
Near-infrared-emitting heteroleptic cationic iridium complexes derived from 2,3-diphenylbenzo[g]quinoxaline as in vitro theranostic photodynamic therapy agents†
Li
Wang
a,
Huimin
Yin
b,
Peng
Cui
ac,
Marc
Hetu
b,
Chengzhe
Wang
a,
Susan
Monro
b,
Richard D.
Schaller
d,
Colin G.
Cameron
e,
Bingqing
Liu
a,
Svetlana
Kilina
 a,
Sherri A.
McFarland
*be and
Wenfang
Sun
*a
a,
Sherri A.
McFarland
*be and
Wenfang
Sun
*a
aDepartment of Chemistry and Biochemistry, North Dakota State University, Fargo, North Dakota 58108-6050, USA. E-mail: Wenfang.sun@ndsu.edu
bDepartment of Chemistry, Acadia University, 6 University Avenue, Wolfville, NS B4P 2R6, Canada
cMaterials and Nanotechnology Program, North Dakota State University, Fargo, North Dakota 58105, USA
dCenter for Nanoscale Materials, Argonne National Laboratory, Argonne, IL 60439, USA
eDepartment of Chemistry and Biochemistry, University of North Carolina at Greensboro, 310 McIver Street, Greensboro, NC 27402-6170, USA
First published on 19th May 2017
Abstract
Five heteroleptic cationic iridium complexes with a π-expansive cyclometalating 2,3-diphenylbenzo[g]quinoxaline (dpbq) ligand (C^N ligand) and different diimine ligands (N^N ligands) (i.e. 2,2′-bipyridine (bpy, 1), phenanthroline (phen, 2), 2-(2-pyridinyl)quinoline (pqu, 3), 2,2′-bisquinoline (bqu, 4), and 2-(quinolin-2-yl)quinoxaline (quqo, 5)) were synthesized and characterized. The lowest-energy singlet electronic transitions (S1 states) were mainly dpbq ligand-centred 1ILCT (intraligand charge transfer)/1MLCT (metal to ligand charge transfer) transitions mixed with some 1π,π* transitions for complexes 1–4 with increased contributions from 1LLCT (ligand to ligand charge transfer) in 3 and 4. For complex 5, the S1 state was switched to the 1LLCT/1MLCT transitions. All five complexes displayed weak near-infrared (NIR) phosphorescence, with maximal emission output spanning 700–1400 nm and quantum yields being on the order of 10−3. The triplet state absorptions of 1–4 all resembled that of the [Ir(dpbq)2Cl]2 dimer with lifetimes of ca. 400 ns, while the TA spectrum of 5 possessed the characteristics of both the quqo ligand and the [Ir(dpbq)2Cl]2 dimer with a bi-exponential decay of ca. 5 μs and 400 ns. While the photophysics of these complexes differ slightly, their theranostic photodynamic therapy (PDT) effects varied drastically. All of the complexes were biologically active toward melanoma cells. Complexes 2 and 3 were the most cytotoxic, with 230–340 nM activity and selectivity factors for melanoma cells over normal skin fibroblasts of 34 to 40 fold. Complexes 2, 3, and 5 became very potent cytotoxins with light activation, with EC50 values as low as 12–18 nM. This potent nanomolar light-triggered activity combined with a lower dark toxicity resulted in 5 having a phototherapeutic index (PI) margin of almost 275. The bpy coligand led to the least amount of dark toxicity of 1, while phen and pqu produced cytotoxic but selective complexes 2 and 3. The quqo coligand produced the most potent complex 5 for in vitro PDT, both in terms of photocytotoxicity and PI. All Ir(III) complexes exhibited very bright NIR phosphorescence in melanoma cells. The wide range of cytotoxicity and photocytotoxicity effects within a relatively small class of complexes highlights the importance of the identity of the coligand in the biological activity of the π-expansive biscyclometalated Ir(III) complexes, and their bright NIR emission in live cells demonstrates their potential as theranostic PDT agents.
Introduction
Photodynamic therapy (PDT) is an underexploited anticancer modality that works by destroying tumors and tumor vasculature and invoking an immune response.1,2 Historically, organic porphyrin-related compounds have been employed as photosensitizers (PSs) for PDT based on a mechanism that involves cytotoxic singlet oxygen (1O2) sensitized by the PS in oxygenated environments.3 More recently, metal complexes based on Ru and Os have been explored as PSs,4–8 and one Ru compound, TLD1433, is currently in a clinical trial for treating bladder cancer with PDT (ClinicalTrials.gov Identifier: NCT03053635).9 The hope is that the metal complexes will overcome some of the limitations of the organic PSs that have hindered the development of PDT, namely, oxygen dependence and the relatively short wavelengths of light used to activate the PS. While well-oxygenated tumors that are superficial may respond well to traditional PDT, some of the most aggressive and drug-resistant tumors,10,11 including solid tumors, have proven to be a challenge that the metal complexes may be able to overcome.It is generally accepted that the triplet excited states of PSs exert their PDT effects through energy or electron transfer to ground state oxygen to produce reactive oxygen species (ROS), most notably 1O2. Thus, PSs with appropriate triplet energy levels, high quantum yields for triplet state formation, and long intrinsic triplet lifetimes are highly desirable for PDT. Intersystem crossing (ISC) is fast in metal complexes due to the heavy atom effect, and this is desirable for efficient triplet state formation. However, fast ISC back to the ground state also limits the intrinsic lifetime of the reactive triplet excited state, giving far less time for efficient bimolecular interactions that are critical for photocytotoxic effects. One way to mitigate this issue while maintaining fast ISC rates for triplet state formation is to utilize spin-forbidden transitions in organic chromophores. For example, we have demonstrated that ruthenium (Ru) complexes equipped with ligands either contiguously fused12,13 or tethered9,14–16 to π-expansive organic chromophores produce very potent in vitro PDT effects that are presumably due to very long intrinsic triplet lifetimes. The idea of a metal–organic dyad construct that installs a spatially-separated pendant organic chromophore in a Ru(II) complex for generating long-lived triplets was first put forward by Ford and Rodgers17 and later expanded by others.18 Turro and coworkers extended this idea further by showing that contiguously fused π-expansive ligands such as benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine (dppn) also produced long-lived triplets in Ru(II) dyads, and demonstrated that these agents act as potent DNA photocleavage agents.19 Inspired by these studies, we have focused on developing a variety of transition-metal complexes with π-expansive ligands as PDT agents using both approaches.
While cyclometalated Ir(III) complexes have been extensively studied in organic light-emitting diodes20,21 and light-emitting electrochemical cells,22,23 PSs24,25 and photocatalysts,26–28 very few Ir(III) complexes as PSs for in vitro PDT have been reported.29–32 This is despite Ir(III) having one of the largest spin–orbit coupling (SOC) constants known (3909 cm−1)33 and consequently very high quantum yields for triplet state formation.34,35 We have recently shown that biscyclometalated Ir(III) complexes with long triplet state lifetimes have favorable properties for both reverse saturable absorption (RSA) and PDT.36 These systems were characterized by π-expansive ligands with systematic variations in the two identical cyclometalating ligands as well as the diimine ligand. Light-enhanced cytotoxicities were as low as 3 nM with phototherapeutic indices (PIs) greater than 400 at relatively soft light doses.
In the search for better PSs that combine therapeutic, imaging and targeting capabilities into a single molecule,37 we have continued our exploration of such promising biscyclometalated Ir(III) complexes as PSs for PDT. Ir(III) complexes have the potential to act simultaneously as therapeutic and diagnostic (theranostic) agents owing to their large luminescence quantum yields38–40 and excitation/emission energies that can be easily tuned by varying the structure and substituents of the cyclometalating and/or the ancillary diimine ligand, thus covering almost the entire visible spectrum.41–43
Near-infrared (NIR) emitting Ir(III) complexes, however, are relatively rare,44–46 but highly desirable for bioimaging and labeling.40 The biscyclometalated Ir(III) complexes reported herein were designed as NIR emitters to add diagnostic capacity to their predicted in vitro PDT effects. Phosphorescent cyclometalated cationic Ir(III) complexes are good candidates as they possess (1) large Stokes shifts (more than 100 nm) to avoid inner filter effects; (2) rapid transmembrane activity (short incubation time and less potential toxicity); (3) long luminescence lifetimes (100 ns to 1 ms) for time-resolved detection; and (4) enhanced photostabilities (less photobleaching). Photostability is particularly important for allowing continuous exposure of the complexes to irradiation and enabling real-time monitoring of the probes. All of these features are highly desirable for a theranostic PDT agent.
In this report we utilize the π-expansive 2,3-diphenylbenzo[g]quinoxaline as the two cyclometalating (C^N) ligands and probe the effects of systematic changes to the identity of the diimine ligand (Chart 1) on the photophysical and photobiological properties of the resulting complexes. We also demonstrate their NIR phosphorescence in live cells, underscoring their theranostic potential.
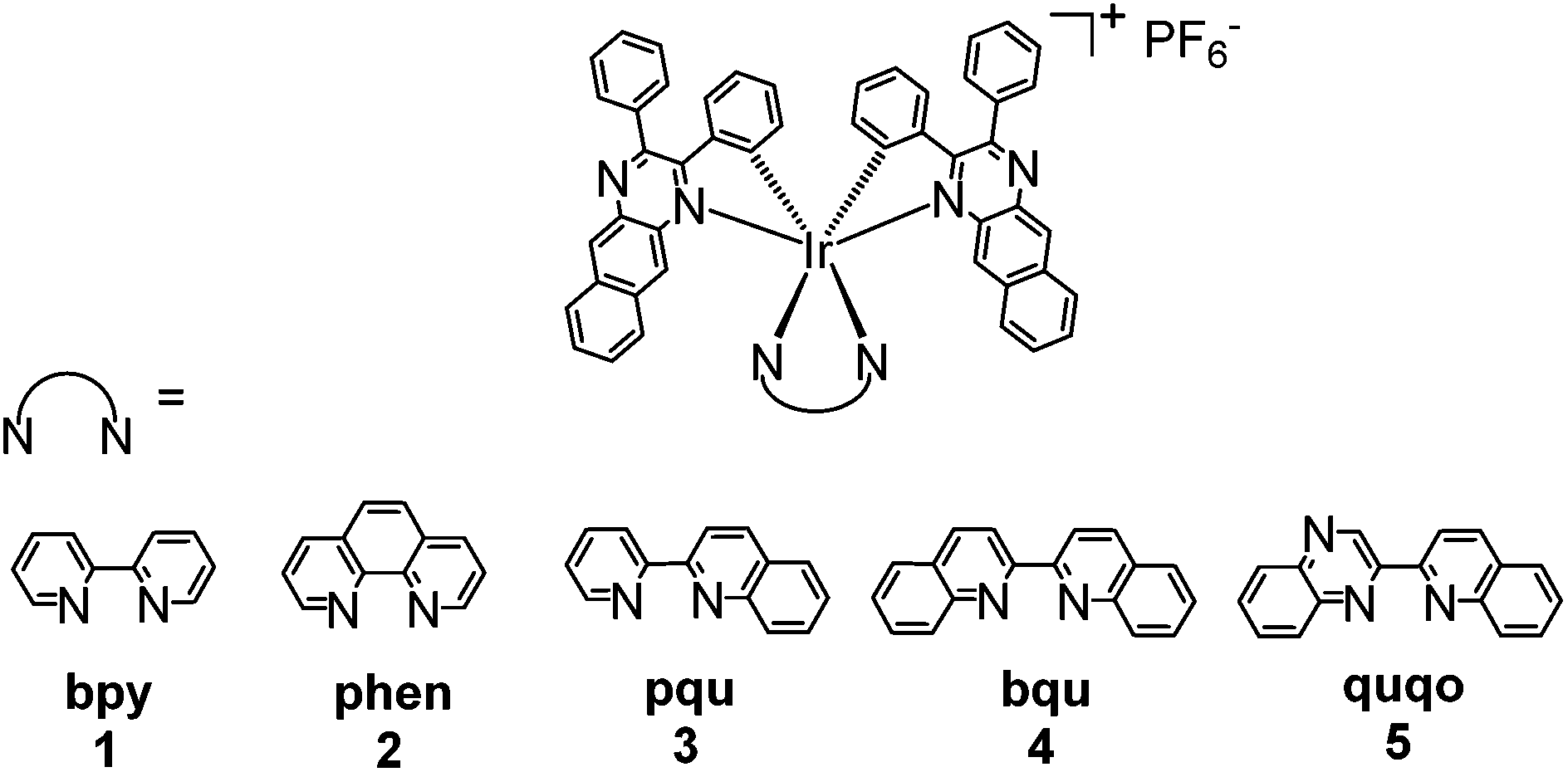 | ||
| Chart 1 Molecular structures of the cationic iridium complexes 1–5. | ||
Experimental section
Synthesis and characterization
All reagents and solvents were purchased from commercial sources and used as is unless otherwise mentioned. The spectroscopic grade solvents used for photophysical studies were purchased from VWR International and used as received. 1H NMR spectra were recorded on a Bruker-400 spectrometer in CDCl3 with tetramethylsilane (TMS) as the internal standard or in d6-DMSO. High resolution mass spectrometry (HRMS) analyses were performed on a Waters Synapt G2-Si mass spectrometer with electrospray ionization (ESI). Elemental analyses were conducted by NuMega Resonance Laboratories, Inc. (San Diego, California). The diimine ligands pqu47 and quqo48 and the cyclometalating dpbq49 ligand were prepared according to the reported procedures while the other ligands (bpy, phen, bqu) were commercially available. The iridium dimer [Ir(dpbq)2Cl]2 was prepared following the Nonoyama method.501. Red solid (85 mg, yield: 74%). 1H NMR (400 MHz, d6-DMSO): δ 9.11 (s, 2H), 8.74 (s, 2H), 8.41–8.38 (d, J = 2.8 Hz, 2H), 8.24–8.16 (m, 6H), 8.03–8.01 (m, 6H), 7.78 (s, 6H), 7.59–7.52 (m, 4H), 7.36 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 6.82 (t, J = 8.0 Hz, 2H), 6.70–6.67 (m, 4H). ESI-HRMS calcd for [C58H38IrN6]+ (M − PF6): 1011.2792, found: 1011.2788. Anal calcd (%) for C58H38IrN6PF6: C, 60.25; H, 3.31; N, 7.27. Found: C, 60.27; H, 3.65; N 6.89.
2. Red solid (65 mg, yield: 69%). 1H NMR (400 MHz, d6-DMSO): δ 9.53–9.52 (d, J = 4.0 Hz, 2H), 8.86–8.83 (d, J = 8.0 Hz, 2H), 8.63 (s, 2H), 8.59–8.55 (m, 2H), 8.08–8.05 (m, 6H), 8.02–8.00 (d, J = 8.0 Hz, 4H), 7.80–7.79 (m, 6H), 7.53–7.46 (m, 4H), 7.34–7.32 (d, J = 8.0 Hz, 2H), 7.27–7.25 (d, J = 8.0 Hz, 2H), 6.89–6.85 (m, 2H), 6.74–6.73 (m, 4H). ESI-HRMS calcd for [C60H38IrN6]+ (M − PF6): 1035.2792, found: 1035.2788. Anal calcd (%) for C60H38IrN6PF6·H2O: C, 60.14; H, 3.36; N, 7.01. Found: C, 60.52; H, 3.33; N, 7.10.
3. Red solid (65 mg, yield: 54%). 1H NMR (400 MHz, d6-DMSO): δ 8.89 (s, 1H), 8.82 (s, 1H), 8.77–8.75 (d, J = 8.0 Hz, 1H), 8.66–8.60 (m, 3H), 8.38–8.30 (m, 3H), 8.19–8.17 (d, J = 8.0 Hz, 2H), 8.13–8.10 (t, J = 4.0 Hz, 1H), 8.05–7.95 (m, 4H), 7.78–7.76 (m, 3H), 7.64–7.45 (m, 8H), 7.38 (s, 1H), 7.28–7.26 (d, J = 8.0 Hz, 1H), 7.24–7.20 (t, J = 8.0 Hz, 1H), 7.09–7.07 (d, J = 8.0 Hz, 1H), 6.90–6.85 (t, J = 8.0 Hz, 1H), 6.80–6.76 (m, 2H), 6.71–6.59 (m, 5H), 6.11–6.09 (d, J = 8.0 Hz, 1H). ESI-HRMS calcd for [C62H40IrN6]+ (M − PF6): 1061.2949, found: 1061.2939. Anal calcd (%) for C62H40IrN6PF6·H2O: C, 60.83; H, 3.46; N, 6.86. Found: C, 60.86; H, 3.36; N 6.67.
4. Red solid (65 mg, yield: 52%). 1H NMR (400 MHz, d6-DMSO): δ 8.86 (m, 4H), 8.56–8.53 (d, J = 8.0 Hz, 2H), 8.28–8.26 (d, J = 8.4 Hz, 2H), 8.16–8.14 (d, J = 8.4 Hz, 2H), 7.98 (s, 2H), 7.85–7.81 (t, J = 8.0 Hz, 2H), 7.70–7.48 (m, 12H), 7.38–7.25 (m, 6H), 6.97–6.95 (d, J = 8.4 Hz, 2H), 6.90–6.87 (t, J = 8.0 Hz, 2H), 6.80–6.78 (d, J = 8.0 Hz, 2H), 6.74–6.70 (t, J = 8.0 Hz, 2H), 6.21–6.19 (d, J = 8.0 Hz, 2H). 13C NMR (101 MHz, d6-DMSO) δ 165.40, 159.54, 154.11, 152.79, 146.95, 144.05, 142.47, 139.41, 137.25, 136.29, 133.46, 133.33, 132.73, 132.58, 132.36, 132.00, 130.61, 130.50, 130.02, 129.32, 129.09, 128.80, 128.65, 128.21, 127.58, 127.24, 122.60, 122.48, 122.44. ESI-HRMS calcd for [C66H42IrN6]+ (M − PF6): 1111.3105, found: 1111.3098. Anal calcd (%) for C66H42IrN6PF6·1.4H2O: C, 60.63; H, 3.49; N, 6.39. Found: C, 60.92; H, 3.89; N 6.09.
5. Red solid (50 mg, yield: 40%). 1H NMR (400 MHz, d6-DMSO): δ 9.90 (s, 1H), 8.94–8.75 (m, 4H), 8.40–8.38 (d, J = 8.0 Hz, 1H), 8.31–8.30 (d, J = 8.0 Hz, 1H), 8.19–8.10 (m, 4H), 7.88 (t, J = 6.0 Hz, 1H), 7.72–7.21 (m, 19H), 6.98 (t, J = 6.0 Hz, 1H), 6.78–6.72 (m, 7H), 6.35–6.33 (d, J = 8.0 Hz, 1H), 6.21–6.19 (d, J = 8.0 Hz, 1H). ESI-HRMS calcd for [C65H41IrN7]+ (M − PF6): 1112.3058, found: 1112.3042. Anal calcd (%) for C65H41IrN7PF6: C, 62.10; H, 3.29; N, 7.80. Found: C, 61.74; H, 3.35; N 7.54.
Photophysical studies
The ultraviolet-visible (UV-vis) absorption spectra were recorded on a Varian Cary 50 spectrophotometer. Steady-state emission spectra were obtained with 473 nm excitation and an InGaAs array (spectral response range: 0.9–1.7 μm) as the detector and a 500 nm long pass filter to block the excitation beam. A NIR dye IR-26 (Φ = 0.0005)51 was used as the reference for the emission quantum yield measurement. The emission spectra upon excitation at shorter wavelengths were recorded on a Horiba Jobin Yvon FluoroMax-4 fluorometer/phosphorometer that is equipped with a Hamamatsu photomultiplier tube (PMT) R928 (spectral response range: 185–900 nm) as the detector. The nanosecond transient difference absorption (TA) spectra and decay characteristics were measured in degassed CH3CN solutions on an Edinburgh LP-920 laser flash photolysis spectrometer. The third harmonic output (355 nm) of a Nd:YAG laser (Quantel Brilliant, pulse width = 4.1 ns, repetition rate = 1 Hz) was used as the excitation source. Each sample solution was purged with argon for 45 min prior to measurement.Singlet oxygen quantum yields
Singlet oxygen emission from dilute solutions (5 μM) of the PF6− salts of the complexes in spectroscopic-grade CH3CN was measured using a PTI Quantamaster equipped with a Hamamatsu R5509-42 near-infrared PMT. Quantum yields for singlet oxygen emission (ΦΔ) were calculated relative to [Ru(bpy)3](PF6)2 as the standard (ΦΔ = 0.56 in aerated CH3CN52) according to eqn (1), where I, A, and η are integrated emission intensity, absorbance at the excitation wavelength, and refractive index of the solvent, respectively. The calculated ΦΔ was reproducible to within <5%.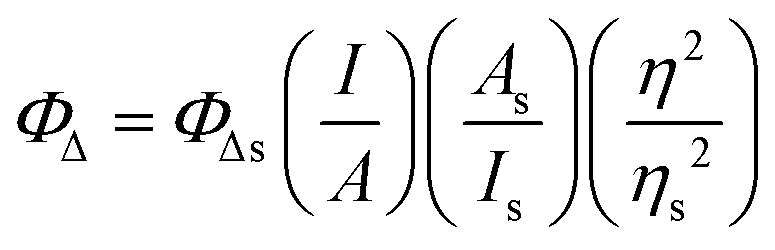 | (1) |
Computational methods
Singlet geometry optimizations of all iridium complexes were performed with density functional theory (DFT) formalism implemented in Gaussian09 software package.53 The time-dependent DFT (TDDFT) method was employed to calculate the absorption spectra of complexes using a Gaussian09 software package as well. The linear response density was calculated within a TDDFT framework,54 from which the excitation energies and oscillator strength can be extracted by iteratively solving the eigenvalue equation problem based on Davidson algorithm.55–58 Forty optical transitions were calculated to obtain the absorption spectra at an energy range comparable to the experimental UV-vis spectra.To obtain the fluorescence emission energies, we optimized the lowest singlet excited state geometry using the TDDFT analytical approach.59 To obtain the phosphorescence emission energies, we first optimized at the lowest triplet ground state geometry using the unrestricted DFT method (ΔSCF approach) within Gaussian09 software. The lowest triplet excitation energy was calculated through the combined scalar relativistic ZORA and TDDFT approach using the NWChem software package. The one-electron energies and orbitals were obtained by solving the one-electron ZORA Kohan–Sham equation.53
The hybrid PBE1 functional60 was used for both the ground and excited state calculations. The LANL2DZ basis set was applied for Ir, while the 6-31G* basis set was applied for the remaining atoms. Both geometry optimization and optical absorption calculations were performed in a solvent medium using a conductor-like polarizable continuum model (CPCM),61,62 as implemented in Gaussian09. Fluorescence and phosphorescence calculations were performed via COSMO continuum solvation63,64 as implemented in NWChem. Dichloromethane (CH2Cl2, εr = 9.08) was chosen as the solvent medium for consistency with experimental studies.
To visualize the excited states that can be represented as hole–electron pairs created upon photoexcitation, natural transition orbitals (NTOs) were provided.62,65 By performing NTO calculations implemented in Gaussian09, an electron–hole pair transition from a ground state to an excited state could be realized through unitary transformation of the transition density matrix of a given excited state.62 For visualization of the lowest singlet and triplet emitting states, the dominant molecular orbitals contributing to the excited state were plotted by performing the eigenvector analysis of this state. Chemcraft-1.7 software66 was used for plotting excited state charge densities by setting the isovalue as 0.02.
Photobiological activity studies
The details of the cell culture, cytotoxicity and photocytotoxicity studies, confocal microscopy, and DNA mobility-shift assays are provided in the ESI.†Results and discussion
Molecular design and synthesis
Previous studies revealed that the degree of π-conjugation of the C^N ligands impacts the emission from Ir(III) complexes dramatically.67–69 Introduction of a highly conjugated C^N ligand is required to achieve the NIR emission of the complexes.44As shown in Scheme 1, the dpbq ligand was synthesized by a condensation reaction between commercially available 2,3-naphthalenediamine and benzil in absolute ethanol with p-TsOH as a dehydration agent in 92% yield. Subsequent reaction of dpbq with IrCl3·3H2O in a refluxed ethoxyethanol/water (v/v = 3/1) mixture resulted in a red precipitate, which was washed with water to give the [Ir(dpbq)2Cl]2 dimer with satisfactory purity. Complexes 1–5 were synthesized by reaction of the [Ir(dpbq)2Cl]2 dimer with the corresponding diimine ligand in mixed CH2Cl2/methanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). All the complexes are readily dissolved in CH2Cl2, CH3CN, and DMSO, but they have limited solubility in nonpolar solvents such as hexane and toluene. All our complexes were purified by column chromatography on silica gel and characterized by 1H NMR, ESI-HRMS (ESI Fig. S1–S5†) and elemental analysis. All complexes were very stable even in coordinating solvents such as DMSO, as reflected by the absence of detectable decomposition by TLC from the d6-DMSO sample solutions kept under ambient conditions in the NMR tubes.
:1). All the complexes are readily dissolved in CH2Cl2, CH3CN, and DMSO, but they have limited solubility in nonpolar solvents such as hexane and toluene. All our complexes were purified by column chromatography on silica gel and characterized by 1H NMR, ESI-HRMS (ESI Fig. S1–S5†) and elemental analysis. All complexes were very stable even in coordinating solvents such as DMSO, as reflected by the absence of detectable decomposition by TLC from the d6-DMSO sample solutions kept under ambient conditions in the NMR tubes.
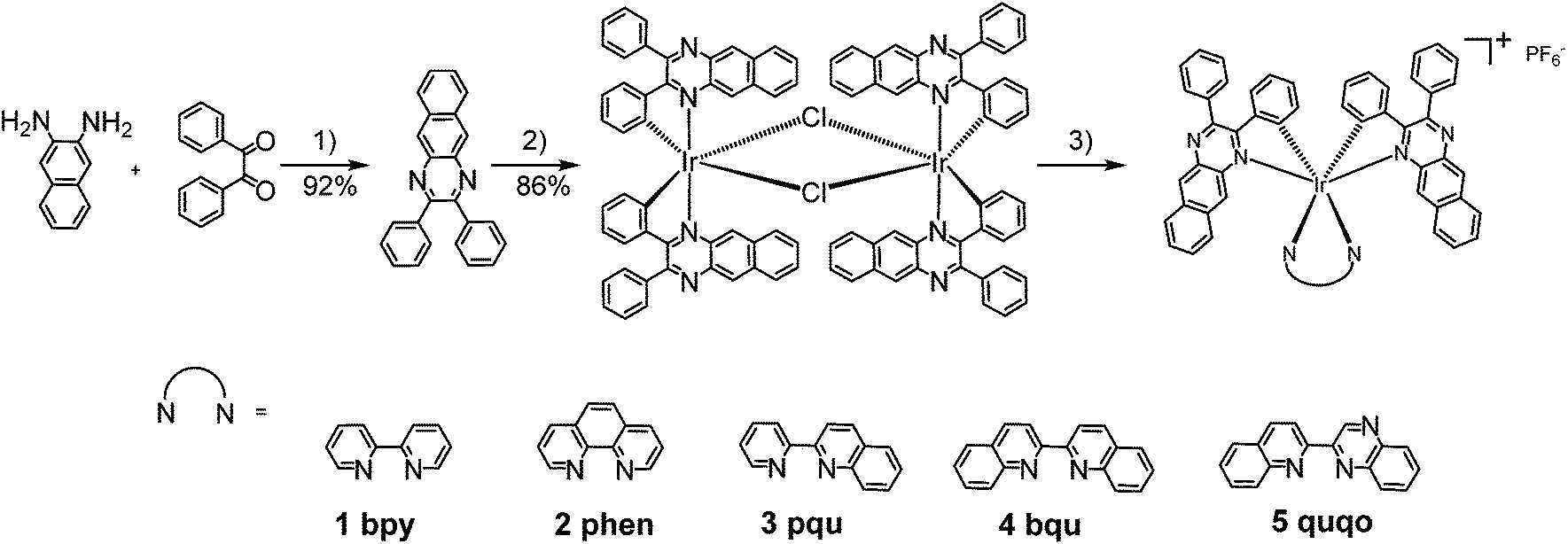 | ||
| Scheme 1 Synthetic route for complexes 1–5. Reaction conditions: (1) p-TsOH, EtOH, reflux; (2) IrCl3·3H2O, 2-ethoxylethanol/H2O, reflux; (3) bpy, AgSO3CF3, CH2Cl2/MeOH, reflux; then NH4PF6, r.t. | ||
For all of the photophysical studies discussed in the following sections, the PF6 salts of the complexes were used. The Cl salts of the complexes were used for the photobiological studies.
Electronic absorption
The UV-vis absorption spectra of complexes 1–5 were collected in CH2Cl2, and the results are shown in Fig. 1 and Table 1. Variable concentration experiments confirmed that no ground state aggregation occurred in the concentration range studied (5 × 10−6 to 1 × 10−4 M−1).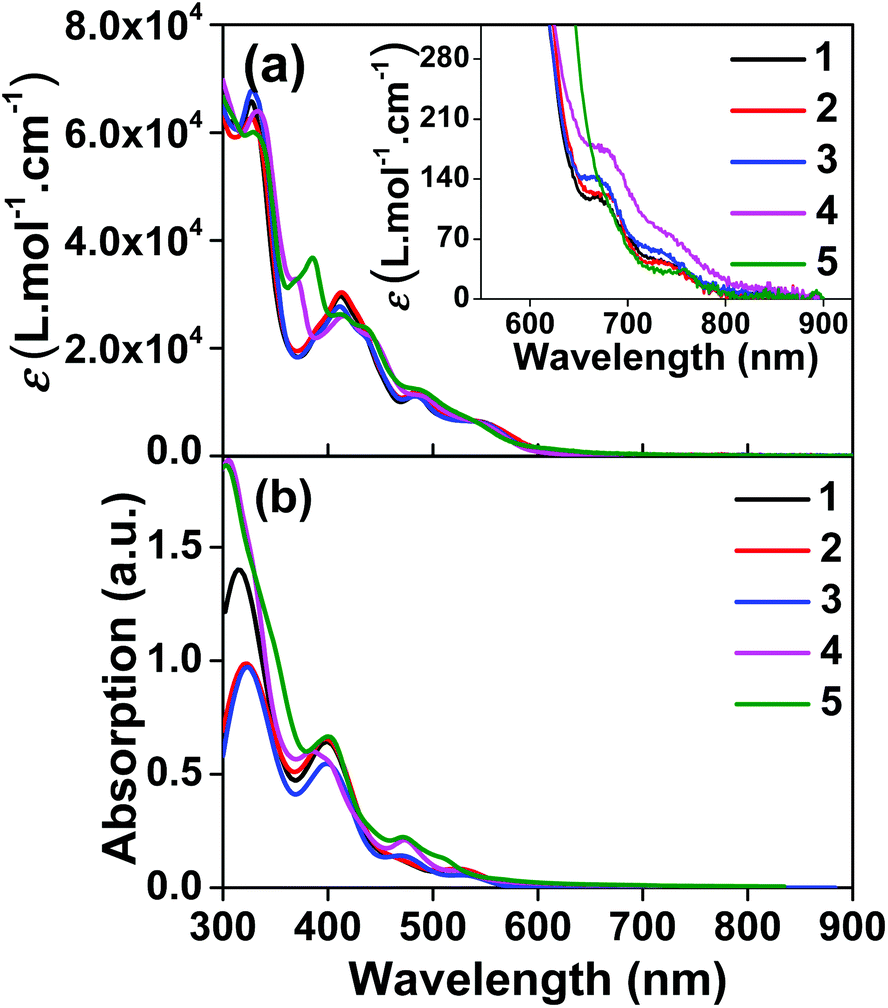 | ||
| Fig. 1 (a) Experimental and (b) calculated absorption spectra of complexes 1–5 in dichloromethane. The inset in panel (a) shows the expansion of the absorption above 550 nm. | ||
| λ abs/nm (ε/104 M−1 cm−1)a | λ em/nm; Φemb | τ 0 /ns | λ T1–Tn/nm (τTA/ns)d | Φ Δ | |
|---|---|---|---|---|---|
| a Absorption band maxima and molar extinction coefficients in CH2Cl2 at room temperature. b Room temperature emission band maxima and emission quantum yields measured in CH2Cl2 with an InGaAs sensor upon 473 nm excitation. IR-26 was used as the reference for the NIR emission quantum yield measurements. c Intrinsic lifetime in CH2Cl2 measured with an Hamamatsu R928 PMT. d Triplet transient absorption band maxima and lifetimes in CH3CN. e Singlet oxygen quantum yields at λex = 550 nm, the values in parenthesis are obtained at λex = 412 nm for 1, 413 nm for 2 and 3, and 417 nm for 4 and 5 in CH3CN. | |||||
| 1 | 327 (6.6), 412 (2.9), 486 (1.1), 550 (0.6), 673 (0.012), 742 (0.004, br) | 794, 911, 965; 0.003 | 440 | 364 (−), 454 (290), 519 (320), 659 (320) | 0.55 (0.42) |
| 2 | 328 (6.3), 413 (3.0), 485 (1.2), 550 (0.7), 681 (0.012), 742 (0.004, br) | 801, 913, 965; 0.0032 | 430 | 374, 450 (350), 522 (350), 668 (350) | 0.47 (0.40) |
| 3 | 328 (6.8), 411 (2.8), 486 (1.1), 546 (0.7), 673 (0.014), 738 (0.005, br) | 794, 910, 964; 0.0025 | 460 | 366, 457 (380), 517 (380), 660 (380) | 0.54 (0.41) |
| 4 | 333 (6.4), 370 (3.3), 416 (2.5), 443 (2.2), 493 (1.1), 545 (0.6), 679 (0.017), 747 (0.007, br) | 801, 916, 968; 0.002 | 370 | 375 (340), 519 (360), 660 (350) | 0.38 (0.33) |
| 5 | 331 (6.0), 385 (3.7), 413 (2.6), 437 (2.4), 490 (1.2), 540 (0.6), 756 (0.003, br) | 800, 915, 970; 0.0017 | 360 | 443 (450 (6%), 4940 (94%)), 508 (420 (14%)), 4820 (86%), 650 (430 (11%), 4960 (89%)) | 0.56 (0.42) |
The absorption spectra of all complexes featured intense bands at wavelengths below 350 nm with molar extinction coefficients of 6.0–6.8 × 104 M−1 cm−1. Based on these large molar extinction coefficients and the natural transition orbitals (NTOs, see ESI Table S1†) obtained from the TDDFT calculations, these bands are assigned to the dpbq ligand-localized 1π,π*/1ILCT transitions with some contributions from the 1LLCT/1MLCT/1LMCT transitions. For complex 5, the 1π,π* transition from the diimine ligand quqo also made a significant contribution. The absorption bands at 400–500 nm are mainly dpbq ligand-centred 1π,π*/1MLCT/1ILCT/1LMCT transitions for all complexes, while complexes 3–5 have 1LLCT transitions contributing to the 400–470 nm band. For complexes 4 and 5, the diimine ligand-based 1π,π* transition also contributed to the 400–470 nm band (see ESI Table S2†). All complexes possess a low-energy absorption band between 500 and 600 nm, which mainly arises from the dpbq ligand-associated 1ILCT/1MLCT transitions combined with some 1π,π* character (see NTOs in Table 2). For complexes 3–5, contributions from the 1LLCT transition systematically increased, with the lowest singlet transition (S1 state) in 5 being switched to the 1LLCT/1MLCT transition. In addition to the aforementioned absorption bands, all complexes displayed very weak but clearly observable absorption bands between 600 and 800 nm (ε < 200 M−1 cm−1). Due to the very small molar extinction coefficients, we attribute these bands to spin-forbidden transitions to the triplet excited states. A similar phenomenon has been reported in other Ir(III) complexes.36,67,68,70,71 Compared with the many other reported Ir(III) complexes that contain fewer π-conjugated C^N ligands, the spin-forbidden transitions in complexes 1–5 are much more red-shifted due to the more π-expansive dpbq ligand. It is apparent that the dpbq ligands played a dominant role in contributing to most of the absorption, while the increased π-conjugation of the diimine ligands gradually increased the diimine ligand related 1LLCT/1MLCT character in the S1 state of complexes 3–5. Additionally, the absorption band at 370 nm and 385 nm in 4 and 5, respectively, should have significant contributions from the diimine ligand centred 1π,π* transition.
| 1 | 2 | 3 | 4 | 5 | ||
|---|---|---|---|---|---|---|
| S1 | S1 | S1 | S1 | S1 | S3 | |
| 531 nm | 531 nm | 533 nm | 528 nm | 565 nm | 512 nm | |
| f = 0.06 | f = 0.05 | f = 0.04 | f = 0.03 | f = 0.005 | f = 0.04 | |
| HOTO |
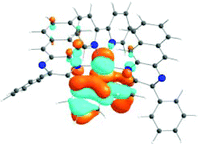
|
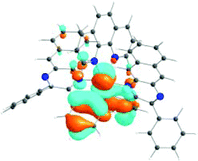
|
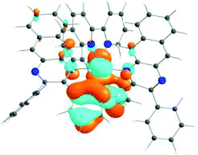
|
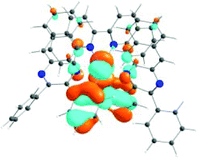
|
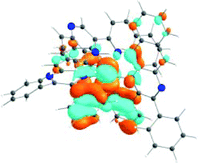
|
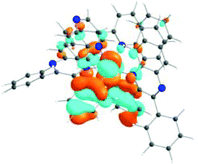
|
| LUTO |
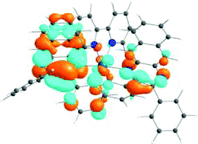
|
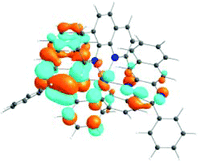
|
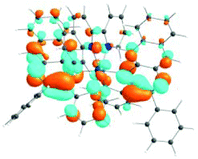
|
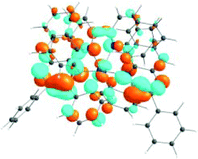
|
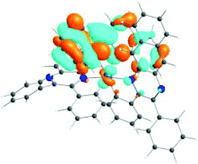
|
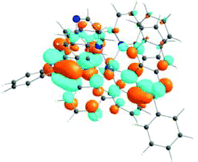
|
Photoluminescence
The room-temperature emission from 1–5 were studied in CH2Cl2 upon excitation at different wavelengths. The spectra are shown in Fig. 2 and ESI Fig. S8,† and the emission data upon excitation at 473 nm are compiled in Table 1. 473 nm light was used as the excitation wavelength because it is the only available blue laser source for our instrument equipped with an InGaAs array detector, and it was capable of exciting both the samples and the IR-26 NIR reference dye with sufficiently bright signals. Upon 473 nm excitation, the emission spectra of all complexes resembled each other and were similar to that of the [Ir(dpbq)2Cl]2 dimer (ESI Fig. S9†). The deoxygenated emission lifetimes were between 360 and 460 ns. These observations point toward a common triplet excited state for all of the complexes, which is localized on the same ligand type, i.e. the C^N ligand. NTOs from the TDDFT calculations (ESI Table S3†) confirmed that both the electrons and holes are predominantly localized on the C^N ligands. Therefore, the emitting triplet excited states have been ascribed to the C^N ligand centred 3π,π* state mixed with some 3ILCT/3MLCT/3LLCT character. The quantum yields for all complexes were very low (near 10−3), which is in agreement with those reported by other groups for similar complexes that contain dpbq as the C^N ligand.49 Small quantum yields are not surprising given that the decreased energy of the emitting state (into the NIR) facilitates radiationless decay to the ground state (energy gap law).72,73 Although the low emission quantum yields of complexes 1–5 may limit their utility in organic light emitting diodes (OLEDs), their luminescence is strong enough for NIR bioimaging applications,74 which will be discussed and demonstrated in the following section.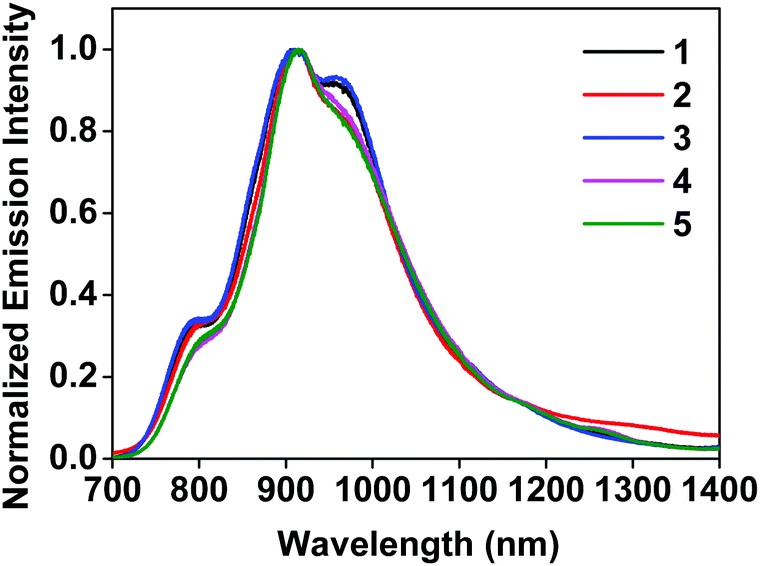 | ||
| Fig. 2 Normalized emission spectra of complexes 1–5 in deoxygenated CH2Cl2 at room temperature using an InGaAs array as the detector and λex = 473 nm. | ||
In contrast, upon excitation at 330 or 370 nm and monitoring with the Hamamatsu PMT R928, complexes 1–4 exhibited dual emission (ESI Fig. S8†). In addition to the NIR emission discussed in the previous paragraph, a broad featureless red emission was observed around 600 nm. This phenomenon resembles that observed from complex 6 in ref. 36 that contains the same dpbq C^N ligand. Considering the structureless feature and the emission energy, we assign this red emission band to the 3MLCT/3LLCT emitting state. Such an attribution is consistent with the 3MLCT/3LLCT emission reported in other Ir(III) complexes with the same or similar diimine ligand.36,69,71 The different natures of the red and NIR emission bands are supported by the difference in their excitation spectra monitored at the band maxima of these two emission bands (ESI Fig. S10†). The observation of the dual emission in complexes 1–4 but not in 5 could be attributed to the larger energy difference between the high-lying 3MLCT/3LLCT state and the lowest-energy dpbq ligand centred 3π,π* state (T1 state) in 1–4.71 With further extended π-conjugation and the stronger electron-withdrawing ability of the quinoxaline group in the diimine ligand of 5, the 3MLCT/3LLCT state in 5 is further stabilized and energetically closer to the dpbq ligand centred 3π,π* state.70 In such a case, dual emission could not be observed. Although dual emission is unusual, it has been reported in other transition-metal complexes including Ir(III) and Ru(II) complexes.14,36,75–84
It is worth pointing out that the feature of the NIR emission bands shown in ESI Fig. S8† for complexes 1–4 appeared to be different from those in Fig. 2. The difference arose from the different spectral responses and sensitivities of the detectors used in these two measurements. The spectra shown in Fig. 2 were measured with an InGaAs array (spectral response range: 0.9–1.7 μm) that is sensitive to the NIR emission and allows the full NIR spectra of these complexes to be recorded. In contrast, the spectral response range of the Hamamatsu PMT R928 is 185–900 nm, which allows both the red emission and part of the NIR emission to be collected but the detector sensitivity dramatically decreases beyond 800 nm and the emission beyond 850 nm cannot be observed.
Transient absorption (TA)
The triplet excited-states of complexes 1–5 were further investigated by nanosecond transient absorption (TA) spectroscopy. The TA spectra of 1–5 at zero-time-delay recorded upon excitation at 355 nm at room temperature in deaerated acetonitrile solution are shown in Fig. 3, and the time-resolved nanosecond TA spectra of 1–5 are shown in Fig. S11†.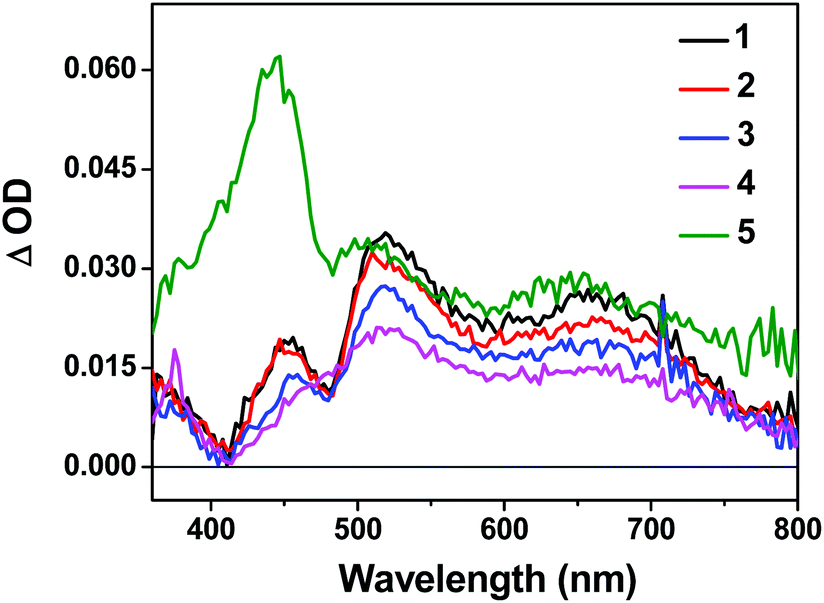 | ||
| Fig. 3 Nanosecond TA spectra of complexes 1–5 in acetonitrile solution (λex = 355 nm, A355 = 0.4 in a 1 cm cuvette) immediately after excitation. | ||
The TA spectra of all complexes were characterized by positive signals at 360–800 nm. The shapes of the TA spectra and the associated decay lifetimes of 1–4 all resemble those of the [Ir(dpbq)2Cl]2 dimer,68 indicating that the absorbing triplets were most likely 3π,π*/3CT states centered on the coordinated dpbq ligand. The agreement between the TA and emission lifetimes implies the same orbital parentage of the TA and emitting states, i.e. the coordinated dpbq ligand centred 3π,π* state combined with an 3ILCT/3MLCT/3LLCT character as discussed in the photoluminescence section. However, on going from complex 1 to 4, the ΔOD values decreased with increasing π-conjugation of the diimine ligand. In fact, the bands near 450 nm gradually decreased and eventually became indistinguishable from the 510 nm band in complex 4. This is likely related to the increased ground-state absorption in this spectral region in 4. The TA spectral features of 5 resembled those of 1–4 in the region of 480–800 nm. However, an intense absorption band appeared near 440 nm, which was distinct from the spectra of 1–4. The shape and energy of this band matched well with the TA band of the quqo ligand (ESI Fig. S12†). In addition, the decay profiles at the TA band maxima of 5 were bi-exponential, with a longer lifetime of approximately 5 μs and a shorter lifetime of 420 ns (see Table 1). The longer lifetime is in agreement with the TA lifetime of the quqo ligand (5.87 μs at 440 nm) while the shorter lifetime is similar to the coordinated dpbq ligand-centred 3π,π*/3CT excited state. Therefore, it appears that the observed TA spectrum of 5 has TA features of both the quqo ligand and the [Ir(dpbq)2Cl]2 dimer. The possibility of the longer-lived species being from a trace amount of non-coordinated quqo ligand was excluded based on the absence of the quqo fluorescence at 480 nm.
Singlet oxygen generation
The complexes were analyzed for their ability to generate 1O2 by direct measurement of 1O2 emission centred at 1270 nm. [Ru(bpy)3](PF6)2, with a reported singlet oxygen quantum yield (ΦΔ) of 0.56 in aerated CH3CN52 was used as the standard. Despite the most intense absorption and excitation maxima appearing at wavelengths shorter than 500 nm, the largest 1O2 quantum yields were produced with excitation at 550 nm for all of the complexes. The calculated values of ΦΔ for the complexes ranged from 38 to 56% (±5%) (Table 1). Complexes 1, 3, and 5 generated 1O2 with about the same efficiency as the standard (54–56%). Complexes 2 and 4 were less efficient, with ΦΔ = 47% and 38%, respectively. These 1O2 quantum yields were dependent on the nature of the initially populated excited states, and were attenuated with shorter wavelength excitation. For example, ΦΔ for all of the complexes except 4 was reduced to 40–42% (Table 1) with excitation between 412 and 417 nm, where the absorption and excitation maxima were most intense. Complex 4 also underwent a reduction in ΦΔ to 33% with 417 nm excitation. Although not exceptionally high, this 1O2 production was anticipated to result in some photocytotoxicity in cellular assays.Cytotoxicity and photocytotoxicity assays
The photobiological activities of the five Ir(III) complexes were assessed in the SK-MEL-28 melanoma cell line. The effective concentration required to reduce cell viability to 50% (EC50) was determined from sigmoidal fits of the dose–response curves between 1 nM and 300 μM PS with (light EC50) or without (dark EC50) a light treatment. The light treatment was broadband visible or single-wavelength red (625 nm) light delivered at a fluence of 100 J cm−2 and irradiance of 35.7 mW cm−2 and 32.3 mW cm−2 for visible and red, respectively. The phototherapeutic index (PI) was calculated as the ratio of dark to light EC50 values and is a measure of the therapeutic margin for in vitro PDT. Additionally, the dark cytotoxicity was measured in normal skin fibroblasts (CCD-1064Sk) to determine any selectivity for cancer cells over normal cells. The selectivity factor (SF) is defined as the ratio of the dark EC50 value measured for CCD-1064Sk cells and the dark EC50 value measured for SK-MEL-28 cells. As long as the dark cytotoxicity toward normal cells is low and the PI large, SF > 1 is not a requirement. PDT is inherently selective with spatial and temporal control of light delivery.The dark cytotoxicities toward melanoma cells for the Ir(III) complexes varied from 230 nM to 18 μM, and increased in the order: 3 > 2 > 4 > 5 > 1 (Table 3, Fig. 4 and ESI Fig. S13†). When compared with noncancerous skin fibroblasts, complexes 2 and 3 were up to 40-fold more cytotoxic toward the melanoma cells while 1, 4, and 5 displayed almost no selectivity for the cancer cells (ESI Table S4†). The relatively simple change in the identity of the diimine ligand from bpy in complex 1 to phen in complex 2 increased the dark cytotoxicity toward melanoma cells by more than 50-fold. Benzannulation at C5–C6 to form pqu produced an Ir(III) complex 3 that was almost 80-times more dark cytotoxic relative to 1. However, a second benzannulation to form the symmetric bqu as in complex 4 increased dark cytotoxicity toward the cancer cells by only 6-fold relative to 1. Replacement of one of the quinoline rings with quinoxaline in complex 5 did not substantially alter the dark toxicity. Increased π-expansion on the diimine ligand in complexes 2 and 3 does appear to increase dark cytotoxicity toward the melanoma cells relative to bpy. However, the dark cytotoxicity toward melanoma cells does not directly correlate with the lipophilicity index of the diimine ligand in this class of complexes. Interestingly, the dark cytotoxicity toward normal skin fibroblasts increased in the order 4 > 5 > 3 > 2 > 1, which did more closely parallel the lipophilicity index of the diimine ligand. The substantial deviation in the trend observed for complexes 2 and 3 in melanoma cells resulted in very large SFs, making these complexes of interest as traditional chemotherapeutics.
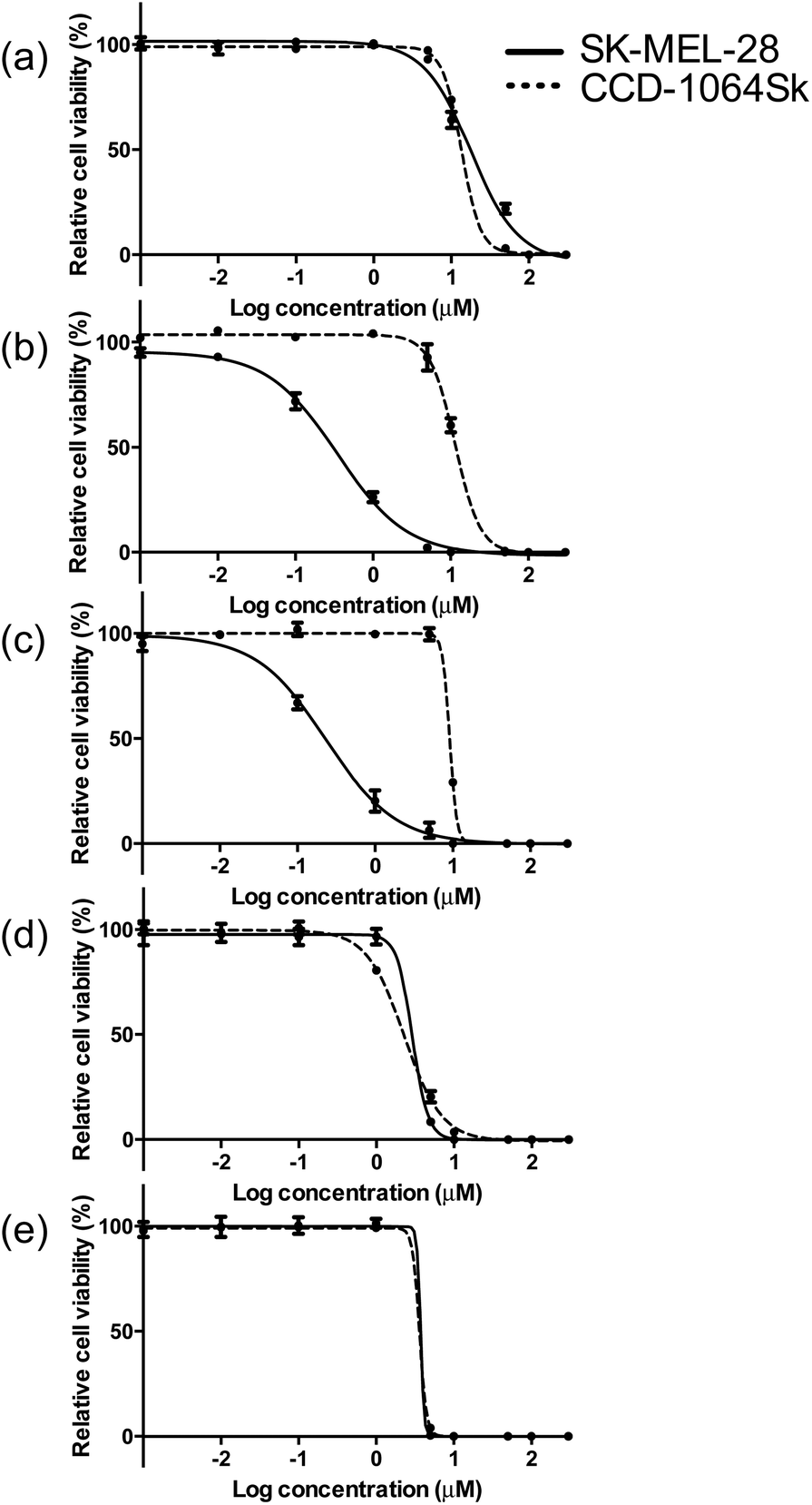 | ||
| Fig. 4 Comparison of dark cytotoxicity for complexes 1 (a), 2 (b), 3 (c), 4 (d) and 5 (e) in SK-MEL-28 (solid line) and CCD-1064Sk cells (dotted line). | ||
| Complexa | Dark | Visible | Red | |||
|---|---|---|---|---|---|---|
| EC50 (μM) | SFb | EC50 (μM) | PIc | EC50 (μM) | PIc | |
| a Complexes screened as their chloride salts. b SF = selectivity factor. c PI = phototherapeutic index. | ||||||
| 1 | 17.9 ± 2.27 | 0.8 | 0.25 ± 0.02 | 71 | 1.71 ± 0.12 | 10 |
| 2 | 0.34 ± 0.03 | 34 | 0.015 ± 0.001 | 23 | 0.16 ± 0.05 | 2.1 |
| 3 | 0.23 ± 0.03 | 40 | 0.018 ± 0.004 | 13 | 0.15 ± 0.01 | 1.5 |
| 4 | 2.92 ± 0.68 | 0.8 | 0.12 ± 0.01 | 24 | 2.11 ± 0.21 | 1.4 |
| 5 | 3.27 ± 0.14 | 1.1 | 0.012 ± 0.001 | 273 | 0.20 ± 0.01 | 16 |
The visible light EC50 values measured in melanoma cells ranged from 12 to 252 nM, with complex 5 being the most photocytotoxic (Table 3, Fig. 5 and ESI Fig. S13†). As expected, in vitro red light PDT was attenuated 10- to 20-fold relative to the more energetically visible light treatment, with red light EC50 values spanning 150 nM to 2.1 μM. Photocytotoxicity increased in the order 5 ≈ 2 ≈ 3 > 4 > 1 for visible PDT and 3 ≈ 2 ≈ 5 > 1 > 4 for red PDT. Generally speaking, complexes 2, 3, and 5 clustered around 15 nM for visible PDT and 170 nM for red PDT, while 1 and 4 clustered around 190 nM for visible PDT and 1.9 μM for red PDT. These results demonstrate that phen, pqu, and quqo as coligands were the most effective at increasing photocytotoxic effects, while bpy and bqu were less effective. Similar to the dark toxicity trends, benzannulation had the effect of increasing phototoxicity. However, replacement of C4 with N on going from 4 to 5 increased photocytotoxicity, whereas this change had only a marginal effect on dark toxicity (with 5 being slightly less dark toxic).
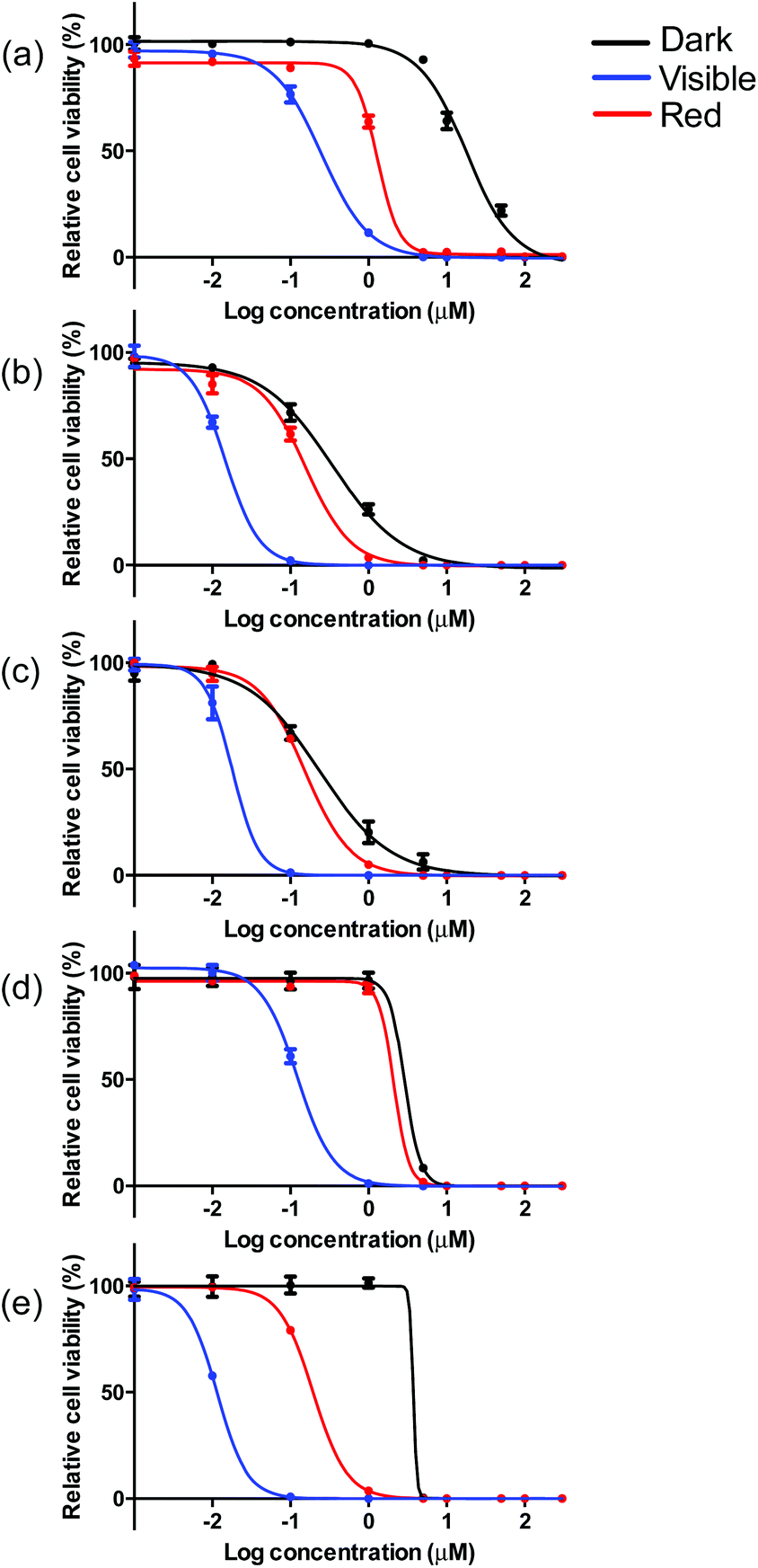 | ||
| Fig. 5 In vitro dose–response curves for complexes 1 (a), 2 (b), 3 (c), 4 (d) and 5 (e) in SK-MEL-28 cells treated in the dark (black) and with visible (blue) or red (red) light activation. | ||
These photocytotoxicity trends could not be directly correlated to differences in absorption coefficients, emission quantum yields, or TA/emission lifetimes. When considering complexes 2–5, larger photocytoxicities generally paralleled 1O2 quantum yields, whereby complex 4 with the smallest value for ΦΔ, was approximately ten-fold less phototoxic. However, complex 1 was the least phototoxic of the series despite being among the more efficient singlet 1O2 sensitizers. While cell-free 1O2 experiments do not directly correlate with in vitro cellular conditions, it appears that factors other than singlet oxygen generation may govern the in vitro PDT effects of this class of Ir(III) complexes.
Taken together, the dark and light cytotoxicities yielded respectable PDT effects for certain members of this class of complexes (Table 3, Fig. 5 and S13†). Complex 5 produced the largest PDT effects in this series, with a PI > 270 for visible light and >15 for red light. Complex 1 was also an effective PDT agent (visible PI > 70, red PI = 10), but was almost four-fold less effective than 5. Complexes 2–4 were the least effective PDT agents, and yielded PIs between 10 and 25 for visible light and up to only 2 for red light. Given that the light EC50 values for 2, 3, and 5 did not differ substantially between the two light treatments, yet 5 had a 20-fold larger phototherapeutic margin, the underlying factor governing the PDT effects in this class of complexes appears to be dark toxicity.
Cellular imaging
Phosphorescence from this class of Ir(III) complexes proved to be a convenient tool for monitoring uptake by SK-MEL-28 melanoma cells with and without a light treatment (Fig. 6). Excitation for confocal imaging was performed by using an argon-krypton laser (458/488 nm) and emission was collected through a 475 nm long pass filter. The images were captured after subjecting the complexes to a brief cellular incubation time (1 h) to ensure sub-lethal conditions. All of the complexes were taken up by cells even in the absence of a light trigger, indicating that the large differences in dark toxicity may not be due to differences in cellular uptake if it is assumed that the intensity of intracellular luminescence is proportional to concentration. This uptake was enhanced with illumination as would be expected with PDT inflicted damage to the cellular membranes. The differences in dark toxicity for the complexes could be easily discerned by changes in cellular morphology. For example, complex 1 was the least toxic in the absence of a light trigger, while complexes 2 and 3 were the most toxic. Cells treated with complex 1 (Fig. 6, diffuse interference contrast (DIC) image) retained their dendritic morphology while only dead/dying cells and debris could be discerned for samples exposed to complex 2. Fig. 6 captured the change in morphology from dendritic to detached spherical, an intermediate stage between viable and dead, quite well for complex 5. It should be noted that the incubation time and light treatment for the confocal imaging was different from that used for the in vitro assays to ensure that some cells would be viable for imaging in each sample. Because the in vitro cell assay conditions and the confocal imaging conditions were different, a quantitative assessment of overall cytotoxicity and photocytotoxicity by visualization of morphological changes was not attempted. Rather the purpose of the cellular imaging experiment was to highlight that the inherent phosphorescence from the complexes can be used for imaging cellular uptake and localization, and that the morphological changes that accompany cell death do parallel the results from the quantitative in vitro measurements regarding toxicity. | ||
| Fig. 6 Confocal luminescence images of SK-MEL-28 cells dosed with complexes 1–5 (50 μM) (a) in the dark and (b) with visible light (50 J cm−2) activation. | ||
DNA interactions
In order to determine whether light-mediated DNA damage could be a factor in the in vitro PDT effects observed for this class of complexes, supercoiled plasmid DNA (20 μM bases) was exposed to increasing concentrations of 1–5 and light treatment (Fig. 7, lanes 3–8) and compared with DNA alone (lanes 1 and 2) or DNA treated with complexes but no light (lane 9). In this gel electrophoretic mobility shift assay,16,84,85 undamaged supercoiled DNA (Form I) migrates the farthest through the gel, while aggregated DNA or condensed DNA (Form IV) hardly moves under the electrophoretic conditions employed. If single strand breaks occur to form relaxed circular DNA (Form II), the DNA migrates farther than form IV but slower than Form I. Frank double strand breaks or single strand breaks on opposing strands (within about 16 base pairs) produce linear DNA (Form III). When plasmid DNA was exposed to 1–5 and light, detectable amounts of Form IV DNA were observed at [MC]:[bases] ratios as low as 1 (lane 4). Increases in Form II (or Form III) DNA were not observed and indicated that the most prominent interaction with DNA is the induction of aggregation rather than strand breaks.
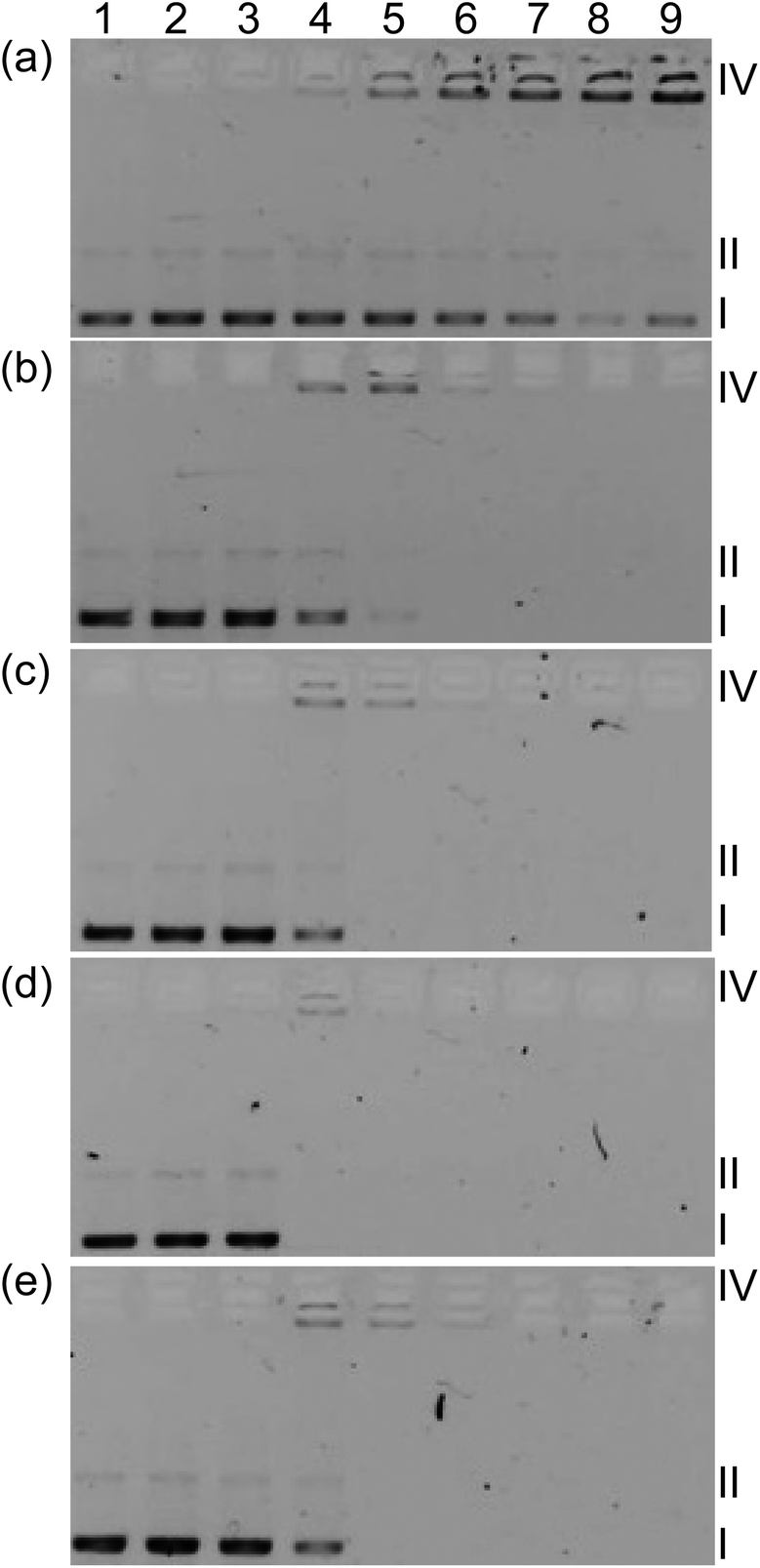 | ||
| Fig. 7 DNA photocleavage of pUC19 DNA (20 μM bases) dosed with metal complex (MC) 1 (a), 2 (b), 3 (c), 4 (d) or 5 (e) and visible light (14 J cm−2). Gel mobility shift assays employed 1% agarose gels (0.75 μg mL−1 ethidium bromide) electrophoresed in 1× TAE at 8 V cm−1 for 30 min. Lane 1, DNA only (−hν); lane 2, DNA only (+hν); lane 3, 5 μM MC (+hν); lane 4, 20 μM MC (+hν); lane 5, 40 μM MC (+hν); lane 6, 60 μM MC (+hν); lane 7, 80 μM MC (+hν); lane 8, 100 μM MC (+hν); lane 9, 100 μM MC (−hν). Forms I, II, and IV DNA refer to supercoiled plasmid, nicked circular plasmid, and aggregated plasmid, respectively. | ||
Disappearance of gel bands at higher complex concentrations with or without light treatment precluded a quantitative comparison of the relative strengths of the complex–DNA interactions, although qualitatively the interactions appeared very similar. A lack of DNA staining could be attributed to quenching of the fluorescence from DNA stain ethidium bromide (EtBr), competition for EtBr intercalation sites, or distortion of the helix (that prevents EtBr binding) caused by complex binding. Interestingly, complex 1 did not cause disappearance of bands even at high concentration but did induce a similar aggregation pattern as observed for the other complexes before the bands became too faint to analyze. The conversion from Form I to Form IV DNA by 1 occurred with no accompanying DNA photocleavage, indicating that DNA damage by singlet oxygen did not occur (normally observed as strand breaks to yield detectable Form II86). The absence of photoinduced Form II DNA was also evident for 1–4 as direct conversion of Form I to Form IV before the signal from Form IV disappeared. Given the similarities in the DNA aggregation profiles produced by 1–5 and the variation in their dark and light cytotoxicities, it might be inferred that DNA is not an important intracellular target. Moreover, the absence of Form II DNA points toward a mechanism for photocytotoxicity that does not involve singlet oxygen. However, the cell-free experiments do not mimic the complexity of the cellular environment and dynamic processes, which involve uptake, efflux, metabolism, and localization. Thus in vitro DNA targeting and singlet oxygen damage cannot be discounted with certainty. Efforts are underway to understand the cellular targets and the underlying mechanism for in vitro PDT.
Conclusions
We have synthesized and characterized five heteroleptic cationic Ir(III) complexes with 2,3-diphenylbenzo[g]quinoxaline as the cyclometalating ligands and diimine ligands with varying degrees of π-conjugation as the coligand. The UV-vis absorption spectra of these complexes exhibited intense absorption bands below 500 nm and spin-forbidden broad absorption bands between 600 nm and 800 nm (ε < 200 M−1 cm−1). The intensities of these bands generally increased as the diimine ligand π-conjugation increased. All of the complexes possessed weak but structured emission in the NIR region, which was attributed to a dpbq ligand centred 3π,π* state combined with some 3ILCT/3MLCT/3LLCT character (supported by DFT calculations). The nanosecond TA spectra of complexes 1–4 all resembled that of the [Ir(dpbq)2Cl]2 dimer, but the TA spectrum of complex 5 possessed features characteristic of both the quqo coligand and the [Ir(dpbq)2Cl]2 dimer and a bi-exponential decay.The Ir(III) complexes of this study were biologically active, with some members (2 and 3) acting as selective chemotherapeutics toward melanoma cells and others acting as potent in vitro PDT agents. These activities were in the nanomolar regime with SFs as large as 40 and PIs of almost 275 (5). The intracellular biological target(s) and mechanism of action are unknown at this time, but all of the Ir(III) complexes induced aggregation of DNA and production of 1O2 in cell-free experiments, and were taken up readily by melanoma cells. The inherent NIR phosphorescence of these complexes translated to a convenient diagnostic tool for viewing cellular uptake and distribution. The systematic variation of the coligand in this class of complexes proved very influential, with very large differences in the resulting cytotoxicity and photocytotoxicity profiles, despite not having comparable differences in DNA interactions, 1O2 quantum yields, and cellular uptake. The anticancer potency and breadth of activity in this small subset warrant further study of this new class of Ir(III) complexes, especially considering their demonstrated potential as in vitro theranostic PDT agents.
Acknowledgements
W. Sun acknowledges financial support from the Army Research Laboratory (W911NF-14-2-0081) for the synthesis and photophysical studies of the complexes, and the support from the Argonne National Laboratory via User Proposal CNM 48389. Use of the Center for Nanoscale Materials was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The computational part of the work was supported by NSF (DMR-1411086 and CNS-1229316) to W. Sun and S. Kilina. S. A. McFarland acknowledges financial support from the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Institutes of Health Research (CIHR), the Canadian Foundation for Innovation (CFI), the Nova Scotia Research and Innovation Trust (NSRIT), Acadia University, and the University of North Carolina at Greensboro.Notes and references
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, 2000 Search PubMed.
- M. R. Hamblin and Y.-Y. Huang, Handbook of Photomedicine, Taylor & Francis, 2014 Search PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- E. C. Glazer, Isr. J. Chem., 2013, 53, 391–400 CrossRef CAS.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 CrossRef CAS PubMed.
- C. Mari and G. Gasser, Chim. Int. J. Chem., 2015, 69, 176–181 CrossRef CAS.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550–25564 RSC.
- M. Stephenson, C. Reichardt, M. Pinto, M. Wächtler, T. Sainuddin, G. Shi, H. Yin, S. Monro, E. Sampson, B. Dietzek and S. A. McFarland, J. Phys. Chem. A, 2014, 118, 10507–10521 CrossRef CAS PubMed.
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS PubMed.
- T. Sainuddin, J. McCain, M. Pinto, H. Yin, J. Gibson, M. Hetu and S. A. McFarland, Inorg. Chem., 2016, 55, 83–95 CrossRef CAS PubMed.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- C. Reichardt, M. Pinto, M. Wächtler, M. Stephenson, S. Kupfer, T. Sainuddin, J. Guthmuller, S. A. McFarland and B. Dietzek, J. Phys. Chem. A, 2015, 119, 3986–3994 CrossRef CAS PubMed.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, J. Inorg. Biochem., 2016, 158, 45–54 CrossRef CAS PubMed.
- W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1992, 96, 2917–2920 CrossRef CAS.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336–1350 CrossRef CAS.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- S. B. Meier, D. Tordera, A. Pertegás, C. Roldán-Carmona, E. Ortí and H. J. Bolink, Mater. Today, 2014, 17, 217–223 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed.
- P. Majumdar, X. Yuan, S. Li, B. Le Guennic, J. Ma, C. Zhang, D. Jacquemin and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC.
- R.-R. Ye, C.-P. Tan, L. He, M.-H. Chen, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2014, 50, 10945–10948 RSC.
- L. He, Y. Li, C.-P. Tan, R.-R. Ye, M.-H. Chen, J.-J. Cao, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- A. Kando, Y. Hisamatsu, H. Ohwada, T. Itoh, S. Moromizato, M. Kohno and S. Aoki, Inorg. Chem., 2015, 54, 5342–5357 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Taylor & Francis Group, LLC, Boca Raton, FL, 3rd edn, 2006, p. 620 Search PubMed.
- R. Gao, D. G. Ho, B. Hernandez, M. Selke, D. Murphy, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 14828–14829 CrossRef CAS PubMed.
- M. K. Nazeeruddin, R. Humphry-Baker, D. Berner, S. Rivier, L. Zuppiroli and M. Graetzel, J. Am. Chem. Soc., 2003, 125, 8790–8797 CrossRef CAS PubMed.
- C. Wang, L. Lystrom, H. Yin, M. Hetu, S. Kilina, S. A. McFarland and W. Sun, Dalton Trans., 2016, 45, 16366–16378 RSC.
- Y. Choi, S. Kim, M.-H. Choi, S.-R. Ryoo, J. Park, D.-H. Min and B.-S. Kim, Adv. Funct. Mater., 2014, 24, 5781–5789 CrossRef CAS.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- Q. Zhao, M. Yu, L. Shi, S. Liu, C. Li, M. Shi, Z. Zhou, C. Huang and F. Li, Organometallics, 2010, 29, 1085–1091 CrossRef CAS.
- G. Zhang, H. Zhang, Y. Gao, R. Tao, L. Xin, J. Yi, F. Li, W. Liu and J. Qiao, Organometallics, 2014, 33, 61–68 CrossRef CAS.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- K. Hasan, A. K. Bansal, I. D. W. Samuel, C. Roldán-Carmona, H. J. Bolink and E. Zysman-Colman, Sci. Rep., 2015, 5, 12325 CrossRef CAS PubMed.
- Ł. Skórka, M. Filapek, L. Zur, J. G. Małecki, W. Pisarski, M. Olejnik, W. Danikiewicz and S. Krompiec, J. Phys. Chem. C, 2016, 120, 7284–7294 Search PubMed.
- R. Tao, J. Qiao, G. Zhang, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2012, 116, 11658–11664 CAS.
- R. Tao, J. Qiao, G. Zhang, L. Duan, C. Chen, L. Wang and Y. Qiu, J. Mater. Chem. C, 2013, 1, 6446–6454 RSC.
- L. Xin, J. Xue, G. Lei and J. Qiao, RSC Adv., 2015, 5, 42354–42361 RSC.
- A.-H. Li, E. Ahmed, X. Chen, M. Cox, A. P. Crew, H.-Q. Dong, M. Jin, L. Ma, B. Panicker, K. W. Siu, A. G. Steinig, K. M. Stolz, P. A. R. Tavares, B. Volk, Q. Weng, D. Werner and M. J. Mulvihill, Org. Biomol. Chem., 2007, 5, 61–64 CAS.
- M. Yu, Q. Zhao, L. Shi, F. Li, Z. Zhou, H. Yang, T. Yi and C. Huang, Chem. Commun., 2008, 2115–2117 RSC.
- H.-Y. Chen, C.-H. Yang, Y. Chi, Y.-M. Cheng, Y.-S. Yeh, P.-T. Chou, H.-Y. Hsieh, C.-S. Liu, S.-M. Peng and G.-H. Lee, Can. J. Chem., 2006, 84, 309–318 CrossRef CAS.
- N. Matsuo, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef.
- O. E. Semonin, J. C. Johnson, J. M. Luther, A. G. Midgett, A. J. Nozik and M. C. Beard, J. Phys. Chem. Lett., 2010, 1, 2445–2450 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford, CT, USA, 2009 Search PubMed.
- E. K. U. Gross and W. Kohn, Phys. Rev. Lett., 1985, 55, 2850–2852 CrossRef CAS PubMed.
- M. E. Casida, in Recent Advances in Computational Chemistry: Vol. 1 Recent Advances in Density Functional Methods, ed. D. P. Chong, 1995, pp. 155–192 Search PubMed.
- L. Künne, Z. Phys. Chem., 1998, 204, 263–264 CrossRef.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- E. R. Davidson, J. Comput. Phys., 1975, 17, 87–94 CrossRef.
- S. J. A. van Gisbergen, J. G. Snijders and E. J. Baerends, Comput. Phys. Commun., 1999, 118, 119–138 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. M. York and M. Karplus, J. Phys. Chem. A, 1999, 103, 11060–11079 CrossRef CAS.
- E. R. Batista and R. L. Martin, Encyclopedia of Computational Chemistry, John Wiley and Sons Ltd., Chichester, U.K., 2004 Search PubMed.
- G. A. Zhurko and D. A. Zhurko, ChemCraft 1.7, http://www.chemcraftprog.com Search PubMed.
- Z. Li, P. Cui, C. Wang, S. Kilina and W. Sun, J. Phys. Chem. C, 2014, 118, 28764–28775 CAS.
- L. Wang, P. Cui, S. Kilina and W. Sun, J. Phys. Chem. C, 2017, 121, 5719–5730 CAS.
- E. E. Langdon-Jones, A. J. Hallett, J. D. Routledge, D. A. Crole, B. D. Ward, J. A. Platts and S. J. A. Pope, Inorg. Chem., 2013, 52, 448–456 CrossRef CAS PubMed.
- W. Sun, C. Pei, T. Lu, P. Cui, Z. Li, C. McCleese, Y. Fang, S. Kilina, Y. Song and C. Burda, J. Mater. Chem. C, 2016, 4, 5059–5072 RSC.
- R. Liu, N. Dandu, J. Chen, Y. Li, Z. Li, S. Liu, C. Wang, S. Kilina, B. Kohler and W. Sun, J. Phys. Chem. C, 2014, 118, 23233–23246 CAS.
- E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, J. Phys. Chem., 1986, 90, 3722–3734 CrossRef CAS.
- L. Della Ciana, W. J. Dressick, D. Sandrini, M. Maestri and M. Ciano, Inorg. Chem., 1990, 29, 2792–2798 CrossRef CAS.
- R. Kumar, T. Y. Ohulchanskyy, I. Roy, S. K. Gupta, C. Borek, M. E. Thompson and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 1474–1481 CAS.
- S.-H. Wu, J.-W. Ling, S.-H. Lai, M.-J. Huang, C. H. Cheng and I.-C. Chen, J. Phys. Chem. A, 2010, 114, 10339–10344 CrossRef CAS PubMed.
- E. C. Glazer, D. Magde and Y. Tor, J. Am. Chem. Soc., 2005, 127, 4190–4192 CrossRef CAS PubMed.
- E. Sakuda, C. Matsumoto, Y. Ando, A. Ito, K. Mochida, A. Nakagawa and N. Kitamura, Inorg. Chem., 2015, 54, 3245–3252 CrossRef CAS PubMed.
- K. A. King and R. J. Watts, J. Am. Chem. Soc., 1987, 109, 1589–1590 CrossRef CAS.
- A. P. Wilde, K. A. King and R. J. Watts, J. Phys. Chem., 1991, 95, 629–634 CrossRef CAS.
- S. Ladouceur, L. Donato, M. Romain, B. P. Mudraboyina, M. B. Johansen, J. A. Wisner and E. Zysman-Colman, Dalton Trans., 2013, 42, 8838–8847 RSC.
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Inorg. Chem., 2015, 54, 6582–6593 CrossRef CAS PubMed.
- Y.-S. Yeh, Y.-M. Cheng, P.-T. Chou, G.-H. Lee, C.-H. Yang, Y. Chi, C.-F. Shu and C.-H. Wang, ChemPhysChem, 2006, 7, 2294–2297 CrossRef CAS PubMed.
- Z. Li, H. Li, B. J. Gifford, W. D. N. Peiris, S. Kilina and W. Sun, RSC Adv., 2016, 6, 41214–41228 RSC.
- D. l. Praseuth, A. Gaudemer, J.-B. Verlhac, I. Kraljic, I. Sissoëff and E. Guillé, Photochem. Photobiol., 1986, 44, 717–724 CrossRef CAS PubMed.
- D. T. Croke, L. Perrouault, M. A. Sari, J. P. Battioni, D. Mansuy, C. Helene and T. Le Doan, J. Photochem. Photobiol., 1993, 18, 41–50 CrossRef CAS.
- E. R. Blazek, J. G. Peak and M. J. Peak, Photochem. Photobiol., 1989, 49, 607–613 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the photobiological activity studies, NMR and mass spectra, solvent dependent UV-vis absorption spectra, comparison of the experimental and calculated UV-vis absorption spectra, natural transition orbitals (NTOs), time-resolved nanosecond TA spectra, in vitro dose–response curves for complexes 1–5 in CCD-1064Sk normal fibroblasts, and the comparison of in vitro dose–response curves for complexes 1–5 in SK-MEL-28 cells. See DOI: 10.1039/c7dt00913e |
| This journal is © The Royal Society of Chemistry 2017 |