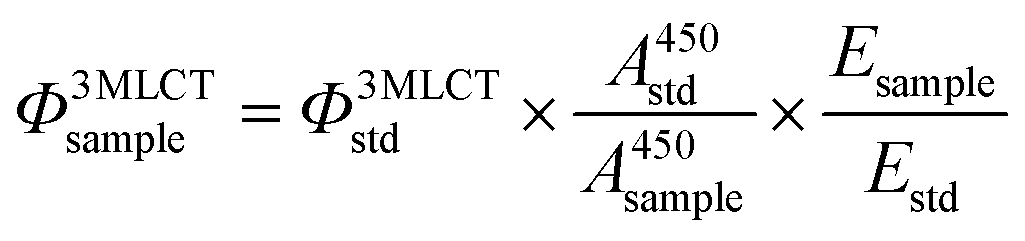DOI:
10.1039/C7DT01540B
(Paper)
Dalton Trans., 2017,
46, 9969-9980
Frontier orbitals of photosubstitutionally active ruthenium complexes: an experimental study of the spectator ligands’ electronic properties influence on photoreactivity†
Received
27th April 2017
, Accepted 12th July 2017
First published on 20th July 2017
Abstract
The synthesis and characterization of [Ru(tpy)(R2bpy)(L)](X)n complexes (tpy = 2,2′:6′,2′′-terpyridine, R2bpy = 4,4′-dimethyl-2,2′-bipyridine (dmbpy), or 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (tfmbpy), X = Cl− or PF6−, and n = 1 or 2) are described. The dmbpy and tfmbpy bidentate ligands allow for investigating the effects of electron-donating and electron-withdrawing ligands, respectively, on the frontier orbital energetics as well as the photoreactivity of these ruthenium polypyridyl complexes for five prototypical monodentate ligands L = Cl−, H2O, CH3CN, 2-(methylthio)ethanol (Hmte), or pyridine. According to spectroscopic and electrochemical studies, the dmbpy analogues displayed a singlet metal-to-ligand charge transfer (1MLCT) transition at higher energy than the tfmbpy analogues. The shift of the 1MLCT to higher energy results from the lowest unoccupied molecular orbital (LUMO) for the dmbpy analogues being tpy-based, whereas for the tfmbpy analogues orbital inversion occurs resulting in a tfmbpy-based LUMO. The energy level of the highest occupied molecular orbital (HOMO) was considerably affected by the nature of the monodentate ligand. Visible light irradiation of the complexes demonstrated that the tfmbpy analogue increased the rate and quantum yields of photosubstitution reactions, compared to the dmbpy analogue, suggesting that the electron-withdrawing substituents allowed better thermal accessibility of the triplet metal-centered (3MC) state from the photochemically generated triplet metal-to-ligand charge transfer (3MLCT) excited state. A correlation between the photolability of the monodentate ligands and the electrochemical reversibility of the metal-based oxidation is also reported.
Over the past several decades, ruthenium photochemistry has been studied for various applications including dye-sensitized solar cells,1 energy storage,2 photochemical synthesis,3 water oxidation catalysis,4 or nitric oxide delivery to biological targets.5 Additionally, ruthenium polypyridyl complexes have received significant attention as light-activated anti-cancer drugs because they can undergo ligand photosubstitution, which sometimes results in an “activated” ruthenium species that is more cytotoxic than the non-irradiated precursor.6–12 Photosubstitution reactions occur under visible light irradiation because of the unique excited state properties of ruthenium polypyridyl complexes. Upon visible light irradiation, Ru-based complexes display low energy metal-to-ligand charge transfer (1MLCT) transitions, which are the result of an electron being excited from the Ru(dπ)-based highest occupied molecular orbital (HOMO) to a ligand(π*)-based lowest unoccupied molecular orbital (LUMO). The 1MLCT excited state then undergoes rapid intersystem crossing to the 3MLCT excited state, from which triplet metal-centered (3MC) states are thermally accessible. Such 3MC states are ligand field excited states where an electron is excited from the t2g to eg orbital, resulting in ligand dissociation due to the anti-bonding character of the eg orbital.13,14 A number of studies have been performed that have increased our understanding of the relationship between the photoreactivity of ruthenium polypyridyl complexes and the energy of their 3MLCT and 3MC states15–21 or the topology of their triplet hypersurface.22 These studies have demonstrated the critical role of steric hindrance on the 3MLCT–3MC energy gap,23–28 showed that extended conjugation of the bidentate ligand affects the population of 3MC state via competition of low-lying bpy-based π–π* excited state,12,25 or explained why thioether ligands are more prone to photosubstitution compared to amines.29 Electrochemical studies of [Ru(tpy)(N,N)(L)]n+ complexes demonstrated that electron-withdrawing bidentate ligands tended to decrease the energy gap between the frontier orbitals (HOMO and LUMO).30 However, one question remains poorly addressed, what is the effect of electron-withdrawing spectator ligands that have no additional steric hindrance on the photosubstitution quantum yields?
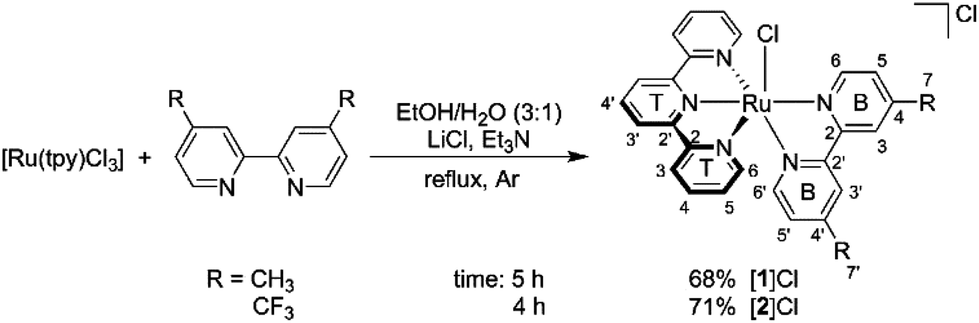
In this work we aimed at answering this question by studying two series of complexes having the general formula [Ru(tpy)(R2bpy)(L)]n+ (tpy = 2,2′:6′,2′′-terpyridine, L = monodentate ligand) with two electronically different bidentate ligands: R2bpy = 4,4′-dimethyl-2,2′-bipyridine (dmbpy) and R2bpy = 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (tfmbpy). Methyl and trifluoromethyl groups are typical isosteres as the hydrogen and fluorine atoms have comparable sizes. The van der Waals radius of fluorine (1.35 Å) is the next smallest after hydrogen (1.20 Å). However, CH3 and CF3 groups have opposite electronic properties because of the large differences in electronegativity and inductive effects of F vs. H:31 dmbpy is electron-donating, whereas tfmbpy is electron-withdrawing. Importantly, these substituents were placed in 4 and 4′ position on the bipyridine spectator ligand, so that both series of complexes have rigorously identical steric requirements. The monodentate ligands L were also systematically varied as their electronic properties strongly influence the photosubstitution reaction efficiency: L was Cl−, H2O, CH3CN, 2-(methylthio)ethanol (Hmte), or pyridine. The synthesis of ten [Ru(tpy)(R2bpy)(L)]n+ complexes is reported, as well as the study of their electrochemical, spectroscopic, and photochemical properties.
Results and discussion
Ligand synthesis
Unlike dmbpy, which is commercially available, 4,4′-bis(trifluoromethyl)-bpy was synthesized using a modified method from Liao et al. based on the nickel catalyst NiCl2·6H2O (Scheme S1†).32 Our modifications included the change of the solvent from DMF to THF, adding twice as much nickel catalyst as reported, and also one equivalent of 2,2′-bpy per mol catalyst. Iodine and acetic acid were not necessary. As a result, the ligand was obtained with much higher yield (56%) compared to the original protocol (10%). The yield was also improved compared to that reported by Chan et al. where 30 mol% of Ni(PPh3)2Cl2, a typical catalyst of Negishi coupling reaction, was used.33 The improvement in yield was attributed to two effects. First, the initial formation of [Ni(bpy)Cl2], which according to Liao et al. facilitates the catalytic cycle,32 was verified by 1H NMR spectroscopy: a paramagnetic bpy-containing species34 was observed instead of free bpy ligand (Fig. S1†). Furthermore, THF being a stronger ligand than DMF, it may stabilize the catalyst by coordinating to nickel throughout the catalytic cycle.35 The Experimental section and Fig. S2–S5† show the full characterization of the tfmbpy ligand by 1H, 13C, 19F{1H} NMR spectroscopy, and electrospray mass spectrometry (ES-MS).
Synthesis of [Ru(tpy)(R2bpy)(L)]n+ complexes
As illustrated in Scheme 1, the dmbpy and tfmbpy ligands were coordinated to [Ru(tpy)Cl3] to afford complexes [1]Cl and [2]Cl (see Fig. 1 for complex numbers).27,36,37 Both complexes had a typical dark violet colour. The 1H NMR spectrum of [2]Cl showed that the protons were de-shielded compared to [1]Cl. As expected, the chemical shift of the alpha proton of the bipyridine (i.e. B6, see Scheme 1) was noticeably shifted downfield because of the electron-withdrawing trifluoromethyl substituents.38 The non-equivalence of the two trifluoromethyl groups was confirmed by two chemical shifts observed in the 19F{1H} NMR spectrum, as well as separate B7 and B7′ quartets by 13C NMR (see spectra in Fig. S6–S10†).
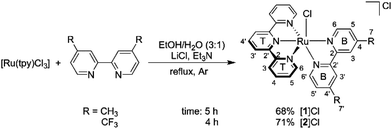 |
| | Scheme 1 Synthesis and atom numbering for [Ru(tpy)(R2bpy)(Cl)]Cl, where R2bpy = dmbpy ([1]Cl) or tfmbpy ([2]Cl). | |
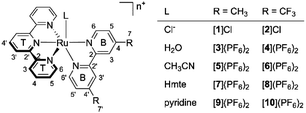 |
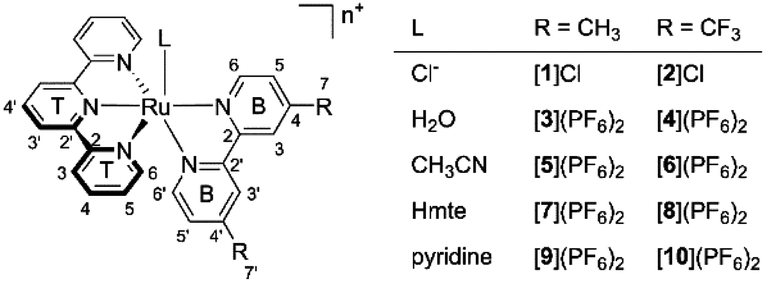
| | Fig. 1 The synthesized and characterized [Ru(tpy)(R2bpy)(L)]n+ complexes in this work. n = 1 when L = Cl−, and n = 2 when L = H2O, CH3CN, Hmte, or py. | |
From the chlorido complexes, the aqua ([3](PF6)2 and [4](PF6)2), acetonitrile ([5](PF6)2 and [6](PF6)2), thioether ([7](PF6)2 and [8](PF6)2), and pyridine ([9](PF6)2 and [10](PF6)2) complexes were synthesized using modified literature procedures.25,27,30 Generally, the syntheses started from the chlorido complex, which was refluxed under argon with AgPF6 and an excess of the target monodentate ligand (see Experimental section). The difference between the dmbpy and tfmbpy analogues was not very significant except for the syntheses of the aqua complexes. Complex [1]Cl fully converted into [3](PF6)2 after refluxing the mixture of [1]Cl and AgPF6 in acetone/water for 3 h, whereas [2]Cl showed minimal conversion to [4](PF6)2 after 16 h under the same reaction conditions. Therefore, a microwave reaction was performed, where acetone was excluded to prevent the potential competition with H2O for coordination to ruthenium. To remove the starting material, dichloromethane extraction was used, yielding [4](PF6)2 in the aqueous phase (Fig. S13†).
1H NMR spectroscopy of all the complexes in acetone-d6, displayed a trend in the chemical shifts of the alpha proton on the bipyridine ligand (B6). For both the dmbpy and tfmbpy series, the B6 proton became gradually less de-shielded following the series Cl− > Hmte ≈ CH3CN ≈ H2O > pyridine (Fig. S20 and S21†). The particularly upfield-shifted B6 proton of the pyridine complexes resulted from the shielding cone of the coordinated pyridine ligand, whereas the large downfield shift for the chloride analogues was interpreted as a consequence of the inductive effect of the chloride.
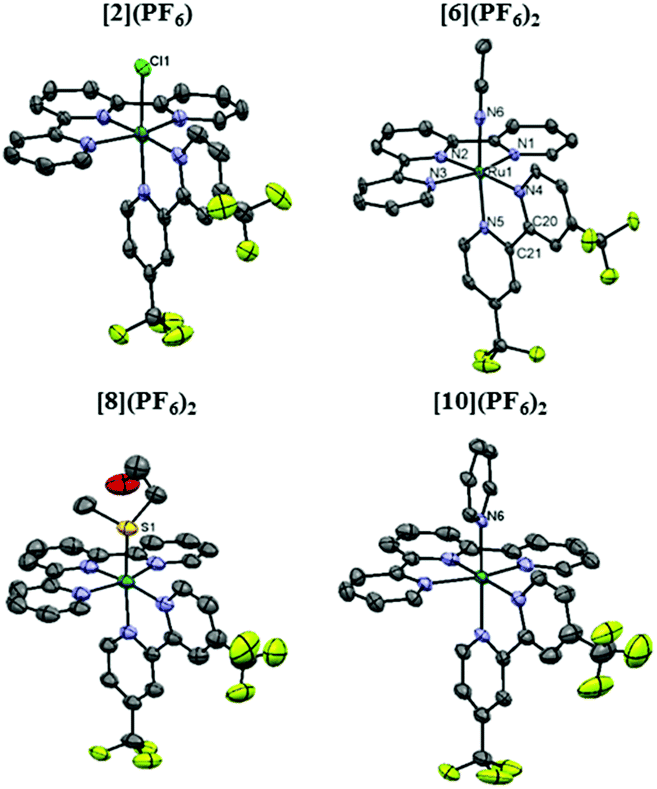
Single crystals of [1]Cl·C2H6O, [2](PF6)·C3H6O, [5](PF6)2·0.70CH3CN, [6](PF6)2·CH3CN, [7](PF6)2·C3H6O, [8](PF6)2, [9](PF6)2·C7H8 and [10](PF6)2·C3H6O suitable for X-ray structure determination were obtained by slow vapor diffusion. Specific solvent conditions for crystallization are described in the Experimental section. The displacement ellipsoid plots of the tfmbpy analogue crystal structures are given in Fig. 2, and the dmbpy analogues are shown in Fig. S34† along with the crystal data in Table S1.† As expected, the complexes display octahedral geometry without significant distortion of the bpy (Fig. 2 and S34† and Table 1). Comparing a dmbpy complex to its tfmbpy analogue (i.e. [1]+vs. [2]+, or [5]2+vs. [6]2+), the bond distances from the Ru1 atom to the bidentate ligand were longer for the dmbpy complexes than their tfmbpy analogues. However, the Ru1–L bond distances were not significantly affected by the property of the bidentate ligands, whereas significant variation occurred depending on the heteroatom bound to ruthenium (Table 1).
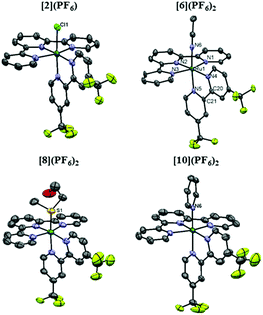 |
| | Fig. 2 Displacement ellipsoid plot (50% probability level) of the crystal structures of [2](PF6), [6](PF6)2, [8](PF6)2, and [10](PF6)2. The counter-anions, disorder, hydrogen atoms, and the uncoordinated lattice solvent molecules were omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) in the crystal structures of [1]Cl·C2H6O, [2](PF6)·C3H6O, [5](PF6)2·0.70CH3CN, [6](PF6)2·CH3CN, [7](PF6)2·C3H6O, [8](PF6)2, [9](PF6)2·C7H8 and [10](PF6)2·C3H6O
| |
[1]Cl·C2H6O |
[2](PF6) ·C3H6O |
[5](PF6)2·0.70MeCNb |
[6](PF6)2·CH3CN |
[7](PF6)2·C3H6O |
[8](PF6)2 |
[9](PF6)2·C7H8 |
[10](PF6)2·C3H6O |
|
L = Cl1 for [1]Cl and [2](PF6); N6 for [5](PF6)2, [6](PF6)2, [9](PF6)2, and [10](PF6)2; or S1 for [7](PF6)2 and [8](PF6)2. See Fig. S34.
A crystal structure of [5](PF6)2 was previously reported39 with comparable parameters as the data in this table.
|
| Ru1–N1 |
2.062(4) |
2.062(3) |
2.076(2) |
2.084(5) |
2.070(2) |
2.078(3) |
2.085(2) |
2.072(3) |
| Ru1–N2 |
1.961(3) |
1.962(3) |
1.974(2) |
1.983(5) |
1.963(2) |
1.963(4) |
1.961(2) |
1.974(3) |
| Ru1–N3 |
2.072(4) |
2.071(3) |
2.073(2) |
2.074(4) |
2.070(2) |
2.074(3) |
2.055(2) |
2.081(3) |
| Ru1–N4 |
2.086(3) |
2.070(3) |
2.093(6) |
2.076(5) |
2.099(2) |
2.092(4) |
2.089(2) |
2.077(3) |
| Ru1–N5 |
2.047(3) |
2.026(3) |
2.044(7) |
2.033(5) |
2.070(2) |
2.061(3) |
2.056(2) |
2.057(3) |
| Ru1–La |
2.3983(9) |
2.4050(9) |
2.032(2) |
2.036(5) |
2.3757(7) |
2.374(2) |
2.100(2) |
2.103(3) |
| N1–Ru1–N3 |
158.95(15) |
159.18(11) |
159.08(9) |
159.04(18) |
159.46(9) |
158.51(15) |
159.49(8) |
158.19(12) |
| N4–Ru1–N5 |
78.46(13) |
78.79(11) |
79.4(6) |
78.84(18) |
78.33(9) |
78.16(13) |
78.34(8) |
78.15(11) |
| Ru1–N4–C20–C21 |
−6.3(4) |
6.9(4) |
−4.8(5) |
−0.6(6) |
2.3(3) |
−3.3(5) |
2.9(3) |
−7(5) |
| Ru1–N5–C21–C20 |
3.7(5) |
−4.8(4) |
4.5(5) |
0.8(6) |
−1.1(3) |
−2.0(5) |
1.8(3) |
−7(3) |
Electrochemical properties
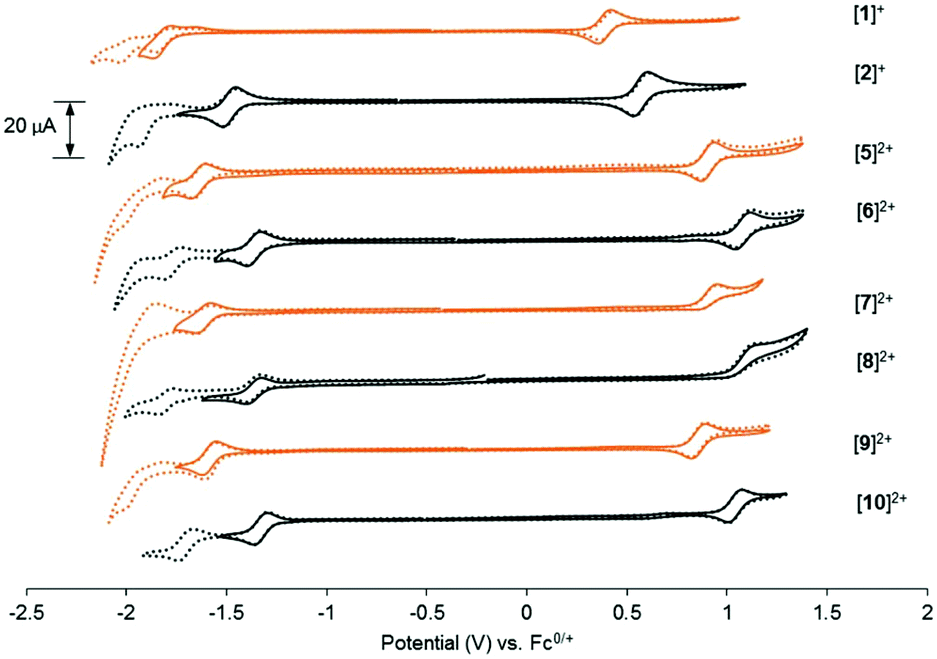
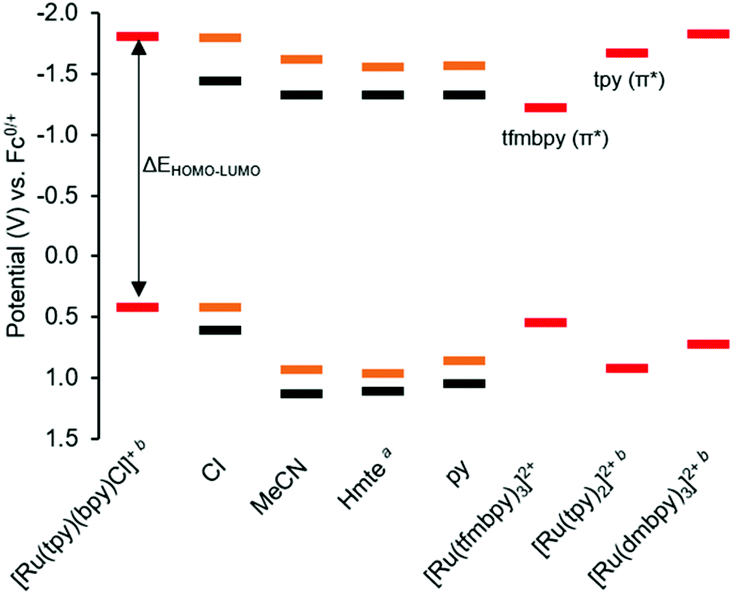
To gain insight into the frontier orbitals of these complexes, the electrochemical properties of the complexes were studied using cyclic voltammetry (CV). As summarized in Table 2 and Fig. 3, the dmbpy and tfmbpy complexes showed clear differences in oxidation and reduction potentials. Irrespective of the monodentate ligand, the half-wave potential of the Ru-based oxidation for the tfmbpy complex was 100–200 mV higher than that of the dmbpy complex. The shift to higher potential for the tfmbpy analogues was expected as the electron-withdrawing substituents decreased the electron density of the metal center, making the RuII to RuIII oxidation more difficult. Similarly, the half-wave potential of the ligand-based reduction for the tfmbpy complex was more negative than that of the dmbpy complexes. This result is consistent with the trifluoromethyl groups stabilizing the extra charge added on the reduced ligand, which makes reduction more favourable. In Fig. 4, an energy diagram derived from Table 2 clearly shows that the stabilization of the LUMO from the dmbpy to the tfmbpy complexes was greater than that of the HOMO. For ruthenium polypyridyl complexes, the LUMO is usually more ligand-based than the HOMO. Here, comparison of the LUMOs for [Ru(tpy)(bpy)(Cl)]+ (−1.81 V vs. Fc0/+),40 [Ru(tpy)2]2+ (−1.67 V vs. Fc0/+),41 and [Ru(tfmbpy)3]2+ (−1.22 V vs. Fc0/+) (Fig. S35†) suggested that the LUMO of the dmbpy complexes was tpy-based, whereas that of the tfmbpy complexes was mostly localized on the tfmbpy ligand.
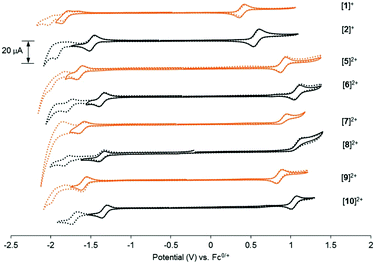 |
| | Fig. 3 Cyclic voltammograms of 1 mM [1]Cl, [2](PF6), or [5](PF6)2 through [10](PF6)2 in 0.1 M Bu4NPF6/CH3CN (ν = 100 mV s−1). The orange lines represent the 4,4′-dimethyl-2,2′-bipyridine analogues and the black lines represent the 4,4′-bis(trifluoromethyl)-2,2′-bipyridine analogues. Dashed lines show the second ligand-based reductions of the complexes. | |
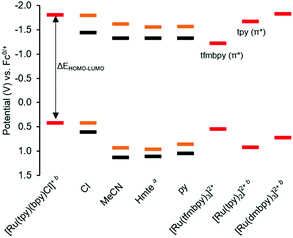 |
| | Fig. 4 HOMO and LUMO orbital energy diagram derived from cyclic voltammetry data (Table 2). The orange lines represent the 4,4′-dimethyl-2,2′-bipyridine analogues [1]+, [5]2+, [7]2+, [9]2+, and the black lines represent the 4,4′-bis(trifluoromethyl)-2,2′-bipyridine analogues [2]+, [6]2+, [8]2+, [10]2+, with the monodentate ligands specified on the x-axis. The red lines represent the E1/2 of the first metal-based oxidation and ligand-based reduction of the references. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) For both Hmte complexes, E1/2 was calculated from the electrochemical data measured with a scan rate of 1000 mV s−1, and used in this chart, instead of the Epa in Table 2. bThe potentials were previously reported.40–42 For both Hmte complexes, E1/2 was calculated from the electrochemical data measured with a scan rate of 1000 mV s−1, and used in this chart, instead of the Epa in Table 2. bThe potentials were previously reported.40–42 | |
Table 2 Redox potentials and ipa/ipc of [1]Cl, [2](PF6), and [5](PF6)2–[10](PF6)2 as measured by cyclic voltammetrya
| Complex |
[complex]{(n+1)/n}+ |
[complex]{n/(n−1)}+ |
|
E
1/2 (V) |
i
pa/ipc |
E
1/2 (V) |
i
pa/ipc |
|
Measurement conditions: 1 mM of the complexes in 0.1 M Bu4NPF6/CH3CN, scan rate 100 mV s−1. n = 1 for [1]+ and [2]+, and n = 2 for the other complexes. The potentials are referenced to Fc0/+.
E
pa.
Ref. 40.
Ref. 41.
Ref. 42.
|
| [1]+ |
0.41 |
1.01 |
−1.80 |
0.94 |
| [2]+ |
0.60 |
1.03 |
−1.45 |
0.96 |
| [5]2+ |
0.93 |
0.96 |
−1.62 |
1.13 |
| [6]2+ |
1.12 |
0.86 |
−1.33 |
1.07 |
| [7]2+ |
0.98b |
— |
−1.57 |
0.60 |
| [8]2+ |
1.16b |
— |
−1.33 |
1.09 |
| [9]2+ |
0.86 |
0.88 |
−1.58 |
1.03 |
| [10]2+ |
1.04 |
0.87 |
−1.33 |
1.00 |
| [Ru(tpy)(bpy)(Cl)]+c |
0.42 |
|
−1.81 |
|
| [Ru(tpy)2]2+d |
0.92 |
|
−1.67 |
|
| [Ru(tfmbpy)3]2+ |
0.54 |
|
−1.22 |
|
| [Ru(dmbpy)3]2+e |
0.72 |
|
−1.83 |
|
In addition to the energy of the frontier orbitals, the electronic effects also influenced the lability of the monodentate ligand. As previously reported, the cyclic voltammogram of [1]+ showed that the Cl− ligand was replaced by CH3CN after the first ligand-based reduction.30 This was indicated by oxidation occurring at about −1.65 V during anodic scanning. In contrast, the ligand substitution was not observed in the CV scan of [2]+. The inertness of the complex was previously attributed to the localization of the extra charge in electron-deficient tfmbpy rather than in the Ru–Cl bond. Unfortunately, it was difficult to discern whether such differences in ligand lability between the dmbpy and tfmbpy complexes was present for the other monodentate ligands due to the significant overlap of the Epa of the first ligand-based reduction.
In addition to the electronic effects observed for the dmbpy and tfmbpy analogues on the LUMO, the monodentate ligands influenced the RuIII/RuII oxidation potentials, hence the HOMO energy level in these complexes. The HOMO of the chloride complexes appeared at lower potentials (∼0.5 V vs. Fc0/+) due to the π-donor character of the Cl− ligand, while the difference among the CH3CN, Hmte, and pyridine complexes was found negligible. The stabilization of the HOMO, for example, from [1]+ to [5]2+ was greater than the stabilization of the HOMO from [1]+ to [2]+, meaning that the monodentate ligands affected the HOMO more than the LUMO. Also of interest are the metal-based oxidations for the Hmte complexes [7]2+ and [8]2+, which were the only ones to show irreversible character. Such deviation was ascribed to the character of the sulphur atom whose affinity for RuIII is weaker than for RuII. Hence, the Ru–S bond was likely to dissociate upon oxidation of the metal center, and thus the Epc of the original sulphur-binding complex was not observed. Though it is possible that the oxygen atom on Hmte, which is a hard base compared to sulphur, binds to Ru, the question whether the S–O linkage isomerization occurred like in the ruthenium-sulfoxide complexes, is unclear from these results.43 When the scan rate was increased to 2000 mV s−1, the cathodic peak for the reduction of RuIII could be detected, i.e. the electrochemical RuIII/RuII couple became more reversible (Fig. S36 and S37†). In this respect, the irreversibility at standard scan rates seems to be an indication of the increased lability of the Hmte ligand when the metal center becomes electron-deficient. This result will be revisited in relation to the photosubstitution results.
Electronic absorption properties
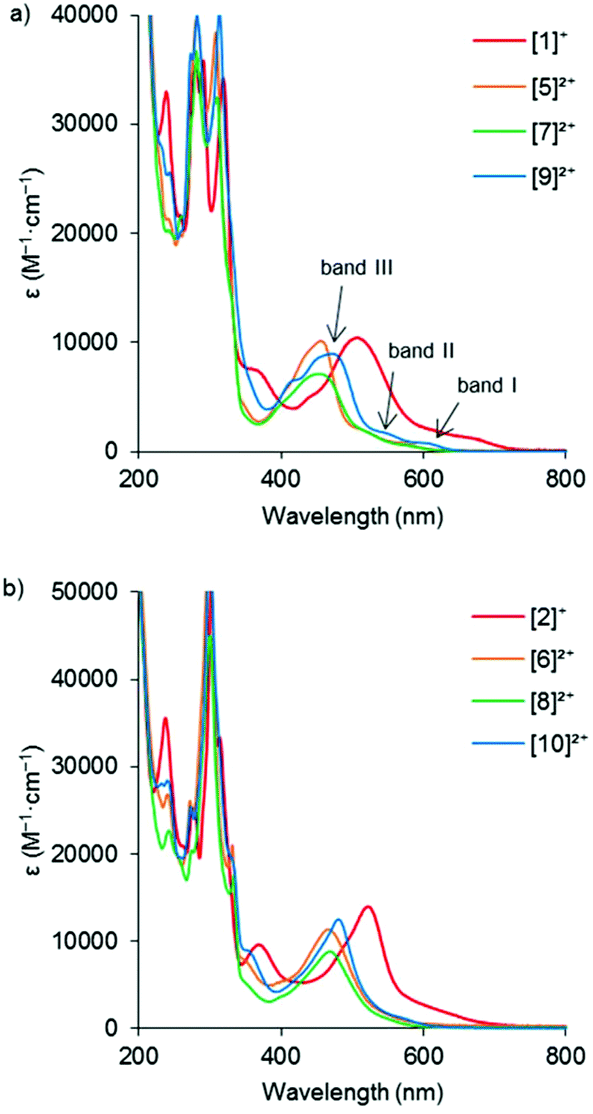
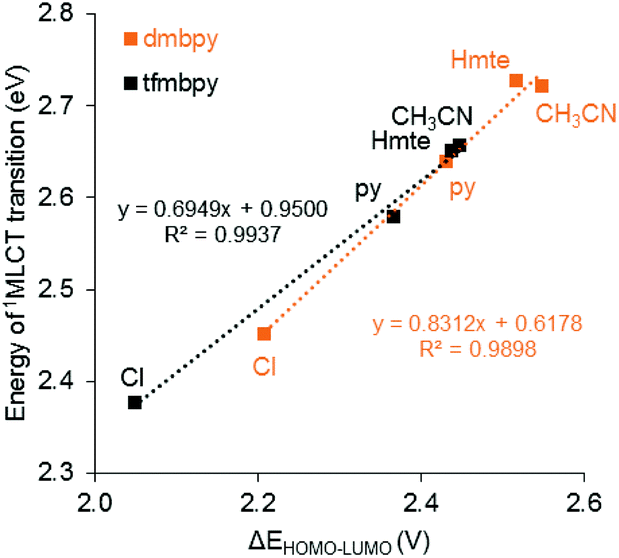
In order to investigate how the changes in the frontier orbital energy levels are reflected in the 1MLCT transitions, the electronic absorption spectra of the Ru complexes were measured in acetonitrile (except for the aqua complexes, which were measured in acetone). Each of the spectra revealed intense MLCT-based transitions between 450–530 nm, summarized in Table 3 and Fig. 5 (see Fig. S38† for the spectra of the aqua complexes). Overall, the wavelength of the 1MLCT transition (λabsmax) for the tfmbpy complexes occurred at lower energy than the dmbpy analogues. Thus, the tfmbpy ligand decreased the HOMO–LUMO gap, compared to dmbpy. The spectra of the dmbpy complexes were analogous to those of the [Ru(tpy)(bpy)(L)]n+ complexes in that the weak absorptions (band I and II) were observed at lower energy than the dominant 1MLCT transition.40 The bands were not as resolved for the tfmbpy complexes. When the energy of the dominant 1MLCT transitions, which was calculated from the λabsmax, was plotted against ΔEHOMO–LUMO, the potential difference between the E1/2 of the HOMO and LUMO (Fig. 6), the eight complexes generally fit a linear trend (R2 = 0.9845). Nevertheless, the R2 slightly improved (0.9937 and 0.9898) when two separate trends were obtained for the respective series of complexes. This divergence between the dmbpy and tfmbpy series presumably results from the different nature of the 1MLCT transition, likely due to the change in the character of the LUMO.
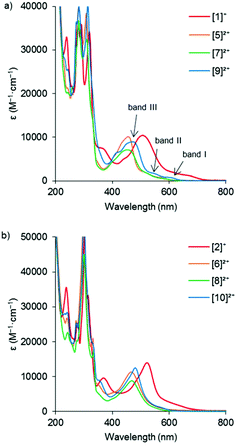 |
| | Fig. 5 Electronic absorption spectra of (a) [Ru(tpy)(dmbpy)(L)]n+, and (b) [Ru(tpy)(tfmbpy)(L)]n+. Three of the 1MLCT transitions were labelled in the spectrum of [9]2+ as an example. | |
 |
| | Fig. 6 Plot of the 1MLCT transition energy calculated from λabsmaxversus the difference between the Ru-based oxidation potential and the first ligand-based reduction potential (ΔEHOMO–LUMO) obtained from cyclic voltammetry. The orange lines represent the 4,4′-dimethyl-2,2′-bipyridine analogues and the black lines represent the 4,4′-bis(trifluoromethyl)-2,2′-bipyridine analogues. | |
Table 3 Wavelength of 1MLCT transition (λabsmax), molar absorptivity (ε (M−1 cm−1)), and quantum yields of ligand photosubstitution (ΦPS) of [Ru(tpy)(R2bpy)(L)](PF6)n complexesa
| L |
[Ru(tpy)(dmbpy)(L)](PF6)n |
[Ru(tpy)(tfmbpy)(L)](PF6)n |
|
λ
absmax (nm) |
ε (M−1 cm−1) |
Φ
PS
|
λ
absmax (nm) |
ε (M−1 cm−1) |
Φ
PS
|
|
Measured in CH3CN at 25 °C unless otherwise specified.
Measured in acetone.
|
| Cl− |
506 |
10500 |
— |
522 |
14100 |
— |
| MeCN |
456 |
10100 |
— |
467 |
10900 |
— |
| Hmte |
455 |
7000 |
0.011 |
468 |
8700 |
0.038 |
| Py |
470 |
9100 |
5.1 × 10−5 |
481 |
12400 |
6.5 × 10−5 |
| H2Ob |
488 |
9600 |
— |
494 |
6700 |
— |
Photochemistry
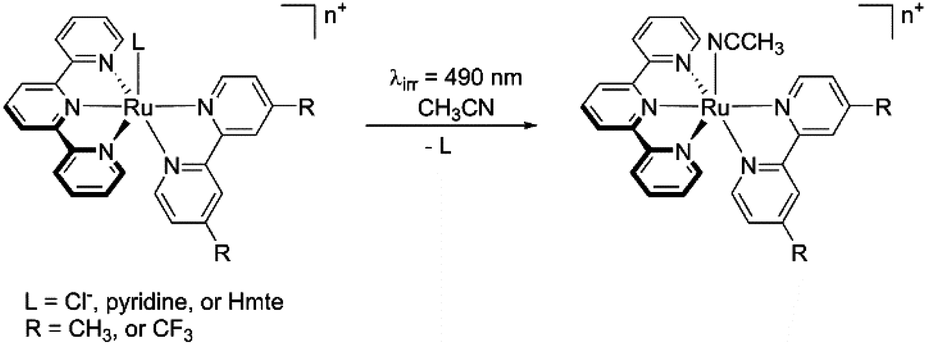
The impact of the bidentate and monodentate ligands on light-induced ligand substitution was studied in acetonitrile as illustrated in Scheme 2. The Cl−, pyridine, or Hmte analogues were irradiated in CH3CN by 490 nm light for 30 minutes at 25 °C. The reactions were monitored by electronic absorption spectroscopy, and the end product was analysed using 1H NMR spectroscopy and electrospray mass spectrometry (ES-MS).
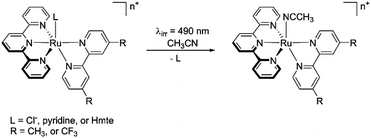 |
| | Scheme 2 Scheme of visible light-induced ligand exchange reaction. | |
The monodentate photosubstitution rate was considerably affected by the choice of coordinated monodentate ligand rather than by that of the bidentate ligand. The most unreactive analogue among the three was the chlorido complexes [1]+ and [2]+: no photoconversion was observed according to electronic absorption spectra (Fig. S39 and S40†). ES-MS at the end of the irradiation only showed the starting complex (Fig. S41 and S42†). The inertness of the Cl− ligand is attributed to the strong π-donor character of the ligand, in addition to a particularly low energy 1MLCT, which theoretically extends the energy gap between the 3MLCT and 3MC states, and thus lowers photosubstitution rates. In the case of the photoreactions of [9]2+ and [10]2+, a slow shift of the λabsmax towards higher energy wavelengths was observed upon visible light irradiation, and isosbestic points were observed at 465 nm and 467 nm on each of the electronic absorption spectra (Fig. S43 and S44†). ES-MS confirmed that the shift resulted from the replacement of the pyridyl ligand by CH3CN, resulting in complexes [5]2+ and [6]2+, respectively (Fig. S45 and S46†). Due to the low conversion of the photoreaction, the photoproducts were difficult to discern by 1H NMR spectroscopy (Fig. S47 and S48†). However, the reaction kinetics showed first-order behaviour, and the rate constant (kΦPS) of the tfmbpy complex was approximately twice larger than that of the dmbpy analogue. The quantum yields of ligand photosubstitution (ΦPS) were estimated using eqn (1),27
| | 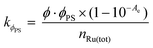 | (1) |
where
Φ is photon flux (mol s
−1),
Ae is absorbance at the irradiation wavelength, and
nRu(tot) is the total number of moles of Ru complexes (mol) (Table S2
†). For complexes [
9]
2+ and [
10]
2+ΦPS was found lower than 10
−4 for both complexes (
Table 3).
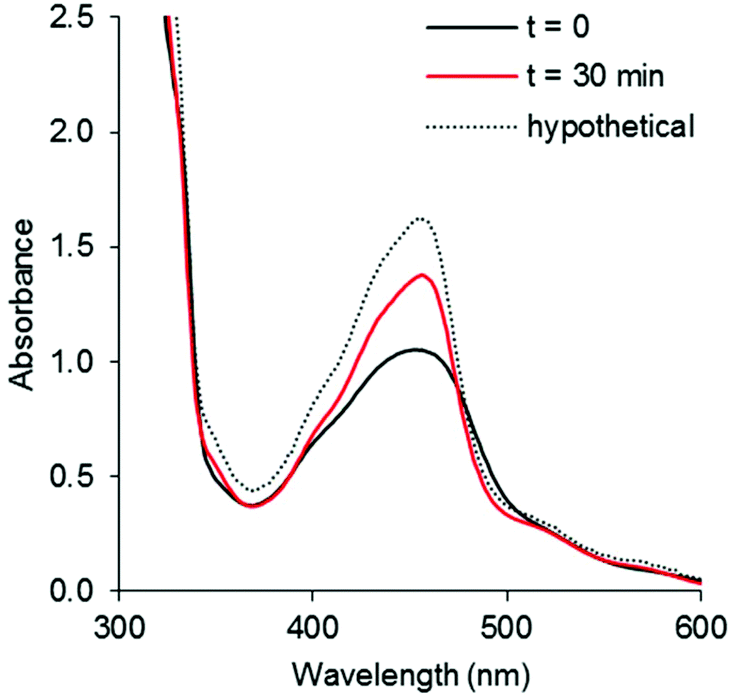
Unlike the chloride and pyridine analogues, the Hmte ligand in complexes [7]2+ and [8]2+ were readily substituted by CH3CN upon visible light irradiation. As shown in Fig. 7 and S50,† the selectivity of the photoreaction was indicated by clear isosbestic points in the electronic absorption spectra at 475 nm for [7]2+, and 497 and 266 nm for [8]2+, despite negligible shift of the absorption maxima λabsmax. Within 10 min, the spectra of [8]2+ became time-independent, suggesting quick conversion to the CH3CN complex (Fig. S49b†). In addition, ES-MS and 1H NMR spectra at the final time point supported the displacement of the Hmte ligand and the full consumption of the starting complexes (Fig. S52–S55†). The tfmbpy complex was noticeably more reactive than the dmbpy analogue (Fig. S51†), with ΦPS of 0.038 for [8]2+vs. 0.011 for [7]2+ (Table 3). These results demonstrate that the electron-withdrawing tfmbpy ligand considerably improved the efficiency of the photosubstitution reaction. Such increased reactivity was not observed in the ground states, as in complexes [7]2+–[10]2+ were all stable at room temperature over 12 h in the dark (Fig. S56–S59†).
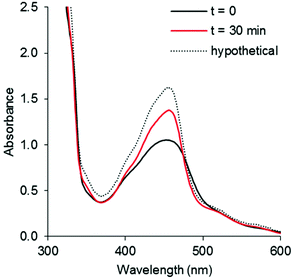 |
| | Fig. 7 UV-vis spectra of (a) [7](PF6)2 irradiated by 490 nm light (photon flux = 1.08 × 10−7 mol s−1) in CH3CN for 30 min, [Ru]tot = 1.6 × 10−4 M. Dashed line shows the spectrum expected for full conversion into [5]2+, black line shows the initial spectrum, and red line shows the spectrum obtained after 30 min irradiation. | |
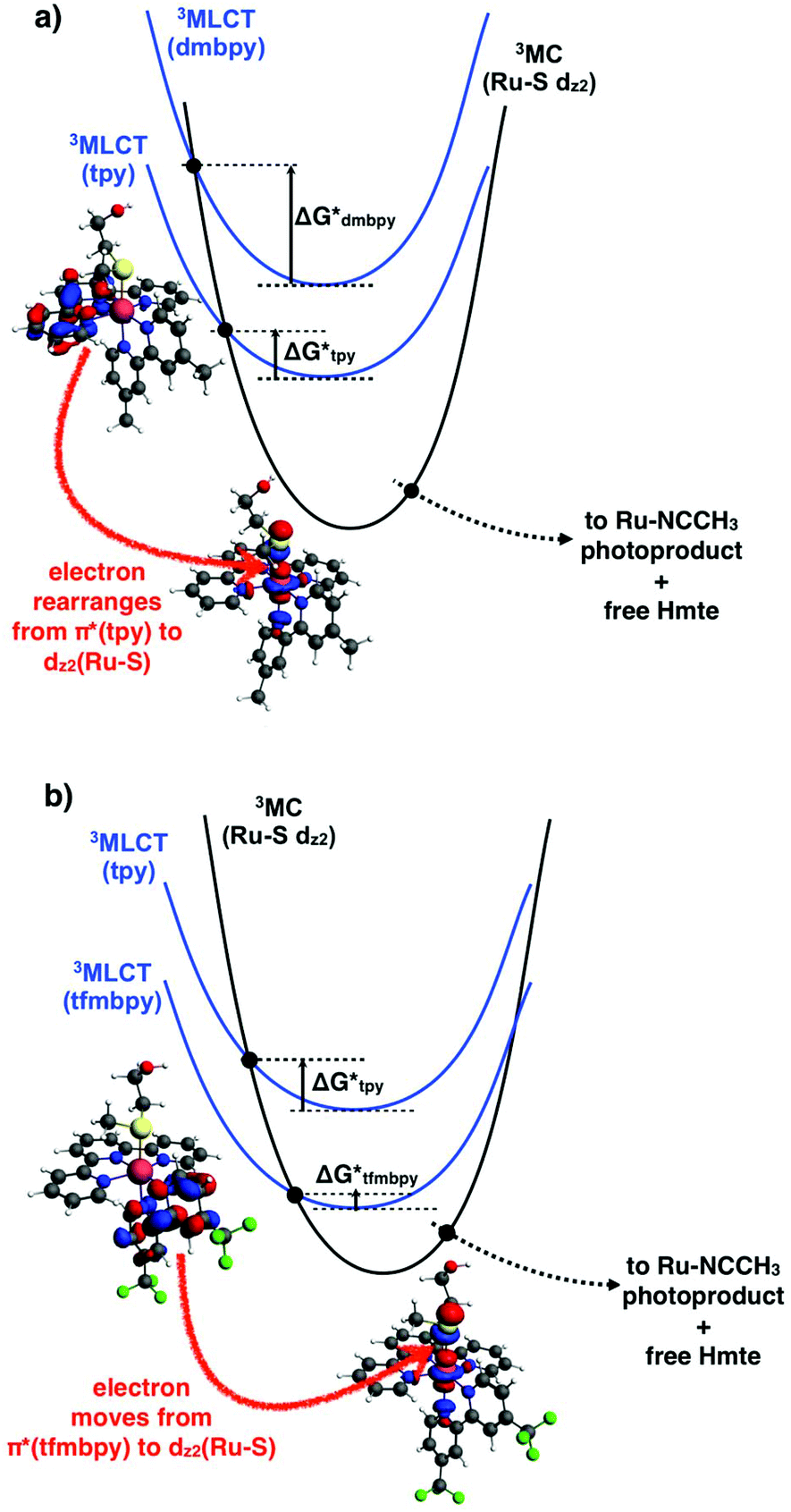
In ruthenium polypyridyl complexes thermal population of the 3MC state from the photochemically generated 3MLCT-based excited state is usually claimed to determine whether photosubstitution or 3MLCT-based phosphorescence will occur. Both processes compete with each other, so that a higher emission quantum yield is usually associated with a lower photosubstitution efficiency. Looking at the particular case of the thioether monodentate ligand (Hmte), the electron-withdrawing properties of tfmbpy are expected to lower the ligand field splitting parameter of [8]2+ and thus the energy level of its 3MC state, compared to [7]2+. If the 3MLCT remains at the same energy, complex [8]2+ would have a smaller 3MC–3MLCT gap and therefore a higher photosubstitution quantum yield, which was supported by the ΦPS for [8]2+ being four times higher than for [7]2+. This interpretation also suggests that the phosphorescence of [8]2+ should be weaker than that of [7]2+. The 3MLCT-based phosphorescence for this family of complexes is typically very weak, with emission quantum yields (Φem) in the range of 10−4 to 10−5, but it can be measured (Table S3†). Unexpectedly, we found that [8]2+ displayed a slightly more intense (Φem = 1.1 × 10−4) and red-shifted emission (699 nm) compared to [7]2+ (640 nm, Φem = 3.5 × 10−5). These facts are consistent with the electrochemical results suggesting that the LUMO is terpyridine-based for [7]2+, but tfmbpy-based for [8]2+. Due to the electron-withdrawing effect of the trifluoromethyl groups, putting an electron in the tfmbpy(π*)-based orbital must contribute to stabilizing the 3MLCT state of [8]2+, which should red shift the emission maximum for this complex and lower the photosubstitution quantum yield. In other words, [8]2+ has both a lower 3MLCT state and a lower 3MC state compared to [7]2+, which makes any comparison of the relative values of phosphorescence and photosubstitution quantum yields difficult.
On the other hand, in the 3MLCT state of [8]2+ the electron promoted in the π*(tfmbpy) orbital lies first in a plane that contains the Ru–S bond (Fig. 8b), and second trans to the Hmte ligand, which presumably elongates the Ru–S bond due to polarization effects.44 Both factors may contribute to enhancing the rate of the 3MLCT → 3MC transition, and indeed the photosubstitution quantum yield for this complex is higher than that of [7]2+ (in the same solvent). In the 3MLCT state of [7]2+ an electron is promoted to the tpy(π*)-based orbital that lies in a plane perpendicular to the Ru–S bond (Fig. 8a), which may lower the coupling with the 3MC excited state, and hence the rate of the 3MLCT → 3MC transition. The fact that for [7]2+ both the emission and the photosubstitution quantum yields are lower than for [8]2+ also suggests that non-radiative decay processes that do not go via the 3MC states may also play a role in the deactivation mechanism of the 3MLCT state of these ruthenium complexes. Overall, the electronic effects of the tfmbpy ligand seem not only to change the relative energy levels of the 3MLCT and 3MC states (Fig. 8), but they also change the geography and the geometry of the electronic density distribution in the 3MLCT excited states, and thus the electronic rearrangements necessary to perform the 3MLCT → 3MC transition.
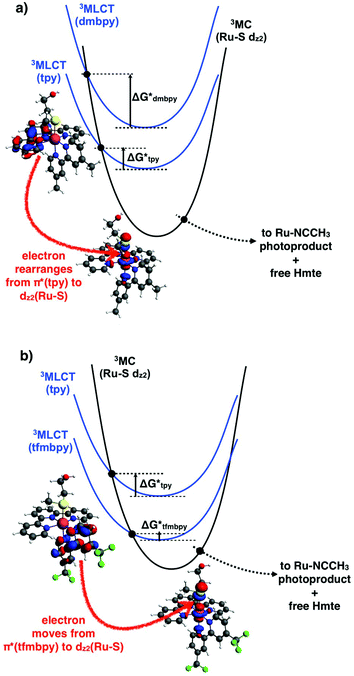 |
| | Fig. 8 Triplet potential energy curves showing interconversion of a terpyridine- or bipyridine-based 3MLCT state (blue) in (a) [7]2+ or (b) [8]2+, respectively, into the 3MC state (black) leading to Ru–S bond breakage. The HOMOs of the 3MLCT and 3MC states are also calculated at the PBE0/TZP/COSMO level and shown for [7]2+ and [8]2+. Black dots represent transition states on the triplet hypersurface. Y-Axis (potential energy), ground states, 1MLCT states, and photoproducts, are not shown for clarity. | |
In the discussion on the irreversibility of the Ru oxidation for the [Ru(tpy)(R2bpy)(Hmte)]2+ complexes, we mentioned that the degree of reversibility of the oxidation peak was a measure of ligand lability in the ground state. It seems that it is also correlated to the variation of ligand lability in the excited state. Indeed, we observe that reversible metal-based oxidation is synonymous with photochemical inertness of the monodentate ligand. For instance, the metal-based redox reaction of the chlorido complexes [1]+ and [2]+ was highly reversible (ipc/ipa ≈ 1.0), and they were found extremely unreactive upon light irradiation. In contrast, the Epc of the Hmte analogues [7]2+ and [8]2+ were not observed, i.e. the oxidation peaks were highly irreversible, but they displayed increased photosubstitution of Hmte, compared to the chloride complexes. The pyridine complexes displayed quasi-reversible behaviour and very slow photoconversion. This correlation is reasonable as the electrochemical oxidation and the photochemical transition to 3MLCT states share great similarities, as in the 3MLCT state an electron is excited from the metal-based HOMO to a ligand-based LUMO orbital, thereby formally increasing the oxidation state of the metal in the excited state to RuIII. In this respect, reversibility of the Ru-based oxidation as measured by CV is a useful way to predict the lability of the monodentate ligand upon light irradiation. This correlation mostly depends on the monodentate ligand and not to be strongly affected by the electronic property of the bipyridine ligand.
Conclusion
This work represents one of the few examples of a series of ruthenium polypyridyl complexes investigating solely the effect of the electronic properties of the bidentate spectator ligand on photosubstitution reactions. Focusing on complexes of the type [Ru(tpy)(R2bpy)(L)]n+, our work clearly demonstrates that the monodentate ligand (L) more strongly affected the Ru(dπ)-based HOMO orbitals than the ligand(π*)-based LUMO: the more π-donating Cl analogue displayed 0.5 V more positive RuIII/RuII redox couple compared to the more π-accepting ligands CH3CN, or pyridine. By contrast, the electron-withdrawing tfmbpy ligand stabilized both the HOMO and LUMO, which changed the electronic distribution in the 3MLCT state, compared to complexes based on dmbpy. The dmbpy complexes displayed typical Ru(dπ) → tpy(π*)-based MLCT states, while the tfmbpy analogues displayed Ru(dπ) → tfmbpy(π*)-based MLCT states. Such changes have counter-intuitive effects on the photochemical properties of the tfmbpy complexes. On the one hand, the 1MLCT absorption band is red-shifted by 8–16 nm and the weak 3MLCT phosphorescence is red-shifted by 52–59 nm. On the other hand, for the Hmte and pyridine adducts, which were the only ones to be photosubstitutionally reactive in acetonitrile, the tfmbpy complexes displayed up to a 4 times higher photosubstitution quantum yield, compared to the dmbpy analogue, although both the 3MLCT and 3MC states are predicted to be stabilized by the electron-withdrawing group. These results open interesting questions, such as whether it is possible to play on the electron donating and accepting properties of the spectator ligands to design ruthenium complexes that can photosubstitute ligands at higher quantum efficiencies using lower energy light.
Experimental section
General specifications
All reagents for which the synthesis is not given were commercially available from Sigma-Aldrich, J. T. Baker, Alfa Aesar, or Actu-All Chemicals, and they were used as received from the suppliers. The ruthenium precursor [Ru(tpy)Cl3] was prepared following literature method.45 Microwave reaction was performed using a Biotage Initiator. 1H, 13C, and 19F{1H} NMR spectra were recorded on a Bruker DPX-300, AV-400, AV-500, or AV-600 spectrometer, all at room temperature. Electronic absorption spectroscopy was recorded using a Varian Cary 50 UV-vis spectrometer. Electrospray mass spectra were obtained by Dionex UltiMate 3000 HPLC.
4,4′-Bis(trifluoromethyl)-2,2′-bipyridine.
Nickel(II) chloride hexahydrate (71 mg, 0.30 mmol) was added to dry THF (50 mL) and the solution was heated to 40 °C to dissolve the solid. To the pale yellow solution, 2,2′-bpy (47 mg, 0.30 mmol) was added, which turned green. Then 2-bromo-4-trifluoromethylpyridine (679 mg, 3.0 mmol), Zn (245 mg, 3.75 mmol), and LiCl (127 mg, 3.0 mmol) were added, and the reaction was heated at 60 °C for 5 h under Ar. When the reaction was complete as monitored by TLC (silica gel, hexane/DCM (6:1)), ∼5 mL of 1 M HCl was added to consume the remaining Zn, followed by the addition of ammonium hydroxide (25% NH3) until the mixture became alkaline. THF was rotary evaporated, and DCM and water were added for extraction (∼70 mL × 3). The organic phase was washed with water, extracted again, and dried with MgSO4. After filtration, the solvent was removed. The crude product was purified by silica gel column chromatography using hexane/DCM (6:1) (Rf = 0.28). The solvent was evaporated to yield a white solid (247 mg, 56%). 1H NMR (400 MHz, CDCl3) agreed with literature.461H NMR (500 MHz, CD3OD): δ 8.95 (d, J = 5.0 Hz, 1H, H6), 8.73 (s, 1H, H3), 7.77 ppm (dd, J = 5.1, 1.1 Hz, 1H, H5). 13C NMR (500 MHz, CD3OD): δ 157.3 (s, C2), 152.1 (s, C6), 140.6 (q, 2JCF = 34.0 Hz, C4), 124.4 (q, 1JCF = 272.3 Hz, C7), 121.3 (q, 3JCF = 3.5 Hz, C5), 117.7 ppm (q, 3JCF = 3.8 Hz, C3). 19F{1H} NMR (500 MHz, CD3OD): δ −66.5 ppm. MS: m/z 293.0, [L + H]+ (calc.: 293.1).
[Ru(tpy)(dmbpy)Cl]Cl ([1]Cl).
[Ru(tpy)Cl3] (500 mg, 1.1 mmol), 4,4′-dmbpy (210 mg, 1.1 mmol), Et3N (250 μL, 1.8 mmol), and LiCl (50 mg, 1.2 mmol) were added and stirred in EtOH/H2O (3:1) mixture (100 mL), and the reaction was heated at reflux for 5 h under Ar. A dark violet solution was hot filtered over Celite to remove insoluble starting material and by-products. The filtrate was rotary evaporated and purified by deactivated alumina column chromatography using 5% MeOH/DCM. The dark violet fraction was collected, and evaporated (465 mg, 69%). 1H NMR (500 MHz, CD3OD): δ 10.00 (d, J = 5.7 Hz, 1H, B6), 8.68 (s, 1H, B3), 8.63 (d, J = 8.2 Hz, 2H, T3′), 8.52 (d, J = 8.0 Hz, 2H, T6), 8.40 (s, 1H, B3′), 8.08 (t, J = 8.1 Hz, 1H, T4′), 7.89 (td, J = 7.8, 1.5 Hz, 2H, T5), 7.86 (dd, J = 5.8, 1.8 Hz, 1H, B5), 7.72 (d, J = 5.5 Hz, 2H, T3), 7.33 (ddd, J = 7.2, 5.5, 1.3 Hz, 2H, T4), 7.11 (d, J = 6.0 Hz, 1H, B6′), 6.87 (dd, J = 6.1, 1.8 Hz, 1H, B5′), 2.81 (s, 3H, B7), 2.37 ppm (s, 3H, B7′). 13C NMR (500 MHz, CD3OD): δ 160.3 (s, T2), 159.7 (s, T2′), 159.2 (s, B2), 157.2 (s, B2′), 153.2 (s, T3), 152.7 (s, B6), 151.9 (s, B6′), 150.5, (s, B4), 149.6 (s, B4′), 138.2 (s, T5), 135.0 (s, T4′), 129.0 (s, B5), 128.5 (s, T4), 128.4 (s, B5′), 125.4 (s, B3), 125.3 (s, B3′), 124.7 (s, T6), 123.6 (s, T3′), 21.4 (s, B7), 20.8 ppm (s, B7′). MS: m/z 554.1, [M − Cl]+ (calc.: 554.1). Crystal growing: vapor diffusion of diethyl ether into a solution of [1]Cl in ethanol.
[Ru(tpy)(tfmbpy)Cl]Cl ([2]Cl).
[Ru(tpy)Cl3] (370 mg, 0.85 mmol), 4,4′-tfmbpy (3) (250 mg, 0.85 mmol), Et3N (188 μL, 1.35 mmol), and LiCl (39 mg, 0.93 mmol) were added and stirred in EtOH/H2O (3:1) mixture (140 mL), and the reaction was heated at 100 °C for 4 h under Ar. A dark violet solution was hot filtered over Celite to remove insoluble starting material and by-products. The filtrate was rotary evaporated and purified by deactivated alumina column chromatography using 5% MeOH/DCM. The solvent was evaporated to yield dark violet solid. It was redissolved in minimal amount of MeOH, and precipitated with ether (417 mg, 71%). The PF6 salt of the complex, [2]PF6, was made by dissolving [2]Cl in a minimal amount of MeOH and stirring with a KPF6 saturated aqueous solution for 3 h. The solution was filtered and washed with water and ether. 1H NMR (300 MHz, CD3OD): δ 10.49 (d, J = 6.0 Hz, 1H, B6), 9.35 (s, 1H, B3), 9.08 (s, 1H, B3′), 8.71 (d, J = 8.1 Hz, 2H, T3′), 8.57 (d, J = 8.1 Hz, 2H, T6), 8.33 (dd, J = 6.0, 1.8 Hz, 1H, B5), 8.25 (t, J = 8.1 Hz, 1H, T4′), 7.97 (td, J = 7.9, 1.5 Hz, 2H, T5), 7.80 (d, J = 6.0 Hz, 1H, B6′), 7.68 (d, J = 5.2 Hz, 2H, T3), 7.44–7.26 ppm (m, 3H, B5′ + T4). 13C NMR (500 MHz, CD3OD): δ 161.4 (s, B2), 159.9 (s, T2), 158.78 (s, T2′), 158.7 (s, B2′), 155.0 (s, B6), 154.6 (s, B6′), 153.7 (s, T3), 139.0 (s, T5), 138.8 (q, 2JCF = 35.2 Hz, B4), 137.6 (q, 2JCF = 35.2 Hz, B4′), 136.8 (s, T4′), 128.7 (s, T4), 125.1 (s, T6), 125.1 (q, 1JCF = 73.2 Hz, B7), 124.2 (q, 3JCF = 3.8 Hz, B5), 124.1 (s, T3′), 123.5 (q, 3JCF = 3.7 Hz, B5′), 122.9 (q, 1J = 72.7 Hz, B7′), 122.0 (q, 3J = 3.8 Hz, B3), 121.6 ppm (q, 3J = 3.8 Hz, B3′). 19F{1H} NMR (500 MHz, CD3OD): δ −65.6, −66.1 ppm. MS: m/z 662.0, [M − Cl]+ (calc.: 662.0). Crystal growing: vapor diffusion of diethyl ether into a solution of [2](PF6) in acetone. Elemental analysis calcd (%) for C27H17Cl2F6N5Ru·1.5 H2O: C 44.76 H 2.78 N 9.67; found: C 44.89 H 2.79 N 9.67.
[Ru(tpy)(dmbpy)(OH2)](PF6)2 ([3](PF6)2).
The complex [1]Cl (25 mg, 0.0424 mmol) was dissolved in 16 mL of acetone/water (3:5), and AgPF6 (23.6 mg, 0.0933 mmol) was added. The reaction was refluxed at 80 °C for 3 h under Ar. It was filtered over Celite and the solvent was rotary evaporated. It was dissolved in minimal amount of acetone and precipitated with ether (24 mg, 69%). 1H NMR (300 MHz, (CD3)2CO): δ 9.39 (d, J = 5.7 Hz, 1H, B6), 8.78–8.65 (m, 3H, T3′ + B3), 8.57 (d, J = 8.1 Hz, 2H, T6), 8.38 (s, 1H, B3′), 8.20 (t, J = 8.1 Hz, 1H, T4′), 8.07–7.92 (m, 3H, T5 + B5), 7.73 (d, J = 5.4 Hz, 2H, T3), 7.37 (t, J = 6.7 Hz, 2H, T4), 7.14 (d, J = 5.9 Hz, 1H, B6′), 6.85 (d, J = 6.0 Hz, 1H, B5′), 2.77 (s, 3H, B7), 2.30 ppm (s, 3H, B7′). MS: m/z 268.4 [M − 2PF6]2+ (calc.: 268.6); 259.5 [M − 2PF6 − H2O]2+ (calc.: 259.6); 538.2 [Ru–O–Ru dimer + Na]2+ (calc.: 538.1).
[Ru(tpy)(tfmbpy)(OH2)](PF6)2 ([4](PF6)2).
The complex [2]Cl (30 mg, 0.0434 mmol), and AgPF6 (33 mg, 0.130 mmol) were added to 5 mL of water in a microwave vial. The vial was purged with Ar for 20 min. The mixture was heated by microwave at 140 °C for 10 min. Some of the starting complex did not react and suspended in the red solution, so the reaction was heated again at 140 °C for another 10 min. DCM and more water were added in order to extract the product. The product dissolved in the water phase and the starting complex dissolved in organic phase. Approximately 170 mL total was used for each phase for extraction. The water phase was filtered over Celite to filter away insoluble by-products. The water was rotary evaporated to yield a dark red solid (15 mg, 36%). 1H NMR (300 MHz, (CD3)2CO): δ 10.15 (d, J = 5.9 Hz, 1H, B6), 9.54 (s, 1H, B3), 9.22 (s, 1H, B3′), 8.91 (d, J = 8.2 Hz, 2H, T3′), 8.75 (d, J = 8.0 Hz, 2H, T6), 8.54 (d, J = 6.0 Hz, 1H, B5), 8.48 (t, J = 8.1 Hz, 1H, T4′), 8.15 (t, J = 7.9 Hz, 2H, T5), 8.11 (d, J = 6.1 Hz, 1H, B6′), 8.06 (dd, J = 5.5 Hz, 2H, T3), 7.53–7.42 ppm (m, 3H, B5′ + T4), 6.25 (s, 2H, OH2). MS: m/z 322.3 [M − 2PF6]2+ (calc.: 322.5); 313.2 [M − 2PF6 − H2O]2+ (calc.: 313.5); 646.1 [Ru–O–Ru dimer + Na]2+ (calc.: 646.1).
[Ru(tpy)(dmbpy)(NCCH3)](PF6)2 ([5](PF6)2).
The complex was synthesized using a modified literature procedure.30 Complex [1]Cl (49 mg, 0.083 mmol) was dissolved in 30 mL of acetonitrile, and the reaction was refluxed at 82 °C under Ar. According to TLC (aluminum oxide, 5% MeOH/DCM), no product was observed after 18 h. To the mixture, 2.2 equivalents (46 mg, 0.182 mmol) of AgPF6 was added and refluxed for an additional 3 h, resulting in a yellow compound on TLC along with starting material. To assist conversion to the aqua, 20 mL of distilled water was added to the reaction and refluxed for an additional 18 hours, which resulted in an orange suspension. The suspension was filtered over Celite, and washed with acetonitrile. The solvent was rotary evaporated. The solid was dissolved in minimal amount of acetonitrile, then precipitated with ether, and filtered (47 mg, 67%). 1H NMR spectrum in CD3CN corresponded to literature.301H NMR (300 MHz, (CD3)2CO): δ 9.69 (d, J = 5.7 Hz, 1H, B6), 8.85 (d, J = 8.1 Hz, 2H, T3′), 8.79 (s, 1H, B3), 8.72 (d, J = 8.1 Hz, 2H, T6), 8.54 (s, 1H, B3′), 8.43 (t, J = 8.1 Hz, 1H, T4′), 8.14 (td, J = 7.9, 1.5 Hz, 2H, T5), 7.97 (dd, J = 5.4, 1.5 Hz, 2H, T3), 7.92 (dd, J = 5.9, 1.7 Hz, 1H, B5), 7.52 (ddd, J = 7.4, 5.6, 1.3 Hz, 2H, T4), 7.39 (d, J = 5.8 Hz, 1H, B6′), 7.06 (d, J = 5.6 Hz, 1H, B5′), 2.79 (s, 3H, B7), 2.40 (s, 3H, B7′), 2.30 ppm (s, 3H, CH3-CN). MS: m/z 279.8 [M − 2PF6]2+ (calc.: 280.1); 705.2 [M − PF6]+ (calc.: 705.1). Crystal growing: vapor diffusion of toluene into a solution of [5](PF6)2 in acetonitrile.
[Ru(tpy)(tfmbpy)(NCCH3)](PF6)2 ([6](PF6)2).
The complex [2]Cl (25 mg, 0.036 mmol) was dissolved in 1:1 mixture of acetonitrile and water (50 mL), and AgPF6 (20 mg, 0.079 mmol) was added. The reaction was refluxed at 82 °C under Ar for 17 h. The orange solution was filtered over Celite, and the solvent was rotary evaporated. The solid was dissolved in minimal amount of acetonitrile, then precipitated with ether, and filtered (25 mg, 73%). 1H NMR (300 MHz, (CD3)2CO): δ 10.24 (d, J = 5.9 Hz, 1H, B6), 9.51 (s, 1H, B3), 9.27 (s, 1H, B3′), 8.90 (d, J = 8.1 Hz, 2H, T3′), 8.73 (d, J = 8.1 Hz, 2H, T6), 8.53 (t, J = 8.1 Hz, 1H, T4′), 8.39 (dd, J = 6.0, 1.8 Hz, 1H, B5), 8.17 (td, J = 7.9, 1.5 Hz, 2H, T5), 8.07 (d, J = 6.0 Hz, 1H, B6′), 8.03 (dd, J = 5.5, 1.4 Hz, 2H, T3), 7.57 (d, J = 6.0, 1.9 Hz, 1H, B5′), 7.49 (ddd, J = 7.2, 5.5, 1.3 Hz, 2H, T4), 2.33 ppm (s, 3H, CH3-CN). MS: m/z 333.7 [M − 2PF6]2+ (calc.: 334.0); 813.2 [M − PF6]+ (calc.: 813.0). Crystal growing: vapor diffusion of diethyl ether into a solution of [6](PF6)2 in acetonitrile. Elemental analysis calcd (%) for C29H20F18N6P2Ru·0.5 H2O·0.5 C3H6O·0.5 CH3CN: C 37.24 H 2.53 N 8.96; found: C 37.16 H 2.55 N 8.97.
[Ru(tpy)(dmbpy)(Hmte)](PF6)2 ([7](PF6)2).
The complex [3](PF6)2 (28 mg, 0.034 mmol) was dissolved in 16 mL of mixture of acetone and water (3:5), and 2-(methylthio)ethanol (30 μL, 0.34 mmol) was added. The reaction was refluxed at 80 °C under Ar for 21 h. The orange solution was rotary evaporated, redissolved in minimal amount of acetone, and then precipitated with ether. It was filtered to yield orange solid (19 mg, 62%). 1H NMR (300 MHz, (CD3)2CO): δ 9.72 (d, J = 5.8 Hz, 1H, B6), 8.89 (d, J = 8.2 Hz, 2H, T3′), 8.80 (s, 1H, B3), 8.74 (d, J = 8.1 Hz, 2H, T6), 8.57 (s, 1H, B3′), 8.47 (t, J = 8.1 Hz, 1H, T4′), 8.17 (td, J = 7.9, 1.5 Hz, 2H, T5), 8.01 (d, J = 5.0 Hz, 2H, T3), 7.96 (dd, J = 5.9, 1.7 Hz, 1H, B5), 7.54 (ddd, J = 7.4, 5.6, 1.3 Hz, 2H, T4), 7.33 (d, J = 5.8 Hz, 1H, B6′), 7.11 (dd, J = 5.5, 1.6 Hz, 1H, B5′), 3.53 (t, 5.7 Hz, 2H, –S–CH2–CH2–), 2.78 (s, 3H, B7), 2.40 (s, 3H, B7′), 1.97 (t, 5.7 Hz, 2H, –S–CH2–CH2–), 1.50 ppm (s, 3H, CH3–S–). MS: m/z 305.3 [M − 2PF6]2+ (calc.: 305.6); 756.2 [M − PF6]+ (calc.: 756.1). Crystal growing: vapor diffusion of diethyl ether into a solution of [7](PF6)2 in acetone. Elemental analysis calcd (%) for C30H31F12N5OP2RuS·2 H2O·0.75 C3H6O·0.25 PF6: C 38.11 H 3.92 N 6.89; found: C 38.19 H 3.92 N 6.97.
[Ru(tpy)(tfmbpy)(Hmte)](PF6)2 ([8](PF6)2).
The complex [2]Cl (27 mg, 0.0383 mmol) was dissolved in 16 mL of mixture of acetone and water (3:5), and 2-(methylthio)ethanol (33 μL, 0.38 mmol), and AgPF6 (21 mg, 0.084 mmol) were added. The reaction was refluxed at 80 °C for 22 h. The reaction mixture was filtered over Celite and the solvent was rotary evaporated. According to 1H NMR, there seemed to be more than one species. Therefore, the crude product was dissolved in 16 mL of acetone/water (3:5), the same amount of AgPF6 and 100 μL of 2-(methylthio)ethanol were added, and the reaction was refluxed at 80 °C under Ar for 24 h. The orange solution was again filtered over Celite, and the solvent was removed (32 mg, 82%). 1H NMR (400 MHz, (CD3)2CO): δ 10.36 (d, J = 5.9 Hz, 1H, B6), 9.53 (s, 1H, B3), 9.33 (s, 1H, B3′), 8.99 (d, J = 8.2 Hz, 2H, T3′), 8.81 (dd, J = 8.1, 1.2 Hz, 2H, T6), 8.57 (t, J = 8.1 Hz, 1H, T4′), 8.52 (dd, J = 6.1, 1.9 Hz, 1H, B5), 8.19 (td, J = 7.9, 1.5 Hz, 2H, T5), 8.10 (dd, J = 5.5, 1.6 Hz, 2H, T3), 7.99 (d, J = 5.9 Hz, 1H, B6′), 7.63 (dd, J = 5.9, 1.9 Hz, 1H, B5′), 7.50 (ddd, J = 7.7, 5.5, 1.3 Hz, 2H, T4), 4.83 (t, J = 5.5 Hz, 1H, –OH), 3.53 (td, J = 5.5 Hz, 2H, –CH2–OH), 2.10 (t, J = 5.7 Hz, 2H, –S–CH2–), 1.52 ppm (s, 3H, CH3–S–). MS: m/z 359.1 [M − 2PF6]2+ (calc.: 359.5); 864.2 [M − PF6]+ (calc.: 864.0). Crystal growing: vapor diffusion of diethyl ether into a solution of [8](PF6)2 in ethanol. Elemental analysis calcd (%) for C30H25F18N5OP2RuS: C 35.73 H 2.50 N 6.94; found: C 35.78 H 2.52 N 6.83.
[Ru(tpy)(dmbpy)(py)](PF6)2 ([9](PF6)2).
The complex [3](PF6)2 (41 mg, 0.05 mmol) was dissolved in 20 mL of ethanol/water (1:1), and excess amount of pyridine (1 mL) was added. The reaction was refluxed at 80 °C under Ar for 24 h. Ethanol was rotary evaporated, and then saturated KPF6 aqueous solution was added. After the mixture was stirred, the dark orange precipitate was filtered and washed with water and ether. According to 1H NMR in (CD3)2CO, the product still had pyridine. The crude product was dissolved in minimal amount of acetone, precipitated with ether, and filtered (23 mg, 51%). 1H NMR spectrum in CD3CN corresponded to literature.471H NMR (300 MHz, (CD3)2CO): δ 8.83 (m, 3H, B3 + T3′), 8.76 (d, J = 5.9 Hz, 1H, B6), 8.73 (d, J = 8.1 Hz, 2H, T6), 8.58 (s, 1H, B3′), 8.34 (t, J = 8.1 Hz, 1H, T4′), 8.17 (td, J = 7.9, 1.5 Hz, 2H, T5), 8.12 (d, J = 5.4, 1.6 Hz, 2H, T3), 8.02 (dd, J = 6.5, 1.6 Hz, 2H, P2), 7.89 (tt, J = 7.7, 1.6 Hz, 1H, P4), 7.84 (d, J = 5.8 Hz, 1H, B5), 7.59 (ddd, J = 7.4, 5.6, 1.3 Hz, 2H, T4), 7.41 (d, J = 5.8 Hz, 1H, B6′), 7.34 (t, J = 6.5 Hz, 2H, P3), 7.06 (dd, J = 5.5, 1.6 Hz, 1H, B5′), 2.77 (s, 3H, B7), 2.41 ppm (s, 3H, B7′). MS: m/z 298.9 [M − 2PF6]2+ (calc.: 299.1); 743.2 [M − PF6]+ (calc.: 743.1). Crystal growing: vapor diffusion of toluene into a solution of [9](PF6)2 in acetone.
[Ru(tpy)(tfmbpy)(py)](PF6)2 ([10](PF6)2).
The complex [4](PF6)2 (20 mg, 0.022 mmol) was dissolved in 20 mL of ethanol/water (1:1), and excess amount of pyridine (1 mL) was added. The reaction was refluxed at 80 °C under Ar for 24 h. After the solvent was rotary evaporated, the orange solid was dissolved in minimal amount of acetone, precipitated with ether, and filtered (19.5 mg, 90%). 1H NMR (300 MHz, (CD3)2CO): δ 9.60 (s, 1H, B3), 9.36 (s, 1H, B3′), 9.34 (d, J = 4.8 Hz, 1H, B6), 8.96 (d, J = 8.1 Hz, 2H, T3′), 8.84 (d, J = 8.3 Hz, 2H, T6), 8.47 (t, J = 8.1 Hz, 1H, T4′), 8.35 (d, J = 6.1 Hz, 1H, B5), 8.27–8.17 (m, 4H, T5 + T3), 8.11 (d, J = 5.8 Hz, 3H, B6′ + P2), 7.96 (t, J = 7.6 Hz, 1H, P4), 7.64–7.50 (m, 3H, B5′ + T4), 7.41 ppm (t, J = 6.7 Hz, 2H, P3). MS: m/z 352.8 [M − 2PF6]2+ (calc.: 353.0). Crystal growing: vapor diffusion of toluene into a solution of [10](PF6)2 in acetone. Elemental analysis calcd (%) for C32H22F12N6P2Ru·1.5 H2O·0.25 C3H6O·0.25 PF6·0.25 C5H5N: C 39.34 H 2.75 N 8.51; found: C 39.40 H 2.72 N 8.54.
[Ru(tfmbpy)3]Cl2.
The complex was synthesized using literature method.481H NMR (400 MHz, (CD3)2CO): δ 9.44 (d, J = 1.9 Hz, 1H, H3), 8.54 (d, J = 5.9 Hz, 1H, H6), 7.89 ppm (dd, J = 5.9, 1.8 Hz, 1H, H5). MS: m/z 489.4 [M − 2Cl]2+ (calc.: 489.0); 1013.0 [M − Cl]+ (calc.: 1013.0).
Electrochemistry
Cyclic voltammetry experiments were performed using a three-electrode cell setup, which included a platinum working electrode, a silver wire pseudo reference electrode, and platinum wire auxiliary electrode. The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in CH3CN. The final complex concentration was 1 mM. Prior to measurement, the solvent was purged with argon and was measured at room temperature using an Autolab PGSTAT10 potentiostat and GPES 4.9 program by Eco Chemie. Each CV experiment was referenced to the ferrocene oxidation potential.
Emission quantum yields
The quantum yield of 3MLCT emission was determined in a custom-built setup. All optical parts were connected using optical fibers from Avantes (Apeldoorn, The Netherlands), with a diameter of 200–600 μm. The samples were measured in a 1 cm path length QS cuvette. For each measurement, 3 mL of sample consisting of the compound in acetonitrile (λ450absnm = 0.1), was placed in a CUV-UV/VIS-TC temperature-controlled cuvette holder from Avantes. The sample was deoxygenated using nitrogen gas for 10 min while also equilibrating to 293 K. Emission spectroscopy was performed with a 450 nm fiber-coupled laser (Laser system LRD-0450 from Laserglow, Toronto, Canada), which was set to 50 mW at the cuvette (4 mm beam diameter; 0.4 W cm−2) at a 90° angle with respect to the spectrometer. The excitation power was measured using a S310C thermal sensor connected to a PM100USB power meter (Thorlabs). The emission spectrum was visualized from 500–800 nm with an Avantes 2048L StarLine UV-Vis spectrometer as detector. No difference in the electronic absorption spectrum (measured using a Varian Cary 50 UV-vis spectrometer) was found due to exposure to the 450 nm laser, showing that emission is from the starting compound. All emission spectra were recorded using Avasoft software from Avantes and further processed using Microsoft Office Excel 2010 and Origin Pro software.
The quantum yield of emission from the 3MLCT excited state was calculated using the relative method with [Ru(bpy)3]Cl2 as the standard (0.06 in deoxygenated CH3CN), according to eqn (2):
| |  | (2) |
where
Φ3MLCT is the quantum yield of
3MLCT emission,
A450 is the absorbance at 450 nm (generally 0.1 for a 1 cm path length),
E is the integrated area of the
3MLCT emission band, and sample and std denote the sample and standard ([Ru(bpy)
3]Cl
2), respectively.
Photochemistry
Photosubstitution experiments were performed using the setup described in literature,27 where a 490 nm LED lamp was fixed on top of a 1 cm quartz cuvette, and the temperature was controlled using a Peltier controller to maintain 25 °C during the 30 min irradiation. The beginning concentration of the solutions was determined such that the absorbance at the irradiation wavelength (λirr = 490 nm) was approximately 0.5. The photon flux of the 490 nm LED lamp was determined to be approximately 1.0 × 10−7 Einstein s−1 by ferrioxalate actinometry.49 The specific photon flux and reaction conditions are referenced in each experiment. The concentrations were calculated using the two-wavelength method.27
Acknowledgements
The European Research Council is kindly acknowledged for a Starting Grant to S. B. This work was also supported by the Dutch Organization for Scientific Research (NWO) via a VIDI grant to S. B. Prof. E. Bouwman is kindly acknowledged for support and input.
References
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- G. Li, W. M. Ward and G. J. Meyer, J. Am. Chem. Soc., 2015, 137, 8321–8323 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- P. C. Ford and S. Wecksler, Coord. Chem. Rev., 2005, 249, 1382–1395 CrossRef CAS.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- B. M. Blunden and M. H. Stenzel, J. Chem. Technol. Biotechnol., 2015, 90, 1177–1195 CrossRef CAS.
- R. N. Garner, J. C. Gallucci, K. R. Dunbar and C. Turro, Inorg. Chem., 2011, 50, 9213–9215 CrossRef CAS PubMed.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603–11613 CrossRef CAS PubMed.
- A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030–10032 CrossRef CAS PubMed.
- B. Siewert, V. H. S. van Rixel, E. J. van Rooden, S. L. Hopkins, M. J. B. Moester, F. Ariese, M. A. Siegler and S. Bonnet, Chem. – Eur. J., 2016, 22, 10960–10968 CrossRef CAS PubMed.
- L. N. Lameijer, S. L. Hopkins, T. G. Brevé, S. H. C. Askes and S. Bonnet, Chem. – Eur. J., 2016, 22, 18484–18491 CrossRef CAS PubMed.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS.
- S. Bonnet and J.-P. Collin, Chem. Soc. Rev., 2008, 37, 1207–1217 RSC.
- P. A. Scattergood, U. Khushnood, A. Tariq, D. J. Cooke, C. R. Rice and P. I. P. Elliott, Inorg. Chem., 2016, 55, 7787–7796 CrossRef CAS PubMed.
- Q. Sun, B. Dereka, E. Vauthey, L. M. Lawson Daku and A. Hauser, Chem. Sci., 2017, 8, 223–230 RSC.
- Q. Sun, S. Mosquera-Vazquez, L. M. Lawson Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. M. Lawson Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 282–283, 87–99 CrossRef CAS.
- C. E. Welby, G. K. Armitage, H. Bartley, A. Wilkinson, A. Sinopoli, B. S. Uppal, C. R. Rice and P. I. P. Elliott, Chem. – Eur. J., 2014, 20, 8467–8476 CrossRef CAS PubMed.
- C. E. Welby, C. R. Rice and P. I. P. Elliott, Angew. Chem., Int. Ed., 2013, 52, 10826–10829 CrossRef CAS PubMed.
- J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2017, 454, 7–20 CrossRef CAS PubMed.
- A. J. Göttle, F. Alary, M. Boggio-Pasqua, I. M. Dixon, J.-L. Heully, A. Bahreman, S. H. C. Askes and S. Bonnet, Inorg. Chem., 2016, 55, 4448–4456 CrossRef PubMed.
- S. Bonnet, J.-P. Collin, J.-P. Sauvage and E. Schofield, Inorg. Chem., 2004, 43, 8346–8354 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani and C. Turro, Chem. Commun., 2015, 51, 8777–8780 RSC.
- J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed.
- B. A. Albani, T. Whittemore, C. B. Durr and C. Turro, Photochem. Photobiol., 2015, 91, 616–623 CrossRef CAS PubMed.
- A. Bahreman, B. Limburg, M. A. Siegler, E. Bouwman and S. Bonnet, Inorg. Chem., 2013, 52, 9456–9469 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- R. N. Garner, L. E. Joyce and C. Turro, Inorg. Chem., 2011, 50, 4384–4391 CrossRef CAS PubMed.
- T. A. White, S. Maji and S. Ott, Dalton Trans., 2014, 43, 15028–15037 RSC.
- K. Mikami, Y. Itoh and M. Yamanaka, Chem. Rev., 2004, 104, 1–16 CrossRef CAS PubMed.
- L.-Y. Liao, X.-R. Kong and X.-F. Duan, J. Org. Chem., 2014, 79, 777–782 CrossRef CAS PubMed.
- K. S. Chan and A. K. S. Tse, Synth. Commun., 1993, 23, 1929–1934 CrossRef CAS.
- J. Broomhead and F. Dwyer, Aust. J. Chem., 1961, 14, 250–252 CAS.
- N. A. Eckert, E. M. Bones, R. J. Lachicotte and P. L. Holland, Inorg. Chem., 2003, 42, 1720–1725 CrossRef CAS PubMed.
- P. A. Adcock, F. R. Keene, R. S. Smythe and M. R. Snow, Inorg. Chem., 1984, 23, 2336–2343 CrossRef CAS.
- S. C. Rasmussen, S. E. Ronco, D. A. Mlsna, M. A. Billadeau, W. T. Pennington, J. W. Kolis and J. D. Petersen, Inorg. Chem., 1995, 34, 821–829 CrossRef CAS.
- E. Sondaz, A. Gourdon, J.-P. Launay and J. Bonvoisin, Inorg. Chim. Acta, 2001, 316, 79–88 CrossRef CAS.
- B. A. Johnson, H. Agarwala, T. A. White, E. Mijangos, S. Maji and S. Ott, Chem. – Eur. J., 2016, 22, 14870–14880 CrossRef CAS PubMed.
- C.-N. Tsai, M. M. Allard, R. L. Lord, D.-W. Luo, Y.-J. Chen, H. B. Schlegel and J. F. Endicott, Inorg. Chem., 2011, 50, 11965–11977 CrossRef CAS PubMed.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. Cargill Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- Y. Kawanishi, N. Kitamura and S. Tazuke, Inorg. Chem., 1989, 28, 2968–2975 CrossRef CAS.
- A. W. King, L. Wang and J. J. Rack, Acc. Chem. Res., 2015, 48, 1115–1122 CrossRef CAS PubMed.
- B. J. Coe and S. J. Glenwright, Coord. Chem. Rev., 2000, 203, 5–80 CrossRef CAS.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404–1407 CrossRef CAS.
- R. M. O'Donnell, R. N. Sampaio, G. Li, P. G. Johansson, C. L. Ward and G. J. Meyer, J. Am. Chem. Soc., 2016, 138, 3891–3903 CrossRef PubMed.
- N. C. Fletcher and F. R. Keene, J. Chem. Soc., Dalton Trans., 1998, 2293–2302 RSC.
- M. Furue, K. Maruyama, T. Oguni, M. Naiki and M. Kamachi, Inorg. Chem., 1992, 31, 3792–3795 CrossRef CAS.
-
J. G. Calvert and J. N. Pitts, in Photochemistry, John Wiley and Sons, New York, 1966, pp. 780–786 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ligand synthesis and characterization, complex/photoproduct characterization and crystal structures. CCDC 1544943–1544950. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01540b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Maxime A.
Siegler
b and
Sylvestre
Bonnet
a,
Maxime A.
Siegler
b and
Sylvestre
Bonnet