
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A ruthenium(II) bis(phosphinophosphinine) complex as a precatalyst for transfer-hydrogenation and hydrogen-borrowing reactions†
R. J.
Newland
a,
M. F.
Wyatt
b,
R. L.
Wingad
*c and
S. M.
Mansell
 *a
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk; Web: http://www.mansellresearch.org.uk
bEPSRC UK National Mass Spectrometry Facility (NMSF), Swansea University Medical School, Grove Building Extn, Singleton Park, Swansea SA2 8PP, UK
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: Rich.Wingad@bristol.ac.uk
First published on 7th April 2017
Abstract
The 2-phosphinophosphinine 2-PPh2-3-Me-6-SiMe3-PC5H2 (2) has been prepared and was shown to act as a κ2-chelating ligand in cis-[RuCl2(2)2] (4). Complex 4 was a competent precatalyst for the room temperature transfer hydrogenation of acetophenone (0.1 mol% 4 and 0.5 mol% KOtBu) and the conversion of methanol/ethanol mixtures to the advanced biofuel isobutanol in 50% yield and 96% selectivity.
Phosphinines (the P-analogue of pyridine) have developed into useful ligands in coordination chemistry1 and catalysis.2 The LUMO of phosphinine (A) contains significant phosphorus p-character that leads to π-accepting behaviour, but can also lead to nucleophiles attacking the electrophilic P atom,1b,3 followed by protonation (Scheme 1).3b,4
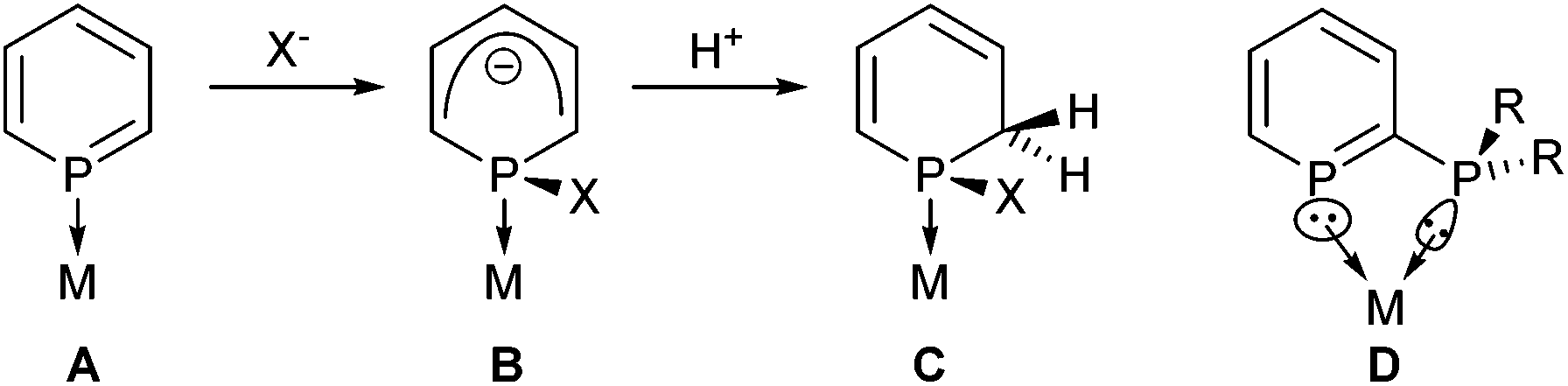 | ||
| Scheme 1 Reactivity of coordinated phosphinines (A–C), and a 2-phosphinophosphinine (D). | ||
Conventionally, the electrophilicity of phosphinines has been viewed as problematic in catalysis,5 however, the heterocyclic species B and C have been shown to be useful ligands in their own right.1a,5,6 This electrophilic reactivity could therefore be used as part of a ligand activation strategy to produce new coordination complexes or turn precatalysts into catalysts.7 Metal–ligand cooperation8 and bifunctional catalysis9 are both important topics in catalysis that have only emerged recently.
Donor-substituted phosphinines10 are useful ligands because they combine the unique properties of phosphinines with more conventional donors, such as pyridine.11 However, there are only a handful of methods for introducing phosphine substituents.12 For example, bromophosphinines were shown to undergo Pd-catalysed coupling to produce 2-phosphinophosphinines.13 In 1996, Mathey, Le Floch and co-workers demonstrated that diazaphosphinine 1 (Scheme 2) reacts in consecutive steps with alkynes to give 2,3,5,6-tetrasubstituted phosphinines, including 2-phosphinophosphinines.14 However, reports of phosphinophosphinines in catalysis are limited to the use of a wide bite-angle phosphinophosphinine in the regioselective hydroformylation of styrene,12b,15 and a Rh-phosphitophosphinine complex used in asymmetric hydrogenation catalysis.16
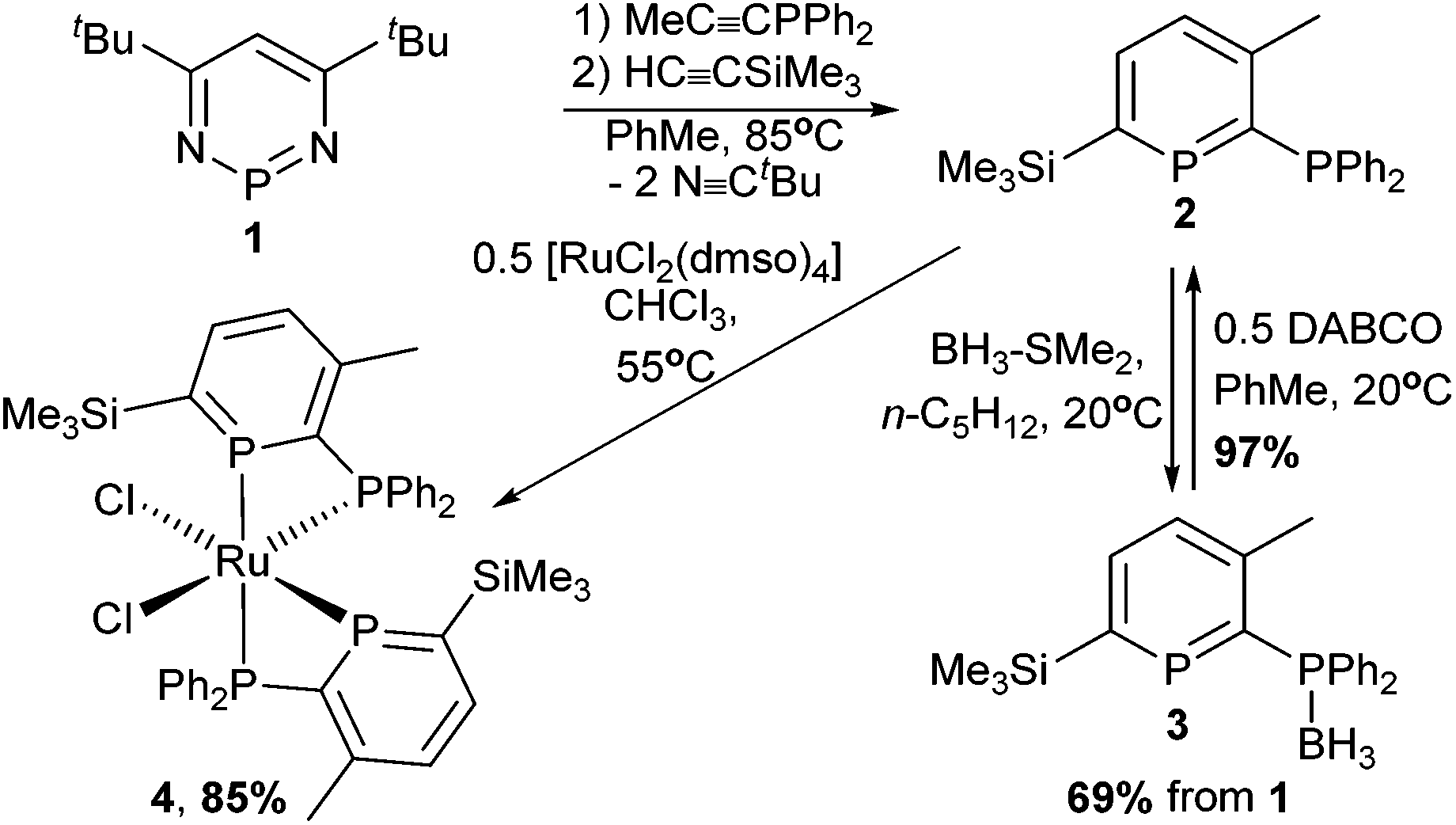 | ||
| Scheme 2 Synthesis of 2-phosphinophosphinine 2 and coordination to Ru. | ||
Transfer hydrogenation holds great potential in chemical synthesis because it replaces hazardous reducing agents, such as hydrogen gas or metal hydrides, with more convenient chemical sources of hydrogen, commonly isopropanol or formic acid/formate.17 Related to this are hydrogen-borrowing processes that involve the oxidation of a saturated substrate by transfer of an equivalent of dihydrogen to a metal centre, thereby facilitating new reactivity, before the borrowed hydrogen is then returned.18 These fields have typically been dominated by Ru and Ir catalysts.17,18 Recently there has been renewed interest in the Guerbet reaction,19 a borrowed hydrogen process in which an alcohol is dehydrogenated to an aldehyde and then undergoes aldol condensation before rehydrogenation of the product to a longer chain alcohol.20 Wass and co-workers reported that a family of Ru complexes with diphosphine21 or aminophosphine22 supporting ligands could selectively convert ethanol to n-butanol, or ethanol/methanol to isobutanol,23via the Guerbet reaction. Of the catalysts tested, trans-[RuCl2(dppm)2] (dppm = 1,1-bis(diphenylphosphino)methane) – containing a small bite-angle diphosphine ligand – proved to be the most active and stable,21 whereas wider bite angle ligands were less active. We became interested in the greater s-character of the formally sp2-hybridised P lone pair of phosphinine (estimated as ca. 61% for PC5H5)1a compared to conventional phosphines because we reasoned that the reduced directional preference would lead to less strained 4-membered chelate rings (D). Additionally, the ligand non-innocence of phosphinines has not been fully explored in catalysis, so we set out to synthesise a suitable chelating 2-phosphinophosphinine precatalyst and then screen this complex for catalytic reactivity in transfer hydrogenation and hydrogen borrowing reactions. Ligated RuCl2 complexes are typical precatalysts for these reactions, and they are converted into the active catalysts by reaction with alkoxides.24 The exact catalytic mechanisms are still under examination,25 but are believed to involve metal hydrides.24,26
Using the methodology of Le Floch and Mathey,14 the 2-phosphinophosphinine 2 was synthesised by sequential reaction of 1 with diphenylprop-1-ynylphosphine27 and trimethylsilylacetylene in toluene at 85 °C over 2 days (Scheme 2). To facilitate product work-up, the borane adduct 3 was generated immediately after extraction of 2 from the crude reaction mixture by reaction with Me2S–BH3 at room temperature, and 3 precipitated as a colourless solid in good yield (69% from 1). 3 was fully characterised by elemental analysis, single crystal X-ray diffraction, mass spectrometry and multinuclear NMR spectroscopy (see ESI†).
Deprotection of 3 by reaction with 0.5 equivalents of DABCO28 (1,4-diazabicyclo[2.2.2]octane) at room temperature followed by removal of the insoluble DABCO·2BH3 by filtration gave the parent phosphinophosphinine 2 as a pure pale-yellow solid. 2 is stable to ROH (R = Me, Et, iPr) as well as atmospheric oxygen and moisture, greatly facilitating storage and handling, and batches of up to 3 g have been prepared from 1 in 5 days. 31P{1H} NMR spectroscopy showed a characteristic resonance at high frequency for the phosphinine P atom located in the aromatic ring (δ 249.8 ppm) coupled to the phosphine P at lower chemical shift (−7.5 ppm, 2JPP 31.6 Hz). These values are similar to the related silyl-substituted phosphinine 2,3-(PPh2)2-6-SiMe3-PC5H2 (δ: 256 and −10 ppm).14b Complete characterisation was achieved using multinuclear NMR spectroscopy, elemental analysis and mass spectrometry. Single crystal X-ray diffraction experiments confirmed the molecular structure of 2 (Fig. 1A and Table S2†). Although the X-ray crystal structures of approximately 30 uncoordinated phosphinines are known,29 compound 2 represents the first structural determination of an unfunctionalised 2-phosphinophosphinine. The P–C bond lengths in the aromatic ring (ca. 1.75 Å) are intermediate between P–C single (ca. 1.83 Å) and double (ca. 1.66 Å) bonds.1b The C–C bond lengths are similar to the C–C bond length in benzene (1.40 Å), although C1–C2 is slightly longer (1.423(4) Å). One P–Ph bond is located in the plane of the phosphinine ring and the other is perpendicular. This has the effect of orienting the phosphine lone pair away from any potential overlap with the phosphinine ring, and the Ph2P–C bond length resembles a standard single bond (1.847(3) Å).
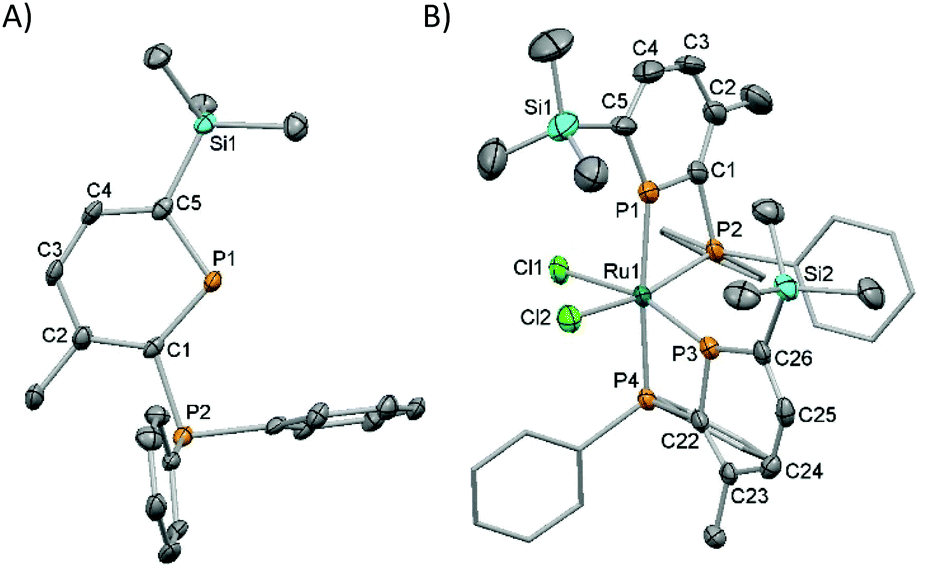 | ||
| Fig. 1 Thermal ellipsoid plots (50%) of the molecular structures of 2 (A) and 4 (B, Ph rings in 4 are displayed without thermal ellipsoids for clarity). Selected bond distances (Å) and angles (°) for 2: P(1)–C(1) 1.754(3), C(1)–C(2) 1.423(4), C(2)–C(3) 1.404(4), C(3)–C(4) 1.393(4), C(4)–C(5) 1.396(4), C(5)–P(1) 1.749(3), C(1)–P(2) 1.847(3), C(1)–P(1)–C(5) 103.6(1), P(1)–C(1)–P(2) 119.9(1); for 4: P(1)–Ru(1) 2.310(2), P(2)–Ru(1) 2.342(2), P(3)–Ru(1) 2.241(2), P(4)–Ru(1) 2.369(2), Ru(1)–Cl(1) 2.430(2), Ru(1)–Cl(2) 2.477(2), P(1)–Ru(1)–P(2) 69.89(6), P(3)–Ru(1)–P(4) 70.32(5). | ||
Coordination to Ru was accomplished by reacting two equivalents of 2 with [RuCl2(dmso)4] at 55 °C in CHCl3 for 5 hours to give cis-[RuCl2(2)2] (4) as a bright orange solid. Monitoring the reaction by 31P{1H} NMR spectroscopy showed consumption of the two doublets observed for 2 and the concomitant appearance of four multiplets, two at high frequency and two around 0 ppm that integrated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio. Large trans-couplings (2JPP 425 Hz) were observed for one phosphinine and one phosphine P atom. The presence of these trans-couplings suggested the selective formation of the C1cis-isomeric structure for 4 (as a racemic mixture of its enantiomers), which was confirmed by single crystal X-ray diffraction experiments (Fig. 1B and Table S2†).
:1:1:1 ratio. Large trans-couplings (2JPP 425 Hz) were observed for one phosphinine and one phosphine P atom. The presence of these trans-couplings suggested the selective formation of the C1cis-isomeric structure for 4 (as a racemic mixture of its enantiomers), which was confirmed by single crystal X-ray diffraction experiments (Fig. 1B and Table S2†).
The solid-state structure of 4 showed the first crystallographic evidence of κ2 coordination of a 2-phosphinophosphinine to a transition metal. The molecular structure of 4 showed a distorted Oh Ru centre with cis-Cl ligands and two inequivalent, bidentate 2-phosphinophosphinine ligands. The shortest Ru–P bond (P(3)–Ru(1) 2.241(2) Å) is to the phosphinine trans to a Cl, the longest (P(4)–Ru(1) 2.369(2) Å) to the phosphine trans to a phosphinine. The Ru–Cl bond lengths are similar, with the Ru–Cl bond length to Cl(1) trans to a phosphinine donor slightly shorter. The reasons behind the selective formation of one isomer are not completely clear, but the C1 isomer does avoid either both phosphinines, or both phosphines, being trans to each other. Whilst a handful of bridging bimetallic complexes of 2-phosphinophosphinines are known (in reactions with CuI, [Ni(cod)2]/CO, Mn2(CO)10 and [M2Cp2(CO)4] (M = Fe, Mo)),30 to the best of our knowledge, there is only one brief mention of mono-metallic κ2 complexes (to Gp 6 carbonyls).12c The bite angles of the two bidentate ligands in 4 are 69.89(6)° and 70.32(5)°, which are smaller than in cis-[RuCl2(dppm)2] (72.1(3)°)31 and trans-[RuCl2(vdpp)2] (73.12(2)°, vdpp = 1,1-bis(diphenylphosphino)ethene),32 but similar to cis-[RuCl2(Ph2PCH2phospholyl)2] (71.12(5) and 70.53(4)°).334 was further characterised by elemental analysis and mass spectrometry. Phosphinines with small ortho-substituents were previously shown to bind to Ru in an η1 fashion in trans-[RuCl2(PC5H5)4]34 or [Ru(η5-C5Me5)(2-Br-4,5-Me2-PC5H2)Cl],35 but η6 coordination was favoured in [Ru(η5-C5Me5)(2,6-(SiMe3)2-PC5H3)][BF4] with larger ortho-substituents.35
Complex 4 was subsequently tested as a precatalyst for the transfer-hydrogenation of substituted acetophenones. All of the substrates were tested using 0.1 mol% 4 and 0.5 mol% KOtBu in isopropanol at room temperature (Table 1). Good conversion to 1-phenylethanol was observed within one hour at room temperature (94%, run 1), and this was also observed for para-Br and -F acetophenone (97 and 96% respectively, runs 2 and 3). Electron-donating groups required heating and substrates with para-Me and ortho-OMe substitution went to excellent conversion at the boiling point of isopropanol (runs 5 and 7). Substrates with para-OMe (runs 8 and 9) and -NO2 substituents (runs 10 and 11) only proceeded to moderate conversion even at 82 °C.‡
|
|
||||
|---|---|---|---|---|
| Run | R1 | R2 | Temperature | Conv.a (%) |
| Conditions: 4 (0.1 mol%), KOtBu (0.5 mol%), iPrOH, (0.4 M [substrate]), 1 h.a Conversion determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene internal standard, average of two runs.b 24 h run, low conversion due to insolubility of substrate.c No increase in conversion after 2 h. | ||||
| 1 | H | H | 20 °C | 94% |
| 2 | H | Br | 20 °C | 97% |
| 3 | H | F | 20 °C | 96% |
| 4 | H | Me | 20 °C | 87%c |
| 5 | 82 °C | 98% | ||
| 6 | OMe | H | 20 °C | 5%c |
| 7 | 82 °C | >99% | ||
| 8 | H | OMe | 20 °C | 48%c |
| 9 | 82 °C | 79%c | ||
| 10 | H | NO2 | 20 °C | 5%b |
| 11 | 82 °C | 72%c | ||
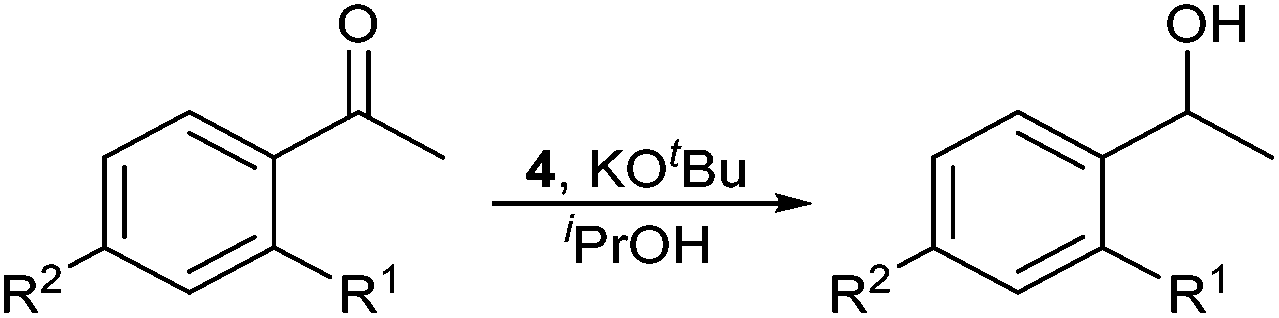
These results show that 4 is an excellent precatalyst for transfer hydrogenation of acetophenones at room temperature. In comparison, 0.1 mol% [RuCl2(PPh3)3] achieved only a 75% conversion of acetophenone after 6 hours at 82 °C.36 Other Ru complexes with phosphorus ligands (including tripodal phosphines and a PCP pincer ligand) also required heating to achieve good conversion.§ [Ru(η6-p-cymene){1-Bu-2-P(S)Ph2-3-Me-5,6-Ph2-PC5H}Cl], a Ru complex of a phosphacyclohexadienyl anion bearing a sulfur donor, was also shown to perform poorly as a catalyst requiring 2.5 days at 80 °C to go to completion.6 A catalyst with a higher activity (full conversion after 1 minute at room temperature) has been demonstrated with a Ru complex bearing NH functionality that undergoes ligand-activation with base,37 and a Ru pyridyl-phosphole complex has shown full conversion at extremely low loadings (5 × 10−6 mol%) at 90 °C.38§
Activation of the precatalyst with KOtBu could affect the phosphinine moieties (leading to B and C) as well as substituting the Cl ligands. This was demonstrated by 31P{1H} NMR spectroscopy that revealed loss of the high frequency phosphinine resonances in 4 and formation of a major species with resonances at 70 and 20 ppm upon reaction with KOtBu in iPrOH at 20 °C, along with a number of other resonances. Analysis of these reaction mixtures re-dissolved in acetone-d6 also demonstrated that many species were present with their chemical shifts indicating complete transformation of the phosphinine moieties. Mass spectrometry experiments similarly revealed multiple new species in the ≈ m/z 700–1200 range.† Clearly, there are many processes involved in the activation of the precatalyst.
Complex 4 was tested for activity in the co-condensation of methanol and ethanol (Scheme 3).19a,23 In this process methanol and ethanol are dehydrogenated to formaldehyde and acetaldehyde, which undergo aldol coupling to yield, after rehydrogenation, n-propanol. A further dehydrogenation, aldol coupling, re-hydrogenation cycle with a second equivalent of methanol yields isobutanol. Using standard conditions (see ESI†) 4 was found to produce isobutanol in 38% yield with good selectivity (88%) after 2 h in the liquid fraction.¶ In comparison, the previously reported catalyst, trans-[RuCl2(dppm)2] gave 65% isobutanol (98% selectivity).23 The reduction in selectivity is due to a larger proportion of the intermediate n-propanol being formed (5% yield). Extending the reaction time to 20 h led to an increase in both isobutanol yield (50%) and selectivity (96%), similar to results obtained using the phosphinoamine based catalyst trans-[RuCl2(Ph2P(CH2)2NH2)2] (51% yield, 90% selectivity after 20 h).23
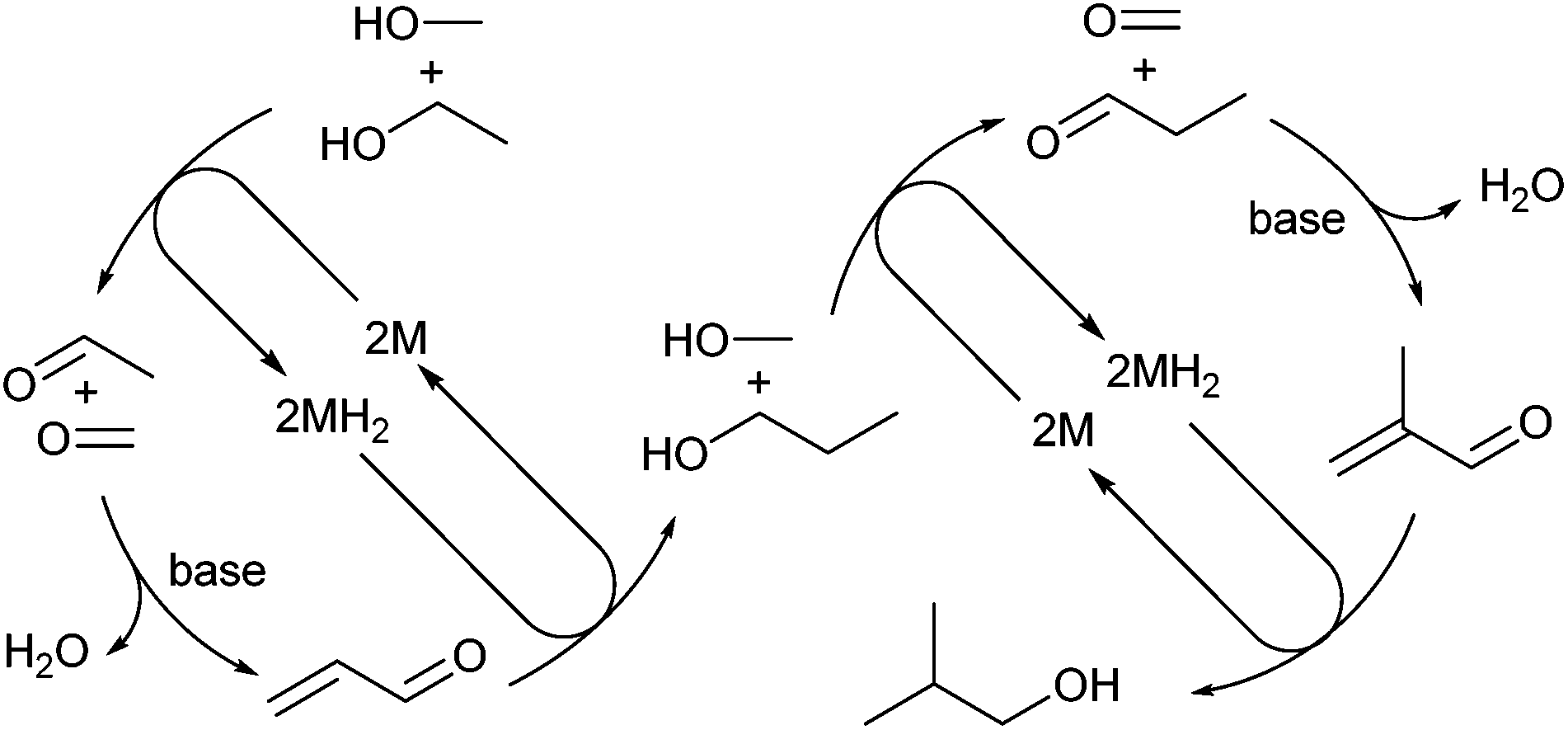 | ||
| Scheme 3 Co-condensation of MeOH and EtOH via a hydrogen borrowing mechanism. | ||
In conclusion, a bidentate 2-phosphinophosphinine ligand has been prepared along with the cis-ruthenium dichloride complex 4, which contains two κ2-chelating phosphinophosphinine ligands. 4 was shown to act as a precatalyst for the transfer hydrogenation of acetophenone at room temperature to 94% conversion in one hour upon activation with KOtBu. Substrates with electron withdrawing groups (para-Br, para-F) also proceeded to good conversion at room temperature, and substrates with electron donating groups (para-Me and ortho/para-OMe) reacted at higher temperatures (82 °C). Complex 4 was also an effective precatalyst for the upgrading of MeOH/EtOH mixtures to isobutanol in 50% yield and 96% selectivity over 20 hours. This demonstrates the first use of 2-phosphinophosphinines in catalysis.
Prof. Duncan Wass (University of Bristol) is thanked for useful discussions. We thank Heriot-Watt University and the EPSRC (DTP funding for RJN) for support, and acknowledge the help of Dr Juraj Bella (University of Edinburgh) and Dr Dave Ellis (Heriot-Watt University) with NMR spectroscopy. UK Catalysis Hub is kindly thanked (RLW) for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1).
Notes and references
- (a) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef CAS; (b) C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187 CrossRef.
- (a) C. Müller and D. Vogt, Dalton Trans., 2007, 5505 RSC; (b) C. Müller and D. Vogt, in Phosphorus Compounds: Advanced Tools in Catalysis and Material Sciences, ed. M. Peruzzini and L. Gonsalvi, 2011, pp. 151–181 Search PubMed; (c) L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257 CrossRef PubMed; (d) L. Weber, Angew. Chem., Int. Ed., 2002, 41, 563 CrossRef CAS.
- (a) A. Moores, L. Ricard, P. Le Floch and N. Mézailles, Organometallics, 2003, 22, 1960 CrossRef CAS; (b) I. de Krom, E. A. Pidko, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 7523 CrossRef CAS PubMed.
- (a) P. Le Floch, D. Carmichael and F. Mathey, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 77, 255 Search PubMed; (b) B. Schmid, L. M. Venanzi, A. Albinati and F. Mathey, Inorg. Chem., 1991, 30, 4693 CrossRef CAS; (c) I. de Krom, L. E. E. Broeckx, M. Lutz and C. Müller, Chem. – Eur. J., 2013, 19, 3676 CrossRef CAS PubMed; (d) A. Campos-Carrasco, L. E. E. Broeckx, J. J. M. Weemers, E. A. Pidko, M. Lutz, A. M. Masdeu-Bultó, D. Vogt and C. Müller, Chem. – Eur. J., 2011, 17, 2510 CrossRef CAS PubMed.
- A. Moores, N. Mézailles, L. Ricard and P. Le Floch, Organometallics, 2005, 24, 508 CrossRef CAS.
- M. Dochnahl, M. Doux, E. Faillard, L. Ricard and P. Le Floch, Eur. J. Inorg. Chem., 2005, 2005, 125 CrossRef.
- M. Doux, N. Mezailles, M. Melaimi, L. Ricard and P. Le Floch, Chem. Commun., 2002, 1566 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS PubMed.
- (a) C. Müller and D. Vogt, C. R. Chim., 2010, 13, 1127 CrossRef; (b) M. H. Habicht, F. Wossidlo, M. Weber and C. Müller, Chem. – Eur. J., 2016, 22, 12877 CrossRef CAS PubMed; (c) X. D. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. S. Li, G. Becker, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641 CrossRef CAS PubMed; (d) X. Chen, Z. Li, F. Yanan and H. Grützmacher, Eur. J. Inorg. Chem., 2016, 2016, 633 CrossRef CAS; (e) F. Mathey and P. Le Floch, in Hetarenes and related ring systems, ed. D. S. Black and M. Alvarez, Georg Thieme Verlag, Stuttgart, 2005, pp. 1097–1156 Search PubMed.
- C. Müller, J. A. W. Sklorz, I. de Krom, A. Loibl, M. Habicht, M. Bruce, G. Pfeifer and J. Wiecko, Chem. Lett., 2014, 43, 1390 CrossRef.
- (a) P. Le Floch, D. Carmichael and F. Mathey, Organometallics, 1991, 10, 2432 CrossRef CAS; (b) B. Breit, J. Mol. Catal. A: Chem., 1999, 143, 143 CrossRef CAS; (c) G. Märkl, C. Dörges, T. Riedl, F. G. Klärner and C. Lodwig, Tetrahedron Lett., 1990, 31, 4589 CrossRef; (d) P. Rosa, P. Le Floch, L. Ricard and F. Mathey, J. Am. Chem. Soc., 1997, 119, 9417 CrossRef CAS; (e) K. Waschbüsch, P. Le Floch and F. Mathey, Organometallics, 1996, 15, 1597 CrossRef.
- P. Le Floch, D. Carmichael, L. Ricard and F. Mathey, J. Am. Chem. Soc., 1993, 115, 10665 CrossRef CAS.
- (a) N. Avarvari, P. Le Floch and F. Mathey, J. Am. Chem. Soc., 1996, 118, 11978 CrossRef CAS; (b) N. Avarvari, P. Le Floch, L. Ricard and F. Mathey, Organometallics, 1997, 16, 4089 CrossRef CAS.
- B. Breit, Chem. Commun., 1996, 2071 RSC.
- C. Müller, L. G. López, H. Kooijman, A. L. Spek and D. Vogt, Tetrahedron Lett., 2006, 47, 2017 CrossRef.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- (a) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed; (b) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS; (c) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS PubMed; (d) F. Huang, Z. Liu and Z. Yu, Angew. Chem., Int. Ed., 2016, 55, 862 CrossRef CAS PubMed; (e) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305 RSC.
- (a) H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6, 7125 CrossRef CAS; (b) D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876 RSC; (c) A. Galadima and O. Muraza, Ind. Eng. Chem. Res., 2015, 54, 7181 CrossRef CAS; (d) J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588 CrossRef CAS.
- M. Guerbet, C. R. Acad. Sci. Paris, 1899, 128, 1002 Search PubMed.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005 CrossRef CAS PubMed.
- R. L. Wingad, P. J. Gates, S. T. G. Street and D. F. Wass, ACS Catal., 2015, 5, 5822 CrossRef CAS.
- R. L. Wingad, E. J. E. Bergström, M. Everett, K. J. Pellow and D. F. Wass, Chem. Commun., 2016, 52, 5202 RSC.
- J.-E. Bäckvall, J. Organomet. Chem., 2002, 652, 105 CrossRef.
- (a) P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604 CrossRef CAS PubMed; (b) J. Toubiana and Y. Sasson, Catal. Sci. Technol., 2012, 2, 1644 RSC.
- (a) J. S. M. Samec, J.-E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC; (b) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS.
- W. Hewertson, I. C. Taylor and S. Trippett, J. Chem. Soc. C, 1970, 1835 RSC.
- H. Brisset, Y. Gourdel, P. Pellon and M. Le Corre, Tetrahedron Lett., 1993, 34, 4523 CrossRef CAS.
- Search of the CSD, version 5.37.
- (a) N. Mézailles, P. Le Floch, K. Waschbüsch, L. Ricard, F. Mathey and C. P. Kubiak, J. Organomet. Chem., 1997, 541, 277 CrossRef; (b) K. Waschbüch, P. Le Floch, L. Ricard and F. Mathey, Chem. Ber., 1997, 130, 843 CrossRef; (c) N. Mézailles, F. Mathey and P. L. Floch, in Prog. Inorg. Chem, John Wiley & Sons, Inc., 2007, pp. 455–550 Search PubMed.
- A. R. Chakravarty, F. A. Cotton and W. Schwotzer, Inorg. Chim. Acta, 1984, 84, 179 CrossRef CAS.
- F. A. Cotton, M. P. Diebold and M. Matusz, Polyhedron, 1987, 6, 1131 CrossRef CAS.
- D. H. Nguyen, J.-C. Daran, S. Mallet-Ladeira, T. Davin, L. Maron, M. Urrutigoity, P. Kalck and M. Gouygou, Dalton Trans., 2013, 42, 75 RSC.
- C. Elschenbroich, J. Six, K. Harms, G. Frenking and G. Heydenrych, Eur. J. Inorg. Chem., 2008, 2008, 3303 CrossRef.
- N. Mézailles, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 2001, 20, 3304 CrossRef.
- R. L. Chowdhury and J.-E. Backvall, J. Chem. Soc., Chem. Commun., 1991, 1063 RSC.
- M. Zhao, Z. Yu, S. Yan and Y. Li, Tetrahedron Lett., 2009, 50, 4624 CrossRef CAS.
- C. Thoumazet, M. Melaimi, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 2003, 22, 1580 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthetic details, analytical data and crystallographic data. CCDC 1519355–1519357. Additional research data supporting this publication are available from Heriot-Watt University's research data repository at DOI: 10.17861/fde98e25-d7ed-459b-9d90-977119ba4562. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01022b |
| ‡ Throughout the catalytic runs, clear, homogeneous, orange solutions were observed with one exception; para-nitro acetophenone formed a dark brown solution and the substrate was poorly soluble in isopropanol. Nanoparticles have been implicated in the transfer hydrogenation catalysis of [RuCl2(PPh3)3], although particularly under boiling conditions.25b |
| § See ESI† for a comparison of selected literature systems used in transfer hydrogenation of acetophenone. |
| ¶ As with previous reports, both solid and gaseous products are also formed during catalysis. 1H and 13C{1H} NMR spectroscopy showed that the solid contains sodium formate. During the reactions a pressure build up is observed, for example, during the 2 h experiment the internal reactor pressure reaches approximately 32 bar. After cooling, approximately 12 bar pressure remains. This gas is presumably hydrogen formed as a by-product of formate synthesis. |
| This journal is © The Royal Society of Chemistry 2017 |