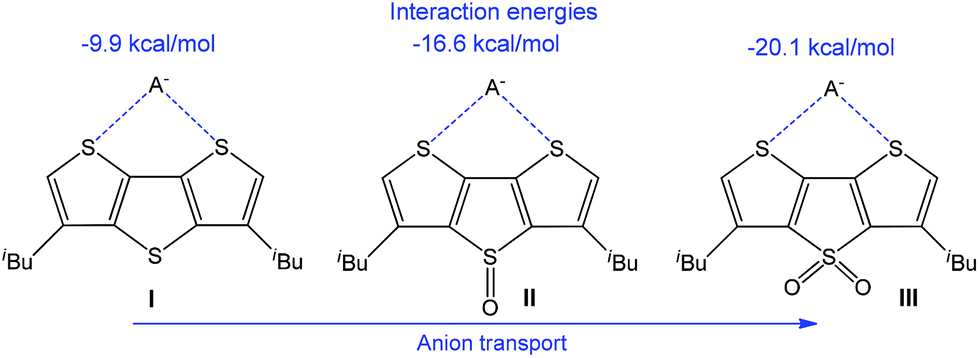 | ||
| Scheme 29 Chalcogen bond assisted anion transport.114 | ||
5. Conclusions
In comparison with the well-established hydrogen and halogen bonding, the chalcogen bond (a family member of σ-hole interactions) is much less explored. The properties of chalcogen bonding (directionality, tunability, hydrophobicity and donor atom size) can be exploited in synthetic operations similar to those of halogen bonding, but the multiplicity of chalcogen bonding provides additional manoeuvering in synthesis and design. For instance, multiple chalcogen bonds between sulfur, selenium and tellurium centres favour the formation of nanotube-like structures in the solid state that are able to host other molecules.41Furthermore, the chalcogen bond can direct asymmetric organic reactions,62 stabilize organic radicals91,92 or support thermodynamic isomers.67 In catalysis, 5-membered chalcogen bond closed intermediates can determine the destination of the reaction.71,72,93–95 Cooperation of the chalcogen bond with other types of noncovalent interactions, e.g. halogen bond, is particularly useful.94 In materials chemistry and crystal engineering, all types of chalcogen bonding, viz. negative charge assisted, positive charge assisted, conventional (or “neutral”) and resonance assisted, have been successfully employed.140,148,154,159 Chalcogen bonds have been also used in the recognition of neutral molecules and anions,113,179,181 as well as in anion transport.114
Chalcogen bonding has mainly been observed in the solid state, but several studies have shown its role in solution, and this area of research is of particular importance due to potential application in analytical chemistry and medicine. Moreover, the role of chalcogen bonding (mainly S⋯O interactions) in biological systems is undeniable78 and the strategic inclusion of chalcogens in artificial drug design and in the development of synthetic transmembrane anion transporters can be provisioned.
We hope the considered examples and their discussion will attract additional attention to this exciting field of chalcogen bond assisted applications. Moreover, as for halogen bonding,61 we may expect the recognition of chalcogen bonding by the International Union of Pure and Applied Chemistry (IUPAC) in the near future.
Acknowledgements
This work has been partially supported by the Baku State University, as well as the Foundation for Science and Technology (FCT), Portugal [UID/QUI/00100/2013 project]. M. N. K. acknowledges FCT for financial support in the form of an “Investigador 2013” contract and the corresponding IF/01270/2013/CP1163/CT0007 project. The work was also supported by the Ministry of Education and Science of the Russian Federation (Agreement number 02.a03.21.0008).References
- J. D. van der Waals, Over de Continuïteit van den Gas-en Vloeistoftoestand (On the continuity of the gaseous and liquid state), Ph.D. Dissertation, University of Leiden, 1873 Search PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press Inc., New York, 1999 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- A. Karshikoff, Non-covalent Interactions in Proteins, Imperial College Press, Singapore, 2006 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 690 CrossRef PubMed.
- Hydrogen bonding in organic synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009 Search PubMed.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond, Oxford University Press, USA, 2009 Search PubMed.
- P. Metrangolo and G. Resnati, Halogen Bonding: Fundamentals and Applications (Stucture and Bonding), Springer, Heidelberg, 2010 Search PubMed.
- P. Hobza and K. Müller-Dethlefs, Non-covalent Interactions: Theory and Experiment, Royal Society of Chemistry, 2010 Search PubMed.
- A. K. Baev, Specific Intermolecular Interactions of Organic Compounds, Springer, 2012 Search PubMed.
- S. Scheiner, Acc. Chem. Res., 2013, 46, 280 CrossRef CAS PubMed.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- A. K. Baev, Specific Intermolecular Interactions of Nitrogenated and Bioorganic Compounds, Springer, 2014 Search PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Cryst. Growth Des., 2014, 14, 2697 CAS.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Rev., 2015, 115, 8867 CAS.
- Noncovalent Forces, ed. S. Scheiner, Springer, Dordrecht, 2015 Search PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7188 CrossRef PubMed.
- S. K. Singh and A. Das, Phys. Chem. Chem. Phys., 2015, 17, 9596 RSC.
- Non-Covalent Interactions in the Synthesis and Design of New Compounds, ed. A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, John Wiley & Sons, Inc., Hoboken, NJ, 2016 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072 CrossRef CAS PubMed.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155 CrossRef PubMed.
- K. T. Mahmudov and A. J. L. Pombeiro, Chem. – Eur. J., 2016, 22, 16356 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2017, 354, 54 CrossRef.
- R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720 RSC.
- N. M. Adcock, Adv. Inorg. Chem. Radiochem., 1972, 15, 1 CrossRef.
- R. Gleiter and R. Gygax, Top. Curr. Chem., 1976, 63, 49 CrossRef CAS.
- G. Angyan, R. A. Poirier, A. Kucsman and I. G. Csizmadia, J. Am. Chem. Soc., 1987, 109, 2237 CrossRef.
- H.-B. Burgi and J. Dunitz, J. Am. Chem. Soc., 1987, 109, 2924 CrossRef.
- S. Scheiner, Int. J. Quantum Chem., 2013, 113, 1609 CrossRef CAS.
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS PubMed.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541 CrossRef CAS PubMed.
- A. Pecina, M. Lepšík, D. Hnyk, P. Hobza and L. Fanfrlík, J. Phys. Chem. A, 2015, 119, 1388 CrossRef CAS PubMed.
- W. Wang, B. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132 CrossRef CAS PubMed.
- D. B. Werz, R. Gleiter and F. Rominger, J. Am. Chem. Soc., 2002, 124, 10638 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- T. P. Tauer, M. E. Derrick and C. D. Sherrill, J. Phys. Chem. A, 2005, 109, 191 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613 CrossRef CAS PubMed.
- M. D. Esrafili and F. Mohammadian-Sabet, Struct. Chem., 2015, 26, 199 CrossRef CAS.
- M. D. Esrafili and N. Mohammadirad, J. Mol. Model., 2015, 21, 176 CrossRef PubMed.
- M. D. Esrafili and F. Mohammadian-Sabet, Chem. Phys. Lett., 2015, 634, 210 CrossRef CAS.
- T. Clark, J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2008, 14, 689 CrossRef CAS PubMed.
- V. D. P. N. Nziko and S. Scheiner, J. Phys. Chem. A, 2014, 118, 10849 CrossRef CAS PubMed.
- P. Chaudhary, J. T. Goettel, H. P. A. Mercier, S. Sowlati-Hashjin, P. Hazendonk and M. Gerken, Chem. – Eur. J., 2015, 21, 6247 CrossRef CAS PubMed.
- F. R. Knight, A. L. Fuller, M. l. Bühl, A. M. Z. Slawin and J. D. Woollins, Inorg. Chem., 2010, 49, 7577 CrossRef CAS PubMed.
- T. Chivers and J. Konu, Angew. Chem., Int. Ed., 2009, 48, 3025 CrossRef CAS PubMed.
- T. Kusamoto, H. M. Yamamoto and R. Kato, Cryst. Growth Des., 2013, 13, 4533 CAS.
- U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2014, 118, 3183 CrossRef CAS PubMed.
- E. Alikhani, F. Fuster, B. Madebenea and S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 2430 RSC.
- P. Sanz, O. Mó and M. Yáñez, Chem. – Eur. J., 2003, 9, 4548 CrossRef CAS PubMed.
- S. J. Grabowski, W. A. Sokalski, E. Dyguda and J. Leszczynski, J. Phys. Chem. B, 2006, 110, 6444 CrossRef CAS PubMed.
- S. J. Grabowski, Chem. Rev., 2011, 11, 2597 CrossRef PubMed.
- S. J. Grabowski, J. Phys. Chem. A, 2011, 115, 12340 CrossRef CAS PubMed.
- E. Arunan1, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637 Search PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- F. V. Gonzalez, A. Jain, S. Rodriguez, J. A. Saez, C. Vicent and G. Peris, J. Org. Chem., 2010, 75, 5888 CrossRef CAS PubMed.
- E. M. Fatila, J. Goodreid, R. Clérac, M. Jennings, J. Assoud and K. E. Preuss, Chem. Commun., 2010, 46, 6569 RSC.
- J. Konu, T. Chivers and H. M. Tuononen, Chem. – Eur. J., 2010, 16, 12977 CrossRef CAS PubMed.
- S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626 CrossRef CAS PubMed.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. A. Jenkins, J. F. Britten, D. R. Morim and I. Vargas Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed.
- A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Yu. Kukushkin, J. Am. Chem. Soc., 2016, 138, 14129 CrossRef CAS PubMed.
- A. J. Karhu, O. J. Pakkanen, J. M. Rautiainen, R. Oilunkaniemi, T. Chivers and R. S. Laitinen, Dalton Trans., 2016, 45, 6210 RSC.
- S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061 CrossRef CAS PubMed.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864 RSC.
- S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812 CrossRef CAS PubMed.
- E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193 RSC.
- S. S. Zade, S. Panda, H. B. Singh, R. B. Sunoj and R. J. Butcher, J. Org. Chem., 2005, 70, 3693 CrossRef CAS PubMed.
- S. K. Tripathi, U. Patel, D. Roy, R. B. Sunoj, H. B. Singh, G. Wolmershãuser and R. J. Butcher, J. Org. Chem., 2005, 70, 9237 CrossRef CAS PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78 RSC.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357 CrossRef CAS PubMed.
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725 RSC.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383 CrossRef CAS PubMed.
- Y. Nagao, T. Hirata, S. Goto, S. Sano, A. Kakehi, K. Iizuka and M. Shiro, J. Am. Chem. Soc., 1998, 120, 3104 CrossRef CAS.
- A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Org. Lett., 2009, 11, 1487 CrossRef CAS PubMed.
- E. M. Takaluoma, T. T. Takaluoma, R. Oilunkaniemi and R. S. Laitinen, Z. Anorg. Allg. Chem., 2015, 641, 772 CrossRef CAS.
- D. Tilly, F. Chevallier and F. Mongin, Synthesis, 2016, 184 CAS.
- M. Iwaoka and N. Isozumi, Molecules, 2012, 17, 7266 CrossRef CAS PubMed.
- C. Enguehard, M. Hervet, H. Allouchi, J.-C. Debouzy, J.-M. Leger and A. Gueiffier, Synthesis, 2001, 595 CrossRef CAS.
- J. S. Dhau, R. Dhir and A. Singh, J. Organomet. Chem., 2011, 696, 2406 CrossRef CAS.
- K. Akiba, T. Tsuchiya, N. Inamoto, K. Onuma, N. Nagashima and A. Nakamura, Chem. Lett., 1976, 723 CrossRef CAS.
- C. Zhu, A. Yoshimura, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Org. Lett., 2012, 14, 3170 CrossRef CAS PubMed.
- C. Sharma, A. K. Singh, J. Joy, E. D. Jemmis and S. K. Awas, RSC Adv., 2016, 6, 91689 RSC.
- R. Gleiter and G. Haberhauer, J. Org. Chem., 2014, 79, 7543 CrossRef CAS PubMed.
- J. F. Britten, A. W. Cordes, R. C. Haddon, M. E. Itkis, R. T. Oakley, R. W. Reed and C. M. Robertson, CrystEngComm, 2002, 4, 205 RSC.
- K. V. Shuvaev, A. Decken, F. Grein, T. S. M. Abedin, L. K. Thompson and J. Passmore, Dalton Trans., 2008, 37, 4029 RSC.
- S. Menichetti, R. Amorati, V. Meoni, L. Tofani, G. Caminati and C. Viglianisi, Org. Lett., 2016, 18, 5464 CrossRef CAS PubMed.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320 CrossRef CAS PubMed.
- D. Manna and G. Mugesh, J. Am. Chem. Soc., 2012, 134, 4269 CrossRef CAS PubMed.
- E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919 RSC.
- P. Metrangolo and G. Resnati, Nat. Chem., 2012, 4, 437 CrossRef CAS PubMed.
- E. I. Carrera and D. S. Seferos, Macromolecules, 2015, 48, 297 CrossRef CAS.
- A. A. Jahnke and D. S. Seferos, Macromol. Rapid Commun., 2011, 32, 943 CrossRef CAS PubMed.
- R. Gleiter, D. B. Werz and B. J. Rausch, Chem. – Eur. J., 2003, 9, 2676 CrossRef CAS PubMed.
- K. Kobayashi, H. Masu, A. Shuto and K. Yamaguchi, Chem. Mater., 2005, 17, 6666 CrossRef CAS.
- M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. Ángyán, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283 CAS.
- M. Bai, S. P. Thomas, R. Kottokkaran, S. K. Nayak, P. C. Ramamurthy and T. N. G. Row, Cryst. Growth Des., 2014, 14, 459 CAS.
- A. Alberola, D. J. Eisler, L. Harvey and J. M. Rawson, CrystEngComm, 2011, 13, 1794 RSC.
- C. P. Constantinides, D. J. Eisler, A. Alberola, E. Carter, D. M. Murphy and J. M. Rawson, CrystEngComm, 2014, 16, 7298 RSC.
- S. Chandra and A. Bhattacharya, J. Phys. Chem. A, 2016, 120, 10057 CrossRef CAS PubMed.
- M. R. Koebel, A. Cooper, G. Schmadeke, S. Jeon, M. Narayan and S. Sirimulla, J. Chem. Inf. Model., 2016, 56, 2298 CrossRef CAS PubMed.
- Y. Yi, S. Fa, W. Cao, L. Zeng, M. Wang, H. Xu and X. Zhanga, Chem. Commun., 2012, 48, 7495 RSC.
- R. L. Melen, R. J. Less, C. M. Pask and J. M. Rawson, Inorg. Chem., 2016, 55, 11747 CrossRef CAS PubMed.
- R. Shukla and D. Chopra, Cryst. Growth Des., 2016, 16, 6734 CAS.
- R. D. McCullough, Adv. Mater., 1998, 10, 93 CrossRef CAS.
- D. A. Haynes, CrystEngComm, 2011, 13, 4793 RSC.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126 CrossRef CAS PubMed.
- G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Chem. Commun., 2016, 52, 9881 RSC.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093 CrossRef CAS PubMed.
- R. J. Fick, G. M. Kroner, B. Nepal, R. Magnani, S. Horowitz, R. L. Houtz, S. Scheiner and R. C. Trievel, ACS Chem. Biol., 2016, 11, 748 CrossRef CAS PubMed.
- Handbook of Chalcogen Chemistry: New Perspectives in Sulfur, Selenium and Tellurium, ed. F. Devillanova and W.-W. D. Mont, The Royal Society of Chemistry, Cambridge, UK, 2nd edn, 2013, vol. 2 Search PubMed.
- J. J. Novoa, M. C. Rovira, C. Rovira, J. Veciana and J. Tarrés, Adv. Mater., 1995, 7, 233 CrossRef CAS.
- S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485 RSC.
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422 RSC.
- R. Gleiter and G. Haberhauer, Coord. Chem. Rev., 2017, 344, 263 CrossRef CAS.
- F. Sun, H. Cheng, J. Chen, N. Zheng, Y. Li and J. Shi, ACS Nano, 2016, 10, 8289 CrossRef CAS PubMed.
- C. Ataca, H. Şahin and S. Ciraci, J. Phys. Chem. C, 2012, 116, 8983 CAS.
- J. F. Corrigan, O. Fuhr and D. Fenske, Adv. Mater., 2009, 21, 1867 CrossRef CAS.
- C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666 CrossRef CAS PubMed.
- M. Al-Hashimi, Y. Han, J. Smith, H. S. Bazzi, S. Y. A. Alqaradawi, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2016, 7, 1093 RSC.
- H.-W. Lin, W.-Y. Lee, C. Lu, C.-J. Lin, H.-C. Wu, Y. W. Lin, B. Ahn, Y. Rho, M. Ree and W.-C. Chen, Polym. Chem., 2012, 3, 767 RSC.
- H.-W. Lin, W.-Y. Lee and W.-C. Chen, J. Mater. Chem., 2012, 22, 2120 RSC.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364 CrossRef CAS PubMed.
- M. Kaur, D. S. Yang, J. Shin, T. W. Lee, K. Choi, M. J. Cho and D. H. Choi, Chem. Commun., 2013, 49, 5495 RSC.
- B. Kim, H. R. Yeom, M. H. Yun, J. Y. Kim and C. Yang, Macromolecules, 2012, 45, 8658 CrossRef CAS.
- A. K. Mahrok, E. I. Carrera, A. J. Tilley, S. Ye and D. S. Seferos, Chem. Commun., 2015, 51, 5475 RSC.
- M. Planells, B. C. Schroeder and I. McCulloch, Macromolecules, 2014, 47, 5889 CrossRef CAS.
- M. Mammi, R. Bardi, G. Traverso and S. Bezzi, Nature, 1961, 192, 1282 CrossRef CAS.
- T. R. Lynch, I. P. Mellor, S. C. Nyburg and P. Yates, Tetrahedron Lett., 1967, 8, 373 CrossRef.
- P. W. Anderson, P. A. Lee and M. Saitoh, Solid State Commun., 1973, 13, 595 CrossRef CAS.
- R. E. Rosenfield, R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860 CrossRef CAS.
- T. N. G. Row and R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477 CrossRef CAS.
- A. Chesney, M. R. Bryce, M. A. Chalton, A. S. Batsanov and J. A. K. Howard, J. Org. Chem., 1996, 61, 2877 CrossRef CAS PubMed.
- T. Chivers, I. Krouse, M. Parvez, I. Vargas-Baca, T. Ziegler and P. Zoricak, Inorg. Chem., 1996, 35, 5836 CrossRef CAS.
- T. Kojima, K. Tanaka, T. Ishida and T. Nogami, J. Org. Chem., 2004, 69, 9319 CrossRef CAS PubMed.
- C. S. Clarke, S. I. Pascu and J. M. Rawson, CrystEngComm, 2004, 6, 79 RSC.
- J. Konu, M. Ahlgrén, S. M. Aucott, T. Chivers, S. H. Dale, M. R. J. Elsegood, K. E. Holmes, S. L. M. James, P. F. Kelly and R. S. Laitinen, Inorg. Chem., 2005, 44, 4992 CrossRef CAS PubMed.
- A. F. Cozzolino, J. F. Britten and I. Vargas-Baca, Cryst. Growth Des., 2006, 6, 181 CAS.
- A. Yu. Makarov, K. Tersago, K. Nivesanond, F. Blockhuys, C. Van Alsenoy, M. K. Kovalev, I. Yu. Bagryanskaya, Y. V. Gatilov, M. M. Shakirov and A. V. Zibarev, Inorg. Chem., 2006, 45, 2221 CrossRef CAS PubMed.
- J. L. Brusso, S. Derakhshan, M. E. Itkis, H. Kleinke, R. C. Haddon, R. T. Oakley, R. W. Reed, J. F. Richardson, C. M. Robertson and L. K. Thompson, Inorg. Chem., 2006, 45, 10958 CrossRef CAS PubMed.
- E. Coronado, S. Curreli, C. Giménez-Saiz, C. J. Gómez-García and A. Alberola, Inorg. Chem., 2006, 45, 10815 CrossRef CAS PubMed.
- D. B. Werz, F. R. Fischer, S. C. Kornmayer, F. Rominger and R. Gleiter, J. Org. Chem., 2008, 73, 8021 CrossRef CAS PubMed.
- J. L. Dutton, C. D. Martin, M. J. Sgro, N. D. Jones and P. J. Ragogna, Inorg. Chem., 2009, 48, 3239 CrossRef CAS PubMed.
- C. Allen, D. A. Haynes, C. M. Pask and J. M. Rawson, CrystEngComm, 2009, 11, 2048 RSC.
- A. A. Leitch, X. Yu, C. M. Robertson, R. A. Secco, J. S. Tse and R. T. Oakley, Inorg. Chem., 2009, 48, 9874 CrossRef CAS PubMed.
- A. F. Cozzolino and I. Vargas-Baca, Cryst. Growth Des., 2011, 11, 668 CAS.
- T. Murata, Y. Yakiyama, K. Nakasuji and Y. Morita, CrystEngComm, 2011, 13, 3689 RSC.
- S. M. Mali, T. F. Schneider, A. Bandyopadhyay, S. V. Jadhav, D. B. Werz and H. N. Gopi, Cryst. Growth Des., 2012, 12, 5643 CAS.
- F. R. Knight, R. A. M. Randall, K. S. A. Arachchige, L. Wakefield, J. M. Griffin, S. E. Ashbrook, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Inorg. Chem., 2012, 51, 11087 CrossRef CAS PubMed.
- Y. Beldjoudi, D. A. Haynes, J. J. Hayward, W. J. Manning, D. R. Prattc and J. M. Rawson, CrystEngComm, 2013, 15, 1107 RSC.
- J. Fanfrlík, A. Přáda, Z. Padělková, A. Pecina, J. Macháček, M. Lepšík, J. Holub, A. Růžička, D. Hnyk and P. Hobza, Angew. Chem., Int. Ed., 2014, 53, 10139 CrossRef PubMed.
- S. P. Thomas, K. Satheeshkumar, G. Mugesh and T. N. G. Row, Chem. – Eur. J., 2015, 21, 6793 CrossRef CAS PubMed.
- V. P. Singh, J. Poon, R. J. Butcher, X. Lu, G. Mestres, M. K. Ott and L. Engman, J. Org. Chem., 2015, 80, 7385 CrossRef CAS PubMed.
- S. P. Thomas, S. P. K. P. Veccham, L. J. Farrugia and T. N. G. Row, Cryst. Growth Des., 2015, 15, 2110 CAS.
- K. Suresh, V. S. Minkov, K. K. Namila, E. Derevyannikova, E. Losev, A. Nangia and E. V. Boldyreva, Cryst. Growth Des., 2015, 15, 3498 CAS.
- S. Mondal, M. Konda, B. Kauffmann, M. K. Manna and A. K. Das, Cryst. Growth Des., 2015, 15, 5548 CAS.
- I. S. Antonijević, G. V. Janjić, M. K. Milčić and S. D. Zarić, Cryst. Growth Des., 2016, 16, 632 Search PubMed.
- K. Eichstaedt, A. Wasilewska, B. Wicher, M. Gdaniec and T. Połoński, Cryst. Growth Des., 2016, 16, 1282 CAS.
- K. Durka, K. Gontarczyk, S. Luliński, J. Serwatowski and K. Woźniak, Cryst. Growth Des., 2016, 16, 4292 CAS.
- S. K. Rajagopal, P. S. Salini and M. Hariharan, Cryst. Growth Des., 2016, 16, 4567 CAS.
- E. S. Yandanova, D. M. Ivanov, M. L. Kuznetsov, A. G. Starikov, G. L. Starova and V. Yu. Kukushkin, Cryst. Growth Des., 2016, 16, 2979 CAS.
- M. M. Karjalainen, C. Sanchez-Perez, J. M. Rautiainen, R. Oilunkaniemi and R. S. Laitinen, CrystEngComm, 2016, 18, 4538 RSC.
- P. R. Prasad, K. Selvakumar, H. B. Singh and R. J. Butcher, J. Org. Chem., 2016, 81, 3214 CrossRef CAS PubMed.
- J. T. Goettel and M. Gerken, Inorg. Chem., 2016, 55, 12441 CrossRef CAS PubMed.
- J. Fanfrlík and D. Hnyk, CrystEngComm, 2016, 18, 8982 RSC.
- K. Dabrowa, M. Ceborska, M. Pawlak and J. Jurczak, Cryst. Growth Des., 2017, 17, 701 CAS.
- J. O’. M. Bockris and A. K. N. Reddy, Modern Electrochemistry: An Introduction to an Interdisciplinary Area, Plenum Press, New York, 1970 Search PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC.
- M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667 RSC.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed.
- S. Scheiner, Chem. – Eur. J., 2016, 22, 18850 CrossRef CAS PubMed.
- T. Suzuki, H. Fujii, Y. Yamashita, C. Kabuto, S. Tanaka, M. Harasawa, T. Mukai and T. Miyashi, J. Am. Chem. Soc., 1992, 114, 3034 CrossRef CAS.
- T. Suzuki, J. Synth. Org. Chem., Jpn., 1995, 53, 403 CrossRef CAS.
- H. Zhao and F. Gabbaï, Nat. Chem., 2010, 2, 984 CrossRef CAS PubMed.
- M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1996, 118, 8077 CrossRef CAS.
- T. Suzuki, Pure Appl. Chem., 1996, 68, 281 CAS.
- J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843 RSC.
- P. A. Gale, Acc. Chem. Res., 2011, 44, 216 CrossRef CAS PubMed.
- J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148 RSC.
- A. V. Jentzsch, D. Emery, J. Mareda, P. Metrangolo, G. Resnati and S. Matile, Angew. Chem., Int. Ed., 2011, 50, 11675 CrossRef PubMed.
- J. T. Davis, Nat. Chem., 2010, 2, 516 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |