
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable AuIII complexes with four N-heterocyclic carbene groups can be prepared in high yield directly from KAuCl4†
Ahmed H.
Mageed
ab,
Brian W.
Skelton
ac and
Murray V.
Baker
 *a
*a
aSchool of Molecular Sciences M310, The University of Western Australia, 35 Stirling Highway, Perth, WA 6009, Australia. E-mail: murray.baker@uwa.edu.au
bDepartment of Chemistry, Faculty of Science, The University of Kufa, P.O. Box 21, Najaf 54001, Iraq
cCentre for Microscopy, Characterisation and Analysis M310, The University of Western Australia, Perth, WA 6009, Australia
First published on 2nd June 2017
Abstract
Gold(III) N-heterocyclic carbene (NHC) complexes of form [Au(NHC)4Cl2]Cl were synthesized by reaction of KAuCl4 with bis- and tetrakis(imidazolium) salts in the presence of a mild base. Treatment of these complexes with KPF6 afforded four-coordinate AuIII complexes of form [Au(NHC)4](PF6)3. X-Ray crystallography showed the [AuIII(NHC)4]3+ cations in the hexafluorophosphate salts to have a square planar Au(NHC)4 moiety [Au⋯CNHC 2.024(4)–2.082(7) Å]. In the [AuIII(NHC)4Cl2]+ cations in the chloride salts, coordination about Au was tetragonally-distorted octahedral, the axial Au–Cl bonds being substantially longer [Au⋯Cl 3.148(2)–3.693(1) Å] than the equatorial Au–CNHC bonds [Au⋯C 2.024(4)–2.082(7) Å]. NMR and conductance studies suggested that the structures of the complexes seen in the solid state persisted in DMSO solution, except in one case where a chlorido ligand dissociated from [AuIII(NHC)4Cl2]+ to form [AuIII(NHC)4Cl]2+. The AuIII(NHC)4 unit was surprisingly robust. An AuIII complex was found to undergo H/D exchange reactions in D2O solution at 100 °C with no signs of decomposition detectable by 1H NMR spectroscopy. 1H NMR studies showed that various complexes containing AuIII(NHC)4 moieties underwent little or no decomposition when heated at 120 °C in DMSO-d6 for extended periods.
Introduction
Interest in gold chemistry has been growing, in part due to the promise of applications in fields of medicine, nanotechnology and catalysis.1–5 For example, many gold compounds have been found to inhibit cancer cell growth,1–3 and so gold complexes are being investigated as potential replacements for or alternatives to cisplatin in cancer chemotherapy. In this context, there has been increased research activity on the synthesis of new gold complexes involving N-heterocyclic carbene (NHC) complexes. These studies have mostly focused on mononuclear AuI–NHC complexes, with considerably less attention being paid to AuIII–NHC complexes.6,7The majority of Au complexes known to date are complexes of AuI. Synthesis of AuIII complexes is often problematic because of the highly oxidizing nature of AuIII. Attempts to synthesize AuIII complexes from AuIII salts frequently results in side-reactions during which reduction of AuIII occurs.8–12 Not surprisingly, the most common synthetic pathway to AuIII–NHC complexes involves oxidative addition of halogens or halogen equivalents to AuI–NHC precursor complexes.8,9,13–31 Other procedures, such as carbene transfer between AgI and AuIII,8 or reaction of imidazolium ions with AuIII in the presence of base,9,10 are usually complicated by reduction of AuIII to AuI. However, Lu et al. synthesized the AuIII–tetracarbene complex 1 by treatment of HAuCl4 with an AgI–NHC complex.32 Most AuIII–NHC complexes reported to date have square-planar AuIII centres with one or two NHC ligands, 1 being the only Au(NHC)4 motif reported to date. While the AuIII centre in 1 is square planar, many complexes of AuIII with other ligands contain square pyramidal and octahedral Au centres (e.g. [Au(phen)(CN)2Br] and [Au(diars)2I2]+), albeit strongly distorted, the axial metal–ligand bonds being longer than the equatorial ones.33,34
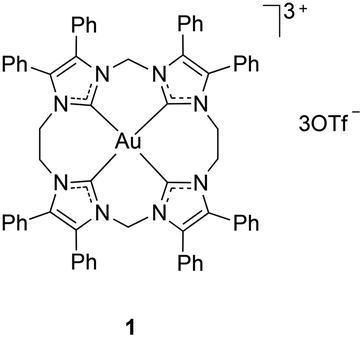
Results and discussion
Synthesis of AuIII–NHC complexes
AuIII–NHC chlorido complexes [Au(L1)2Cl2]Cl 2, [Au(L2)2Cl2]Cl 3, and [AuL3Cl2]Cl 4 (Scheme 1) were synthesized by heating KAuCl4 with the appropriate imidazolium salt and LiOAc in DMF, following procedures we used previously to synthesize AuI–NHC cyclophane complexes.1 The complexes [Au(L1)2Cl2]Cl 2, [Au(L2)2Cl2]Cl 3 and [AuL3Cl2]Cl 4 precipitated from the mixture and were purified by recrystallisation; yields after recrystallisation were 81, 79, and 48% respectively. These yields are surprisingly high, as previous attempts so synthesize AuIII–NHC complexes by reaction of imidazolium salts with AuIII salts in the presence of base were accompanied by a substantial amount of reduction of AuIII to AuI,9,10 the problem being exacerbated by long reaction times.10 We attribute these high yields in part to remarkable stability of the AuIII–tetrakis(NHC) complexes (see NMR studies, below). Interestingly, the [Au(NHC)4Cl2]Cl structure seems to be the preferred product even when the stoichiometry is unfavourable—when we allowed L1·2HCl and KAuCl4 to react in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio in the presence of excess LiOAc, the only product obtained was [Au(L1)2Cl2]Cl 2 (86% yield based on L1·2HCl). The structure of the starting imidazolium salt (i.e., whether it contains a mono-, bis-, or tetrakis(imidazolium) ion) is also an important factor in determining the outcome of the reactions. The use bis- or tetrakis(imidazolium) salts as precursors makes formation of the AuIII(NHC)4 moiety kinetically favourable compared to competing processes in which Au(III) participates in redox reactions. When we attempted syntheses using simple monoimidazolium salts (1,3-dimethylimidazolium iodide and 1,3-diisopropylimidazolium bromide), we did not obtain any AuIII(NHC)4-type complex, instead obtaining small amounts of [(NHC)AuX3], AuI–NHC complexes, and the urea-type product of oxidation of the imidazolium ion at C2. Other researchers have also reported poor results from attempts to synthesize AuIII complexes directly from monoimidazolium ions and AuIII salts in the presence of base.9,10
:1 mole ratio in the presence of excess LiOAc, the only product obtained was [Au(L1)2Cl2]Cl 2 (86% yield based on L1·2HCl). The structure of the starting imidazolium salt (i.e., whether it contains a mono-, bis-, or tetrakis(imidazolium) ion) is also an important factor in determining the outcome of the reactions. The use bis- or tetrakis(imidazolium) salts as precursors makes formation of the AuIII(NHC)4 moiety kinetically favourable compared to competing processes in which Au(III) participates in redox reactions. When we attempted syntheses using simple monoimidazolium salts (1,3-dimethylimidazolium iodide and 1,3-diisopropylimidazolium bromide), we did not obtain any AuIII(NHC)4-type complex, instead obtaining small amounts of [(NHC)AuX3], AuI–NHC complexes, and the urea-type product of oxidation of the imidazolium ion at C2. Other researchers have also reported poor results from attempts to synthesize AuIII complexes directly from monoimidazolium ions and AuIII salts in the presence of base.9,10
 | ||
| Scheme 1 Synthesis of AuIII–NHC complexes. | ||
The PF6− salts [Au(L1)2](PF6)35, [Au(L2)2](PF6)36, and [AuL3](PF6)37 were obtained from the corresponding chlorides [Au(L1)2Cl2]Cl 2, [Au(L2)2Cl2]Cl 3, and [AuL3Cl2]Cl 4 respectively, by metathesis with KPF6 in methanol, and were obtained as analytically-pure, white powders, in quantitative yields.
X-ray studies
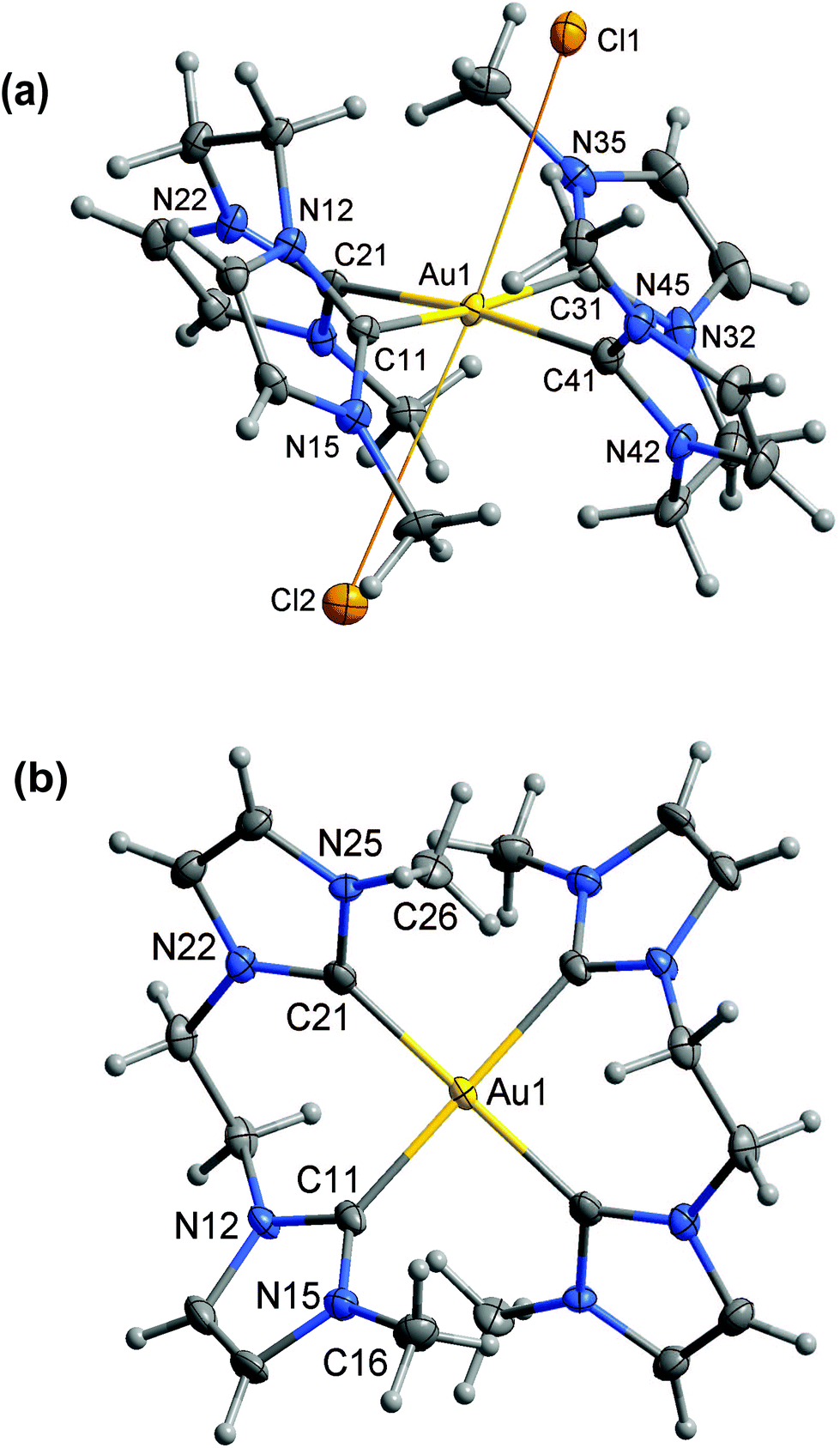
Results of X-ray diffraction studies of the AuIII complexes are summarized in Tables 1 and 2 and Fig. 1–3. The chlorido complexes [Au(L1)2Cl2]+, [Au(L2)2Cl2]+ and [AuL3Cl2]+ each contain an AuIII centre in a strongly distorted octahedral environment, and are examples of rarely-encountered 20-electron complexes. Removal of the chlorido ligands results in [Au(L1)2]3+, [Au(L2)2]3+ and [AuL3]3+, which might be thought of as structures containing commonly encountered four-coordinate AuIII centres (16-electron complexes), but the picture is complicated by fluorines from PF6− anions occupying axial coordination sites. | ||
| Fig. 1 (a) Crystal structure (50% probability level for the displacement ellipsoids) of the cation of [Au(L1)2Cl2]Cl 2. Selected bond lengths (Å) and angles (°): Au(1)–C(11) 2.040(9), Au(1)–C(41) 2.043(9), Au(1)–C(31) 2.046(9), Au(1)–C(21) 2.050(9), Au(1)–Cl(1) 3.148(2), Au(1)–Cl(2) 3.192(2), C(11)–Au(1)–C(41) 179.3(3), C(41)–Au(1)–C(31) 85.0(4), Cl(1)–Au(1)–Cl(2) 176.78(6). (b) Crystal structure (50% probability level for the displacement ellipsoids) of the cation of [Au(L1)2](PF6)35. PF6 anions have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au(1)–C(21) 2.041(2), Au(1)–C(11) 2.045(2), C(21)–Au(1)–C(11) 83.31(9). | ||
 | ||
| Fig. 2 (a) Crystal structure (50% probability level for the displacement ellipsoids) of the cation of [Au(L2)2Cl2]Cl 3. Selected bond lengths (Å) and angles (°): Au(1)–C(11) 2.024(4), Au(1)–C(21) 2.054(4), Au(1)–Cl(1) 3.433(1), Au(1)–Cl(2) 3.693(1), C(11)–Au(1)–C(31) 174.87(17), C(11)–Au(1)–C(21) 89.84(17), Cl(1)–Au(1)–Cl(2) 172.17(3). (b) Crystal structure (50% probability level for the displacement ellipsoids) of the cation of [Au(L2)2](PF6)36. PF6 anions have been omitted for clarity. Selected bond lengths (Å) and angles (°): Au(1)–C(11) 2.0402(18), Au(1)–C(21) 2.062(2), C(11)–Au(1)–C(21) 88.22(8). | ||
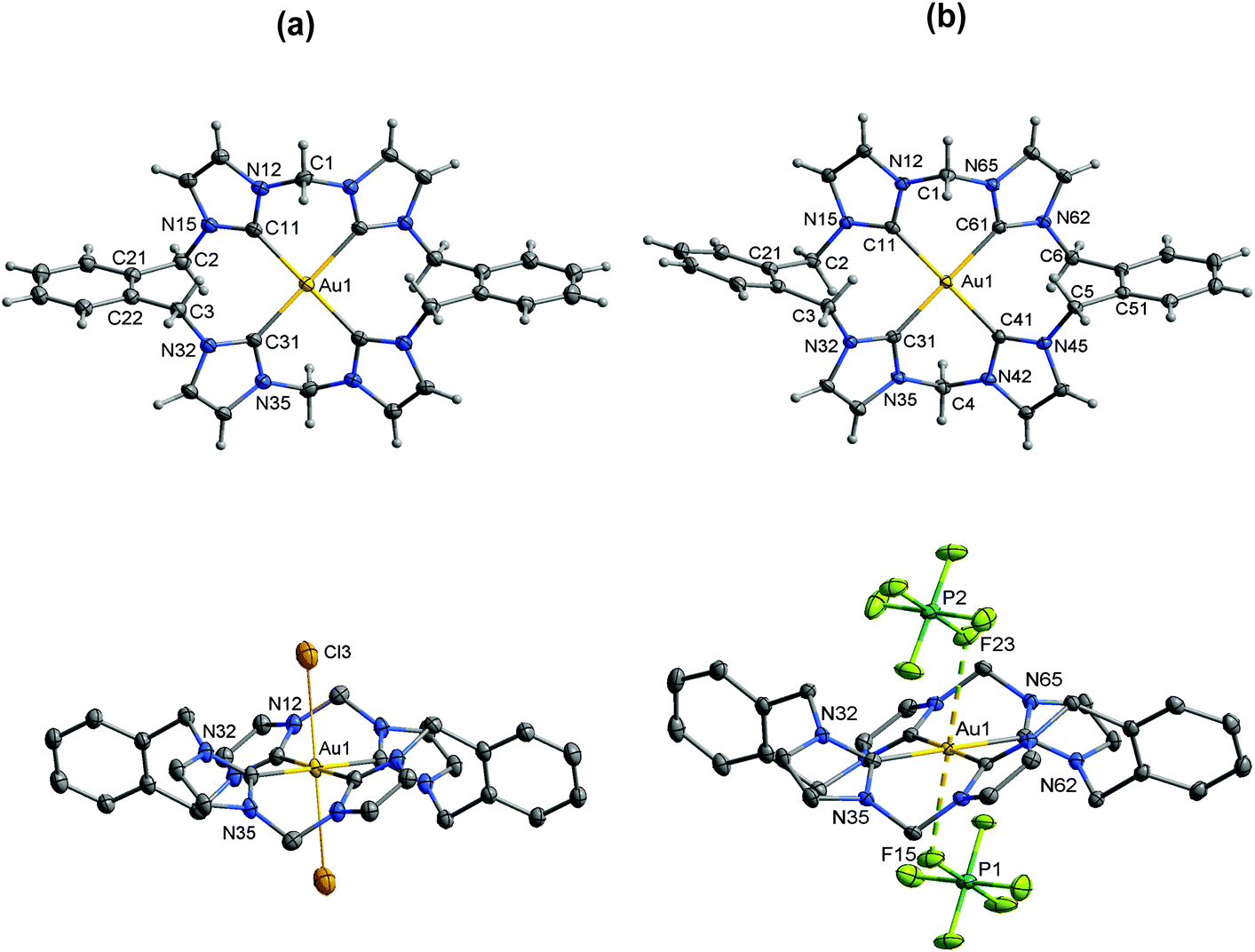 | ||
| Fig. 3 (a) Crystal structure (30% probability level for the displacement ellipsoids) of the cation of [AuL3Cl2]Cl 4. Selected bond lengths (Å) and angles (°): Au(1)–C(11) 2.048(8), Au(1)–C(31) 2.082(7), Au(1)–Cl(3) 3.017(4), C(11)–Au(1)–C(31) 95.5(3). (b) Crystal structure (50% probability level for the displacement ellipsoids) of the cation 1 of [AuL3](PF6)37. (Cation 2 is similar.) Selected bond lengths (Å) and angles (°): Au(1)–C(41) 2.051(2), Au(1)–C(31) 2.052(2), Au(1)–C(11) 2.056(2), Au(1)–C(61) 2.056(2), Au(2)–C(71) 2.050(2), Au(2)–C(91) 2.055(2), Au(1)–F(15) 3.004(5), Au(1)–F(23) 3.089(2), Au(2)–F(33) 3.903(2), C(41)–Au(1)–C(31) 84.77(8), C(41)–Au(1)–C(11) 178.57(8), F(15)–Au(1)–F(23) 179.47(8), C(71)–Au(2)–C(91) 93.94(8). | ||
| Complex | [AuL1Cl2]Cl·(MeOH)22 | [AuL2Cl2]Cl·(MeOH) 3 | [AuL3Cl2]Cl·(MeOH)3.54 | [AuL1](PF6)3·(MeCN)25 | [AuL2](PF6)3·(MeCN)26 | [AuL3](PF6)3·(MeCN)27 |
|---|---|---|---|---|---|---|
| Empirical formula | C20H32AuCl3N8O2 | C21H32AuCl3N8O | C33.50H42AuCl3N8O3.50 | C22H30AuF18N10P3 | C24H34AuF18N10P3 | C34H34AuF18N10P3 |
| Formula weight | 719.85 | 715.86 | 916.07 | 1066.43 | 1094.49 | 1214.59 |
| Wavelength/Å | 0.71073 | 0.71073 | 1.54184 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P21/c | I2/m | C2/c | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 11.2465(4) | 8.4520(3) | 11.5286(4) | 19.6697(5) | 19.5075(5) | 9.50080(10) |
| b/Å | 12.5742(5) | 21.5282(5) | 18.4128(5) | 11.0832(2) | 11.1838(2) | 15.6009(2) |
| c/Å | 18.5100(7) | 14.1226(4) | 16.9015(6) | 17.0857(6) | 18.0374(4) | 21.6100(3) |
| α/° | 95.646(1) | |||||
| β/° | 100.893(4) | 101.964(3) | 98.985(3) | 113.018(4) | 113.442(3) | 96.197(1) |
| γ/° | 94.683(1) | |||||
| V/Å3 | 2570.44(17) | 2513.88(13) | 3543.7(2) | 3428.18(19) | 3610.39(16) | 3155.08(7) |
| Z | 4 | 4 | 4 | 4 | 4 | 3 |
| ρ(calc)/Mg m−3 | 1.860 | 1.891 | 1.717 | 2.066 | 2.014 | 1.918 |
| μ/mm−1 | 6.070 | 6.203 | 10.284 | 4.565 | 4.338 | 3.734 |
| Crystal size/mm3 | 0.638 × 0.230 × 0.069 | 0.186 × 0.127 × 0.080 | 0.147 × 0.116 × 0.057 | 0.48 × 0.31 × 0.22 | 0.40 × 0.285 × 0.245 | 0.355 × 0.247 × 0.191 |
| θ range for data collection/° | 2.455 to 27.0 | 2.463 to 32.562 | 3.574 to 67.329 | 2.276 to 37.068 | 2.147 to 37.625 | 1.708 to 30.066 |
| Reflections collected | 24788 |
31630 |
17415 |
31084 |
43319 |
71746 |
| Independent reflections | 14559 |
8553 | 3272 | 8533 | 9243 | 18510 |
| R(int) | 0.1079 | 0.0591 | 0.0482 | 0.0436 | 0.0423 | 0.0379 |
| Max./min. transmission | 0.665 and 0.204 | 0.667 and 0.429 | 0.630 and 0.369 | 0.644 and 0.417 | 0.504 and 0.357 | 1.000/0.741 |
| Restraints/parameters | 0/315 | 0/313 | 166/247 | 0/267 | 12/415 | 0/971 |
| Goodness-of-fit on F2 | 1.105 | 1.092 | 1.117 | 1.124 | 1.047 | 1.060 |
| R 1 [I > 2σ(I)] | 0.0628 | 0.0460 | 0.0546 | 0.0302 | 0.0263 | 0.0252 |
| wR2 [I > 2σ(I)] | 0.1842 | 0.0765 | 0.1430 | 0.0660 | 0.0531 | 0.0552 |
| R 1 (all data) | 0.0712 | 0.0681 | 0.0691 | 0.0437 | 0.0475 | 0.0316 |
| wR2 (all data) | 0.1887 | 0.0819 | 0.1541 | 0.0711 | 0.0604 | 0.0576 |
| Δρ(max/min)/e Å−3 | 4.765 and −2.329 | 2.330 and −0.797 | 2.749 and −2.259 | 2.244 and −1.084 | 1.443 and −1.440 | 2.804 and −0.744 |
| CCDC number | 1528857 | 1528858 | 1528859 | 1528860 | 1528861 | 1528862 |
| Complexes | Au–C | Au–Cl or Au–FPF5 | C–Au–Cb | C–Au–Cc | Cl–Au–Cl | φ | φ′ | θ | θ′ |
|---|---|---|---|---|---|---|---|---|---|
| a φ = NNHC–C–NNHC; φ′ = NNHC–C–Carene; θ = AuC4/NHC inter-planar angle; θ′ = AuC4-arene inter-planar angle. b Angle between NHCs directly linked by CH2 or (CH2)2 group. c Angle between NHCs not directly linked by CH2 or (CH2)2 group. | |||||||||
| [AuL1Cl2]Cl 2 | 2.040(9) | 3.148(2) | 85.0(4) | 179.3(3) | 176.78(6) | 108.6(7) | — | 40.6(3) | — |
| 2.050(9) | 3.192(2) | 84.5(4) | 178.9(4) | 109.0(7) | 41.1(3) | ||||
| 2.043(9) | 96.0(3) | 39.8(3) | |||||||
| 2.046(9) | 94.5(4) | 38.7(3) | |||||||
| [AuL1]3PF65 | 2.045(2) | 3.380(2) | 83.31(9) | 180 | — | 108.0(2) | — | 43.0(1) | — |
| 2.041(2) | 43.7(1) | ||||||||
| [AuL2Cl2]Cl 3 | 2.024(4) | 3.433(1) | 89.84(17) | 172.65(18) | 172.17(2) | — | — | 54.5(2) | — |
| 2.054(4) | 3.693(1) | 91.40(18) | 174.87(17) | 46.7(2) | |||||
| 2.037(4) | 90.52(17) | 53.5(2) | |||||||
| 2.061(4) | 88.88(17) | 46.3(2) | |||||||
| [AuL2]3PF66 | 2.0402(18) | 3.394(3) | 88.22(8) | 180 | — | — | — | 57.6(1) | — |
| 2.062(2) | 48.8(1) | ||||||||
| [AuL3Cl2]Cl 4 | 2.048(8) | 3.016(4) | 84.5(3) | 180 | 180 | 110.6(3) | 109.9(7) | 36.4(4) | 64.3(3) |
| 2.082(7) | 95.5(3) | 109.8(7) | 36.4(3) | ||||||
| [AuL3]3PF67 | 2.056(2) | 3.004(5) | 84.77(8) | 178.57(8) | — | 107.9(2) | 113.0(2) | 34.6(1) | 74.1(1) |
| (Molecule 1) | 2.052(2) | 3.089(2) | 85.63(8) | 179.75(8) | 107.6(2) | 112.1(2) | 39.4(1) | 63.4(1) | |
| 2.051(2) | 94.52(8) | 109.3(2) | 36.8(1) | ||||||
| 2.056(2) | 95.08(8) | 107.8(2) | 35.7(1) | ||||||
| (Molecule 2) | 2.050(2) | 2.903(4) | 86.06(8) | 180 | — | 108.1(2) | 111.2(2) | 36.7(1) | 63.6(1) |
| 2.055(2) | 93.94(8) | 109.92(2) | 36.4(1) | ||||||
| 1 32 | 2.017(9) | — | 86.0(4) | 174.5(4) | — | 109.6(7) | — | 29.8(3) | — |
| 2.042(10) | 86.0(4) | 174.5(4) | 111.4(8) | 45.3(3) | |||||
| 2.052(9) | 94.4(3) | 42.7(3) | |||||||
| 2.030(11) | 94.3(3) | 33.7(4) | |||||||
In each of the six complexes, the NHC units comprise an approximate square planar array around the AuIII centre. Au–C distances are in the range 2.024(4)–2.082(7) Å across the series of complexes, and are similar to the values seen for other complexes of form [AuIII(NHC)4]3+ (1 in Table 2),32 [AuIII(NHC)(halide)3],8,35 and [AuIII(NHC)2(halide)2]+.8,13,35 In [Au(L1)2Cl2]Cl·(MeOH)22·(MeOH)2, [Au(L2)2Cl2]Cl·(MeOH) 3·(MeOH), and [AuL3Cl2]Cl·(MeOH)3.54·(MeOH)3.5, two Cl− ligands occupy additional (axial) coordination sites, Au–Claxial being in the range 3.148(2)–3.693(1) Å, so that overall coordination geometry is distorted octahedral. These Au–Claxial bond distances are about 0.9 Å longer than Au–Clequatorial (∼2.27 Å) bond lengths seen in [Au(HHCT)Cl2]AuCl4 (HHCT = 1,8-bis(hydroxyethyl)-1,3,6,8,10,13-hexaazacyclotetradec-ane), [Au(dien)C13] (dien = diethylenetriamine), etc.,36–38 but are much shorter than the sum of the van der Waals radii of the atoms (∼4.1 Å for Au⋯Cl).39 This type of distorted octahedral structural motif has been reported34,36,38,40 in only a few cases for AuIII (Table S1†), and is presumably a consequence of these complexes being 20 electron complexes. For [AuL3Cl2]Cl·(MeOH)3.54·(MeOH)3.5, the coordination of AuIII is broadly similar to that seen in [Au(L1)2Cl2]Cl·(MeOH)22·(MeOH)2 and [Au(L2)2Cl2]Cl·(MeOH) 3·(MeOH), but is complicated by the effects of solvation, the axial sites modelled as being partially occupied by chlorido ligands and solvent (methanol) molecules.
[Au(L1)2](PF6)3·(MeCN)25·(MeCN)2 and [AuL3](PF6)3·(MeCN)37·(MeCN)3 have similar coordination environments around Au to those seen in the chlorido series, but with fluorine atoms from PF6− groups occupying the axial coordination sites (AuIII–F 2.92(3)–3.380(2) Å). For [Au(L2)2](PF6)3·(MeCN)26·(MeCN)2, the coordination environment about AuIII is more strictly square planar, there being only one close contact outside the square planar array (AuIII–F(PF5−), 3.394(2) Å), that contact not in a position that could be considered “axial”.
In the complexes containing an Au(L)2 core (Fig. 1 and 2), the two bis(NHC) ligands (L1 or L2) are “inverted” with respect to each other (i.e., for one ligand the methylene/ethylene linker is above the AuC4 plane while for the other ligand the linker is below the AuC4 plane), presumably to avoid unfavourable intramolecular interactions between the N–CH3 groups of each ligand. A similar arrangement is seen in [AuL3]3+ and [AuL3Cl2]+ (Fig. 3), but here the N–CH3 groups are replaced by o-xylylene moieties that complete the (NHC)4 macrocycle. Not surprisingly, the bite angle (C(11)–Au(1)–C(21)) in the complexes of L2 (NHC moieties linked by CH2CH2) is larger than for the complexes of L1 (NHC moieties linked by CH2).
In each complex, the imidazolyl rings are tilted with respect to the AuC4 coordination plane. For all complexes of L1 and L3, in which pairs of imidazolyl units are linked by a methylene bridge, the AuC4-imidazolyl interplanar angle is about 40°. For complexes of L2, in which the imidazolyl units are linked by a longer (ethylene) bridge, the AuC4-imidazolyl interplanar angle is higher (about 50°). This steeper inclination of the imidazolyl rings to the AuC4 plane may be the reason why the axial Au–L bonds are longer in [Au(L2)2Cl2]Cl·(MeOH) 3·(MeOH) than in the other chloride salts, and absent in [Au(L2)2](PF6)3·(MeCN)26·(MeCN)2—steric hindrance from the endo hydrogens of the ethylene bridge may inhibit approach of axial ligands to the Au centre.
UV-vis electronic absorption spectroscopy and conductance measurements
The UV-vis absorption spectra of all the AuIII–NHC complexes displayed intense absorption in the region λ = 230–290 nm (Fig. 4 and Table 3). We attribute these high energy absorption bands to π–π* intraligand transitions involving the NHC ligands.35 In the spectra of [Au(L1)2Cl2]Cl 2 and [Au(L2)2Cl2]Cl 3 (but not for [AuL3Cl2]Cl 4) a weaker absorbance is observed in the region 300–350 nm. These weaker absorption bands have energies and molar extinction coefficients (ε on the order of 103–104 M−1 cm−1) similar to those of ligand-to-metal-charge-transfer (LMCT) bands of the [AuCl4]− ion, which has been studied in detail by Mason et al.41 Similar absorptions were also displayed by other NHC adducts of AuIII halides.17,35 Therefore, we tentatively attribute these low energy absorption bands seen for [Au(L1)2Cl2]Cl 2 and [Au(L2)2Cl2]Cl 3 to LMCTs from the chlorido ligands to the Lewis acidic AuIII centres. A distinct LMCT band is not seen for [AuL3Cl2]Cl 4, but may be overlapped with the high energy absorption band below 300 nm. Similar observations were also reported by Huynh et al. for UV-vis studies of [AuIIICl2(iPr2Bim)2]BF4 (Bim = 1,3-benzimidazol-2-ylidene).35 | ||
| Fig. 4 UV-vis absorption spectra of (a) [Au(L1)2Cl2]Cl 2, [Au(L2)2Cl2]Cl 3, [AuL3Cl2]Cl 4 and (b) [Au(L1)2](PF6)35, [Au(L2)2](PF6)36, [AuL3](PF6)37 in MeCN. | ||
| Complex | λ max/nm (ε/M−1 cm−1)a |
Λ
Mb (S cm2 mol−1) |
|---|---|---|
| a Measured in 0.05 mM MeCN solution at 298 K. b Measured in 1 mM DMSO solution at 298 K. | ||
| [Au(L1)2Cl2]Cl 2 | 302 (3600), 235 (14100) |
41.6 |
| [Au(L1)2](PF6)35 | 250 (14600) |
114.6 |
| [Au(L2)2Cl2]Cl 3 | 301 (4100), 245 (15200) |
83.5 |
| [Au(L2)2](PF6)36 | 241 (15000) |
118.7 |
| [AuL3Cl2]Cl 4 | 275 (8400) | 41.8 |
| [AuL3](PF6)37 | 256 (11600) |
113.4 |
The molar conductance of solutions of the complexes in DMSO (Table 3) was measured to gain insight into possible dissociation of the chlorido ligands from the Au centres. Solutions of each of the salts [Au(L1)2](PF6)35, [Au(L2)2](PF6)36 and [AuL3](PF6)37, showed conductivity in the range 113–119 S cm2 mol−1. These values are in the range expected for 1:3 electrolytes in DMSO.42,43 By contrast, [Au(L1)2Cl2]Cl 2 and [AuL3Cl2]Cl 4 showed a molar conductivity of 44.5 and 41.8 S cm2 mol−1 respectively, in the range expected for a 1:1 electrolyte, while [Au(L2)2Cl2]Cl 3 showed a molar conductivity of 83.5 S cm2 mol−1, which falls in the range expected for 1:2 electrolytes. These results suggest that for [Au(L1)2Cl2]Cl 2 and [AuL3Cl2]Cl 4 in DMSO solution, the complex cation is of the form the [Au(NHC)4Cl2]+ (the same as seen in the solid state), while for [Au(L2)2Cl2]Cl 3, the complex cation is of the form [Au(NHC)4Cl]2+ in DMSO solution. Compared to [Au(L1)2Cl2]+ and [AuL3Cl2]+, the higher propensity of [Au(L2)2Cl2]+ to undergo dissociation of Cl− may be consequence of increased steric congestion around the axial coordination sites, as suggested by the steeper inclination of the imidazolyl rings relative to the AuC4 plane in [Au(L2)2Cl2]+ (see above).
NMR spectroscopy
The 1H and 13C NMR spectra for AuIII tetracarbene complexes (Fig. 5 and S1–S12†) are broadly consistent with the structures seen in the solid state, but show affects of dissociation of chlorido in some cases. | ||
| Fig. 5 1H NMR spectra (600 MHz, DMSO-d6) for: (a) [Au(L1)2Cl2]Cl 2, [Au(L1)2](PF6)35; (b) [Au(L2)2Cl]Cl23, [Au(L2)2](PF6)36; and (c) [AuL3Cl2]Cl 4, [AuL3](PF6)37. | ||
The 1H NMR spectrum of [Au(L1)2Cl2]Cl 2 in DMSO-d6 solution shows a single signal due to the methyl protons (3.69 ppm), two doublet signals in the range 7.5–8.5 ppm, corresponding to the two non-equivalent protons of each imidazolyl ring, and a pair of doublets (AX pattern) centred at 8.09 and 6.79 ppm, corresponding to two non-equivalent protons in the methylene bridge linking the imidazolyl groups in each ligand. The non-equivalence of the methylene protons indicates that the “puckered” conformation adopted by the two L1 ligands in the complex in the solid state persists in solution and is rigid on the NMR timescale. The 1H NMR spectrum of [Au(L1)2](PF6)35 in DMSO-d6 solution shows the same number of signals and splitting patterns, but the signals are at different chemical shifts, markedly so for the signals of the methylene protons. This observation is consistent with the change in coordination of AuIII from Au(NHC)4Cl2 for [Au(L1)2Cl2]Cl 2 to Au(NHC)4 for [Au(L1)2](PF6)35. We tentatively assign the doublet at 8.09 ppm to the endo protons of the CH2 groups, and suggest that its markedly downfield chemical shift is a consequence of an intramolecular H⋯Cl hydrogen bonding interaction with a Cl ligand. Interestingly, in the solid state, the Cl⋯H distances for the endo hydrogens are 2.55 and 2.60 Å, which are within the range of ∼2.2 Å–∼3.0 Å seen for C–H⋯Cl hydrogen bonds.44
It is interesting to note that the 1H NMR chemical shift of the signal for the endo CH2 protons of [Au(L1)2Cl2]Cl 2 in D2O solutions is close to that seen for [Au(L1)2](PF6)35 in DMSO-d6, and markedly different from that seen for [Au(L1)2Cl2]Cl 2 in DMSO-d6. This observation suggests in D2O solution, the cation [Au(L1)2Cl2]+ undergoes solvolysis to give [Au(L1)2]3+. Consistent with this suggestion, we found that when NaCl was dissolved in solutions prepared by dissolving [Au(L1)2Cl2]Cl 2 in D2O, the signal for the endo CH2 protons shifted downfield (i.e., towards the chemical shift seen for the endo protons in [Au(L1)2Cl2]+), and moved back upfield (i.e., towards the chemical shift seen for the endo protons in [Au(L1)2]3+) when Cl− was precipitated from the sample as AgCl (Fig. S13†).
As noted above, conductance measurements showed that dissolution of [Au(L2)2Cl2]Cl 3 in DMSO results in dissociation of one Cl− ligand from the Au centre and formation of [Au(L2)2Cl]2+ in solution. The 1H NMR spectra of solutions prepared by dissolving [Au(L2)2Cl2]Cl 3 in DMSO-d6 can thus be considered to be spectra of the cation [Au(L2)2Cl]2+. These spectra again show a single signal (3.35 ppm) due to the methyl protons and two doublet signals (now in the range 7.7–7.9 ppm) corresponding to the two non-equivalent protons of each imidazole ring, with an apparent AA′XX′ pattern of signals centred at 4.91 and 5.46 ppm, corresponding to the protons in each ethylene bridge. These signals and splitting patterns suggest that the “puckered” conformation adopted by the two L2 ligands of [Au(L2)2Cl2]+ in the solid state persists in solution. In the solid state, there are four non-equivalent environments for the hydrogen atoms in each ethylene bridge, so the observation of an AA′XX′ pattern for the ethylene protons suggests that some twisting within each L2 ligand causes exo and endo protons on each carbon in an ethylene bridge to be rendered equivalent on the NMR timescale (Fig. 6).
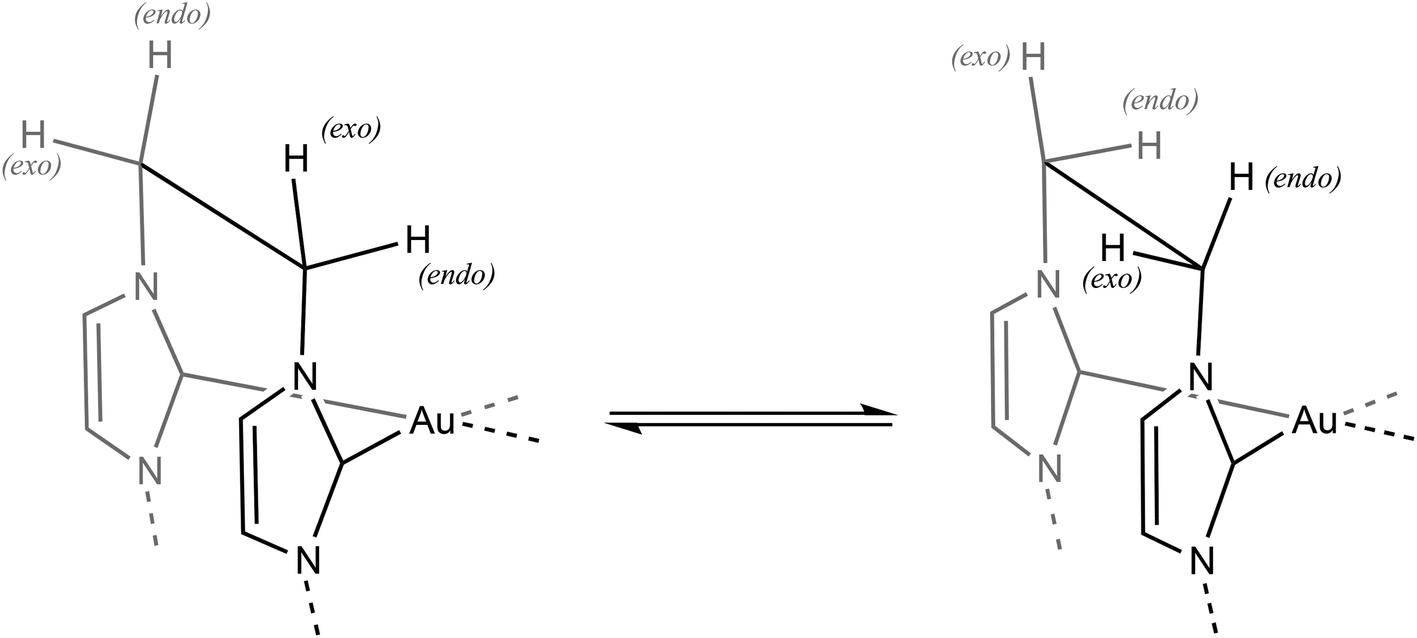 | ||
| Fig. 6 Proposed twisting of the ethylene bridge to account for symmetry observed for 1H NMR signals of [Au(L2)2Cl2]Cl 3 in DMSO-d6 solution. | ||
There are small differences between the 1H NMR signals seen for [Au(L2)2Cl]2+ and [Au(L2)2]3+ (solutions prepared from [Au(L2)2](PF6)3) 6 in terms chemical shift, but the number of signals and splitting patterns are the same in each case. For [Au(L2)2Cl]2+, we note that the existence of an Au(NHC)4Cl coordination motif (approximately square pyramidal) must render the two L2 ligands inequivalent if the ligands are puckered in the same way as seen in the solid state for [Au(L2)2Cl2]Cl 3. Since the number of NMR signals is consistent with the L2 ligands being equivalent on the NMR timescale, it may be that the two L2 ligands are rendered equivalent on the NMR timescale by rapid dissociation and re-association of the chlorido ligand (Fig. 5(b)).
The 1H NMR spectra for both [AuL3Cl2]Cl 4 and [AuL3](PF6)37 in DMSO-d6 solution are analogous to those seen for [Au(L1)2Cl2]Cl 2 and [Au(L1)2](PF6)35. Major differences between the two samples in terms of in chemical shifts of the protons of the methylene linkers, and to a lesser extent those of the benzylic protons, are consistent with a change in coordination of AuIII, from Au(NHC)4Cl2 in [AuL3Cl2]Cl 4 to Au(NHC)4 (or a solvated form) in [AuL3](PF6)37. The number of signals and splitting patterns are consistent with the ligand L3 adopting a “puckered” conformation that does not invert on the NMR timescale.
In the 13C NMR spectra of the new AuIII–NHC complexes having an Au(NHC)4 motif, the signal for the carbene carbon occurs near 146 ppm, which is close to that reported for the related [Au(NHC)4]+ complex 132 but approximately 35 ppm upfield of that for the carbene carbons in AuI–NHC complexes.8 For comparison, imidazolium-derived AuIII complexes of form [Au(R2Im)Cl3] (R2Im = 1,3-dialkylimidazol-2-ylidene) show a carbene signal in the range 134–141 ppm,14,45 and [Au(R2Im)2Cl2]+ and [Au2(L1)2Cl4]2+ exhibit carbene signals near 150–155 ppm,46,47 while [Au(R2Bim)Cl3] and [Au(R2Bim)2Cl2]+ (R2Bim = 1,3-dialkylbenzimidazol-2-ylidene) exhibit carbene signals35 about 10 ppm downfield of those of their imidazolium-derived counterparts.
H–D exchange reactions
Imidazolium ions are well-known to undergo H/D exchange reactions.48–50 We have explored H/D exchange reactions for the imidazolium salt L1·2HCl and the AuIII complex [Au(L1)2]3+ (formed from [Au(L1)2Cl2]Cl 2, see above) in D2O solutions (Fig. 7). For L1·2HCl, the imidazolium H2 protons exchange quickly (<5 min at room temperature) with deuterium from the solvent. This finding is consistent with previous observations that the 1H NMR signal for the H2 protons of imidazolium salts generally “underintegrates” in spectra recorded in D2O and methanol-d4 solutions at room temperature.51–53 When the sample was heated at 100 °C, the imidazolyl H4/H5 protons were fully exchanged over the course of about 48 h, while the CH2 protons were about 40% exchanged over the same period, and the CH3 protons underwent little or no exchange. The analogous exchange reactions were markedly faster for [Au(L1)2]3+ under similar conditions: the imidazolyl H4/H5 protons (δ 7.91, 7.61 ppm) were fully exchanged within 17 h at 100 °C, as indicated by disappearance of the H4/H5 signals from the 1H NMR spectrum, and the CH2 protons were fully exchanged within 48 h at 100 °C (Fig. 7b). Experiments conducted using t-butanol as an internal standard showed negligible H/D exchange of CH3 protons in [Au(L1)2]3+ even after heating for 48 h (Fig. S14†). Not surprisingly, the H/D exchange reactions of [Au(L1)2]3+ were catalysed by base—in the presence of two equivalents of K2CO3, the imidazolyl H4/H5 protons (but not the CH2 protons) underwent partial exchange over 48 h at room temperature (Fig. S15†), and in the presence of two equivalents of NaOH, both the imidazolyl H4/H5 protons and the CH2 protons were almost completely exchanged overnight at room temperature (Fig. S16†). The high charge on the [Au(L1)2]3+ cation is likely a factor contributing to the fast H/D exchange seen in D2O (formation of a deprotonated intermediate [Au(L1)2–H]2+ would be favoured by the concomitant decrease in concentration of positive charge on the complex). | ||
| Fig. 7 1H NMR spectra (600.13 MHz, D2O) for solutions of (a) the imidazolium salt L1·2HCl and (b) the AuIII complex [Au(L1)2]3+ (prepared from [Au(L1)2Cl2]Cl) 2, showing disappearance of signals due to H/D exchange reactions. For L1·2HCl, no signal for the H2 protons was detected even from freshly-prepared solutions. | ||
Stability studies
The new AuIII(NHC)4 complexes reported in this work were stable in the solid state at room temperature under air for at least 1 year. The D2O exchange experiment described above suggests surprisingly high stability for the AuIII(NHC)4 complexes, in that they can survive extended periods in solution at 100 °C, albeit with H/D exchange occurring. To further explore the stability of the new AuIII(NHC)4 complexes, we have used 1H NMR spectroscopy to assessed the longevity of various samples in DMSO-d6 solutions at elevated temperatures. After a DMSO-d6 solution containing [Au(L1)2](PF6)35 was stored at room temperature for 6 months and then heated at 120 °C for 26 h, there was no significant change to the 1H NMR spectrum (Fig. S17†). The same applied for a DMSO-d6 solution containing [Au(L3)](PF6)37 that was maintained at 120 °C for 7 days (Fig. S19†). The chlorido complex [Au(L2)2Cl2]Cl 3 was somewhat less stable; after a DMSO-d6 solution containing this compound was heated at 120 °C for 7 days, integration of the imidazolyl H4/H5 region of the 1H NMR spectrum indicating that ca. 20% of the initial AuIII complex [Au(L2)2Cl2]+ had decomposed, the major decomposition product being the known54 dinuclear AuI complex [Au2(L2)2]2+ (Fig. S18†). The slightly lower stability of [Au(L2)2Cl2]+ may be a consequence of slightly higher strain in the chelate rings (e.g., in the ethylene link joining the NHC units in [Au(L2)2Cl2]+, N–C–C angles are 113.4(3), 113.8(4), 115.7(4) and 116.4(4)°; in the methylene linker in [Au(L1)2Cl2]+, N–C–N angles are 108.5(7) and 109.1(7)°, closer to the ideal tetrahedral angle, 109.5°). For comparison, salts of the dinuclear AuIII complex [Au2(L1)2Cl4]2+ and its bromido counterpart,8,47 which contain AuIII(NHC)2X2 moieties rather than AuIII(NHC)4 moieties, were far less stable. At room temperature in the solid state, ca. 50% of the AuIII/AuIII complex [Au2(L1)2Br4]2+ had decomposed to the AuI/AuIII complex [Au2(L1)2Br2]2+ over 6 months (Fig. S20 and S21†), and no AuIII species could be detected after a DMSO-d6 solution containing [Au2(L1)2Br4]2+ and [Au2(L1)2Br2]2+ was heated at 120 °C for 2 h (Fig. S21†). Similar results were seen for solutions containing [Au2(L1)2Cl4]2+ (Fig. S22†) and [Au2(L2)2Br4]2+ (Fig. S23†).The high stability of AuIII(NHC)4 complexes compared to their AuIII(NHC)2(halide)2 counterparts may reflect the strong σ-donating ability of the NHC ligand, which is better able to stabilise the AuIII centre than the weaker σ-donating halide ligands. The robustness of the metal–NHC bond and steric protection exerted by the chelating NHC ligands must also contribute to the enhanced stability of the AuIII(NHC)4 complexes compared to their AuIII(NHC)2X2 counterparts.
Cyclovoltammetric studies showed that the AuIII complexes did not undergo reversible redox reactions at room temperature in acetonitrile with Bu4NClO4 as electrolyte (see ESI†).
Conclusion
Three new AuIII complexes of form [Au(NHC)4Cl2]Cl may be synthesized in good yield directly from bis- and tetrakis(imidazolium) salts and KAuCl4 in the presence of base. These formally 20 electron complexes have tetragonally distorted octahedral coordination of the AuIII centre, the NHC groups making an approximately square planar array and the chlorido ligands occupying more distant axial positions. The AuIII(NHC)4 motif is remarkably stable, resisting decomposition in DMSO solution at 120 °C or D2O at 100 °C for extended periods. The axial (chlorido) ligands are labile, and metathesis of the [Au(NHC)4Cl2]Cl salts with KPF6 in methanol/water results in removal of the chlorido ligands and formation of three new, formally 16 electron complexes of form [Au(NHC)4](PF6)3. The [Au(NHC)4]3+ ion is sufficiently stable to undergo base-catalysed H/D exchange reactions at most sites on the NHC ligands in D2O at 100 °C without evidence of decomposition.Experimental section
General procedures
Nuclear magnetic resonance spectra were recorded using Bruker ARX400 (400.13 MHz for 1H), Bruker ARX500 (500.13 MHz for 1H and 125.77 MHz for 13C), or Bruker ARX 600 (600.13 MHz for 1H, 150.90 MHz for 13C) spectrometers at ambient temperature. 1H and 13C NMR chemical shifts were referenced to signals of the solvent (DMSO-d6: 1H 2.50 ppm; 13C 39.52). When necessary, assignments were made with the aid of 1H–13C HSQC (heteronuclear single quantum coherence) and 1H–13C HMBC (heteronuclear multiple bond correlation) spectra. Conductance measurements were performed by using a TPS Aqua-CP/A conductivity meter. Cyclic voltammetry experiments were recorded using an eDAQ e-corder 401 system in a three-electrode cell with a glassy carbon (1 mm diameter) working electrode, a platinum (1 mm diameter) counter electrode, and a platinum wire reference electrode. Measurements were taken at room temperature (25 °C) in acetonitrile with 0.1 M Bu4NClO4 as a supporting electrolyte. Microanalyses were performed by The School of Chemistry & Molecular Bioscience, University of Queensland, Australia, and the Instrument Center of National Chung Hsing University, Taiwan. High resolution mass spectra were measured using Agilent LCMS 6510 Q-TOF and Waters LCT Premier XE spectrometers, using the ESI method, with MeCN:H2O (9:1) as solvent. All organometallic compounds were prepared under a nitrogen atmosphere. The imidazolium salts L1·2HCl, L2·2HCl,55,56 and L3·4HCl57 were prepared according to literature procedures, and the AuIII complexes [Au2(L1)2X4]X2 (X = Cl, Br) and [Au2(L2)2Br4]Br2 were prepared in the same way as the corresponding hexafluorophosphate salts.8,47
Synthesis of AuIII–NHC complexes
X-Ray crystal structure determinations
Crystals of [Au(L1)2](PF6)3·(MeCN)25·(MeCN)2, [Au(L2)2](PF6)3·(MeCN)26·(MeCN)2 and [AuL3](PF6)3·(MeCN)37·(MeCN)3 were grown by diffusion of vapours between neat diethyl ether and a solution of the complex in acetonitrile, and crystals of [Au(L1)2Cl2]Cl·(MeOH)22·(MeOH)2, [Au(L2)2Cl2]Cl·(MeOH) 3·(MeOH) and [AuL3Cl2]Cl·(MeOH)3.54·(MeOH)3.5 were grown by diffusion of vapours between neat diethyl ether and a solution of the complex in methanol. Crystallographic data were collected at 100(2) K on either an Oxford Diffraction Gemini or an Oxford Diffraction Xcalibur diffractometer using Mo Kα or Cu Kα radiation. Following analytical absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the program SHELXL-2014.58 Unless stated differently below, all hydrogen atoms were added at calculated positions and refined by use of riding models with isotropic displacement parameters based on those of the parent atoms. Except for those atoms mentioned below, anisotropic displacement parameters were employed throughout for the non-hydrogen atoms. For the structure of [AuL3Cl2]Cl·(MeOH)3.54·(MeOH)3.5, one chlorido ligand was modelled as being disordered with a molecule of methanol with site occupancies constrained to 0.75 and 0.25 from trial refinement and as required for charge balance. Remaining solvent molecules were modelled as three molecules of methanol situated on a crystallographic mirror plane. Geometries and displacement parameters of the solvent were restrained to reasonable values. Solvent hydrogen atoms were not included in the model. For the structure of [Au(L1)2](PF6)3·(MeCN)25·(MeCN)2, the solvent acetonitrile molecule was found to be disordered over two sites with the methyl atom common to both components. In [Au(L2)2](PF6)3·(MeCN)26·(MeCN)2, the ligand and one hexafluorophosphate anion are both disordered over two sets of sites with occupancies refined to 0.765(4) and its complement after trial refinement showed no significant differences in the refined values. The atoms of the minor component of the cation were refined with isotropic displacement parameters. The remaining hexafluorophosphate anion was modelled as being rotationally disordered with site occupancies were constrained to 0.5 after trial refinement. The solvent was modelled as acetonitrile, disordered over two sets of sites. For the structure of [AuL3](PF6)3·(MeCN)37·(MeCN)3, the fluorine atoms of two hexafluorophosphate anions were modelled as being disordered over two sites; hexafluorophosphate anion (1) where site occupancies were refined to 0.75(2) and its complement and hexafluorophosphate anion (3) where fluorine occupancies were constrained to 0.5 after trial refinement.Acknowledgements
The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia. Ahmed H. Mageed thanks The Higher Committee for Education Development in Iraq (HCED) for financial support.References
- P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038–1047 RSC.
- M. P. Rigobello, A. Folda, B. Dani, R. Menabò, G. Scutari and A. Bindoli, Eur. J. Pharmacol., 2008, 582, 26–34 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- H. Schmidbaur and A. Schier, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford UK, 2007, vol. 2, pp. 252–308 Search PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- I. J. Lin and C. S. Vasam, Can. J. Chem., 2005, 83, 812–825 CrossRef CAS.
- M. Baron, C. Tubaro, M. Basato, A. Biffis, M. M. Natile and C. Graiff, Organometallics, 2011, 30, 4607–4615 CrossRef CAS.
- P. de Frémont, R. Singh, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2007, 26, 1376–1385 CrossRef.
- S. Zhu, R. Liang and H. Jiang, Tetrahedron, 2012, 68, 7949–7955 CrossRef CAS.
- I. G. Santos, A. Hagenbach and U. Abram, Dalton Trans., 2004, 677–682 RSC.
- S. D. Khanye, N. B. Báthori, G. S. Smith and K. Chibale, Dalton Trans., 2010, 39, 2697–2700 RSC.
- R. Jothibasu, H. V. Huynh and L. L. Koh, J. Organomet. Chem., 2008, 693, 374–380 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2009, 29, 394–402 CrossRef.
- M. C. Jahnke, T. Pape and F. E. Hahn, Z. Anorg. Allg. Chem., 2010, 636, 2309–2314 CrossRef CAS.
- M. Pažický, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jäkel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448–4458 CrossRef.
- C. Hirtenlehner, C. Krims, J. Hölbling, M. List, M. Zabel, M. Fleck, R. J. Berger, W. Schoefberger and U. Monkowius, Dalton Trans., 2011, 40, 9899–9910 RSC.
- S. Gaillard, X. Bantreil, A. M. Slawin and S. P. Nolan, Dalton Trans., 2009, 6967–6971 RSC.
- M. Kriechbaum, D. Otte, M. List and U. Monkowius, Dalton Trans., 2014, 43, 8781–8791 RSC.
- A. Collado, J. Bohnenberger, M. J. Oliva-Madrid, P. Nun, D. B. Cordes, A. M. Slawin and S. P. Nolan, Eur. J. Inorg. Chem., 2016, 2016, 4111–4122 CrossRef CAS.
- J. Gil-Rubio, V. Cámara, D. Bautista and J. Vicente, Inorg. Chem., 2013, 52, 4071–4083 CrossRef CAS PubMed.
- W. Liu, K. Bensdorf, M. Proetto, A. Hagenbach, U. Abram and R. Gust, J. Med. Chem., 2012, 55, 3713–3724 CrossRef CAS PubMed.
- H. Sivaram, J. Tan and H. V. Huynh, Organometallics, 2012, 31, 5875–5883 CrossRef CAS.
- C. Topf, C. Hirtenlehner, M. Zabel, M. List, M. Fleck and U. Monkowius, Organometallics, 2011, 30, 2755–2764 CrossRef CAS.
- M. Muuronen, J. E. Perea-Buceta, M. Nieger, M. Patzschke and J. Helaja, Organometallics, 2012, 31, 4320–4330 CrossRef CAS.
- J. P. Reeds, A. C. Whitwood, M. P. Healy and I. J. Fairlamb, Organometallics, 2013, 32, 3108–3120 CrossRef CAS.
- C. Topf, C. Hirtenlehner, M. Fleck, M. List and U. Monkowius, Z. Anorg. Allg. Chem., 2011, 637, 2129–2134 CrossRef CAS.
- C. Hemmert, R. Poteau, M. Laurent and H. Gornitzka, J. Organomet. Chem., 2013, 745, 242–250 CrossRef.
- F. dit Dominique, H. Gornitzka, A. Sournia-Saquet and C. Hemmert, Dalton Trans., 2009, 340–352 RSC.
- M. Baron, C. Tubaro, M. Basato, A. A. Isse, A. Gennaro, L. Cavallo, C. Graiff, A. Dolmella, L. Falivene and L. Caporaso, Chem. – Eur. J., 2016, 22, 10211–10224 CrossRef CAS PubMed.
- C. Tubaro, M. Baron, M. Costante, M. Basato, A. Biffis, A. Gennaro, A. A. Isse, C. Graiff and G. Accorsi, Dalton Trans., 2013, 42, 10952–10963 RSC.
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087 RSC.
- G. Marangoni, B. Pitteri, V. Bertolasi, G. Gilli and V. Ferretti, J. Chem. Soc., Dalton Trans., 1986, 1941–1944 RSC.
- V. Duckworth and N. Stephenson, Inorg. Chem., 1969, 8, 1661–1664 CrossRef CAS.
- H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591–4600 CrossRef CAS.
- G. Nardin, L. Randaccio, G. Annibale, G. Natile and B. Pitteri, J. Chem. Soc., Dalton Trans., 1980, 220–223 RSC.
- L. S. Hollis and S. J. Lippard, J. Am. Chem. Soc., 1983, 105, 4293–4299 CrossRef CAS.
- M. P. Suh, I. S. Kim, B. Y. Shim, D. Hong and T.-S. Yoon, Inorg. Chem., 1996, 35, 3595–3598 CrossRef CAS.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- R. Elder and J. Watkins, Inorg. Chem., 1986, 25, 223–226 CrossRef CAS.
- H. Isci and W. R. Mason, Inorg. Chem., 1983, 22, 2266–2272 CrossRef CAS.
- S. Ramalingam and S. Soundararajan, J. Inorg. Nucl. Chem., 1967, 29, 1763–1768 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- C. B. Aakeröy, T. A. Evans, K. R. Seddon and I. Pálinkó, New J. Chem., 1999, 23, 145–152 RSC.
- B. Jacques, D. Hueber, S. Hameury, P. Braunstein, P. Pale, A. l. Blanc and P. de Frémont, Organometallics, 2014, 33, 2326–2335 CrossRef CAS.
- J. L. Hickey, PhD thesis, The University of Western Australia, 2008.
- M. Baron, C. Tubaro, M. Basato, A. Biffis and C. Graiff, J. Organomet. Chem., 2012, 714, 41–46 CrossRef CAS.
- J. L. Wong and J. H. Keck Jr., J. Org. Chem., 1974, 39, 2398–2403 CrossRef CAS.
- Y. Takeuchi, H. J. Yeh, K. L. Kirk and L. A. Cohen, J. Org. Chem., 1978, 43, 3565–3570 CrossRef CAS.
- C. Hardacre, J. D. Holbrey and S. J. McMath, Chem. Commun., 2001, 367–368 RSC.
- M. V. Baker, B. W. Skelton, A. H. White and C. C. Williams, Organometallics, 2002, 21, 2674–2678 CrossRef CAS.
- M. V. Baker, M. J. Bosnich, D. H. Brown, L. T. Byrne, V. J. Hesler, B. W. Skelton, A. H. White and C. C. Williams, J. Org. Chem., 2004, 69, 7640–7652 CrossRef CAS PubMed.
- M. V. Baker, D. H. Brown, C. H. Heath, B. W. Skelton, A. H. White and C. C. Williams, J. Org. Chem., 2008, 73, 9340–9352 CrossRef CAS PubMed.
- M. Baron, C. Tubaro, A. Biffis, M. Basato, C. Graiff, A. Poater, L. Cavallo, N. Armaroli and G. Accorsi, Inorg. Chem., 2012, 51, 1778–1784 CrossRef CAS PubMed.
- Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Dalton Trans., 2009, 4786–4794 RSC.
- O. Sanchez, S. González, M. Fernández, A. R. Higuera-Padilla, Y. Leon, D. Coll, A. Vidal, P. Taylor, I. Urdanibia and M. C. Goite, Inorg. Chim. Acta, 2015, 437, 143–151 CrossRef CAS.
- T. Pape and F. E. Hahn, Dalton Trans., 2013, 42, 7330–7337 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1528857–1528862. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01272a |
| This journal is © The Royal Society of Chemistry 2017 |