DOI:
10.1039/C7DT00972K
(Paper)
Dalton Trans., 2017,
46, 7506-7514
Electrocatalytic proton reduction by a model for [NiFeSe] hydrogenases†
Received
17th March 2017
, Accepted 24th May 2017
First published on 25th May 2017
Abstract
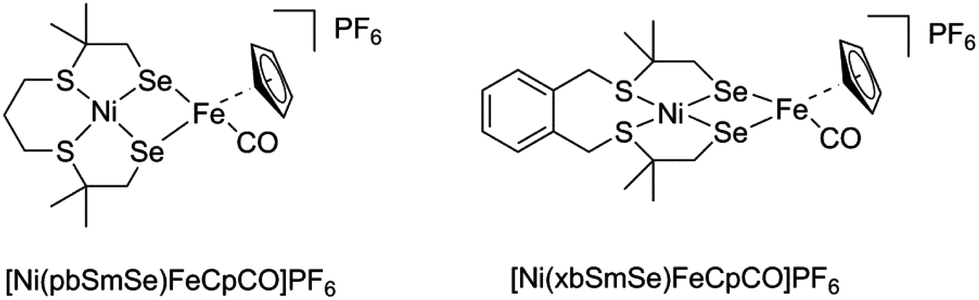
Two new heterodinuclear nickel–iron complexes [Ni(pbSmSe)FeCpCO]PF6 and [Ni(xbSmSe)FeCpCO]PF6 were synthesized as mimics of the [NiFeSe] hydrogenase active site (HCp = cyclopentadiene; H2pbSmSe = 1,9-diselenol-3,7-dithia-2,2,8,8-tetramethylnonane; H2xbSmSe = 1,2,-bis(2-thiabutyl-3,3-dimethyl-4-selenol)benzene). The compounds were characterized by single crystal X-ray diffraction and cyclic voltammetry. X-ray structure determinations showed that in both NiFe complexes the nickel(II) center is in a square-planar S2Se2 environment; the two selenolate donors are bridging to the iron(II) center that is further coordinated to an η5-cyclopentadienyl group and a carbon monoxide ligand. Electrochemical studies showed that the complex [Ni(pbSmSe)FeCpCO]PF6 is an electrocatalyst for the production of H2 in DMF in the presence of acetic acid at −2.1 V vs. Fc+/Fc; a foot-of-the-wave (FOWA) analysis of the catalytic currents yielded an estimation of kobs of 24 s−1.
Introduction
Hydrogenase enzymes, catalyzing the reversible oxidation of dihydrogen, play an important role in the metabolism of bacteria.1 In the past decades, hydrogenases have attracted the attention of synthetic chemists, since dihydrogen gas may be used as a sustainable source of energy. In order to produce dihydrogen gas for the application in fuel cells, new efficient electrocatalysts for the hydrogen-evolving reaction (HER) may be developed by using biomimetic, functional models of hydrogenases.2
Three types of hydrogenases are known, which are classified based on the metal center in the active site. The [FeFe] hydrogenases contain a dinuclear iron center linked to an Fe4S4cluster, comprising CO, CN− and a dithiolate ligand. These [FeFe] hydrogenases catalyze both H2 evolution and uptake, but their predominant activity is in H2 evolution. However, the [FeFe] hydrogenases generally are highly air sensitive. The [Fe] hydrogenases have a mononuclear iron catalytic center and do not contain Fe–S clusters. These enzymes catalyze the transfer of hydride groups and H2 activation.3 The third class of hydrogenases comprises the [NiFe] hydrogenases containing a heterodimetallic Ni–Fe active site with a nickel center bound to four cysteine thiolates with two of the cysteines bridging between the nickel and an iron center (Fig. 1a).4 Although this enzyme is mostly involved in the uptake of H2, it is also able to catalyze the production of H2.3 The [NiFeSe] hydrogenases form a subclass of the [NiFe] hydrogenases, in which one of the non-bridging cysteines (Cys) in the active site of the enzyme is replaced by selenocysteine (Sec) (Fig. 1b).5 [NiFeSe] hydrogenases show interesting properties for H2 production such as their high catalytic rates and their activity at low overpotentials; as they are less air-sensitive they produce H2 even in the presence of low concentrations of O2.6 Compared to their cysteine homologues the [NiFeSe] hydrogenases have higher catalytic activity in the hydrogen evolution reaction.3,7 This difference in activity may be explained by the differences in the physical properties of selenium compared to those of sulfur, such as its higher acidity and higher nucleophilicity, in addition to the lower redox potential of the selenocysteine redox couple. The pKa of Sec is 5.3 whereas the pKa of Cys is 8.3, which may help in the rapid exchange of protons. Selenium is also a softer donor atom than sulfur; the polarizable volume of selenium is 3.8 Å in comparison to 2.9 Å for sulfur. Although these different properties can be the potential causes for the higher catalytic activity of the [NiFeSe] hydrogenases, the exact role of selenocysteine in the [NiFeSe] hydrogenases is still not completely clear.8,9
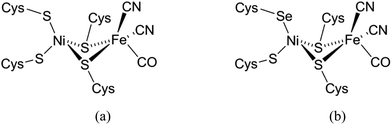 |
| | Fig. 1 Schematic representation of the active site in (a) [NiFe] and (b) [NiFeSe] hydrogenases.4,5 | |
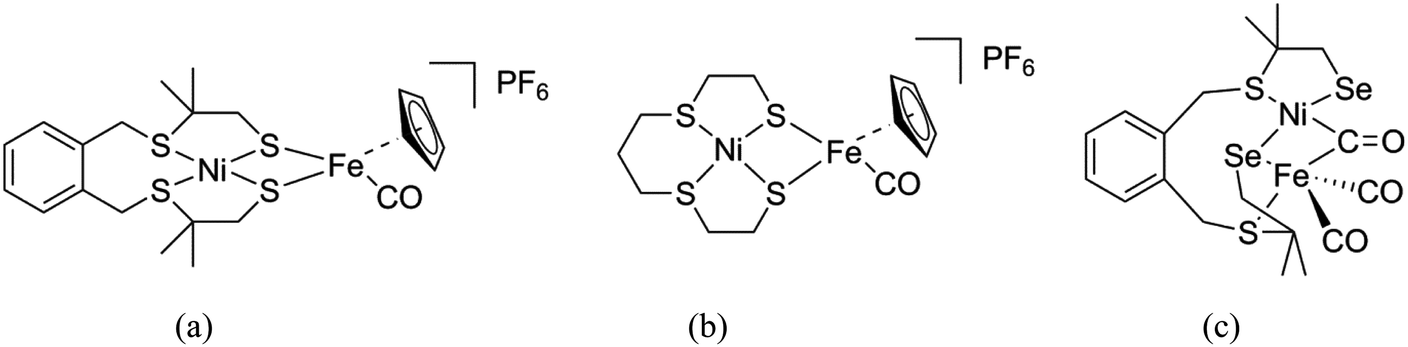
In the past decades a large number of structural and functional models for the active site in [NiFe] hydrogenases have been reported with overpotentials as low as 50 mV. From these studies, it was found that the addition of a cyclopentadienyl (Cp−) ligand resulted in increased catalytic rates and stability of the catalyst.10–14 Apart from these models, a number of mononuclear Ni/Co/Fe complexes and several heterodimetallic [NiRu] complexes have been reported as functional models of the [NiFe] hydrogenases active site.15–19 Two heterodinuclear compounds related to our work described in this paper have been reported by Artero and Schröder, comprising NiS4 centers bound to a {FeCpCO} group (Fig. 2a and b).11,20 However, so far only one heterodimetallic nickel–iron complex was reported comprising a selenolate ligand coordinated to the nickel center, as a potential model of the active site in [NiFeSe] hydrogenases (Fig. 2c).7 In this paper, we describe the synthesis and characterization of two new heterodimetallic nickel–iron complexes (Fig. 3) as mimics of the [NiFeSe] hydrogenase active site. The electrochemical properties and electrocatalytic activity for H2 production of these NiFe complexes are reported.
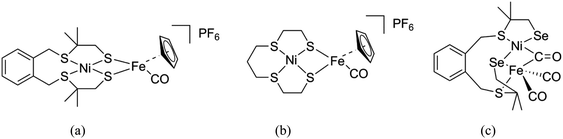 |
| | Fig. 2 Schematic drawings of the [NiFe] hydrogenase models reported by Artero (a)11 and Schröder (b),20 and the first structural model of [NiFeSe] hydrogenases reported by Reisner (c).7 | |
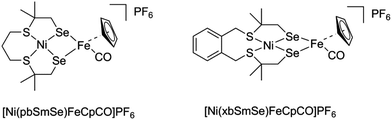 |
| | Fig. 3 Schematic drawing of the heterodimetallic compounds described in this paper. | |
Results and discussion
Synthesis and characterization
The novel heterodinuclear compounds [Ni(pbSmSe)FeCpCO]PF6 and [Ni(xbSmSe)FeCpCO]PF6 were synthesized following the procedure shown in Scheme 1. The precursors to two different selenouronium ligands were synthesized based on reported procedures. The compounds 1,9-dichloro-3,7-dithia-2,2,8,8-tetramethylnonane and bis(3-chloro-2,2-methyl-1-thiapropyl)-o-xylene were treated with two equivalents of selenourea in ethanol to give the selenouronium ligand precursors (1) and (2) as white powders in good purities in 86% and 90% yield, respectively. The ligand precursor (2) has been reported earlier.7 The compounds [Ni(pbSmSe)] (3) and [Ni(xbSmSe)] (4) were synthesized by heating the selenouronium ligand precursors in refluxing ethanol with [Ni(acac)2] in the presence of NMe4OH. The compounds [Ni(pbSmSe)] (3) and [Ni(xbSmSe)] (4) were obtained as green solids in 64% and 83% yield, respectively. The compounds were characterized with 1H and 13C NMR spectroscopy, mass spectrometry, elemental analysis and single crystal X-ray diffraction for compound (3); the X-ray structure of compound (4) has been reported.7 The nickel compounds (3) and (4) give rise to sharp resonances in the 1H NMR spectra indicating that the nickel(II) centers in these compounds are in low-spin, square-planar geometries, which are retained in solution. The clear NMR spectra are in contrast with those of the corresponding sulfur-based compounds [Ni(pbSmS)]21 and [Ni(xbSmS)],22 which generally show broad signals. Such broadening of the NMR signals is ascribed to fluxional behavior of the ligand surrounding the nickel ion; as a result part of the time the nickel centers are in more or less tetrahedral geometries giving rise to the paramagnetic high-spin state. Reaction of the mononuclear nickel complexes (3) and (4) with one equivalent of commercially available [FeCp(CO)2I] in dichloromethane provided the corresponding [Ni(L)FeCpCO]I complexes; subsequently the counter anion was exchanged by the addition of NH4PF6 in acetonitrile resulting in the compounds [Ni(pbSmSe)FeCpCO](PF6) (5) and [Ni(xbSmSe)FeCpCO](PF6) (6). These heterodinuclear complexes were characterized with NMR and FTIR spectroscopy, mass spectrometry, elemental analysis and single crystal X-ray diffraction. The 1H NMR spectra of the NiFe complexes recorded in dichloromethane are weak and poorly resolved, but show the expected signals of the cyclopentadienyl, aromatic and methyl protons; however, the methylene groups which are in close proximity to nickel center are not clearly discernible. Several attempts have been undertaken to obtain better quality 1H NMR spectra of the NiFe complexes; spectra were recorded in another solvent (DMSO) and were recorded at different temperatures, but unfortunately to no avail (ESI, Fig. S12–16†). Again, the broadening of these signals might be explained by tetrahedral distortions of the square-planar geometry, resulting in a fraction of the nickel centers to be in the high-spin state. In contrast to the mononuclear compounds this distortion is not fluxional, but immobilized by the bridging of the two selenolate donor atoms between the Ni(II) and Fe(II) centers (see below). In addition, the results of the mass analysis and IR spectra (see below) indicate that a dynamic equilibrium may exist of compounds that are the result of a disproportionation reaction of (5) or (6) [Ni(L)FeCpCO]+ to form [Ni(L)FeCp]+ and [Ni(L)FeCp(CO)2]+, which would also give rise to broad signals.
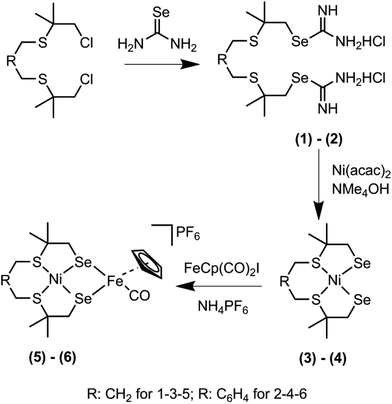 |
| | Scheme 1 Synthesis scheme of the selenouronium salts (1) and (2), the mononuclear Ni complexes (3) and (4) and the heterodinuclear NiFe complexes (5) and (6). | |
Description of the structures
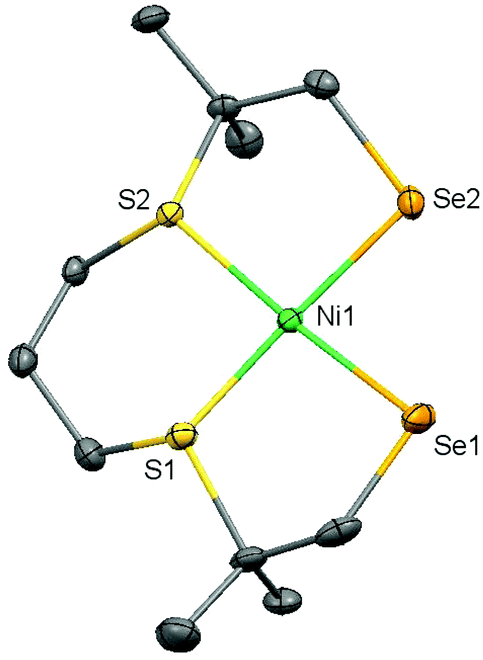
Single crystals of (3) suitable for X-ray structure determination were obtained by vapor diffusion of pentane into a dichloromethane solution of the complex; crystallographic and refinement data are provided in Table S1.† A projection of the molecular structure is given Fig. 4 and selected bond distances and angles are listed in Table 1. Complex (3) crystallizes in the space group P21; the asymmetric unit contains one molecule of the mononuclear compound [Ni(pbSmSe)]. The Ni(II) center is in a square-planar geometry by the coordination of two selenolate and two thioether donor atoms. The Ni–Se bond distances are 2.2898(6) and 2.2910(6) Å; as expected for the larger ionic radius of Se these distances are longer than the Ni–S bond distances of 2.180(8) Å in the thiolate analog [Ni(pbSmS)].21 As a result, the Ni–S thioether bond distances at 2.1707(10) and 2.1608(11) Å are slightly shorter than those of 2.1711(3) and 2.1668(3) Å in [Ni(pbSmS)].21 The square-planar geometry reveals a slight tetrahedral distortion with a dihedral angle of 8.91°, defined by the planes S1–Ni1–S2 and Se1–Ni1–Se2, which is slightly larger than in the thiolate analog having a dihedral angle of 5.17°.21 This larger tetrahedral distortion in the solid state of the selenolate compound is rather surprising, as the NMR spectra of the thiolate compound are broadened due to the fluxionality of the ligand, which results in the nickel ion in the low-spin square-planar structure to be in equilibrium with a nickel center in a more tetrahedral high-spin state. In contrast, the NMR spectra show the selenolate compound to be clearly low-spin and diamagnetic, which may indicate that the tetrahedral distortion in the solid state is merely due to packing effects.
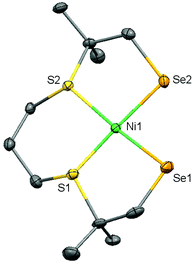 |
| | Fig. 4 Displacement ellipsoid plot (50% probability level) of (3) at 110(2) K. Hydrogen atoms are omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (°) for the complexes (3), (5) and (6)
| |
(3) |
(5) |
(6) |
|
Distances (Å) |
| Ni1–Se1 |
2.2898(6) |
2.2768(8) |
2.2837(5) |
| Ni1–Se2 |
2.2910(6) |
2.2978(7) |
2.2933(5) |
| Ni1–S1 |
2.1608(11) |
2.1996(11) |
2.1835(7) |
| Ni1–S2 |
2.1707(10) |
2.1817(12) |
2.1820(8) |
| Fe1–Se1 |
|
2.3859(7) |
2.4043(5) |
| Fe1–Se2 |
|
2.4018(8) |
2.3923(5) |
| Fe1–Cp(centroid) |
|
1.71(4) |
1.70(3) |
| Fe1–C12/C22 |
|
1.741(4) |
1.773(3) |
| |
|
Angles (°) |
| S1–Ni1–Se1 |
89.79(3) |
92.75(4) |
91.31(2) |
| S2–Ni1–Se2 |
90.79(3) |
93.00(4) |
91.74(2) |
| Se1–Ni1–Se2 |
87.62(2) |
82.52(3) |
80.550(17) |
| S1–Ni1–S2 |
91.80(4) |
91.27(4) |
95.02(3) |
| S2–Ni1–Se1 |
170.94(4) |
173.92(4) |
167.66(3) |
| S1–Ni1–Se2 |
177.40(4) |
172.33(4) |
169.19(3) |
| Se1–Fe1–Se2 |
|
78.11(2) |
76.174(15) |
| Ni1–Se1–Fe1 |
|
85.40(3) |
81.074(17) |
| Ni1–Se2–Fe1 |
|
84.58(3) |
81.139(17) |
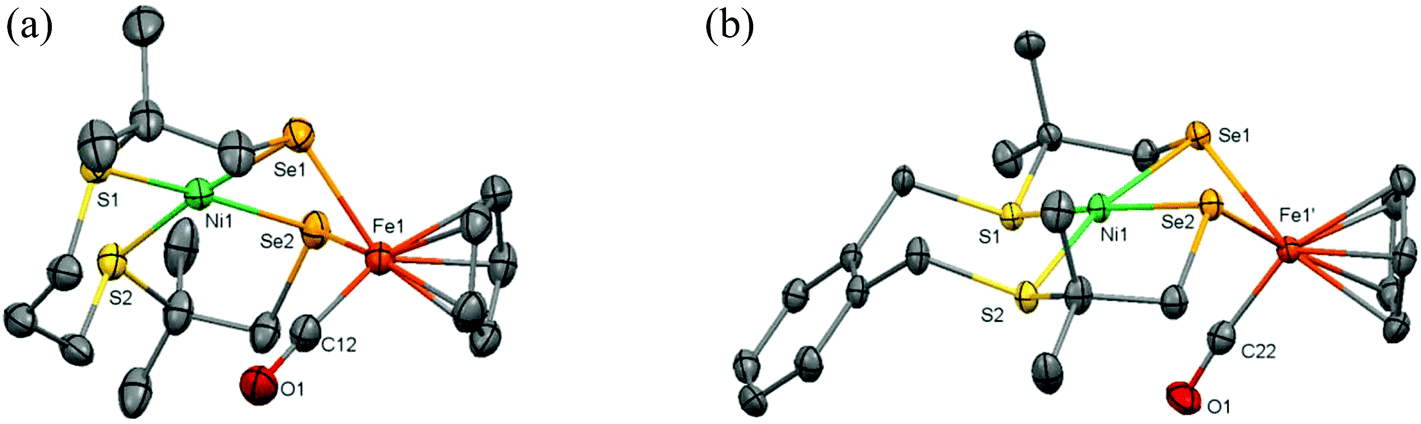
Single crystals of the complexes (5) and (6) were obtained by vapor diffusion of diethyl ether into dichloromethane solutions of the complexes; crystallographic and refinement data are provided in Table S1.† Projections of the molecular structures of the heterodinuclear complexes are shown in Fig. 5; selected bond distances and angles are listed in Table 1. Complex (5) crystallizes in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) and the crystal lattice contains some amount of significantly disordered solvents molecules. Complex (6) crystallizes in the space group P21/c; the coordination spheres around Ni1 and the CO coordinated to Fe1 are found to be slightly disordered over two orientations. The nickel(II) ions in the complexes (5) and (6) are in square-planar geometries bound to two thioether and two selenolate donor atoms. In both compounds the two Se donor atoms are bridging to the Fe(II) ion, resulting in a Ni–Se–Fe–Se ‘butterfly’ core with a ‘hinge’ angle (defined by the angle between the planes through NiSe2 and FeSe2) of 120.87° for (5) and 108.36° for (6). The Fe(II) ion in both complexes is further coordinated to a symmetrically bound η5-cyclopentadienyl ligand and a CO ligand. The Ni–Se distances of 2.2837(5) and 2.2933(5) Å in [Ni(xbSmSe)FeCpCO]PF6 are longer than the corresponding Ni–S thiolate distances of 2.1670(9) and 2.1717(8) Å found in [Ni(xbSmS)FeCpCO]BF4,11 as a result of the larger ionic radius of the selenolate donor atom. For both NiFe complexes the square-planar geometry of the nickel centers is slightly distorted, with dihedral angles of 7.39° and 12.63° for complexes (5) and (6) respectively. This distortion seems to be caused by the bridging of both selenolate atoms between the Ni(II) and Fe(II) centers, resulting in larger S–Ni–Se and significantly smaller Se–Ni–Se angles. In both complexes the molecule of CO is directed towards the Ni center with a Ni–C(O) distance of 3.1 Å for complex (5) and 2.9 Å for complex (6). The major difference between the two heterodinuclear compounds is the relative orientation of the {FeCpCO} group. Whereas in complex (5) the {FeCpCO} group and the bridge between the sulfur atoms are on the same side of the Ni square plane, in complex (6) they are on opposite sides.
and the crystal lattice contains some amount of significantly disordered solvents molecules. Complex (6) crystallizes in the space group P21/c; the coordination spheres around Ni1 and the CO coordinated to Fe1 are found to be slightly disordered over two orientations. The nickel(II) ions in the complexes (5) and (6) are in square-planar geometries bound to two thioether and two selenolate donor atoms. In both compounds the two Se donor atoms are bridging to the Fe(II) ion, resulting in a Ni–Se–Fe–Se ‘butterfly’ core with a ‘hinge’ angle (defined by the angle between the planes through NiSe2 and FeSe2) of 120.87° for (5) and 108.36° for (6). The Fe(II) ion in both complexes is further coordinated to a symmetrically bound η5-cyclopentadienyl ligand and a CO ligand. The Ni–Se distances of 2.2837(5) and 2.2933(5) Å in [Ni(xbSmSe)FeCpCO]PF6 are longer than the corresponding Ni–S thiolate distances of 2.1670(9) and 2.1717(8) Å found in [Ni(xbSmS)FeCpCO]BF4,11 as a result of the larger ionic radius of the selenolate donor atom. For both NiFe complexes the square-planar geometry of the nickel centers is slightly distorted, with dihedral angles of 7.39° and 12.63° for complexes (5) and (6) respectively. This distortion seems to be caused by the bridging of both selenolate atoms between the Ni(II) and Fe(II) centers, resulting in larger S–Ni–Se and significantly smaller Se–Ni–Se angles. In both complexes the molecule of CO is directed towards the Ni center with a Ni–C(O) distance of 3.1 Å for complex (5) and 2.9 Å for complex (6). The major difference between the two heterodinuclear compounds is the relative orientation of the {FeCpCO} group. Whereas in complex (5) the {FeCpCO} group and the bridge between the sulfur atoms are on the same side of the Ni square plane, in complex (6) they are on opposite sides.
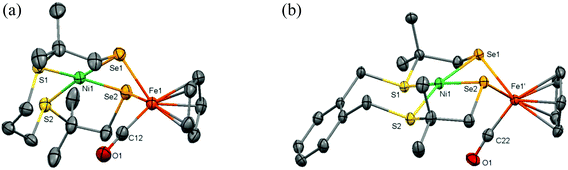 |
| | Fig. 5 Displacement ellipsoids plots (50% probability level) of the cationic complex in (a) (5) and (b) (6) at 110(2) K. Hydrogen atoms, PF6− anions, lattice solvent molecules, and disorder (in (6)) are omitted for clarity. | |
IR spectroscopy of the NiFe complexes
The carbonyl stretching bands in the solid state IR spectra of compound (5) appear at 1918 cm−1 (m), 1987 (s) and 2035 (s), whereas the IR spectrum of compound (6) reveals one strong band at 1923 cm−1 in addition two weaker bands at 1989 and 2037 cm−1 (ESI, Fig. S7 and 8†). Based on its structure complex (5) should show only a single CO band. Indeed, the compound [Ni(xbSmS)FeCpCO]BF4 has been reported to show a single carbonyl stretching band in the IR spectrum at 1939 cm−1.11 However, the compound [Ni(xbSmS)FeCp(CO)2]BF4 was reported to be an intermediate in the formation of [Ni(xbSmS)FeCpCO]BF4, showing two CO stretching bands at 2008 and 2054 cm−1. Interestingly, the mass spectra recorded of crystals of compound (5) show three major fragments: two of them are assigned to the expected monocationic compound [M − PF6]+ and the compound after loss of the CO ligand [M − (PF6) − (CO)]+ (ESI Fig. S19†). Unexpectedly, also a fragment that can be assigned to the monocationic compound with two CO ligands ([M − (PF6) + (CO)]+) is observed. This fragment must be the result of disproportionation of [Ni(pbSmSe)FeCpCO]+ to form [Ni(pbSmSe)FeCp]+ and [Ni(pbSmSe)FeCp(CO)2]+, the former compound would contain a 16-electron FeII center, whereas the second species would be asymmetrically bridged by only one of the selenolate atoms. In contrast, whereas the IR of compound (6) indicates that a product containing two molecules of CO bound to iron may be present, the mass spectrum of (6) does not show a peak that can be assigned to a fragment [M − (PF6) + (CO)]+. We therefore have to conclude that whereas for compound (6) the product with one CO ligand bound to iron is the most stable one, for compound (5) a mixture of [Ni(pbSmSe)FeCpCO](PF6) and [Ni(pbSmSe)FeCp(CO)2](PF6) is obtained, from which a single crystal of the monocarbonyl complex was picked. Because of the small mass difference of one molecule of CO the elemental analysis is not conclusive.
Similar values of IR stretching bands have been reported for the carbonyl ligands in the active sites of the [NiFe] and [NiFeSe] hydrogenases as well as for another structural model of [NiFeSe] hydrogenases.7 The relatively lower energy of the CO stretching frequencies in the selenolate compounds has been attributed to an increase of electron density at the Fe center, as the selenolate donor atoms are more electron-donating than thiolate donor atoms.7
Electrochemical analyses
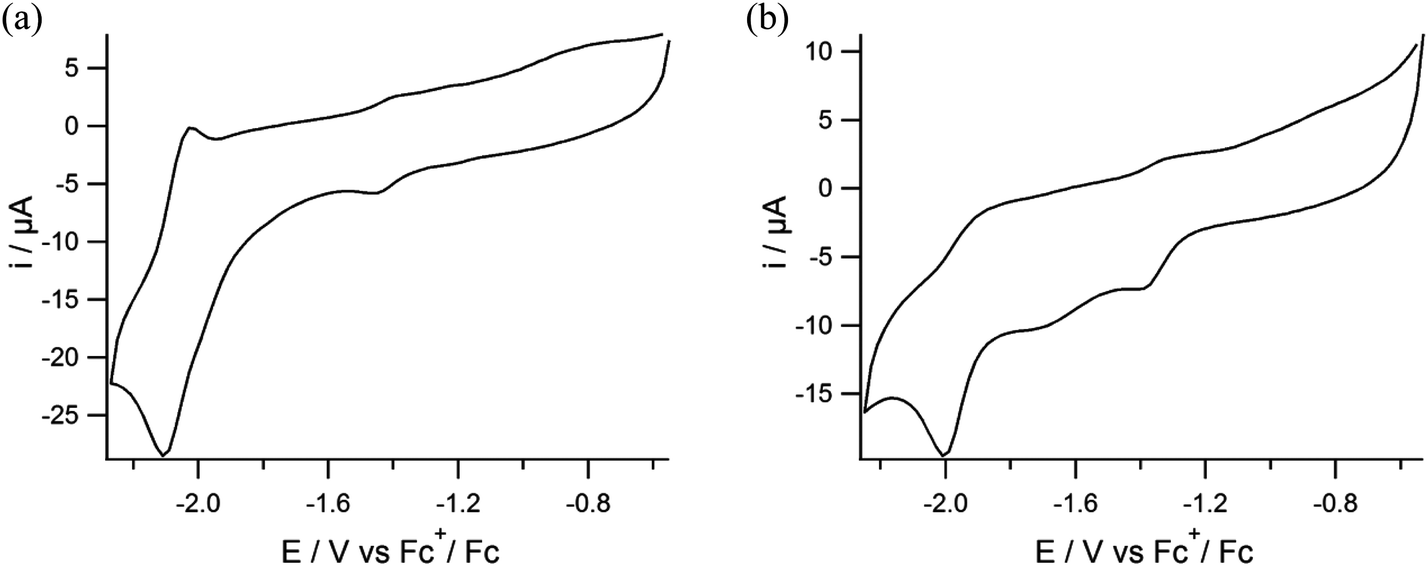
The electrochemical properties of the nickel and nickel–iron complexes were investigated using cyclic voltammetry; the relevant data are presented in Table 2. For the mononuclear complex (3) one reversible reduction wave is observed with an Epc at −2.1 V vs. Fc+/Fc at a scan rate of 200 mV s−1 in DMF (Fig. 6a), which is tentatively ascribed to the NiII/NiI redox couple. In contrast, the corresponding thiolate-containing compound [Ni(pbSmS)] has been reported to show two irreversible waves at −1.05 V and −1.5 V vs. Fc+/Fc at a scan rate of 100 mV s−1 in dichloromethane solution.21 We could not readily find an explanation for the large difference of nearly 1 V between the observed reduction potential of (3) and the values reported for [Ni(pbSmS)]. Therefore a CV of complex (3) was also recorded in dichloromethane solution (ESI, Fig. S3†). Although the reduction of (3) appeared to be irreversible in dichloromethane, the reduction potential of (3) was found to be similar in both dichloromethane and in DMF solution. For the nickel complex (4) one irreversible wave is observed with an Epc at −1.99 V vs. Fc+/Fc at a scan rate of 200 mV s−1 in DMF (Fig. 6b), similar to the irreversible electrochemical behavior reported for the analogous thiolate compound [Ni(xbSmS)] at −2.03 V vs. Fc+/Fc in DMF solution.11 The slightly less negative reduction potential for the nickel center in compound (4) relative to that in (3) may tentatively be ascribed to larger flexibility of the 7-membered chelate ring of the xylyl backbone, facilitating a change in redox state of the nickel center. We do not have an explanation for the differences in reversibility of the reduction wave of the nickel centers in (3) and (4), nor for the observation that the reduction wave for (3) is reversible in DMF, whereas it is irreversible in DCM. Furthermore, we cannot give a reason for the apparent 1 V difference in the reduction potentials of [Ni(pbSmSe)] (3) and [Ni(pbSmS)], especially as the difference in reduction potentials for the xylene-bridged compounds [Ni(xbSmS)] and [Ni(xbSmSe)] (4) is negligible.
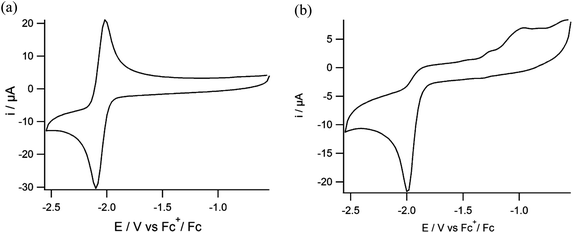 |
| | Fig. 6 Cyclic voltammograms of (a) compound (3) and (b) compound (4) in DMF solutions containing TBAPF6 (0.1 M) as the supporting electrolyte on a glassy carbon electrode at 200 mV s−1. | |
Table 2 Electrochemical data of the Ni and NiFe complexes (vs. Fc+/Fc)a
| Compound |
E
pa (V) |
E
pc (V) |
|
Experimental conditions: 1 mM solutions of complexes in DMF containing 0.1 M TBAPF6 as the supporting electrolyte. Glassy carbon working electrode, platinum counter electrode, Ag/AgCl reference electrode and scan rate 200 mV s−1. The values have been calculated using Fc/Fc+ as an internal reference, which was found to have E1/2 of 0.54 V vs. Ag/AgCl in our conditions.
|
| (3) |
−2.02 |
−2.10 |
| (4) |
|
−1.99 |
| (5) |
−2.03 |
−2.10 |
| (6) |
|
−1.99 |
The same electrochemical conditions with a scan rate of 200 mV s−1 were also used to study the electrochemical behavior of the NiFe complexes (5) and (6) in DMF solutions. For complex (5) one quasi-reversible wave is observed with an Epc at −2.1 V vs. Fc+/Fc (Fig. 7a) with an ipc that is nearly two times higher than the ipa. For complex (6) one quasi-reversible wave is observed with an Epc at −1.99 V vs. Fc+/Fc (Fig. 7b). At first sight it thus seems that the presence of the cyclopentadienyl-iron moiety does not influence the reduction potential of the nickel(II) ion. In addition for both complexes one small wave is observed at around −1.4 V vs. Fc+/Fc.
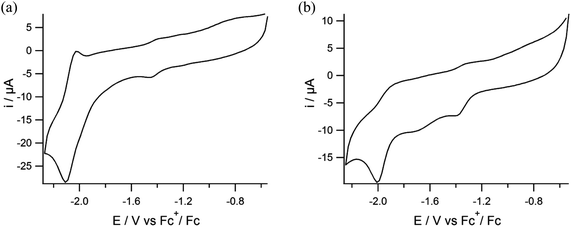 |
| | Fig. 7 Cyclic voltammograms of (a) compound (5) and (b) compound (6) in DMF solutions containing TBAPF6 (0.1 M) as the supporting electrolyte on a glassy carbon electrode at 200 mV s−1. | |
In order to better understand the electrochemical properties of the nickel–iron complexes, the electrochemical behavior of [FeCp(CO)2I] was also examined (Fig. S4†). For this compound one irreversible reduction is observed with an Epc at −2.05 V vs. Fc+/Fc at a scan rate of 200 mV s−1 in DMF, which is tentatively ascribed to the FeII/FeI redox couple. Similar to the nickel–iron complexes, an additional small wave is observed at −1.3 V vs. Fc+/Fc. This feature thus indicates that the small wave around −1.4 V vs. Fc+/Fc in the voltammograms of the nickel–iron complexes is related to the presence of the [FeCp(CO)I] moiety. It seems that the quasi-reversible reduction of complex (5) with an Epc at −2.1 V vs. Fc+/Fc is an overlap of the reduction of the nickel ion in (3) (Epc at −2.1 V vs. Fc+/Fc) with the reduction wave of iron center in [FeCp(CO)2I] (at −2.05 V vs. Fc+/Fc); upon closer inspection of the reduction wave for compound (5) it looks as if this wave indeed reveals a shoulder around −2.0 V. This overlap of two redox events may also explain why the reversibility of this reductive peak changed from reversible in the mononuclear complex [Ni(pbSmSe)] to quasi-reversible in the heterodinuclear compound (5). In comparison, the complex [Ni(xbSmS)FeCpCO]BF4 has been reported to show one reversible redox couple at −1.43 V vs. Fc+/Fc and one irreversible wave at −2.01 V vs. Fc+/Fc in DMF.11 In contrast to the CVs of the complexes (5) and (6), in this report the reduction wave at −1.43 V vs. Fc+/Fc has the same intensity as the one at −2.01 V vs. Fc+/Fc. The reduction wave of complex (6) appears to become more reversible compared to that of the corresponding mononuclear nickel complex [Ni(xbSmSe)].
Electrocatalytic hydrogen evolution in the presence of HOAc
The activity of the new compounds in electrocatalytic proton reduction was studied using cyclic voltammetry with addition of varying amounts of HOAc to DMF solutions of the Ni and NiFe complexes. The reversible reduction observed for complex (3) with an Epc at −2.1 V vs. Fc+/Fc becomes irreversible with the addition of HOAc (ESI, Fig. S1†). On the other hand, for complex (4) the irreversible reduction peak with an Epc at −1.99 V does not change upon addition of HOAc and no catalytic current is observed (ESI, Fig. S2†). The quasi-reversible reduction peak of complex (5) with an Epc at −2.1 V vs. Fc+/Fc becomes irreversible with increasing concentrations of HOAc while the Epc shifts to more negative potentials indicating electrocatalytic activity (Fig. 8). Again, for complex (6) no catalytic wave is observed upon the addition of different equivalents of acid.
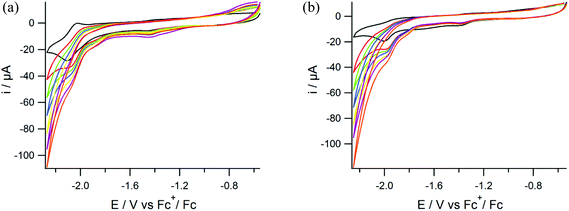 |
| | Fig. 8 Cyclic voltammograms of (a) compound (5) and (b) compound (6) in DMF solutions of TBAPF6 (0.1 M) on a glassy carbon electrode at 200 mV s−1 in the presence of 0 (black), 10 (red), 20 (green), 30 (blue), 40 (yellow), 50 (purple), 60 (orange) mM of acetic acid. | |
To quantify the rate of the hydrogen evolution reaction the foot-of-the-wave (FOWA) analysis was applied.23,24 The FOWA analysis was developed by Costentin and Savéant and can be used for the analysis of voltammograms that do not show an S-shaped curve with a fixed plateau current.23 Using the FOWA analysis a kobs of 24 s−1 was calculated for complex (5) (ESI, Fig. S5†). In order to confirm that indeed dihydrogen gas is formed in the catalytic reaction, a controlled-potential coulometry (CPC) experiment was carried out on a 0.5 mM solution of complex (5) in DMF (5 ml) in the presence of 17.5 μl of HOAc (50 equivalents of H+ per NiFe compound) at −2.1 V vs. Fc+/Fc. The produced dihydrogen gas was quantified volumetrically by GC analysis. The CPC experiment was run for 50 min, while the solution was stirred continuously. Using complex (5) as the electrocatalyst for proton reduction, a total of 64 μl H2was produced by 0.5 mM complex in 50 min with 72% faradaic yield. In a control experiment at this potential formation of H2 is not observed in the absence of the catalyst. In order to compare the activity of the mononuclear [Ni(pbSmSe)] and the dinuclear compound [Ni(pbSmSe)FeCpCO]PF6 a CPC experiment was also run for complex (3). After 50 min the amount of H2 produced by compound (3) appeared to be very low compared to the NiFe complex (5); the formed H2 was barely detectable with a concentration falling out of the lower range of the calibration line. A CPC experiment was also carried out using complex (6) in DMF solution in the presence of HOAc at −1.9 V vs. Fc+/Fc. In this case dihydrogen evolution was not observed and it can be concluded that this compound is not active as an electrocatalyst for proton reduction with a weak acid such as HOAc. In contrast, the complex [Ni(xbSmS)FeCpCO]BF4, the thiolate analogue of complex (6), has been reported to be an electrocatalyst for H2 evolution, which achieved 20 turnovers in 4 h in the presence of the stronger acid trifluoroacetic acid.11
Conclusion
Two novel NiFe complexes are reported as mimics of the [NiFeSe] hydrogenase active site. Both [Ni(pbSmSe)FeCpCO]PF6 and [Ni(xbSmSe)FeCpCO]PF6 complexes have structural similarities with active site of [NiFeSe] hydrogenase. The compounds contain nickel and iron centers coordinated with two selenolate and two thioether donors. The bond distances between the nickel centers and the selenolate donors are 2.29 Å for complexes (5) and (6), whereas the Ni–Se distance in the active site of [NiFeSe] hydrogenase has been reported to be 2.46 Å.5 The major drawback of the selenium complexes compared to the reported sulfur analogues is their higher air sensitivity. The compound [Ni(pbSmSe)FeCpCO]PF6 catalyzes the electrocatalytic reduction of protons in the presence of acetic acid, as shown by CV and CPC experiments, whereas [Ni(xbSmSe)FeCpCO]PF6 does not. The reason for this difference in activity may lie in the disproportionation reaction that seemingly occurs for compound (5) but not for (6), generating an active 16-electron species for (5). To the best of our knowledge compound (5) is thus the first functional model of the [NiFeSe] hydrogenase active site. Although this NiFe compound is just a very modest catalyst, this work is an initial step for the development of more efficient mimics of the [NiFeSe] hydrogenase active site.
Experimental
Materials
All experiments were performed using standard Schlenk techniques or in a glovebox under an argon atmosphere unless otherwise noted. Chemicals were purchased from Acros or Aldrich and were used without further purification. Organic solvents were deoxygenated by the freeze–pump–thaw method and were dried over molecular sieves prior to use. The NMR solvent CD2Cl2 for the metal complexes was deoxygenated by the freeze–pump–thaw method and was stored over molecular sieves in a glovebox. The compounds 1,9-dichloro-3,7-dithia-2,2,8,8-tetramethylnonane,21 bis(3-chloro-2,2-methyl-1-thiapropyl)-o-xylene,22 and [Ni(xbSmSe)]7 were synthesized according to published methods. [FeCp(CO)2I] was purchased from Aldrich. The synthesis of the [NiFe] complexes is based on a method described in literature.20
Physical measurements
NMR spectra were recorded on a 300 MHz Bruker DPX 300 spectrometer and chemical shifts were referenced against the solvent peaks. Mass spectra were obtained with a Finnigan TSQ quantum instrument using ESI. HRMS was recorded on a Thermo Scientific LTQ Orbitrap XL high resolution FT-MS system. Elemental analyses were performed by the Microanalytical Laboratory Kolbe in Germany. IR spectra were recorded on a PerkinElmer UATR Two FT-IR spectrometer. Electrochemical measurements were performed at room temperature under argon using an Autolab PGstat10 potentiostat controlled by GPES4 software. A three-electrode cell system was used with a glassy carbon working electrode, a platinum counter electrode and an Ag/AgCl reference electrode. All electrochemistry measurements were done in DMF solution with tetrabutylammonium hexafluoridophosphate as the supporting electrolyte. All potentials are referenced to the internal reference system Fc+/Fc, which under these conditions was found at 0.54 V vs. Ag/AgCl in DMF. Electrocatalysis experiments were carried out by adding different concentrations of acetic acid to the DMF solution of complexes. Controlled-potential coulometry (CPC) experiments were done with the same three-electrode cell system and electrodes. CPC experiments were recorded with an Autolab PGstat10 potentiostat controlled by GPES4 software. Gas chromatographic analysis was performed on a Shimadzu gas chromatograph GC-2010 at 35 °C fitted with a Supelco Carboxen 1010 molecular sieve column. Helium was used as the carrier gas, and compounds were detected using a thermal conductivity detector operated at 80 mA. The total volume of H2 produced during the reaction was calculated using a calibration line, which was obtained using the external reference method by injection of known amounts of H2 into the GC using a Hamilton gas-tight syringe (see ESI Fig. S6†). A solution of complexes (5) or (6) in DMF (5 ml, 0.5 mM) was placed into a three-electrode cell and prior to each measurement the system was deaerated by bubbling with helium for 10 min. The system was closed, and the headspace was pumped through the solution for 1 min. Afterward, the headspace pumping was temporarily stopped to allow equilibration of the pressure, then the GC measurement was started with a 0.5 mL sample of the headspace injection. The GC valve and the pump (KNF NMS 010 L micro diaphragm pump) were enclosed in a helium-purged housing to prevent air from leaking into the system.
Crystal structure determinations
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Mo Kα radiation (λ = 0.71073 Å) for (3) and Cu Kα radiation (λ = 1.54178 Å) for (5) and (6) under the program CrysAlisPro (Version 1.171.36.32 Agilent Technologies, 2013). The same program was used to refine the cell dimensions and for data reduction. The structures were solved with the program SHELXS-2014/7 and were refined on F2 with SHELXL-2014/7.25 Numerical absorption correction based on gaussian integration or Analytical numeric absorption correction over a multifaceted crystal model was applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions using the instructions AFIX 23, AFIX 43 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 Ueq of the attached C atoms.
Additional notes on the structure determinations
(3) The structure was refined in the space group P21. The absolute configuration was established by anomalous-dispersion effects in diffraction measurements on the crystal. The Flack parameter refines to −0.002(5). CCDC 1537790 contains the supplementary crystallographic data for [Ni(pbSmSe)].
(5) The crystal lattice contains some amount of significantly disordered solvent molecules found in ‘channels’ along the c direction. Their contribution has been taken out using the SQUEEZE (Spek, 2009) procedure in the final refinement.26
(6) The coordination sphere around the Ni center in the complex as well as the CO coordinated to the iron center are found to be slightly disordered over two orientations; the occupancy factor of the major component of the disorder refines to 0.9585(6). Disorder may occur as both orientations have very similar space-filling requirements. CCDC 1537791 and 1537792 contain the supplementary crystallographic data for [Ni(pbSmSe)Fe(CO)Cp](PF6) and [Ni(xbSmSe)Fe(CO)Cp](PF6).
Synthesis of ligand precursor (1)
A solution of selenourea (1.2 g, 9.6 mmol) in 10 ml ethanol was added to a solution of 1,9-dichloro-3,7-dithia-2,2,8,8-tetramethylnonane (1,4 g, 4.83 mmol) in 5 ml ethanol; the reaction mixture was refluxed for 30 min. The solution was allowed to cool, and the solid product was isolated by filtration. The product was washed with cold ethanol and diethyl ether, and dried in vacuo yielding a white powder. Yield: 2.2 g (86%). 1H NMR (300 MHz, (CD3)2SO) = δ (ppm): 9.41 (d, J = 11.3 Hz, 8H, NH), 3.61 (s, 4H, CH2), 2.66 (t, J = 7.2 Hz, 4H, CH2), 1.73 (q, J = 7.1 Hz, 2H, CH2), 1.66 (s, 12H, CH3). 13C NMR (75 MHz, (CD3)2SO): δ (ppm) = 167.02 (C(NH)NH2), 46.31 (CH2–Se), 40.57 (CH2–S), 29.67 (CH2–CH2S), 28.07 (CH3). ESI-MS (H2O): 233.1, calcd: 233.01 [M − 2Cl]2+.
Synthesis of [Ni(pbSmSe)] (3)
A solution of NMe4OH (558 mg, 3.08 mmol), ligand precursor (1) (824 mg, 1.54 mmol) and Ni(acac)2 (396 mg, 1.54 mmol) in 170 ml ethanol was refluxed for 1 h. The solvent was evaporated until approximately 30 ml solvent remained, resulting in a green precipitate. The solid was collected by filtration and washed with ethanol. Yield: 429 mg (64%). 1H NMR (300 MHz, CD2Cl2) δ (ppm): 2.64 (t, J = 7.1 Hz, 4H, CH2–S), 2.41 (s, 4H, CH2–Se), 2.22 (m, 2H, CH2–CH2S), 1.57 (s, 12H, CH3). 13C NMR (75 MHz, CD2Cl2):δ (ppm): 29.19 (CH2, CH2–CH2S), 28.80 (CH2, CH2–Se), 26.70 (CH3), 25.39 (CH2–CH2S). HR-MS (CH2Cl2): 436.89255, calcd: 436.89196 [M + H]+. Elemental analysis calcd (%) for C11H22NiS2Se2·0.2 N(CH3)4Cl: C: 31.02 H: 5.38; found C: 30.73 H: 5.86.
Synthesis of [Ni(pbSmSe)Fe(CO)Cp](PF6) (5)
A solution of [Ni(pbSmSe)] (400 mg, 0.91 mmol) and [FeCp(CO)2I] (279 mg, 0.91 mmol) in 25 ml dichloromethane was stirred at RT for 2 days. The mixture was filtered to remove an insoluble precipitate and the solvent was evaporated using the Schlenk line. The resulting solid was washed with diethyl ether resulting in a brown precipitate which was dried in vacuo. A solution of NH4PF6 (300 mg, 1.84 mmol) in 10 ml acetonitrile was added to the brown solid and the mixture was stirred for 4 h. The solvent was evaporated until dryness, the remaining solid was dissolved in dichloromethane (5 ml) and the solution was filtered to remove NH4I. A large amount (∼30 ml) of diethyl ether was added into the dichloromethane solution, and the mixture was cooled at −35 °C overnight. The resulting brown precipitate was collected by filtration and dried in vacuo. Yield: 105 mg (20%). 1H NMR (300 MHz, CD2Cl2) δ (ppm): 4.75 (s, Cp). ESI-MS (CH3CN): 584.9, calcd: 584.9 [M − PF6]+, 612.9, calcd: 612.9 [M − (PF6) + (CO)]+ and 556.9, calcd: 556.9 [M − (PF6) − (CO)]+. Elemental analysis calcd (%) for C17H27F6FeNiOPS2Se2·0.25(C2H5)2O: C: 28.92 H: 3.98; found C: 29.00 H: 3.93. IR (neat): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2035 (s), 1987 and 1918 (CO stretch) cm−1, 830 (PF6 stretch) cm−1.
= 2035 (s), 1987 and 1918 (CO stretch) cm−1, 830 (PF6 stretch) cm−1.
Synthesis of [Ni(xbSmSe)Fe(CO)Cp](PF6) (6)
A solution of [Ni(xbSmSe)] (200 mg, 0.40 mmol) and [FeCp(CO)2I] (122 mg, 0.40 mmol) in 15 ml dichloromethane was stirred at RT for 2 days. The mixture was filtered to remove an insoluble precipitate and the solvent was evaporated using the Schlenk line. The residue was washed with diethyl ether, resulting in a brown powder which was dried in vacuo. A solution of NH4PF6 (132 mg, 0.81 mmol) in 5 ml acetonitrile was added to the brown solid and the resulting mixture was stirred for 4 h. The solvent was evaporated until dryness, the remaining solid was dissolved in dichloromethane (5 ml) and the solution was filtered to remove NH4I. A large amount (∼30 ml) of diethyl ether was added into the dichloromethane solution, and the mixture was cooled at −35 °C overnight. The resulting brown precipitate was collected by filtration and dried in vacuo. Yield: 18 mg (6%). 1H NMR (300 MHz, CD2Cl2) δ (ppm): 7.93 (m, Ar), 7.31 (m, Ar), 4.74 (s, Cp), 1.76 (s, –CH3), 1.66 (s, –CH3). ESI-MS (CH3CN): 619.0, calcd: 618.9 [M − CO − PF6]+, 647.0, calcd: 646.9 [M − PF6]+. Elemental analysis calcd (%) for C22H29F6FeNiOPS2Se2·0.3CH2Cl2: C: 32.56 H: 3.64; found C: 32.33 H 3.77. IR (neat): = 1923 (CO stretch) cm−1, 828 (PF6 stretch) cm−1.
Acknowledgements
Gerwin K. Spijksma is gratefully acknowledged for HRMS measurement and Hanan Al Habobe is gratefully acknowledged for ESI-MS measurements.
References
- D. Sellmann, F. Lauderbach and F. W. Heinemann, Eur. J. Inorg. Chem., 2005, 371 CrossRef CAS.
- H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577 RSC.
-
L. De Gioia, Bioinorganic Fundamentals and Applications: Metals in Natural Living Systems and Metals in Toxicology and Medicine, Elsevier, 2013, vol. 3 Search PubMed.
- A. Volbeda, M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580 CrossRef CAS PubMed.
- E. Garcin, X. Vernede, E. Hatchikian, A. Volbeda, M. Frey and J. Fontecilla-Camps, Structure, 1999, 7, 557 CrossRef CAS PubMed.
- M. C. Marques, C. Tapia, O. Gutierrez-Sanz, A. R. Ramos, K. L. Keller, J. D. Wall, A. L. De Lacey, P. M. Matias and I. A. C. Pereira, Nat. Chem. Biol., 2017, 13, 2335 CrossRef PubMed.
- C. Wombwell and E. Reisner, Chem. – Eur. J., 2015, 21, 8096 CrossRef CAS PubMed.
- D. Steinmann, T. Nauser and W. H. Koppenol, J. Org. Chem., 2010, 75, 6696 CrossRef CAS PubMed.
- C. S. Baltazar, M. C. Marques, C. M. Soares, A. M. DeLacey, I. A. Pereira and P. M. Matias, Eur. J. Inorg. Chem., 2011, 948 CrossRef CAS.
- M. Fontecave and V. Artero, C. R. Chim., 2011, 14, 362 CrossRef CAS.
- S. Canaguier, M. Field, Y. Oudart, J. Pécaut, M. Fontecave and V. Artero, Chem. Commun., 2010, 46, 5876 RSC.
- S. Canaguier, V. Artero and M. Fontecave, Dalton Trans., 2008, 315 RSC.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682 CrossRef CAS PubMed.
- B. E. Barton and T. B. Rauchfuss, J. Am. Chem. Soc., 2010, 132, 14877 CrossRef CAS PubMed.
- T. Liu, S. Chen, M. J. O'Hagan, M. R. DuBois, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 134, 6257 CrossRef CAS PubMed.
- Y. Oudart, V. Artero, J. Pécaut, C. Lebrun and M. Fontecave, Eur. J. Inorg. Chem., 2007, 2613 CrossRef CAS.
- S. Canaguier, L. Vaccaro, V. Artero, R. Ostermann, J. Pecaut, M. J. Field and M. Fontecave, Chem. – Eur. J., 2009, 15, 9350 CrossRef CAS PubMed.
- Y. Oudart, V. Artero, L. Norel, C. Train, J. Pécaut and M. Fontecave, J. Organomet. Chem., 2009, 694, 2866 CrossRef CAS.
- G. M. Chambers, R. Angamuthu, D. L. Gray and T. B. Rauchfuss, Organometallics, 2013, 32, 6324 CrossRef CAS.
- W. Zhu, A. C. Marr, Q. Wang, F. Neese, D. J. Spencer, A. J. Blake, P. A. Cooke, C. Wilson and M. Schröder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18280 CrossRef CAS PubMed.
- K. Weber, I. Heise, T. Weyhermüller and W. Lubitz, Eur. J. Inorg. Chem., 2014, 148 CrossRef CAS.
- J. A. Verhagen, D. D. Ellis, M. Lutz, A. L. Spek and E. Bouwman, Dalton Trans., 2002, 1275 RSC.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235 CrossRef CAS PubMed.
- N. Elgrishi, B. M. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1537790–1537792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00972k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a