
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe isomeric structure of pentacoordinate chiral spirophosphoranes in solution by the combined use of NMR experiments and GIAO DFT calculations of NMR parameters†
Fedor M.
Polyancev
 ,
Kirill E.
Metlushka
,
Dilyara N.
Sadkova
,
Zilya R.
Khisametdinova
,
Olga N.
Kataeva
,
Vladimir A.
Alfonsov
,
Shamil K.
Latypov
* and
Oleg G.
Sinyashin
,
Kirill E.
Metlushka
,
Dilyara N.
Sadkova
,
Zilya R.
Khisametdinova
,
Olga N.
Kataeva
,
Vladimir A.
Alfonsov
,
Shamil K.
Latypov
* and
Oleg G.
Sinyashin
A.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, Arbuzov str. 8, Kazan, 420088, Russian Federation. E-mail: lsk@iopc.ru
First published on 13th June 2017
Abstract
The interplay of NMR experiments and DFT calculations of NMR parameters is a reliable method for determining the relative configurations of pentacoordinate chiral spirophosphoranes bearing two six- or five-membered rings at the phosphorus atom in solution. The major product of the Betti based derivatives corresponds to the isomers with both substituents at chiral carbons being opposite to the P–H proton. The next populated product corresponds to the isomer with different chiralities at carbons. The least populated isomer is one with both substituents being at the same side of the heterocycle as the P–H bond.
Introduction
Chiral phosphorus compounds are widely used in stereoselective synthesis.1–5 For example, chiral phosphorus ligands are of importance in the design of catalysts for asymmetric homogeneous catalysis.6 Pentacoordinate phosphorus compounds have drawn special attention for their role as intermediates in some biological processes (e.g. enzymatic phosphoryl transfer reactions,7–29 hydrolysis/formation of DNA, RNA, cyclic AMP).30,31Their structural and dynamic properties strongly correlate with the reactivity/selectivity of the processes they are involved in. Although the knowledge about the stereochemistry (the absolute and relative configurations) of these compounds is important in understanding these processes, very few structural studies of pentacoordinate phosphorus systems have been published so far.32–38
Pentacoordinate phosphoranes with an asymmetric phosphorus atom and with several additional chiral chelate ligands are especially difficult to analyze, since these chiral centers may generate a variety of stereoisomers and the determination of their fine structure is not straightforward. To this end X-ray single crystal investigations could be helpful, but obtaining good single crystals is often time consuming and sometimes impossible. This point, coupled with the fact that most chemical reactions and biological processes occur in solution, makes it highly desirable to find an alternative, reliable, and easy way to determine the relative configurations of such hypervalent chiral phosphorus compounds directly in solution.33–38
Progress in NMR correlation techniques and the state-of-the-art density functional theory (DFT) calculations of NMR parameters has emerged in recent years as a very powerful tool for fine structural analysis of organic compounds and biomolecules. There are a number of examples when DFT calculations of NMR 1H, 13C and 15N parameters in the frame of the GIAO method allowed the establishment of tautomeric, isomeric and conformational structures as well.39–53 However applications of similar approaches to analyze phosphorus compounds by DFT calculations of 31P NMR parameters are very rare.54–56
The study here shows that the interplay of the NMR experiment and theory is a powerful method for determining the relative configurations of pentacoordinate chiral spirophosphoranes. For the illustration several pentacoordinate spirophosphoranes bearing two six-membered (Betti based derivatives) and five-membered (α-aminocarbonic acid derivatives) rings at the phosphorus atom were used.
Results
Synthesis
Three chiral elements (two centres and one axis) lead to the four possible combinations. In order to obtain the full set of stereoisomers we used racemic Betti bases. The synthesis of P–H-spirophosphoranes was carried out by substituting the amino groups in the hexaethyltriamidophosphite by 1-(α-aminobenzyl)-2-naphthols (Scheme 1). For example, in the 31P{1H} NMR spectra of the reaction mixture of 4 there are two signals with the chemical shifts (CS) characteristic of phosphoranes (−75.4 (65%) and −76.7 (35%) ppm) that may be attributed to the formation of two isomers.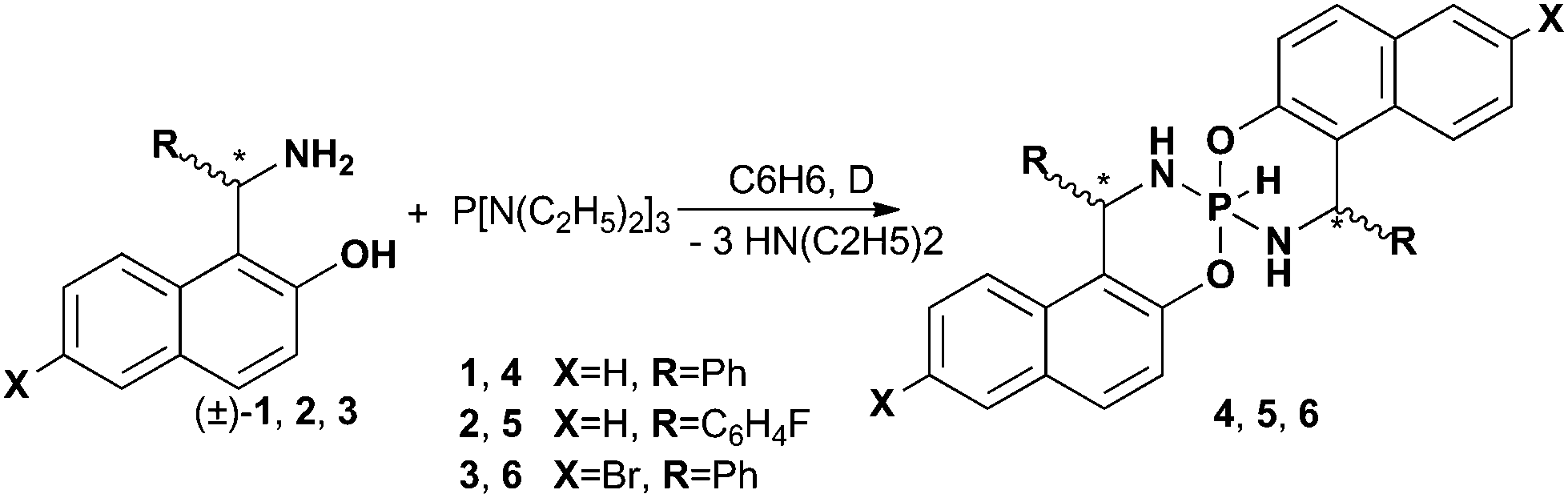 | ||
| Scheme 1 | ||
In contrast to the dominant product (δ31P = −75.4 ppm), the 1H spectra of which correspond to the high symmetry structure, the second component apparently has no symmetry, and two spiro halves of phosphorane 4 differ in the 1H NMR spectrum. When diethylamine hydrochloride is used as the catalyst, these two isomers could be obtained in an about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and their isolation in individual form becomes possible via crystallization. It is interesting that after about 4–6 hours in the NMR spectrum of the individual product that was major in the initial reaction mixture (δ31P = −75.4 ppm), additional signals appeared which can also be attributed to the structure of high symmetry (δ31P = −80.7 ppm) with two spiro-connected six-membered heterocycles being equivalent in the 1H NMR spectra.
:1 ratio and their isolation in individual form becomes possible via crystallization. It is interesting that after about 4–6 hours in the NMR spectrum of the individual product that was major in the initial reaction mixture (δ31P = −75.4 ppm), additional signals appeared which can also be attributed to the structure of high symmetry (δ31P = −80.7 ppm) with two spiro-connected six-membered heterocycles being equivalent in the 1H NMR spectra.
In a similar manner for 5 the formation of two isomers was observed which were separated and analyzed (Fig. 1a and b). In this case the 19F NMR is particularly useful and allows distinguishing symmetrical (one singlet, δ19F = −116 ppm) and non-symmetrical (two singlet's of equal intensity, δ19F = −115.8 and δ19F = −116.6 ppm) isomers. The monitoring of the major isomer 5avia the 1H and 31P NMR showed that after some time an additional product appeared (10%) in solution (δ19F = −115.5 and δ31P = −81.0 ppm) which presumably corresponds to another symmetrical isomer (Fig. 1c).
 | ||
| Fig. 1 1H spectra of 5 in CDCl3 at 303 K. (a) Major product (fresh, 5a); (c) the same product after 24 h; (b) second product; (d) structure of compound 5a with principal NMR correlations: 1H–1H COSY and 1H–13C HMBC (black arrows), 1H–31P HMBC (red arrows), 1H–15N HMBC (blue arrows), 19F–1H HETCOR (orange arrows). | ||
For compound 6 very similar spectra and their evolution were observed (ESI†).
Structure determination
| Comp. | X at C11 | R at C4 | Isomer | ΔEb | δ 31P | δH6 | δH16,20 | δH17,19 | δH4 | δH5 | 1 J PH | 3 J PH4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Chemical shifts in ppm, spin–spin couplings in Hz. b Relative energy, in kcal mol−1. c Experimental data. d No data. | ||||||||||||
| 4 | H | Ph | I | Expc (a) | −75.4 | 7.08 | 7.32 | 7.19 | 5.95 | 4.18 | 788.5 | 34.1 |
| 0 | −75.2 | 7.66 | 7.64 | 7.41 | 5.98 | 3.77 | 715.4 | 36.1 | ||||
| II | Exp (b) | −76.7 | 7.34 | 7.02, 7.45 | 6.93, 7.29 | 5.81, 5.87 | 3.80, 4.00 | 823.5 | 26.3, 33.1 | |||
| 0.53 | −76.4 | 7.80 | 7.42, 7.88 | 7.21, 7.58 | 5.81, 5.84 | 3.46, 3.83 | 742.6 | 27.6, 35.2 | ||||
| III | Exp (c) | −80.7 | 7.54 | n/dd | n/d | 5.74 | 3.58 | 848.6 | 25.6 | |||
| 1.33 | −82.5 | 7.75 | 7.87 | 7.58 | 5.65 | 3.36 | 762.7 | 25.2 | ||||
| 5 | H | FC6H4 | I | Exp (a) | −75.2 | 7.09 | 7.26 | 6.88 | 5.91 | 4.17 | 792.1 | 34.1 |
| 0 | −74.4 | 7.63 | 7.52 | 7.04 | 5.94 | 3.76 | 713.9 | 36.1 | ||||
| II | Exp (b) | −76.6 | 7.32 | 6.91, 7.39 | 6.58, 6.95 | 5.76, 5.82 | 3.77, 3.95 | 824.0 | 27.7, 33.6 | |||
| 0.47 | −75.7 | 7.75 | 7.37, 7.82 | 6.81, 7.17 | 5.77, 5.86 | 3.48, 3.78 | 738.3 | 27.6, 34.6 | ||||
| III | Exp (a) | −80.5 | 7.52 | n/d | n/d | 5.70 | 3.56 | 849.0 | 25.0 | |||
| 1.33 | −82.1 | 7.74 | 7.82 | 7.15 | 5.63 | 3.33 | 764.2 | 24.7 | ||||
| 6 | Br | Ph | I | Exp (a) | −76.4 | 7.08 | 7.27 | 7.19 | 5.89 | 4.17 | 790.4 | 34.1 |
| 0 | −75.6 | 7.62 | 7.58 | 7.41 | 5.92 | 3.74 | 718.3 | 36.0 | ||||
| II | Exp (b) | −77.7 | 7.33 | 6.96, 7.28 | 6.96, 7.28 | 5.74, 5.78 | 3.77, 3.95 | 828.0 | 27.5, 33.7 | |||
| 0.44 | −76.5 | 7.77 | 7.20, 7.78 | 7.18, 7.59 | 5.68, 5.74 | 3.34, 3.81 | 742.6 | 27.6, 35.2 | ||||
| III | Exp (c) | −81.4 | 7.54 | n/d | n/d | 5.71 | 3.58 | 852.3 | 25.1 | |||
| 0.67 | −80.5 | 7.69 | 7.78 | 7.59 | 5.60 | 3.25 | 769.9 | 24.1 | ||||
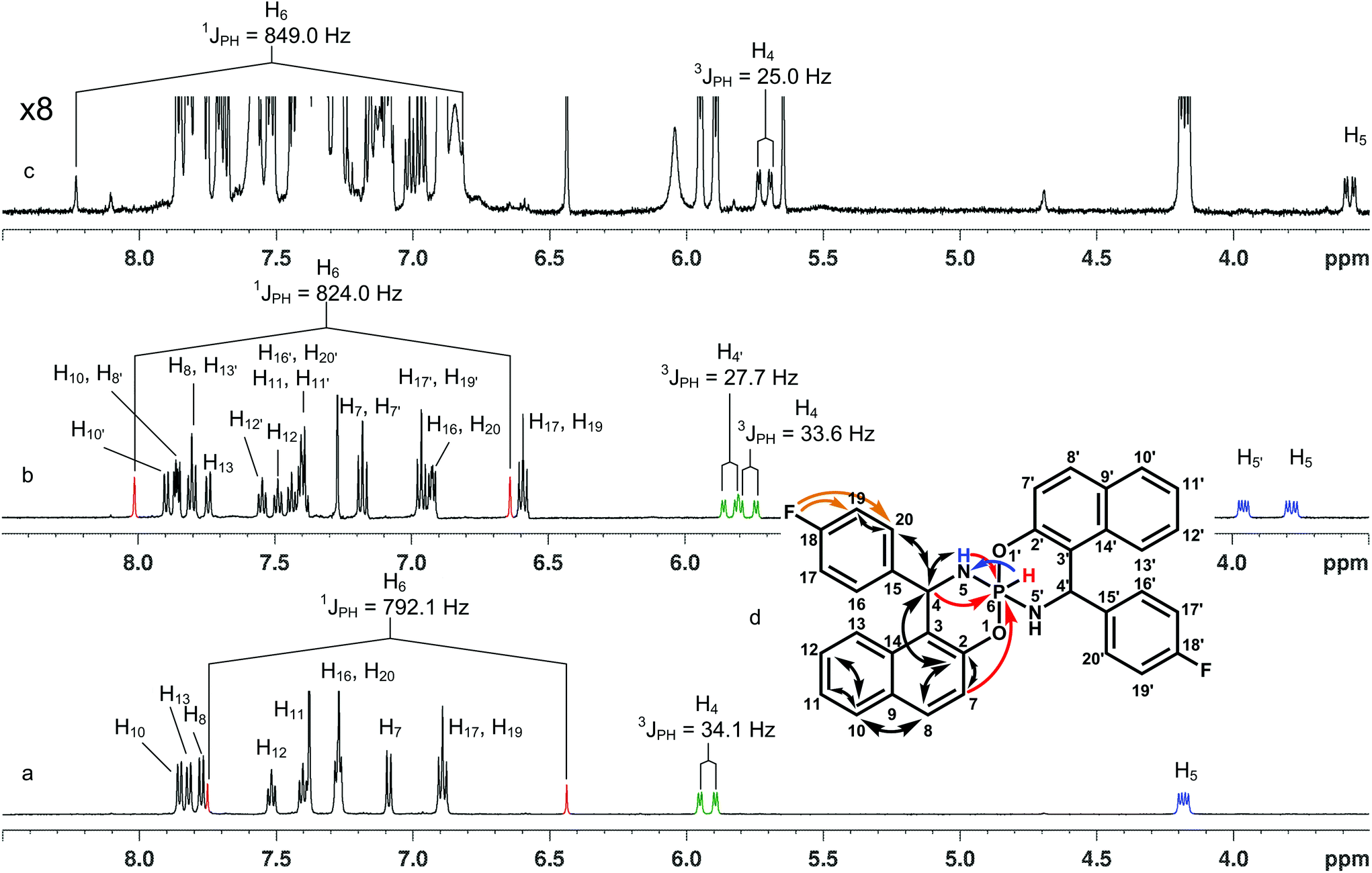
In general, three chiral centers (at P, C4 and C4′) may generate four diastereomers: ΔPRcRc/ΛPScSc, ΛPRcRc‡/ΔPScSc, ΔPRcSc/ΛPScRc and ΔPScRc/ΛPRcSc. However, when both aryls in chiral centers are the same the last two isomers become equivalent in terms of NMR. Thus three main isomers were considered in details: with both aryl substituents being opposite to the P–H bond (ΔPRcRc/ΛPScSc, I), with one aryl opposite to the P–H bond and another one being on the same side of the cycle with the P–H bond (ΔPRcSc/ΛPScRc, II), in the third isomer both aryls are at the same side of the cycle with the P–H bond (ΛPRcRc/ΔPScSc, III) (Fig. 2).
 | ||
| Fig. 2 Energy minimized structures of the main isomers of the title spirophosphorane 4. | ||
Qualitative consideration of these structures predicts some stereospecific features. There are different dihedral angles between H4–C and N–P in isomers I and III, that may lead to different three bond SSCs. As to the II-nd isomer, the geometries of its two spiro-halves are very similar to the halves of the I-st and III-rd isomers, therefore two sets of signals are expected for its 1H (13C, 19F) spectra.
Thus, preliminary identification can be based on NOEs and symmetry considerations. Presumably, the “a” (major) products can be ascribed to the I isomer because there is no NOE between the ortho-aryl and P–H protons. Lack of symmetry in the second product (“b”) may be indicative of the II-isomer. As to the co-product (“c”) that appeared after some time in solution of the major isomer (“a”), there are no clear NOEs that can be used due to its low population and the signal overlapping with the main product.
To develop a well-argued relationship between the NMR parameters and the fine isomeric structure, DFT calculations of the NMR parameters were carried out for these structural hypotheses. It is well demonstrated that modern GIAO CS calculations on 13C, 15N and 1H nuclei achieved experimental accuracy and can be used as an additional tool for chemical structure elucidation. There are a number of examples of application of such calculations on these nuclei to obtain fine information on the isomeric, tautomeric and conformational structure.39–53,57–60 At the same time the examples of application of these approaches on phosphorus are rather sparse.54–56
We used a combination (PBE1PBE/6-311G(2d,2p)//PBE1PBE/6-31+G(d)) that proved to be reliable and effective for 31P NMR CS calculations. First, the geometry optimization in the frames of the DFT (PBE1PBE/6-31+G(d)) method predicts that these three main isomers are close in energy (Table 1). Additional calculations with the inclusion of solvent effects (in the frames of the PCM model, Table S1†) do not change notably the energy distribution. In general, these results are in qualitative agreement with experimental findings that all three isomers are present in the reaction mixture after some time.
The results of the CS and SSC calculations (GIAO PBE1PBE/6-311G(2d,2p)) for optimized structures are also in agreement with preliminary conclusions (some essential data are summarized in Table 1). According to the calculations, the PH, H4 and NH protons and 31P CSs should be notably different, particularly, for the I-st and the III-rd isomers. Namely, for the I-isomer the PH proton should resonate at a higher field than for the III-isomer, while for the H4 and the NH protons the reverse should be observed. Likewise the 31P resonance signal of the I-isomer should be at a lower field than in the III-isomer. For the II-isomer, intermediate values are expected (Table 1). Thus, according to the CS data the major (“a”) product should be assigned to the I-isomer for all title compounds. The second non-symmetrical product corresponds to the II-diastereomer while the minor product may be assigned to the III-isomer.
Some SSCs also demonstrate spectacular distinctions for three isomers (Table 1) that correlate well with experimental data. As can be seen the 1JHP constants are characteristic of these isomers and can be used to assign the isomeric structure. The smallest values (715.4 Hz) are predicted for the I-st isomers while for the III-rd forms these values are notably larger (762.7 Hz). For the II-isomers the PH direct SSCs are expected to have intermediate values (742.6 Hz). The calculations slightly underestimate (by ca. 1.1) the experimental data although the correlation is quite good. Thus, the comparison of the calculated versus experimental SSCs allows the assignment of the major product to the I-st isomer, the second product to the II-nd isomer and the least populated product to the III-rd isomer.
The 3JHP between P and H4 is also very stereospecific (Table 1). First, the calculations predict SSCs to have large absolute values and be essentially different, at least, for the I-st and III-rd isomers. For the II-nd isomers there are two different values for each half but these SSC are close to the corresponding SSCs of the I-st and III-rd isomers, respectively. In spite of the fact that theory slightly underestimates these SSCs, the calculated values and tendencies are in good agreement with the experimentally observed data (Table 1). Thus, the comparison of the experimental versus calculated data also allows the assignment that is in accordance with the above conclusion.
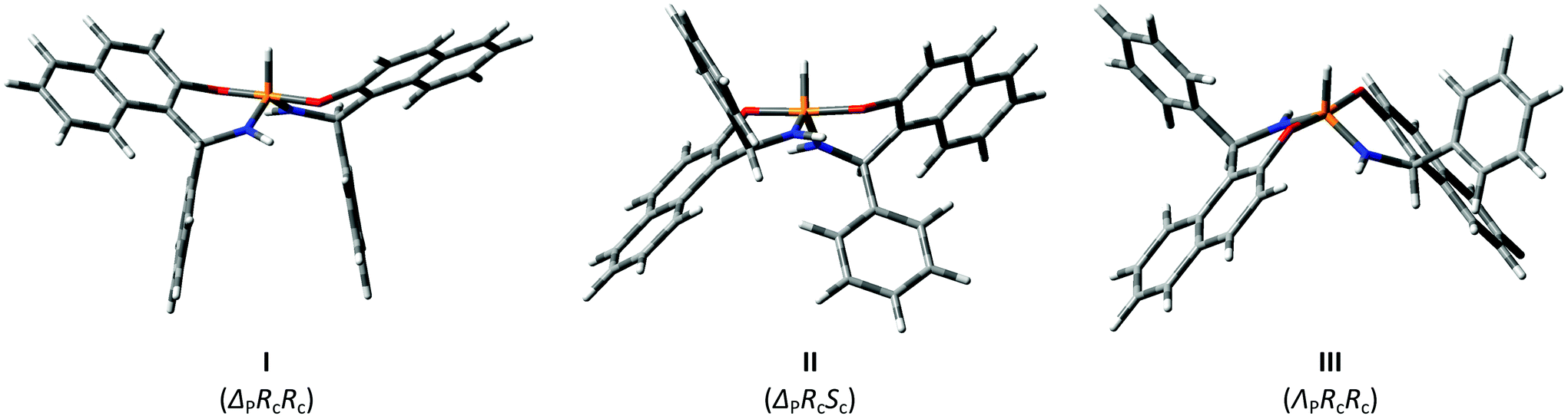
These conclusions were supported by solid-state data in some cases. Namely, good crystals suitable for X-ray analysis were grown for the second isomer of racemic 4, which is crystallized as a conglomerate, and for the major product of racemic 5. The results of X-ray single crystal diffraction turned out to be in full agreement with the NMR data. In crystals the major product of 5 is realized in the I form (see Fig. 3) with both aryls directed opposite to the P–H bond. In general, the solid-state geometry is close to the DFT optimized one.
 | ||
| Fig. 3 Molecular geometry of diastereomers 4b (left) and 5a (right) as determined from X-ray single crystal diffraction. | ||
It is interesting that the crystallized product of 4 corresponded to the asymmetrical II-form that is also in agreement with the conclusion based on the NMR data. Thus for the title chiral pentacoordinate spirophosphoranes there are several NMR parameters that can be used to establish the isomeric structure. These relationships are summarized in Fig. 4.
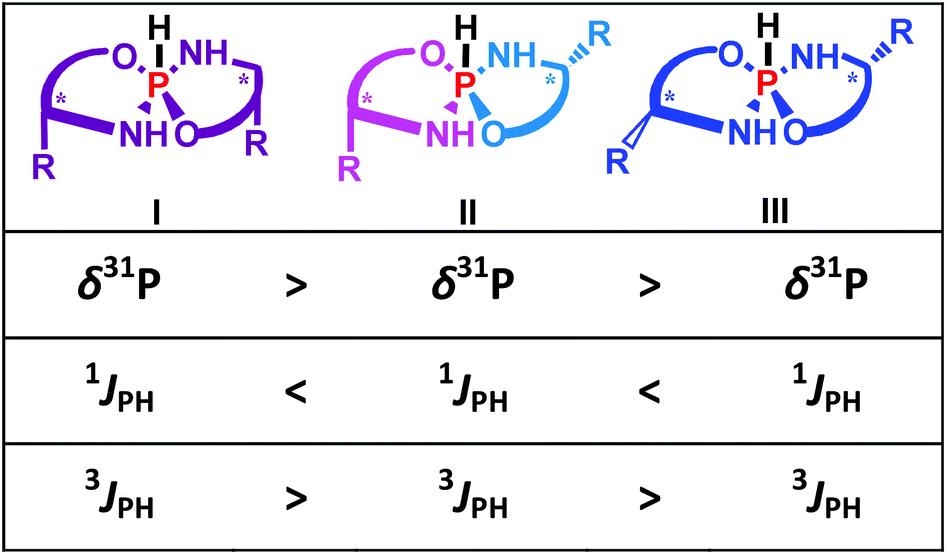 | ||
| Fig. 4 Schematic representation of three isomers of 4–6 and the main relationships between the NMR parameters and isomeric structure. Magnetically equivalent fragments are colored identically. | ||
Correlation of NMR parameters with the isomeric structure for other chiral pentacoordinate spirophosphoranes
The interplay of the NMR experiment and theory can be safely used to determine the isomeric structure of chiral pentacoordinate spirophosphoranes with six-membered heterocycles. To this end it is of interest to see if this approach could also be applied to determine the isomeric structure of other similar chiral pentacoordinate spirophosphoranes, e.g. with five-membered heterocycles. For example, there are experimental data for a series of chiral pentacoordinate spirophosphoranes obtained from α-amino carbonic acids (Scheme 2).33–37 These compounds were extensively analyzed by CD and X-ray single diffraction. When enantiomerically pure reagents were being used, the reactions produced only two diastereomers.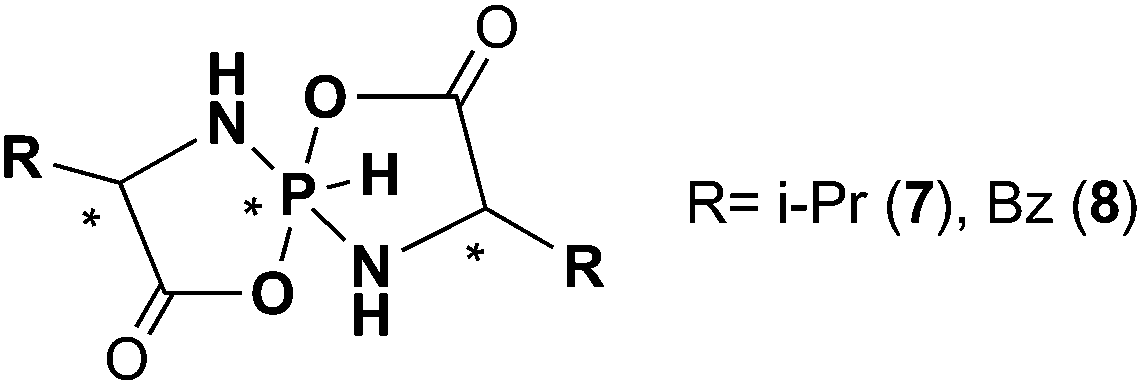 | ||
| Scheme 2 | ||
A brief survey of available NMR data demonstrates that there are also some specific features of experimental CSs and SSCs characteristic of two diastereomers although somewhat distinct from those observed for the title compounds. First, for five-membered spirophosphoranes the reverse 31P CS distribution is observed, i.e. the “I” isomers resonate at a higher field than “III” isomers (some data are included in Table 2).§ Second, the 1JPH values are smaller for the “I” isomers than for the “III”-forms. As to the 3JPH constants they are notably smaller (<13 Hz). Third, there are substantial long range 4J![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) PH4 SSCs (2.4 Hz) in the “III” forms that were not seen in the “I” isomers presumably due to their small values.
PH4 SSCs (2.4 Hz) in the “III” forms that were not seen in the “I” isomers presumably due to their small values.
| Comp. | R at C4 | Isomer | ΔEb | δ 31P | δH6 | δH4 | δH5 | 1 J PH | 3 J PH4 | 4 J H6H4 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Chemical shifts in ppm, spin–spin couplings in Hz. b Relative energy, in kcal mol−1. c Experimental data. d No data. | ||||||||||
| 7 | i-Pr | I | Expc (a)37 | −65.0 | 7.18 | 3.71 | 5.76 | 798.5 | 13.0 | n/dd |
| 0.19 | −65.6 | 7.82 | 3.73 | 2.82 | 697.2 | 8.8 | 0.5 | |||
| III | Exp (b)37 | −63.7 | 7.43 | 3.80 | 3.47 | 824.4 | n/d | 2.4 | ||
| 0 | −62.1 | 7.79 | 3.70 | 2.84 | 724.0 | 5.9 | 2.0 | |||
| 8 | Bz | I | Exp (a)36 | −63.0 | 7.09 | n/d | n/d | 804.9 | n/d | n/d |
| 0 | −68.5 | 7.95 | 4.16 | 3.02 | 710.3 | 10.8 | 0.4 | |||
| III | Exp (b)36 | −60.0 | 5.64 | n/d | n/d | 810.2 | n/d | 2.4 | ||
| 3.58 | −64.5 | 6.62 | 3.81 | 2.67 | 733.6 | 9.4 | 2.1 | |||
These differences are reproduced by theory. According to our calculations performed for the simplest compounds from this series with i-Pr (7) or Bz (8) groups at chiral carbons (Table 2), indeed, the “I” isomer has to resonate at a higher field in the 31P NMR spectra than the “III” form.
The calculated SSCs are also qualitatively consistent with the experimental values, although some underestimation is also seen. First, a larger one bond SSC is predicted for the “III” isomers (Table 2) than for the “I” form. According to the calculations, the 3JPH SSCs should be notably smaller than for the title compounds. Moreover, for the “III”-isomers one may expect a long range four bond SSC between HP and H4 (2.0 versus 0.5 Hz) larger than for the “I” isomer. Thus, several NMR parameters can also be used to determine the relative configuration at chiral carbons in these pentacoordinate spirophosphoranes.
Discussion
Let us consider why some relationships are different in these two series of spirophosphoranes. There are two essential NMR features that distinguish 5-membered spirophosphoranes from 6-membered compounds: (a) 3JPH – small absolute value for the 5-cycle and large for the 6-cycle; (b) δ31PI > δ31PIII for 6-membered and δ31PI < δ31PIII for 5-membered.To reveal key factors that determine these differences we analyzed the influence of substituents at carbon chiral centers and the nature of the fused aromatic fragments theoretically. To simplify the task and to exclude the specific influence of fused aromatic fragments, the models with the benzo fragment instead of the naphthyl group were analyzed (Fig. 5a and b, Table 3). According to these calculations, the relationships between the NMR parameters and isomeric structure are expected to be essentially similar to the title compounds. Thus, there is no specific effect of the naphthyl moiety on the NMR parameters’ distribution in these isomers.
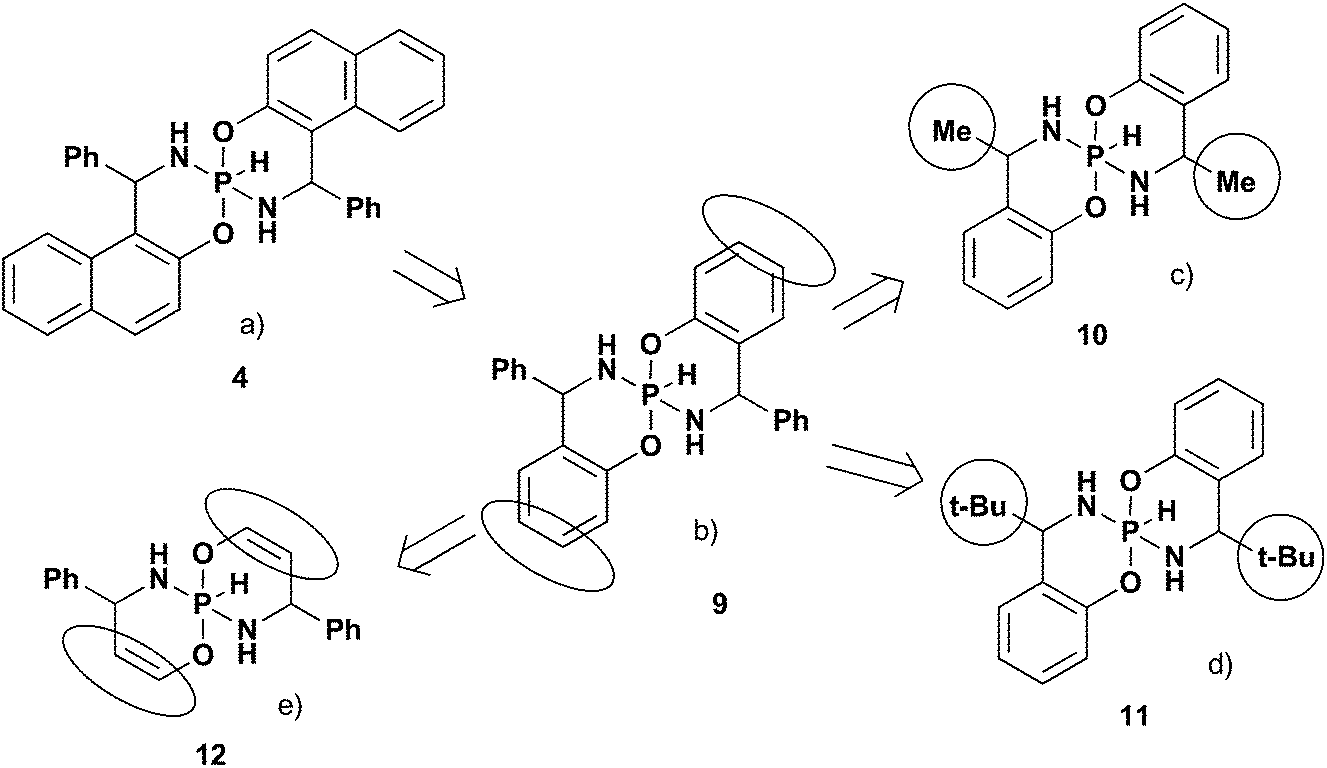 | ||
| Fig. 5 Step-by-step simplification of the title pentacoordinate spirophosphoranes. | ||
| Comp. | R at C4 | Isomer | ΔEb | δ 31P | δH6 | δH4 | δH5 | 1 J PH | 3 J PH4 |
|---|---|---|---|---|---|---|---|---|---|
| a Chemical shifts in ppm, spin–spin couplings in Hz. b Relative energy, in kcal mol−1. | |||||||||
| 9 | Ph | I | 0 | −72.7 | 7.41 | 5.11 | 3.56 | 717.5 | 35.4 |
| III | 0.92 | −80.6 | 7.90 | 4.87 | 3.24 | 770.5 | 20.8 | ||
| 10 | Me | I | 0 | −72.8 | 7.19 | 4.04 | 3.41 | 707.9 | 34.5 |
| III | 1.02 | −78.5 | 7.62 | 3.84 | 2.78 | 762.2 | 25.3 | ||
| 11 | t-Bu | I | 0 | −73.5 | 7.11 | 3.77 | 3.30 | 687.4 | 30.1 |
| III | 0.84 | −76.7 | 7.67 | 3.44 | 2.74 | 771.5 | 21.1 | ||
| 12 | Ph | I | 0 | −93.8 | 7.71 | 4.86 | 3.45 | 729.1 | 21.2 |
| III | 0.30 | −88.1 | 7.30 | 5.28 | 2.60 | 826.4 | 0.6 | ||
The next step to simplify the model is to check whether there is a specific influence of substituents at chiral carbons (e.g. steric interactions). For this purpose we analyzed models with small (Me, 10) and large (t-Bu, 11) alkyl groups, as well as aromatic (Ph, 9) fragments at chiral centers (Fig. 5c and d). According to calculations (Table 3), the main tendencies are also the same as for the title compounds. That is there is no specific influence of the R at the chiral carbon atoms on the δ31P and 3JPH difference in the main isomers.
In order to reveal the impact of the fused aromatic unit on NMR parameters, the benzo fragment was replaced by a double bond (12, Fig. 4e) in the next step. In this case the reverse distribution of 31P CSs for the two main isomers and notably smaller 3JPH SSCs are expected. This is very similar to the trends found in 5-membered compounds.
Thus, in both series the nature of the substituent at the chiral center is not principal. The nature of the fused aromatic fragment is also not crucial. But NMR parameters are considerably different for compounds with the aromatic system and those without the aromatic system. In this case, it is the geometry of the heterocycle which plays the crucial role: while in 6-membered spirophosphoranes with the fused aromatic system the heterocycle adopts boat conformation, in 5- and 6-membered compounds without the aromatic fragment the heterocycle is essentially flat (or envelope) (Fig. 6). Therefore, in the first case the H4–C4–N–P dihedral angle is about 156° and results in a large 3JPH value, in the latter cases these angles are ca. 126° and 107°, thus small 3JPH should be observed. The same difference in the conformation leads to a different distribution of 31P CSs in the two main isomers.
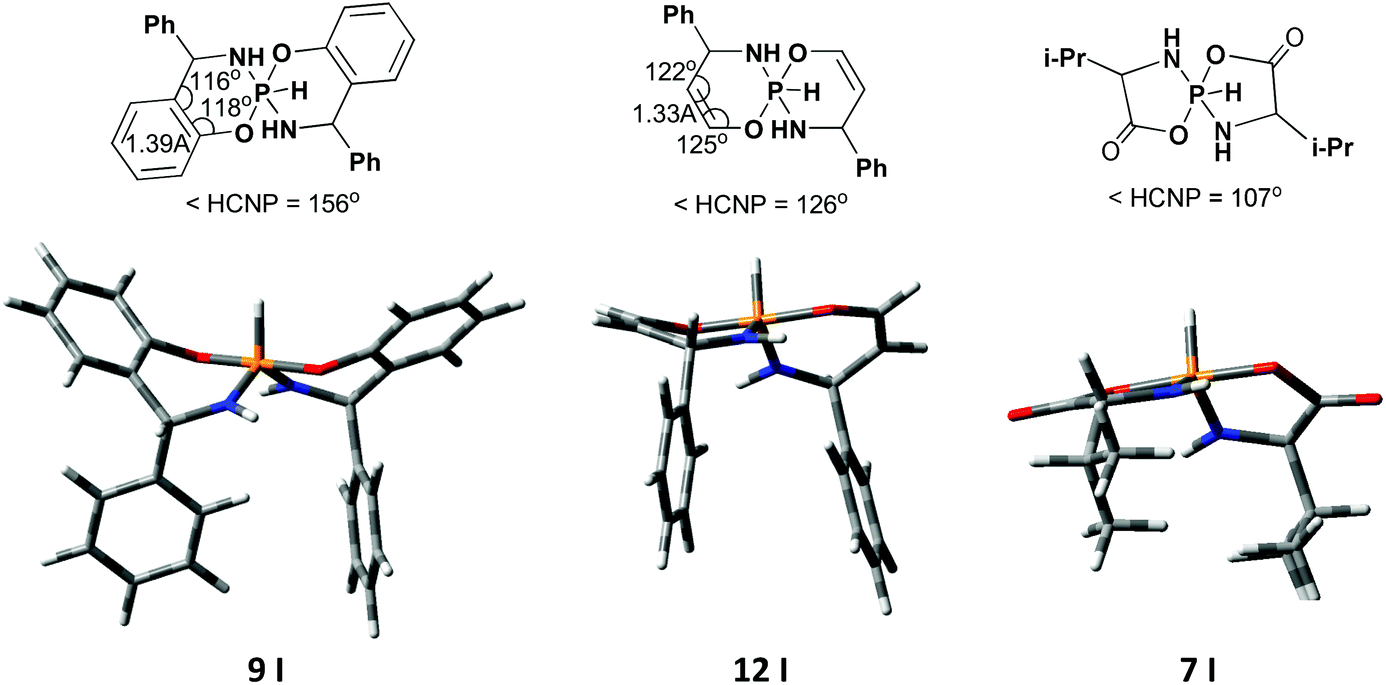 | ||
| Fig. 6 Some geometrical parameters and calculated 3D structures for different 5- and 6-membered pentacoordinate spirophosphoranes. | ||
Conclusions
The major product of the Betti based derivatives corresponds to the isomers with both substituents at chiral carbons being opposite to the P–H proton. The next populated product corresponds to the isomer with different chiralities at carbons. The least populated isomer is one with both substituents being at the same side of the heterocycle as the P–H bond.For such pentacoordinate chiral spirophosphoranes bearing two six-membered rings at the phosphorus atom the distinct NMR features, which allow establishing the isomeric structure, can be formulated as follows: the ΔPRcSc/ΛPRcSc isomer has two nonequivalent sets of signals in 1H (13C) NMR; the ΔPRcRc/ΛPScSc isomer resonances at a lower field in 31P NMR, the PH proton resonances at a higher field, 1JPH is smaller, 3JPH is larger than similar parameters for the ΔPScSc/ΛPRcRc isomer.
For similar spirophosphoranes bearing two five-membered rings at the phosphorus atom the NMR indicators are slightly different, in full agreement with the theory.
This study shows that the interplay of NMR experiments and DFT calculations of NMR parameters is a powerful and reliable method for determining the relative configurations of the pentacoordinate P–H-spirophosphoranes in solution.
Experimental section
NMR spectroscopy
All NMR experiments were performed with 600, 500 and 400 MHz (600.1, 500.1 and 400.1 MHz for 1H NMR; 150.9, 125.8 and 100.6 MHz for 13C NMR; 242.9, 202.5 and 162.0 MHz for 31P NMR, 60.81 MHz for 15N NMR, 376.5 for 19F respectively) spectrometers equipped with a 5 mm diameter probehead and a pulsed gradient unit capable of producing magnetic field pulse gradients in the z-direction of 53.5 G cm−1. For 1H–13C correlations the HSQC experiment is optimized for J = 145 Hz. For 1H–13C long range correlations the HMBC experiment is optimized for J = 8 Hz. For 1H–31P long range correlations the HMBC experiment is optimized for J = 8 Hz. For 1H–15N correlations the HSQC experiment is optimized for J = 90 Hz. For 1H–15N long range correlations the HMBC experiment is optimized for J = 6 Hz. For 19F–1H long range correlations the HETCOR experiment is optimized for J = 3 Hz. DOSY experiments were performed with ledbpgp2s, using a stimulated echo sequence and two spoil gradients. NOE experiments were performed with 1D DPFGNOE techniques. CSs (δ in ppm) were referenced to the solvent CDCl3 (δ = 7.27 ppm for 1H and 77.0 ppm for 13C NMR) and to external H3PO4 (0.0 ppm) for 31P NMR spectra, to external CH3CN (235.5 ppm) for 15N NMR spectra, to external C6F6 (−164.9 ppm) for 19F NMR spectra.Calculations
The quantum chemical calculations were performed using the Gaussian 03 software package.61 Full geometry optimizations have been carried out within the framework of the DFT (PBE1PBE) method using 6-31+G(d) basis sets. 31P CSs were calculated at the PBE1PBE/6-311G(2d,2p) level of theory.5631P CSs were referred to H3PO4 calculated at the same level of theory. The linear scaling procedure was applied.561H CSs were referred to TMS.Synthesis of 1-(α-aminobenzyl)-2-naphthols (1–3)
Compounds 1–3 were synthesized in several steps. First, the corresponding 1,3-diaryl-2,3-dihydro-1H-naphth[1,2-e][1,3]oxazines were obtained.62–64 which were then hydrolyzed by trifluoroacetic acid in the presence of water.65 The free bases were isolated by treating the resulting trifluoroacetates with sodium carbonate.66Synthesis of P–H-spirophosphoranes (4–6)
Spirophosphoranes (4–6) were synthesized by the reaction of the Betti bases and hexaethyltriamidophosphite: (1) without the addition of the catalyst (method A, only 4) resulting in the predominant formation of the (ΔPRcRc/ΔPScSc)-isomer; (2) with the addition of diethylamine hydrochloride (5 mol%) as the catalyst (method B), which allows obtaining (ΔPRcRc/ΔPScSc)- and (ΔPRcSc/ΔPScRc≡ΔPScRc/ΔPRcSc)-isomers in a ratio close to 1:1 by influencing the rate of P–Z bond cleavage (Z = O, N).67
Method A: 1-(α-Aminobenzyl)-2-naphthol (1) (4 mmol) was dissolved in dry benzene (12 mL) under heating. Then the solution of hexaethyltriamidophosphite (2 mmol) in dry benzene (3 mL) was added and the reaction mixture was kept at reflux under argon for 8 hours. After that the solvent was removed and the residue was analyzed by 1H and 31P NMR spectroscopy for the diastereomers’ (4a and 4b) ratio determination ((ΔPRcRc/ΛPScSc):(ΔPRcSc/ΛPScRc) = 65:35). Pure (ΔPRcRc/ΛPScSc)-P–H-phosphorane (4a) was obtained by recrystallization from benzene.
Method B: 1-(α-Aminobenzyl)-2-naphthols (1–3) (4 mmol) were dissolved in dry benzene (12 mL) under heating. Then dry diethylamine hydrochloride (0.1 mmol, 5 mol%) was added followed by the addition of hexaethyltriamidophosphite solution (2 mmol) in dry benzene (3 mL). The resulting reaction mixture was kept at reflux under argon for 5 hours. After that the solvent was removed and the residue was analyzed by 1H and 31P NMR spectroscopy for the diastereomers’ (a and b) ratio determination ((ΔPRcRc/ΛPScSc):(ΔPRcSc/ΛPScRc) = 1:1.1 for 4, 1:1 for 5, 1:0.9 for 6). Pure (ΔPRcRc/ΛPScSc)-4a–6a and (ΔPRcSc/ΛPScRc)-4b–6b diastereomers of P–H-phosphoranes were obtained by fractional crystallization from benzene.
Physical data
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cnaphth), 1665 (COO−), 3066, 3288 cm−1 (NH3+); elemental analysis calcd (%) for C19H15F4NO3: C59.85, H3.96, N 3.67; found: C 59.68, H 3.77, N 3.55.
Cnaphth), 1664 (COO−), 3066, 3254 cm−1 (NH3+); elemental analysis calcd (%) for C19H15BrF3NO3: C51.60, H3.42, Br 18.07, N 3.17; found: C 51.78, H 3.57, Br 17.89, N 3.35.
Cnaphth), 3292, 3362 cm−1 (NH2); elemental analysis calcd (%) for C17H14BrNO: C62.21, H4.30, Br 24.35, N 4.27; found: C62.04, H4.47, Br 24.16, N4.35.
Cnaphth), 2354 (PH), 3414 cm−1 (NH); elemental analysis calcd (%) for C34H27N2O2P: C 77.55, H 5.17, N 5.32, P 5.88; found: C77.35, H 5.01, N 5.49, P 6.02.
Cnaphth), 2358 (PH), 3416 cm−1 (NH); elemental analysis calcd (%) for C34H27N2O2P: C 77.55, H 5.17, N 5.32, P 5.88; found: C77.38, H4.98, N 5.42, P 6.05.
Cnaphth), 2364 (PH), 3420 cm−1 (NH); elemental analysis calcd (%) for C34H25F2N2O2P: C 72.59, H 4.48, N 4.98, P 5.51; found: C 72.66, H 4.31, N 4.87, P 5.32.
Cnaphth), 2358 (PH), 3422 cm−1 (NH); elemental analysis calcd (%) for C34H25F2N2O2P: C 72.59, H 4.48, N 4.98, P 5.51; found: C 72.74, H 4.59, N 4.83, P 5.41.
Cnaphth), 2353 (PH), 3419 cm−1 (NH); elemental analysis calcd (%) for C34H25Br2N2O2P: C 59.67, H 3.68, Br 23.35, N 4.09, P 4.53; found: C 59.86, H 3.49, Br 23.54, N 3.89, P 4.48.
Cnaphth), 2370 (PH), 3422 cm−1 (NH); elemental analysis calcd (%) for C34H25Br2N2O2P: C 59.67, H 3.68, Br 23.35, N 4.09, P 4.53; found: C 59.75, H 3.57, Br 23.12, N 4.27, P 4.65.
Cnaphth), 1665 (COO−), 3066, 3288 cm−1 (NH3+); elemental analysis calcd (%) for C19H15F4NO3: C59.85, H3.96, N 3.67; found: C 59.68, H 3.77, N 3.55.
Cnaphth), 1664 (COO−), 3066, 3254 cm−1 (NH3+); elemental analysis calcd (%) for C19H15BrF3NO3: C51.60, H3.42, Br 18.07, N 3.17; found: C 51.78, H 3.57, Br 17.89, N 3.35.
Cnaphth), 3292, 3362 cm−1 (NH2); elemental analysis calcd (%) for C17H14BrNO: C62.21, H4.30, Br 24.35, N 4.27; found: C62.04, H4.47, Br 24.16, N4.35.
Cnaphth), 2354 (PH), 3414 cm−1 (NH); elemental analysis calcd (%) for C34H27N2O2P: C 77.55, H 5.17, N 5.32, P 5.88; found: C77.35, H 5.01, N 5.49, P 6.02.
Cnaphth), 2358 (PH), 3416 cm−1 (NH); elemental analysis calcd (%) for C34H27N2O2P: C 77.55, H 5.17, N 5.32, P 5.88; found: C77.38, H4.98, N 5.42, P 6.05.
Cnaphth), 2364 (PH), 3420 cm−1 (NH); elemental analysis calcd (%) for C34H25F2N2O2P: C 72.59, H 4.48, N 4.98, P 5.51; found: C 72.66, H 4.31, N 4.87, P 5.32.
Cnaphth), 2358 (PH), 3422 cm−1 (NH); elemental analysis calcd (%) for C34H25F2N2O2P: C 72.59, H 4.48, N 4.98, P 5.51; found: C 72.74, H 4.59, N 4.83, P 5.41.
Cnaphth), 2353 (PH), 3419 cm−1 (NH); elemental analysis calcd (%) for C34H25Br2N2O2P: C 59.67, H 3.68, Br 23.35, N 4.09, P 4.53; found: C 59.86, H 3.49, Br 23.54, N 3.89, P 4.48.
Cnaphth), 2370 (PH), 3422 cm−1 (NH); elemental analysis calcd (%) for C34H25Br2N2O2P: C 59.67, H 3.68, Br 23.35, N 4.09, P 4.53; found: C 59.75, H 3.57, Br 23.12, N 4.27, P 4.65.
X-ray structure determination
Data sets for single crystals of 4b and 5a were collected on Bruker AXS Kappa APEX and Smart Apex diffractometers respectively with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) using complete sphere mode. Programs used: data collection APEX2,68 data reduction ‘SAINT’69 absorption correction SADABS version 2.10,70 structure solution SHELXS97,71 structure refinement by full-matrix least-squares against F2 using SHELXL-9771, hydrogen atoms calculated and refined as riding atoms, except the hydrogens attached to phosphorus and nitrogen atoms which were located from a difference Fourier map and refined isotropically. The crystals contain solvent channels, modeled by disordered ethanol and acetone molecules. Crystals 5b contain a solvate benzene molecule, which is disordered and was refined with geometry and vibrational parameter constraints.Crystal data for 4b
Formula C34H27N2O2P, crystal size 0.39 × 0.39 × 0.15 mm3, M = 526.54, orthorhombic, space group P212121, a = 8.6029(2), b = 15.2183(3), c = 20.4343(4) Å, V = 2675.3(1) Å3, Z = 4, ρc = 1.307 g cm−3, μ = 0.138 mm−1, T = 150(2) K, θ range = 1.67 to 31.17°, reflections collected: 49141; independent: 8492 (Rint = 0.0354) and 7309 observed reflections [I ≥ 2σ(I)], 364 refined parameters, R1 = 0.0406, wR2 = 0.0941 [I > 2σ(I)]; flack parameter −0.03(2), maximal residual electron density: 0.400/−0.229 e Å−3.
Crystal data for 5a
Formula C37H28F2N2O2P, crystal size 0.24 × 0.17 × 0.12 mm3, M = 601.58, monoclinic, space group C2/c, a = 38.86(2), b = 8.799(5), c = 17.269(9) Å, β = 95.512(7)°, V = 5878(6) Å3, Z = 8, ρc = 1.360 g cm−3, μ = 0.144 mm−1, T = 293(2) K, θ range = 2.11 to 26.60°, reflections collected: 28391; independent: 6119 (Rint = 0.1310) and 1891 observed reflections [I ≥ 2σ(I)], 406 refined parameters, R1 = 0.0440, wR2 = 0.0786 [I > 2σ(I)]; maximal residual electron density: 0.185/−0.193 e Å−3. Due to positional disorder of the solvate benzene molecule the ratio of the observed to collected reflections is low ∼31%.
CCDC 1542633 and 1542634 contain the supplementary crystallographic data for this paper.
Notes and references
- K. M. Pietrusiewicz and M. Zabłocka, Chem. Rev., 1994, 94, 1375 CrossRef CAS.
- R. Noyori, in Asymmetric Catalysis in Organic Synthesis, John Wiley & Sons, New York, 1994 Search PubMed.
- L. D. Quin, in A Guide to Organophosphorus Chemistry, Wiley-Interscience, New York, 2000 Search PubMed.
- I. Ojima, in Catalytic Asymmetric Synthesis, Wiley-VCH, New York, 2000 Search PubMed.
- J. Drabowicz, P. Łyźuwa, J. Omelańczuk, K. M. Pietrusiewicz and M. Mikołajczyk, Tetrahedron: Asymmetry, 1999, 10, 2757 CrossRef CAS.
- F. L. Lam, F. Y. Kwong and A. S. C. Chan, Top. Organomet. Chem., 2011, 36, 29 CrossRef CAS.
- F. H. Westheimer, Acc. Chem. Res., 1968, 1, 70 CrossRef CAS.
- S. L. Buchwald, D. H. Pliura and J. R. Knowla, J. Am. Chem. Soc., 1984, 106, 4916 CrossRef CAS.
- J. R. Knowles, Annu. Rev. Biochem., 1980, 49, 877 CrossRef CAS PubMed.
- P. J. J. M. van Ool and H. M. Buck, Recl. Trav. Chim. Pays-Bas, 1984, 103, 119 CrossRef CAS.
- R. R. Holmes, in Pentacoordinated Phosphorus, American Chemical Society, Washington, DC, 1980, vol. 11, p. 87 Search PubMed.
- D. E. C. Corbridge, in Phosphorus, Chemistry, Biochemistry and Technology, Elsevier, Amsterdam, 4th edn, 2000 Search PubMed.
- S. D. Lahiri, G. F. Zhang, D. Dunaway-Mariano and K. N. Allen, Science, 2003, 299, 2067 CrossRef CAS PubMed.
- G. M. Blackburn, N. H. Williams, S. J. Gamblin and S. J. Smerdon, Science, 2003, 301, 1184 CrossRef CAS PubMed.
- K. N. Allen and D. Dunaway-Mariano, Trends Biochem. Sci., 2004, 29, 495 CrossRef CAS PubMed.
- N. J. Baxter, G. M. Blackburn, J. P. Marston, A. M. Hounslow, M. J. Cliff, W. Bermel, N. H. Williams, F. Hollfelder, D. E. Wemmer and J. P. Waltho, J. Am. Chem. Soc., 2008, 130, 3952 CrossRef CAS PubMed.
- W. W. Cleland and A. C. Hengge, Chem. Rev., 2006, 106, 3252 CrossRef CAS PubMed.
- J. A. Adams, Chem. Rev., 2001, 101, 2271 CrossRef CAS PubMed.
- M. Eder, M. Stolz, T. Wallimann and U. Schlattner, J. Biol. Chem., 2000, 275, 27094 CAS.
- B. E. Bernstein and W. G. J. Hol, Biochemistry, 1998, 37, 4429 CrossRef CAS PubMed.
- K. Taira, T. Uchimaru, J. W. Storer, A. Yliniemela, M. Uebayashi and K. Tanabe, J. Org. Chem., 1993, 58, 3009 CrossRef CAS.
- D. M. Perreault and E. V. Anslyn, Angew. Chem., Int. Ed. Engl., 1997, 36, 432 CrossRef.
- R. T. Raines, Chem. Rev., 1998, 98, 1045 CrossRef CAS PubMed.
- Y. Takagi, M. Warashina, W. J. Stec, K. Yoshinari and K. Taira, Nucleic Acids Res., 2001, 29, 1815 CrossRef CAS PubMed.
- A. C. Hengge, Acc. Chem. Res., 2002, 35, 105 CrossRef CAS PubMed.
- Z. Y. Zhang, Acc. Chem. Res., 2003, 36, 385 CrossRef CAS PubMed.
- S. D. Lahiri, G. F. Zhang, D. Dunaway-Mariano and K. N. Allen, Science, 2003, 299, 2067 CrossRef CAS PubMed.
- K. N. Allen and D. Dunaway-Mariano, Trends Biochem. Sci., 2004, 29, 495 CrossRef CAS PubMed.
- L. W. Tremblay, G. F. Zhang, J. Y. Dai, D. Dunaway-Mariano and K. N. Allen, J. Am. Chem. Soc., 2005, 127, 5298 CrossRef CAS PubMed.
- R. R. Holmes, Acc. Chem. Res., 2004, 37, 746 CrossRef CAS PubMed.
- R. R. Holmes, Acc. Chem. Res., 1998, 31, 535 CrossRef CAS.
- N. V. Timosheva, A. Chandrasekaran and R. R. Holmes, J. Am. Chem. Soc., 2005, 127, 12474 CrossRef CAS PubMed.
- H. Fu, J. H. Xu, R. J. Wang, Z. Z. Chen, G. Z. Tu, Q. Z. Wang and Y. F. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 1963 CrossRef CAS.
- G. Yang, Y. Xu, J. Hou, H. Zhang and Y. Zhao, Dalton Trans., 2010, 39, 6953 RSC.
- G. Yang, Y. Xu, J. Hou, H. Zhang and Y. Zhao, Chem. – Eur. J., 2010, 16, 2518 CrossRef CAS PubMed.
- J. Hou, H. Zhang, J. Guo, Y. Liu, P. Xu, Y. Zhao and G. M. Blackburn, Org. Biomol. Chem., 2009, 7, 3020 CAS.
- J. Hou, G. Tang, J. Guo, Y. Liu, H. Zhang and Y. Zhao, Tetrahedron: Asymmetry, 2009, 20, 1301 CrossRef CAS.
- S. Cao, Z. Zhou, W. Dai, P. Zhao, Y. Guo and Y. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 925 CrossRef CAS.
- I. Alkorta and J. Elguero, Struct. Chem., 1998, 9, 187 CrossRef CAS.
- M. G. Chini, C. R. Jones, A. Zampella, M. V. D'Auria, B. Renga, S. Fiorucci, C. P. Butts and G. Bifulco, J. Org. Chem., 2012, 77, 1489 CrossRef CAS PubMed.
- G. Saielli, K. C. Nicolaou, A. Ortiz, H. Zhang and A. Bagno, J. Am. Chem. Soc., 2011, 133, 6072 CrossRef CAS PubMed.
- Sh. Latypov, A. Balandina, M. Boccalini, A. Matteucci, K. Usachev and S. Chimichi, Eur. J. Org. Chem., 2008, 4640 CrossRef CAS.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946 CrossRef CAS PubMed.
- G. Bifulco, P. Dambruoso, L. Gomez-Paloma and R. Riccio, Chem. Rev., 2007, 107, 3744 CrossRef CAS PubMed.
- A. Balandina, V. Mamedov, X. Franck, B. Figadere and Sh. Latypov, Tetrahedron Lett., 2004, 45, 4003 CrossRef CAS.
- A. Bagno, F. Rastrelli and G. Saielli, Chem. – Eur. J., 2006, 12, 5514 CrossRef CAS PubMed.
- B. Mikhova, B. Stamboliyska, A. Koch, H. Duddeck and E. Kleinpeter, Magn. Reson. Chem., 2008, 46, 1153 CrossRef CAS PubMed.
- F. Blanco, I. Alkorta and J. Elguero, Magn. Reson. Chem., 2007, 45, 797 CrossRef CAS PubMed.
- S. Chimichi, M. Boccalini, A. Matteucci, S. V. Kharlamov, Sh. K. Latypov and O. G. Sinyashin, Magn. Reson. Chem., 2010, 48, 607 CAS.
- S. Klod, A. Koch and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2002, 1506 RSC.
- A. E. Aliev, Z. A. Mia, M. J. M. Busson, R. J. Fitzmaurice and S. Caddick, J. Org. Chem., 2012, 77, 6290 CrossRef CAS PubMed.
- R. M. Claramunt, D. Sanz, C. López, J. A. Jiménez, M. L. Jimeno, J. Elguero and A. Fruchier, Magn. Reson. Chem., 1997, 35, 35 CrossRef CAS.
- A. Bagno, Chem. – Eur. J., 2001, 7, 1652 CrossRef CAS.
- C. van Wüllen, Phys. Chem. Chem. Phys., 2000, 2, 2137 RSC.
- B. Maryasin and H. Zipse, Phys. Chem. Chem. Phys., 2011, 13, 5150 RSC.
- Sh. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976 RSC.
- S. Böhm, J. Tomaščiková, J. Imrich, I. Danihel, P. Kristian, A. Kochc, E. Kleinpeter and K. D. Klika, THEOCHEM, 2009, 916, 105 CrossRef.
- S. G. Smith and J. M. Goodman, J. Org. Chem., 2009, 74, 4597 CrossRef CAS PubMed.
- M. J. Bartlett, P. T. Northcote, M. Lein and J. E. Harvey, J. Org. Chem., 2014, 79, 752 CrossRef PubMed.
- S. Di Micco, M. G. Chini, R. Riccio and G. Bifulco, Eur. J. Org. Chem., 2010, 1411 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision A.1, Gaussian, Inc., Pittsburgh, PA, 2003.
- I. Szatmári, T. A. Martinek, L. Lázár, A. Koch, E. Kleinpeter, K. Neuvonen and F. Fülöp, J. Org. Chem., 2004, 69, 3645 CrossRef PubMed.
- V. F. Zheltukhin, K. E. Metlushka, D. N. Sadkova, C. E. McKenna, B. A. Kashemirov and V. A. Alfonsov, Mendeleev Commun., 2007, 17, 239 CrossRef CAS.
- K. E. Metlushka, D. N. Sadkova, L. N. Shaimardanova, A. I. Tufatullin, V. F. Zheltukhin, O. N. Kataeva and V. A. Alfonsov, J. Heterocycl. Chem., 2015, 52, 80 CrossRef CAS.
- G. Bian, S. Yang, H. Huang and L. Song, Synthesis, 2013, 899 CAS.
- C. Cardellicchio, G. Ciccarella, F. Naso, E. Schingaro and F. Scordari, Tetrahedron: Asymmetry, 1998, 9, 3667 CrossRef CAS.
- E. S. Batyeva, V. A. Alfonsov, G. U. Zamaletdinova and A. N. Pudovik, Russ. J. Gen. Chem., 1976, 46, 2204 CAS.
- Bruker, APEX2 Software Suite for Crystallographic Programs, Bruker AXS, Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- Bruker, Area detector control and integration software. Version 5.x. In: SMART and SAINT, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 1996 Search PubMed.
- G. M. Sheldrick, SHELX-97: Programs for Crystal Structure Analysis, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D/2D NMR spectra and calculated data for 4–6. CCDC 1542633 and 1542634. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01605k |
| ‡ The isomer III might be produced from isomer ΔPRcRc (ΛPScSc) by the Berry pseudorotation process, so its configuration was assigned as ΛPRcRc (ΔPScSc). |
| § Unfortunately, 1H CS's data cannot be analyzed in this case as different solvents were used in experiments for different diastereomers. |
| This journal is © The Royal Society of Chemistry 2017 |